

Background: SMURF1 ubiquitin ligase controls ubiquitination and stability of diverse cellular protein substrates.
Results: Deubiquitinase USP9X interacts with SMURF1 and stabilizes SMURF1 through deubiquitination.
Conclusion: USP9X is novel regulator of SMURF1 and is required for SMURF1-dependent cellular physiology.
Significance: Association between deubiquitinase and ubiquitin ligase may serve as a common strategy to control the cellular protein dynamics through modulating ubiquitin ligase activity.
Keywords: Deubiquitination, Proteasome, Protein Degradation, Ubiquitin, Ubiquitin Ligase
Abstract
Ubiquitination is an essential post-translational modification that mediates diverse cellular functions. SMAD-specific E3 ubiquitin protein ligase 1 (SMURF1) belongs to the Nedd4 family of HECT ubiquitin ligases that directly catalyzes ubiquitin conjugation onto diverse substrates. As a result, SMURF1 regulates a great variety of cellular physiologies including bone morphogenetic protein (BMP) signaling, cell migration, and planar cell polarity. Structurally, SMURF1 consists of a C2 domain, two WW domain repeats, and a catalytic HECT domain essential for its E3 ubiquitin ligase activity. This modular architecture allows for interactions with other proteins, which are either substrates or adaptors of SMURF1. Despite the increasing number of SMURF1 substrates identified, current knowledge regarding regulatory proteins and their modes of action on controlling SMURF1 activity is still limited. In this study, we employed quantitative mass spectrometry to analyze SMURF1-associated cellular complexes, and identified the deubiquitinase FAM/USP9X as a novel interacting protein for SMURF1. Through domain mapping study, we found the second WW domain of SMURF1 and the carboxyl terminus of USP9X critical for this interaction. SMURF1 is autoubiquitinated through its intrinsic HECT E3 ligase activity, and is degraded by the proteasome. USP9X association antagonizes this activity, resulting in deubiquitination and stabilization of SMURF1. In MDA-MB-231 breast cancer cells, SMURF1 expression is elevated and is required for cellular motility. USP9X stabilizes endogenous SMURF1 in MDA-MB-231 cells. Depletion of USP9X led to down-regulation of SMURF1 and significantly impaired cellular migration. Taken together, our data reveal USP9X as an important regulatory protein of SMURF1 and suggest that the association between deubiquitinase and E3 ligase may serve as a common strategy to control the cellular protein dynamics through modulating E3 ligase stability.
Introduction
Protein ubiquitination is a versatile post-translational modification that regulates various aspects of cellular physiology, including protein degradation and cell signaling (1). A highly conserved enzymatic pathway, involving E1, E2, and E3 enzymes, orchestrates ubiquitin conjugation onto protein substrates (2). Most E3s fall into two big protein families, containing either a RING domain or a HECT4 domain. A RING E3 serves as a “bridging factor” that connects ubiquitin-E2 with a substrate to facilitate ubiquitin transfer (3). A HECT (homologous to E6AP carboxyl terminus) E3, however, utilizes its HECT domain to bind ubiquitin-E2 and forms a thioester bond between its active cysteine and the ubiquitin molecule. Ubiquitin is then transferred from the active cysteine in the HECT domain to a substrate bound next to the HECT domain (4). As a result, HECT E3s bear intrinsic catalytic activity and directly participate in the ubiquitin relay.
The HECT E3 superfamily can be further classified according to their different N-terminal extensions. Nedd4-like E3s constitute an important subfamily of HECT ligases with highly conserved modular architecture. Besides a carboxyl-terminal HECT domain, Nedd4 family E3s consist of an N-terminal C2 domain followed by several WW domains (5). The C2 domain binds to Ca2+, and is crucial for membrane association and trafficking (6, 7). The WW domain, between C2 and HECT domains, encompasses two conserved tryptophan residues and can recognize the “Pro-Pro-X-Tyr” (P/Y) motif commonly found on the substrates or adaptors of the Nedd4 family E3s (8, 9). Nine members of the Nedd4 E3 family are found in the human genome. They play critical roles in various biological pathways, including endocytosis, destruction of membrane proteins, regulation of cell growth, and virus budding (5, 10). SMURF1 is an important member of the Nedd4-like E3 family (11). It was originally identified as a negative regulator of TGFβ/BMP (bone morphogenetic proteins) signaling pathway by degrading SMAD proteins and TGFβ/BMP receptors (12, 13). To date, over a dozen SMURF1 substrates have been identified, including Runx2, MEKK2, RhoA, Par6, JunB, Talin head, Prickle1, TRAF family proteins, and STAT1 (14–22). This suggests a pleiotropic role for SMURF1 in various cellular functions such as bone homeostasis, embryonic development, cell movement and polarity control, and immune response (20, 23). More recently, SMURF1 was identified as a component regulating autophagy activity (24).
SMURF1 itself is also subject to different forms of regulation in cells. Like many other E3s, SMURF1 undergoes E3-activity-dependent autoubiquitination and is rapidly degraded by the proteasome. Mutation of a highly conserved cysteine within the SMURF1 HECT domain (Cys-699 to Ala) abolishes its E3 activity and significantly stabilizes the protein (16). SMURF1 was also shown to be a substrate by other ubiquitin ligases including SMURF2 and SCFFBXL15, which lead to SMURF1 degradation independent of its own E3 activity (25, 26). More recently, a list of SMURF1-interacting proteins were identified that either modulate its enzyme-substrate interaction or control the E3 activity of the protein (see “Discussion”). However, our knowledge about the regulation mechanism of SMURF1 itself is still limited. To identify regulators of SMURF1 and elucidate their modes of action remains a challenging but exciting undertaking for the study of SMURF1-related biology.
Substrate ubiquitination is also reversible. A group of ubiquitin-specific proteases, de-ubiquitinating enzymes (DUBs), can cleave ubiquitin from its substrates (27). Most DUBs carry out this activity through a highly conserved cysteine in the catalytic domain (28). The major consequence of DUB-initiated deubiquitination includes ubiquitin biogenesis/recycling, substrate stabilization, and cell signaling modulation through ubiquitin-chain editing (29, 30). Among the ∼100 DUBs encoded by the human genome, the ubiquitin-specific peptidase 9, X-linked (USP9X/FAM), is implicated in multiple physiological pathways through targeting a variety of substrates, including β-catenin, AF-6, and EFA6 (31–33). More recently, pro-survival factor MCL-1 and Parkinson disease pathogenic protein α-Synuclein were also reported as substrates for USP9X, expanding the role of USP9X into tumor cell apoptosis and neurodegenerative diseases (34, 35).
In this study, we identified the deubiquitinase USP9X as an interacting protein for SMURF1. By domain mapping, we isolated the WW2 domain of SMURF1 that is sufficient and necessary for the interaction. USP9X-SMURF1 association does not lead to USP9X degradation; instead, USP9X helps to stabilize SMURF1 by antagonizing its autoubiquitination activity. In breast cancer cells, USP9X stabilizes endogenous SMURF1 and is required for the SMURF1-dependent cell motility.
MATERIALS AND METHODS
cDNA Constructs and Mutagenesis
WT human SMURF1 cDNA with a FLAG tag at its N terminus was cloned into the HindIII/EcoRI site of the pcDNA4/TO vector. The SMURF1-C699A mutant was generated by site-directed mutagenesis (Agilent Technologies). To make GST-fused SMURF1 fragments, primers were designed to PCR amplify specific domains from a full-length SMURF1 cDNA, generating WW-(236–312), WW1-(236–267), Linker-(268–282), and WW2-(283–312) domains. The resulting fragments were first cloned into pDONR221 vector by BP reaction and then transferred to pDEST15 vector by LR recombination (Invitrogen). To generate GST-fused SMURF1 truncations (ΔC2, ΔWW, ΔWW1, ΔWW2, ΔHECT), primers were designed to anneal to the nucleotide sequence flanking the region to be deleted. These primers were used to delete the desired domains of SMURF1 by QuikChange mutagenesis according to Ref. 36 (Agilent Technologies). GST-fused USP9X fragments were generated from a full-length USP9X cDNA in a similar fashion as above.
Cell Lines and Culture Conditions
Human embryonic kidney 293T (HEK293T) cells were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Invitrogen). Human breast cancer MDA-MB-231 cells were purchased from ATCC and cultured in RPMI1640 (ATCC). Doxycycline-inducible SMURF1 cell lines were generated by transiently transfecting pCDNA4/TO-SMURF1 (WT/C699A) into the T-RExTM-293 cells that stably express the tetracycline repressor protein (Invitrogen). Inducible-SMURF1 cell lines were maintained in regular DMEM supplemented with 10% tetracycline-free FBS and antibiotics (5 μg/ml of Blasticidin, 250 μg/ml of Zeocin). To induce SMURF1 expression, cells were treated with doxycycline at 100 ng/ml overnight before harvest. All cells were cultured in 37 °C incubator with 5% CO2.
Antibodies
Polyclonal antibody against FLAG tag and monoclonal antibody against tubulin were purchased from Sigma. Monoclonal antibody against USP9X, polyclonal antibody against SMURF1, monoclonal antibody against cyclin D1, and polyclonal antibody against ubiquitin were obtained from Santa Cruz. Monoclonal antibodies against SMAD5 and SMURF1 were purchased from Abcam and were used to detect endogenous proteins by immunoblotting. Monoclonal antibody against V5 tag was purchased from Invitrogen.
FLAG-SMURF1-based Affinity Enrichment and Quantitative Mass Spectrometry
Inducible SMURF1 cells were grown in medium with or without doxycycline overnight, eluted materials were solved by SDS-PAGE. Complete gel lanes were excised and samples were subjected to in-gel tryptic digestion and isobaric labeling using iTRAQ reagents (AB/Sciex). Peptide extracts from untreated cells were labeled with iTRAQ reagent 116 and combined with extracts from corresponding samples of material from doxycycline-treated cells labeled with iTRAQ reagent 117. Peptide sequencing was performed by liquid chromatography-tandem mass spectrometry on an Eksigent 1D+ high-pressure liquid chromatography system coupled to a LTQ-Orbitrap XL mass spectrometer (Thermo Scientific). Peptide mass and fragmentation data acquired by pulsed-Q-dissociation were searched against a combined forward-reverse IPI database using Mascot (Matrix Science). Peptide and protein validation was done using Transproteomic pipeline v3.3sqall (Institute for Systems Biology; tools.proteomecenter.org/software.php) using a false-positive threshold of <1% for protein identifications. For each peptide sequence and modification state, reporter ion signal intensities from all spectral matches were summed for each reporter ion type and corrected according to the isotope correction factors given by the manufacturer. Only peptides unique to a given protein within the total dataset of identified proteins were used for relative protein quantification. Peptide fold-changes were calculated (no doxycycline treatment over doxycycline treatment) and subsequently renormalized using the median fold-change of all quantified peptides to compensate for differences in total protein yield for each affinity purification. Protein fold-changes were derived as median peptide fold-change. p values were calculated using a one-way t test (arbitrarily set to 1 for nonsignificant single peptide quantifications) and adjusted using the Benjamini-Hochberg false discovery rate. Data were visualized for further analysis using Spotfire DXP. All identified proteins are shown in supplemental Table S1.
siRNA-directed Gene Knockdown
For gene knockdown in a SMURF1 stable cell line, cells were seeded at 1× 10E6/well density in a 6-well plate format. After a 48-h incubation, cells reached a confluence of 90%. 6 μl of Dharmafect 1 (Dharmacon) was added into 159 μl of OptiMEM (Invitrogen) and incubated at room temperature (RT) for 5 min. 2.5 μl of specific siRNA (20 μm stock concentration) was added into 162.5 μl of OptiMEM. The two transfection mixtures were combined and incubated at RT for 30 min before adding to the 1.67 ml of cell culture after media change, making the final siRNA concentration 25 nm. Cells were harvested 48 h after siRNA treatment for further analysis. siRNA knockdown in MDA-MB-231 cells were the same except that cells were seeded at 4× 10E6/well before knockdown. Target sequences for siRNA knockdown are as follows: nonsilencing/pGL2, 5′-CGTACGCGGAATACTTCGA-3′; USP9X-1, 5′-AGAAATCGCTGGTATAAAT-3′; USP9X-2, 5′-ACACGATGCTTTAGAATTT-3′; USP9X-3, 5′-GTACGACGATGTATTCTCA-3′; USP9X-4, 5′-GAAATAACTTCCTACCGAA-3′; USP9X-5, 5′-CTACATAAGCAGACAAAAT-3′; and SMURF1, 5′-AACCTTGCAAAGAAAGACTTC-3′.
Immunoprecipitation and Pulldown Assays
For cultured cells, in a 10-cm dish format, HEK293T cells transfected with empty vector or FLAG-SMURF1 were washed in 1× PBS and resuspended in 1 ml of 1× RIPA buffer (10 mm sodium phosphate, pH 7.2, 150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm EDTA)(Boston Bioproducts), supplemented with 1× serine/threonine phosphatase inhibitor, 1× tyrosine phosphatase inhibitor (Millipore), and 1× protease inhibitor mixture (Fisher Scientific). After rotating at 4 °C for 30 min, the cell lysate was collected and precleared by spinning at 14,800 rpm for 10 min. For each pulldown, 30 μl of EZview anti-FLAG affinity gel (Sigma) was added to the normalized lysate (8 mg of total protein by DC Protein Assay, Bio-Rad) and the mixture was incubated overnight at 4 °C. Beads were washed four times with RIPA buffer and immunoprecipitated samples were resolved by SDS-PAGE for immunoblotting. For GST fusion pulldown, GST proteins were expressed in BL21-AI cells (Invitrogen) and extracted by Qproteome Bacterial Protein Prep Kit (Qiagen). For each GST protein, 300 μl of pre-washed glutathione-agarose beads (Thermo Fisher Scientific) was added to the cell lysate and incubated at 4 °C for 2 h. After four washes with GST purification buffer (0.5% Triton X-100, 50 mm Tris-Cl, pH 7.4, 150 mm NaCl, 1 mm EDTA), immobilized GST fusion proteins were resuspended in 300 μl of GST purification buffer supplemented with 1× protease inhibitor (Roche Applied Science) and 10% glycerol. GST fusion proteins were resolved by SDS-PAGE. For USP9X pulldown, 2× 10E9 HEK293T cells were harvested and lysed in 10 ml of lysis buffer (0.5% Nonidet P-40, 20 mm HEPES, pH 7.4, 125 mm NaCl, 1× serine/threonine phosphatase inhibitor, 1× tyrosine phosphatase inhibitor, 1× protease inhibitor). 1 ml of pre-cleared lysate (∼6 × 10E7 cells) was mixed with 10 μm of immobilized GST fusion proteins overnight at 4 °C. After washing with GST-binding buffer, samples were resuspended in 2× sample buffer, boiled, and resolved by SDS-PAGE. USP9X was revealed by immunoblotting using anti-USP9X antibodies. For SMURF1 pulldown, FLAG-SMURF1 was expressed in the FLAG-SMURF1 stable cell line induced by doxycycline. Cell lysate was incubated with immobilized GST-USP9X fragments in a similar way as above. Precipitates were resolved by SDS-PAGE and SMURF1 was identified by immunoblotting using anti-SMURF1 antibodies (Abcam).
Pulse-Chase Analysis
The pulse-chase experiment was done according to Ref. 37. Briefly, inducible SMURF1 cells were seeded at 1× 10E6/well in 6-well format. 48 h after a double siRNA knockdown and 12 h after doxycycline induction, the original media was depleted and cells were starved in 2 ml of DMEM (10% FBS) without l-methionine and l-cystine (Mediatech) for 1 h. Cells were then pulse labeled by addition of 20 μl of 35S-EasyTag (PerkinElmer, 11 μCi/μl in stock) for 30 min. After removal of pulse media, cells were washed once with 1× PBS and re-incubated with chase buffer (DMEM complete media supplemented with 100-fold access of cold l-methionine and l-cystine). Cells were harvested at different time points and resuspended in RIPA lysis buffer supplemented with Ser/Thr/Tyr phosphatase and protease inhibitors as above. Cells were then lysed and equal amounts of radiolabeled lysates were incubated with 30 μl of EZview anti-FLAG affinity gel overnight at 4 °C to pull down FLAG-SMURF1. Precipitates were washed four times with RIPA buffer, resolved by SDS-PAGE, and 35S-labeled SMURF1 was revealed by autoradiography.
Cellular Ubiquitination Assay
Inducible SMURF1 (WT) cells were seeded at 4× 10E5/well in 6-well format and treated with nontargeting or USP9X-specific siRNA, respectively, for 48 h. 12 h after doxycycline induction, cells were harvested in 500 μl of lysis buffer (1% SDS, 25 mm Tris-Cl, pH 7.0). Lysates were denatured by boiling for 15 min and diluted 10-fold in IP buffer (10 mm HEPES, KOH pH 7.5, 150 mm NaCl, 1 mm EDTA, 0.4% Nonidet P-40, 1× protease inhibitor, 1× Ser/Thr and Tyr phosphatase inhibitor) according to Ref. 34. Cellular FLAG-SMURF1 was pulled down by incubating with a EZview anti-FLAG affinity gel overnight at 4 °C. After several washes, precipitates were resuspended in 2× SDS sample buffer and resolved by SDS-PAGE. Ubiquitinated SMURF1 was revealed by antiubiquitin (Santa Cruz) immunoblotting. 1% total cell lysate was also included as Input control.
Cell Migration Assay
Cell migration assay was done according to Ref. 38. Briefly, MDA-MB-231 cells were seeded at 5× 10E5/well in 6-well format and transfected twice by specific siRNAs as above. 36 h after the second transfection, cells were starved in serum-free RPMI1640 media overnight. The next morning, cells were resuspended by trypsinization in serum-free media with 0.1% bovine serum albumin (BSA). 3× 10E5 cells were added to the pre-wet transwell inserts (BD BioCoatTM 8 μm Control insert) and complete culture media with 10% FBS was added to the bottom well. 24 h later, cells were washed, fixed with 10% formaldehyde for 15 min, and stained with crystal violet. Cells remaining in the top wells were removed by swiping with cotton swabs. To quantify cell migration, the insert membrane was cut, solubilized in 10% acetic acid, and absorbance at 596 nm was measured (16). All migration experiments were performed in triplicates and repeated three times.
RESULTS
FAM/USP9X Is a Novel Interacting Protein of SMURF1
To identify novel association factors of SMURF1, a cell line inducibly expressing FLAG-tagged SMURF1 was established. Cells with, or without, SMURF1 induction were harvested and lysed under native conditions. Total cell extract was immunoprecipitated with agarose-conjugated anti-FLAG antibodies followed by elution with FLAG peptide. Eluates were resolved by SDS-PAGE (Fig. 1A). Eluted samples were then excised from the gel, trypsinized, and chemically modified before being subjected to quantitative mass spectrometry analysis. Of all the proteins enriched in the SMURF1-induced population (supplemental Table S1), deubiquitinase FAM/USP9X, among others (e.g. PPAT and NUDT5), was identified as a top hit (Fig. 1B), suggesting USP9X could be a novel interacting protein with SMURF1. To confirm that endogenous USP9X associates with SMURF1, the wild type (WT) or catalytically inactive (C699A) (39) SMURF1 construct, or empty vector, was transfected into the HEK293T cells. After SMURF1 pulldown by agarose-conjugated anti-FLAG antibody, precipitates were resolved by SDS-PAGE followed by anti-USP9X immunoblotting. USP9X was identified only in SMURF1-expressing cells (Fig. 1C), suggesting that USP9X interacts with both SMURF1-WT and SMURF1-C699A. Note that SMURF1-C699A was expressed at a higher level than SMURF1-WT, consistent with the notion that SMURF1 undergoes autoubiquitination and degradation (39). Under this condition, an increased amount of USP9X was precipitated with SMURF1-C699A (Fig. 1C, compare lane 2 with lane 3).
FIGURE 1.
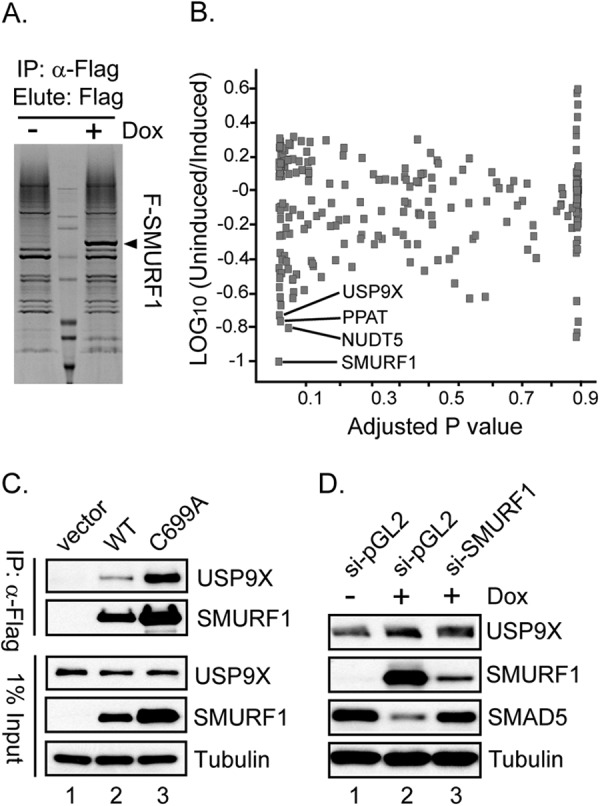
USP9X is identified as an interacting protein of SMURF1. A, Tet-inducible 293T-REx cells integrated with FLAG-tagged SMURF1 were treated overnight with or without doxycycline (Dox). Cell lysates were immunoprecipitated (IP) with anti-FLAG antibodies followed by elution with FLAG peptide. Eluates were resolved by 4–20% SDS-PAGE and stained with Coomassie Brilliant Blue. SMURF1 enrichment was only detected in the doxycycline-treated samples and indicated by the arrowhead. B, scatter plot depicting proteins identified and quantified by quantitative mass spectrometry in the FLAG-SMURF1 pulldown experiment. LOG10 value of fold-reduction in the absence of FLAG-SMURF1 (fold-change: no doxycycline over doxycycline) is plotted on the y axis, and adjusted p value, which represents false discovery rate-corrected statistical significance, is plotted on the x axis. Proteins most significantly reduced in the absence of FLAG-SMURF1 (no doxycycline) are highlighted as potential interactors of SMURF1. C, WT or mutant (C699A) SMURF1, or empty vector, was transfected into HEK293T cells, respectively. 48 h post-transfection, cells were harvested and cell lysates were immunoprecipitated by anti-FLAG affinity gel. Precipitates were resolved by 4–12% SDS-PAGE followed by immunoblotting with anti-USP9X and anti-FLAG (for FLAG-SMURF1) antibodies. Protein levels of USP9X, FLAG-SMURF1, and Tubulin in 1% of the total cell lysate were also shown. D, inducible FLAG-SMURF1 cells were transfected with SMURF1 or nontargeting (pGL2 luciferase) siRNA 48 h before the experiment. 12 h before the experiment, doxycycline was added to selected wells to induce SMURF1 expression. After harvesting the cells, total protein lysate was resolved by 4–12% SDS-PAGE, followed by immunoblotting with anti-USP9X, anti-FLAG, anti-SMAD5, and anti-Tubulin antibodies, respectively.
To test if USP9X could be a substrate for SMURF1, inducible SMURF1 cells were first transfected with SMURF1-targeting siRNA followed by overnight doxycycline treatment. Although the cellular level of SMAD5, a well characterized proteolytic substrate for SMURF1 (12), was sensitive to SMURF1 expression, endogenous USP9X protein remained unchanged (Fig. 1D). Our results indicated that USP9X is a novel interacting protein, but not a substrate, for SMURF1. The SMURF1-USP9X interaction is also independent of the enzymatic activity of SMURF1.
Mapping of the Interacting Domains between SMURF1 and USP9X
Similar to other Nedd4-like E3 family members, SMURF1 can be structurally defined into three functional domains, an N-terminal phospholipid/Ca2+-binding C2 domain, two WW domains, and a carboxyl-terminal HECT domain essential for ubiquitin ligase activity (12). To identify the structural elements of SMURF1 important for USP9X association, various fragments of SMURF1 were generated and expressed as GST fusion proteins (Fig. 2A). To test their interactions with endogenous USP9X, bacterially expressed SMURF1 fragments were immobilized on glutathione beads and incubated with HEK293T cell extracts under native conditions. Precipitates were resolved by SDS-PAGE and immunoblotted with anti-USP9X antibodies. USP9X was readily pulled down by full-length SMURF1, as well as SMURF1 fragments deleted of C2 (ΔC2) or HECT (ΔHECT) domain. However, deletion of the two WW domains of SMURF1 (ΔWW) abolished USP9X interaction (Fig. 2B, upper panel). In particular, deletion of the second (ΔWW2) but not the first (ΔWW1) WW domain significantly impaired USP9X association. Furthermore, GST fusion with the second WW domain (WW2) alone was able to pulldown USP9X (Fig. 2B, lower panel). Our data thus suggested that the second WW domain of SMURF1 is essential and sufficient for USP9X interaction. Besides the ubiquitin carboxyl-terminal hydrolase (UCH) domain essential for its deubiquitinase activity, USP9X has no other apparent structural elements (40). To identify the domain on USP9X required for SMURF1 binding, different fragments of USP9X (33) were generated as GST fusions (Fig. 2C, upper panel), immobilized, and incubated with cell extracts containing exogenous SMURF1. The carboxyl terminus of the USP9X (C-2 domain) exhibited the strongest binding toward SMURF1, whereas the C-1 domain, which harbors the deubiquitinase activity, only weakly interacted with SMURF1 (Fig. 2C, lower panel). Our data support that SMURF1 and USP9X directly interact with each other through the second WW domain of SMURF1 and the carboxyl terminus of USP9X.
FIGURE 2.
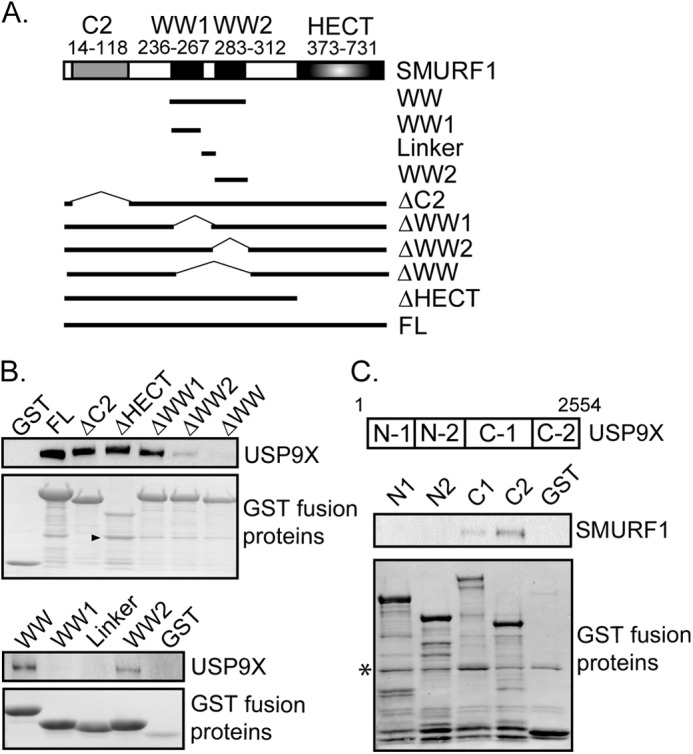
Mapping of interaction domains of SMURF1 and USP9X. A, schematic of SMURF1 protein with C2, WW1/2, and HECT domains highlighted. Schematic of mutants of SMURF1 tested in this study is also shown. All fragments were generated with a GST fusion at the N terminus of the protein. FL, full-length. B, fresh HEK293T cell lysate was incubated with the indicated SMURF1 mutants immobilized on the glutathione-agarose. Precipitates were resolved by 4–12% SDS-PAGE and co-precipitation of USP9X was detected by anti-USP9X immunoblotting. Immobilized SMURF1 fragments and GST control were detected by the Gelcode Blue staining reagent. The arrowhead indicates a major proteolytic product from the SMURF1-ΔHECT fragment. C, schematic of the USP9X protein highlighted with its N-1, N-2, C-1, and C-2 domains tested in this study. All USP9X fragments also contain a GST fusion at the N terminus. Inducible FLAG-SMURF1 cells were treated with doxycycline overnight before harvest under native conditions. The cell lysate was incubated with N-1, N-2, C-1, or C-2 USP9X fragments immobilized by glutathione-agarose. Precipitates were resolved by 4–12% SDS-PAGE and pulldown of SMURF1 was analyzed by anti-FLAG immunoblotting. Immobilized USP9X fragments and GST control were confirmed by the Gelcode Blue staining reagent. The asterisk indicates a nonspecific protein contaminant from the GST pulldown.
USP9X Deubiquitinates SMURF1 and Protects It from Proteasomal Degradation
Previous studies have characterized USP9X as a potent deubiquitinase that can deconjugate ubiquitin chains from a plethora of protein substrates (31–35). In this study, we identified USP9X as an interacting protein for SMURF1, which undergoes active autoubiquitination and self-degradation (12, 39). We hypothesized that USP9X could deubiquitinate SMURF1 and regulate SMURF1 cellular stability. To this end, we utilized the doxycycline-inducible SMURF1 cell line to test our hypothesis. SMURF1-expressing cells were, respectively, transfected with control or USP9X-targeting siRNAs before doxycycline treatment to induce exogenous SMURF1 expression. As shown in Fig. 3A, knockdown of USP9X significantly down-regulated the SMURF1 protein level. Upon SMURF1 induction, the endogenous SMAD5 level was significantly decreased, consistent with a role of SMURF1 in SMAD5 degradation (Fig. 3A, compare lane 1 and lane 2). However, USP9X siRNA partially rescued the endogenous SMAD5 level in cells with overexpressed SMURF1, consistent with a decreased cellular SMURF1 activity. Knockdown of USP9X alone did not affect the SMAD5 level (Fig. 3B), suggesting USP9X only modulates SMAD5 through SMURF1.
FIGURE 3.
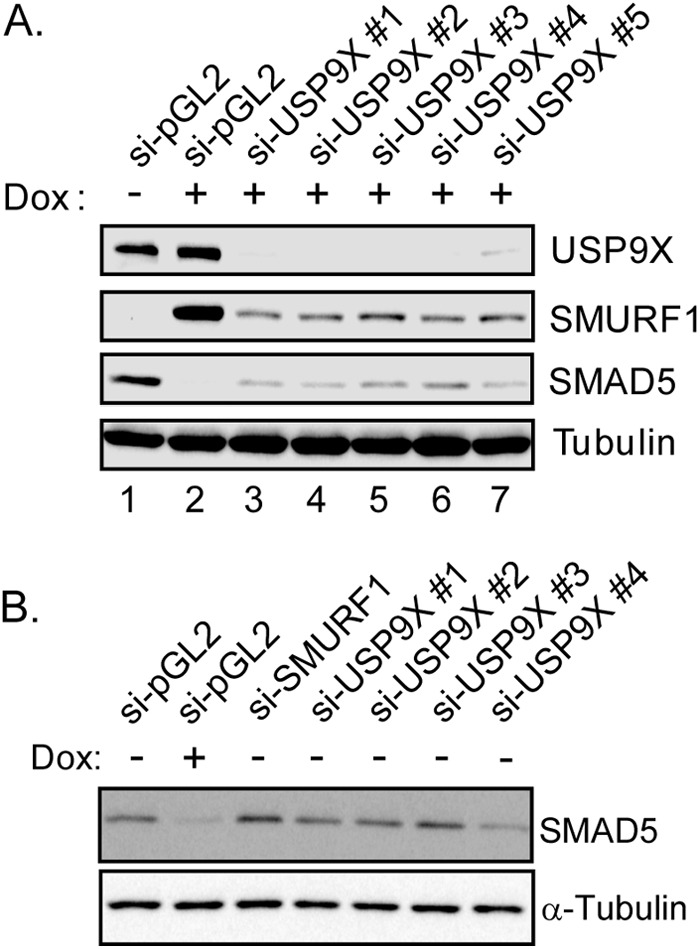
Depletion of USP9X down-regulates SMURF1 level. A, inducible FLAG-SMURF1 cells were transfected with USP9X-specific or nontargeting (pGL2) siRNAs 72 h before the experiment. Selected cells were treated with doxycycline (Dox) overnight to induce SMURF1 expression. Cells were harvested and total cell lysate was resolved by 4–12% SDS-PAGE followed by immunoblotting with the indicated antibodies. B, inducible SMURF1 cells were transfected with siRNAs against SMURF1, USP9X, or pGL2, after overnight treatment with or without doxycycline, cells were harvested and SMAD5 levels were detected by anti-SMAD5 immunoblotting.
USP9X knockdown did not affect SMURF1 mRNA (Fig. 4A), suggesting USP9X controls the SMURF1 protein level possibly through proteolytic regulation. To confirm a role for USP9X in regulating SMURF1 degradation, we employed a cycloheximide-based protein degradation assay. After protein synthesis was blocked by cycloheximide, SMURF1 degraded more rapidly in the USP9X-depleted cells (Fig. 4B). In contrast, the degradation kinetics of Cyclin D1, a short-lived protein (41), was not affected (Fig, 4B). Similarly, by pulse-chase analysis, 35S-labeled SMURF1 exhibited a faster turnover rate in USP9X siRNA-treated than nontargeting siRNA-treated cells (Fig. 4C). Treatment with MG132, a potent proteasome inhibitor, completely rescued the down-regulation of SMURF1 in USP9X-depleted cells (Fig. 4D). This result suggests that USP9X regulates the SMURF1 level post-translationally through the ubiquitin/proteasome pathway.
FIGURE 4.
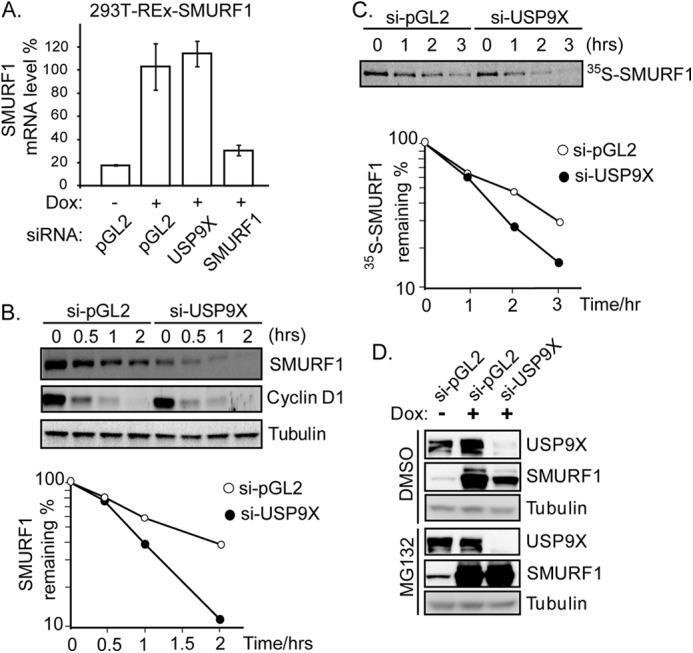
Depletion of USP9X accelerates degradation of SMURF1. A, USP9X knockdown (by si-USP9X#3) does not change the SMURF1 mRNA level. The SMURF1 mRNA level after gene-specific knockdown was determined by quantitative PCR analysis (quantified as percentage of the SMURF1 mRNA in control (si-pGL2) sample (n = 3)). B, inducible FLAG-SMURF1 cells were transfected with USP9X-specific (#3) or nontargeting (pGL2 luciferase) siRNA 48 h before the experiment. To start with the chase, cycloheximide was added to the cell culture at 100 μg/ml final concentration. Cells were harvested 0, 0.5, 1, or 2 h after cycloheximide addition and the cell lysate was resolved by 4–12% SDS-PAGE followed by immunoblotting with specific antibodies. Plot of SMURF1 degradation rates was shown in the lower panel. C, SMURF1 degradation was measured by pulse-chase analysis (details in “Materials and Methods”) and plot of SMURF1 degradation rates was shown in the lower panel. D, FLAG-SMURF1 cells were transfected with USP9X-specific or nontargeting siRNAs, followed by overnight treatment of doxycycline. Cells were then treated with MG132 or DMSO for 6 h before harvest. Total cell lysate was resolved by 4–12% SDS-PAGE and immunoblotted with the corresponding antibodies.
SMURF1 autoubiquitination and self-degradation are dependent on its E3 activity. Mutagenesis of a conserved cystine to alanine (C699A) in the HECT domain of SMURF1 abolished its E3 activity and significantly promoted SMURF1 stability (39). SMURF1 is also reported to be targeted by other E3s and undergo proteolysis independent of its own E3 activity (25, 26). To determine the mechanism by which USP9X mediates SMURF1 stability, we measured the level of SMURF1-WT and -C699A in cells treated with USP9X-targeting siRNAs. Interestingly, knockdown of USP9X only decreased the level of SMURF1-WT (Fig. 5A, compare lane 2 and lane 6), without affecting SMURF1-C699A (Fig. 5A, compare lane 4 and lane 8). This result indicates that enhanced degradation of SMURF1 in USP9X-depleted cells depends on the ligase activity of SMURF1. Because WT SMURF1 can be autoubiquitinated through its own E3 activity, we reasoned that USP9X is likely to stabilize SMURF1 by inhibiting the autoubiquitination of SMURF1. To test this hypothesis, cellular SMURF1 ubiquitination was analyzed with or without USP9X siRNA treatment. As shown in Fig. 5B, after SMURF1 overexpression, ubiquitination of WT SMURF1 was readily detected. Knockdown of USP9X remarkably increased the SMURF1 ubiquitination level, suggesting a role of USP9X in antagonizing SMURF1 autoubiquitination.
FIGURE 5.
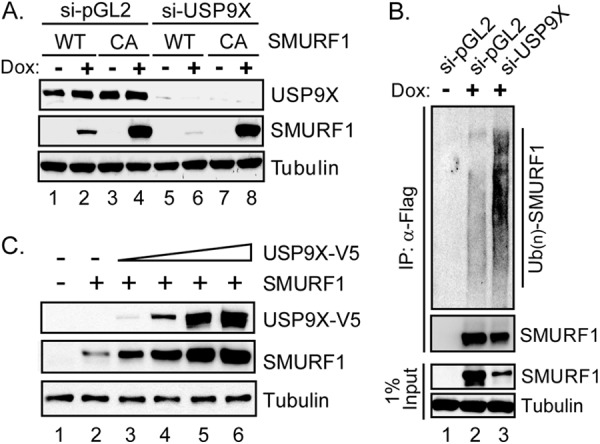
USP9X inhibits autoubiquitination and degradation of SMURF1. A, inducible SMURF1 (WT or enzyme-dead (CA)) cells were transfected with USP9X-specific (#3) or nontargeting (pGP2) siRNAs 48 h before the experiment. After overnight treatment with or without doxycycline (Dox), cell lysates were prepared, resolved by 4–12% SDS-PAGE, and immunoblotted with anti-USP9X and anti-FLAG (for FLAG-SMURF1) antibodies, respectively. B, status of cellular SMURF1 ubiquitination with or without USP9X knockdown. Inducible FLAG-SMURF1 cells were transfected with USP9X-specific (#3) or control (pGL2) siRNAs. After overnight treatment with or without doxycycline, cell lysates were harvested, denatured, and subject to anti-FLAG immunoprecipitation. Precipitates were then resolved by 4–12% SDS-PAGE and immunoblotted with anti-ubiquitin and anti-SMURF1 antibodies, respectively. SMURF1 and Tubulin levels were also shown by immunoblotting in 1% Input. C, 0.5 μg of pcDNA4/TO-SMURF1 cDNA was co-transfected with increasing amount of V5-tagged USP9X construct (0, 1, 2, and 5 μg) into the HEK293T cells. 48 h after transfection, the cell lysate was prepared and subject to SDS-PAGE and immunoblotting analysis with the indicated antibodies. Overexpressed USP9X is detected by anti-V5 immunoblotting.
In the presence of endogenous USP9X, WT SMURF1 is still degraded with a half-life of ∼1 h (Fig. 4, B and C), suggesting that USP9X, at its physiological concentration, cannot fully block SMURF1 degradation. We reasoned that an increased amount of USP9X should promote more SMURF1 association and lead to increased SMURF1 stability. To test that hypothesis, the same amount of SMURF1 was transfected along with an increasing dosage of USP9X cDNA into the HEK293T cells. As predicted, the SMURF1 level increased in a dose-dependent manner with increasing amounts of USP9X expression (Fig. 5C), consistent with a role of USP9X in promoting SMURF1 stability by antagonizing SMURF1 autoubiquitination and self-degradation. Taken together, our data suggest that SMURF1 is a substrate for USP9X-mediated deubiquitination, and is protected by USP9X against autoubiquitination and proteasomal degradation.
Depletion of USP9X Decreased the Level of Endogenous SMURF1 and Inhibited SMURF1-dependent Cancer Cell Migration
Expression of endogenous SMURF1 in HEK293T cells was low and cannot be detected by current antibodies (data not shown). To assess the function of USP9X on endogenous SMURF1, we chose to study this in a MDA-MB-231 cell line of breast cancer origin due to its high levels of endogenous SMURF1 (38). Endogenous SMURF1 was detected by anti-SMURF1 immunoblotting in MDA-MB-231 cells, and the SMURF1 level was drastically decreased by SMURF1 knockdown (Fig. 6A, compare lane 1 and lane 6). Significantly, independent USP9X-targeting siRNAs all led to remarkable reduction of the endogenous SMURF1 protein level (Fig. 6A, compare lane 1 and lanes 2–5). This result suggests that, in MDA-MB-231 cells, USP9X is able to protect endogenous SMURF1 degradation through deubiquitination.
FIGURE 6.
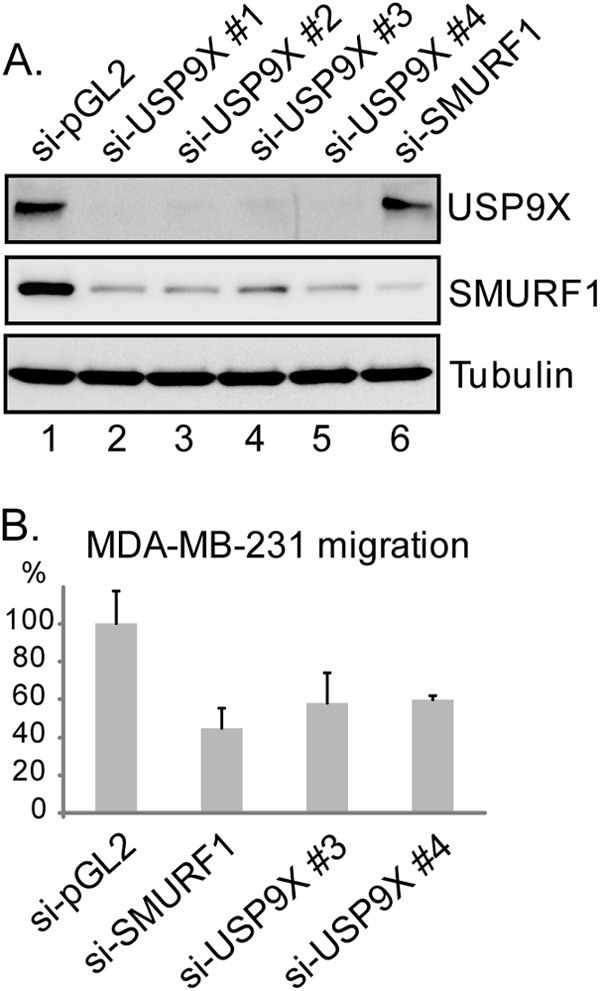
Depletion of USP9X destabilizes SMURF1 and blocks SMURF1-dependent cell migration in MDA-MB-231 cells. A, MDA-MB-231 cells were transfected with USP9X-specific or nontargeting (pGL2) siRNAs. Cells lysates were resolved by 4–12% SDS-PAGE followed by immunoblotting with anti-USP9X and anti-SMURF1 antibodies, respectively. Equal loading was demonstrated by anti-Tubulin immunoblotting. B, MDA-MB-231 cells were treated with USP9X-specific (3 and 4), SMURF1-specific or control (pGL2) siRNAs before the initiation of migration assay. Cell migration efficiency was measured according to Ref. 16 and quantified as percentage of the total migrating cells in control (si-pGL2) sample (n = 3).
MDA-MB-231 cells are highly metastatic breast cancer cells, characteristic of active migration and elevated invasiveness (42). SMURF1 regulates cancer cell migration and invasion by targeting RhoA, a member of Rho family of small GTPases (16, 39). Mechanistically, SMURF1 can complex with RhoA at the leading edge of the lamellipodium and target the RhoA protein for proteasomal degradation. Inhibition of SMURF1 stabilizes RhoA at the leading edge of the cell and significantly inhibits cell motility and plasticity (16, 38, 43). We identified USP9X as a SMURF1-interacting protein that controls the endogenous SMURF1 level through deubiquitination. Therefore, we speculated that depletion of USP9X should down-regulate SMURF1 and phenocopy the effect of SMURF1 inhibition. To test the physiological significance of USP9X-SMURF1 interaction, we tested the role of USP9X in MDA-MB-231 cell migration. A transwell cell migration assay was set up and cells were transfected with respective siRNAs prior to migration assay. Treatment with SMURF1-specific siRNA resulted in ∼60% reduction of cell motility, consistent with the results from previous studies (38). When cells were treated with independent USP9X-target siRNAs, more than 40% reduction in cell migration was observed, underscoring a physiological role for USP9X in controlling the cellular SMURF1 level (Fig. 6B).
DISCUSSION
SMURF1 plays crucial physiological roles in TGF-β/BMP signaling, cell migration, and polarity determination, mainly by targeting corresponding cellular substrates for ubiquitination and proteasomal degradation (12, 13, 20, 23). More recently, SMURF1 was also identified as a component regulating autophagy activity and immune response (21, 24). Reported substrates for SMURF1 include SMAD proteins, TGF-β receptors, Runx2, MEKK2, RhoA, Par6, JunB, Talin head, Prickle1, TRAF family members, and STAT1. To achieve substrate specificity and cope with the dynamic cellular environment, it is imperative that SMURF1 itself is subject to intricate controls by other protein regulators. Increasing evidence has suggested that multiple proteins can interact and modulate SMURF1; however, either the exact controlling mechanism or the direct consequence of those regulations remains to be clarified. In this study, we employed a quantitative mass spectrometry approach and identified a deubiquitinating enzyme, FAM/USP9X, as a novel interacting factor for SMURF1. By domain mapping, we confirmed that the second WW domain (WW2) of SMURF1 is necessary and sufficient for USP9X association. As for USP9X, its carboxyl terminus (C-2 domain) provides the major binding affinity toward SMURF1. Interestingly, the USP9X-SMURF1 interaction does not lead to USP9X degradation. Instead, USP9X can deconjugate ubiquitin from SMURF1 and protect SMURF1 from self-degradation. In MDA-MB-231 breast cancer cells, inhibition of USP9X activity led to down-regulation of endogenous SMURF1 protein. Under that condition, SMURF1-dependent cell motility was significantly impaired, consistent with a role of USP9X in maintaining regular SMURF1-mediated cellular physiologies.
SMURF1 belongs to the Nedd4-like HECT E3 family, which consists of nine members in the human genome (10). Like other Nedd4-like HECT E3s, SMURF1 adopts a highly conserved modular architecture, consisting of an N-terminal C2 domain, two WW domains, and an enzymatic HECT domain at the carboxyl terminus (Fig. 2A)(5). The short stretch of WW domains are usually found important for substrate targeting (8, 9). A typical WW domain is 35–40 amino acids long and harbors two conserved tryptophan residues critical for substrate binding. On the SMURF1 protein, there is only one conserved tryptophan in each of the WW domains, and the two WW domains are believed to work cooperatively to bind SMURF1 substrates (44, 45). On SMURF1 substrates, a PY motif (Pro-Pro-X-Tyr) is commonly found to associate with the WW domains of SMURF1. Similarly, a “PPIKY” sequence was identified in the C-2 domain of USP9X, which is sufficient for SMURF1 binding (Fig. 2C). It is, therefore, possible that USP9X interacts with SMURF1 by the similar mechanism as other SMURF1 substrates. Interestingly, LIM mineralization protein-1 (LMP-1), a regulator of SMURF1, also utilizes its PY motifs to associate with the WW2 domain of SMURF1 (46). LMP-1-SMURF1 interaction effectively competes with SMURF1 binding with its substrates SMAD1/5, and leads to stabilization of SMADs and an increase of BMP signaling (47). Because USP9X also binds to the WW2 domain of SMURF1, it is plausible that USP9X and SMURF1 substrates interact with SMURF1 in a competitive manner. It remains to be investigated if such a mechanism could exist and if that bears any functional consequences.
Besides LMP-1, multiple other SMURF1-interacting proteins have been identified to date that can regulate SMURF1 activity by different mechanisms. Casein kinase 2 interacting protein-1, interacts with the linker region of WW domains of SMURF1, and augments SMURF1 activity and substrate ubiquitination (48). Cdh1, a subunit of the Anaphase promoting complex, was reported to bind C2 and the WW1 domain of SMURF1. Such an interaction also leads to SMURF1 activation, possibly by disrupting SMURF1 homodimer formation (49). Cerebral cavernous malformation 2 recognizes the HECT domain of SMURF1 and helps to localize SMURF1 to the plasma membrane to enhance RhoA ubiquitination and degradation (50). Unique from the abovementioned SMURF1 regulators, USP9X regulates SMURF1 by editing its ubiquitination status and controlling its cellular stability. Ubiquitin ligases SMURF2 and SCFFBXL15 are reported to target SMURF1 for ubiquitination and degradation, independent of the E3 activity of SMURF1 (25, 26). Although USP9X can associate with both WT and enzymatically inactive SMURF1 (Fig. 1C), it only controls the stability of WT, but not mutant, SMURF1 (Fig. 5A). This suggests that USP9X may constitutively associate with SMURF1 to antagonize its autoubiquitination and self-degradation.
Growing evidence suggests that the deubiquitinase/ubiquitin ligase (DUB/E3) complex may represent a common strategy to modulate cellular protein dynamics by mediating E3 stability and activity (51–54). Rsp5, a yeast homolog of Nedd4-like E3, is found to associate with DUB Upb2, which antagonizes Rsp5 activity and limits substrates ubiquitination (55, 56). On the other hand, DUB USP19 associates with and stabilizes KPC1, a RING E3, to promote degradation of KPC1 substrates like p27Kip1 (52). Interestingly, ITCH, another member of the Nedd4-like E3 family, was previously identified to bind and be protected by USP9X. A fragment of ITCH encompassing its four WW domains was also found crucial for USP9X binding (57). Given that all Nedd4 family E3s share similar domain structure, it remains to be tested whether USP9X can interact with other E3s of the Nedd4 family, and utilize a similar molecular mechanism to modulate a broader range of cellular functions.
From studies of different human neoplasias, USP9X has been implicated as both a tumor suppressor and a factor necessary for tumor cell survival (34, 58). Such could be explained by the fact that USP9X targets multiple substrates and plays a tissue-specific function in vivo. SMURF1 activity positively regulates cancer cell motility, and SMURF1 amplification leads to promotion of tumor invasiveness (43, 59). In this study, we discovered that USP9X and SMURF1 exist in a complex. USP9X stabilizes endogenous SMURF1 through deubiquitination and positively regulates breast cancer cell motility. Further study into the mechanism of USP9X-SMURF1 interaction is warranted to advance our knowledge of its physiological roles and may guide for novel cancer therapies.
Acknowledgments
We thank Dr. Stephen A. Wood for generously providing the USP9X cDNA construct for this study. We also thank Estee Toole for help with mass spectrometry analysis.

This article contains supplemental Table S1.
- HECT
- homologous to E6AP carboxyl terminus
- BMP
- bone morphologenetic proteins
- DUB
- de-ubiquitinating enzymes
- USP9X
- ubiquitin-specific peptidase 9, X-linked
- LMP-1
- LIM mineralization protein-1.
REFERENCES
- 1. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 2. Kerscher O., Felberbaum R., Hochstrasser M. (2006) Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22, 159–180 [DOI] [PubMed] [Google Scholar]
- 3. Joazeiro C. A., Weissman A. M. (2000) RING finger proteins. Mediators of ubiquitin ligase activity. Cell 102, 549–552 [DOI] [PubMed] [Google Scholar]
- 4. Bernassola F., Karin M., Ciechanover A., Melino G. (2008) The HECT family of E3 ubiquitin ligases. Multiple players in cancer development. Cancer Cell 14, 10–21 [DOI] [PubMed] [Google Scholar]
- 5. Ingham R. J., Gish G., Pawson T. (2004) The Nedd4 family of E3 ubiquitin ligases. Functional diversity within a common modular architecture. Oncogene 23, 1972–1984 [DOI] [PubMed] [Google Scholar]
- 6. Nalefski E. A., Falke J. J. (1996) The C2 domain calcium-binding motif. Structural and functional diversity. Protein Sci. 5, 2375–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Plant P. J., Yeger H., Staub O., Howard P., Rotin D. (1997) The C2 domain of the ubiquitin protein ligase Nedd4 mediates Ca2+-dependent plasma membrane localization. J. Biol. Chem. 272, 32329–32336 [DOI] [PubMed] [Google Scholar]
- 8. Kanelis V., Farrow N. A., Kay L. E., Rotin D., Forman-Kay J. D. (1998) NMR studies of tandem WW domains of Nedd4 in complex with a PY motif-containing region of the epithelial sodium channel. Biochem. Cell Biol. 76, 341–350 [DOI] [PubMed] [Google Scholar]
- 9. Rotin D. (1998) WW (WWP) domains: from structure to function. Curr. Top. Microbiol. Immunol. 228, 115–133 [DOI] [PubMed] [Google Scholar]
- 10. Rotin D., Kumar S. (2009) Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10, 398–409 [DOI] [PubMed] [Google Scholar]
- 11. Inoue Y., Imamura T. (2008) Regulation of TGF-β family signaling by E3 ubiquitin ligases. Cancer Sci. 99, 2107–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu H., Kavsak P., Abdollah S., Wrana J. L., Thomsen G. H. (1999) A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 400, 687–693 [DOI] [PubMed] [Google Scholar]
- 13. Ebisawa T., Fukuchi M., Murakami G., Chiba T., Tanaka K., Imamura T., Miyazono K. (2001) Smurf1 interacts with transforming growth factor-β type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 276, 12477–12480 [DOI] [PubMed] [Google Scholar]
- 14. Zhao M., Qiao M., Oyajobi B. O., Mundy G. R., Chen D. (2003) E3 ubiquitin ligase Smurf1 mediates core-binding factor α1/Runx2 degradation and plays a specific role in osteoblast differentiation. J. Biol. Chem. 278, 27939–27944 [DOI] [PubMed] [Google Scholar]
- 15. Yamashita M., Ying S. X., Zhang G. M., Li C., Cheng S. Y., Deng C. X., Zhang Y. E. (2005) Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 121, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang H. R., Zhang Y., Ozdamar B., Ogunjimi A. A., Alexandrova E., Thomsen G. H., Wrana J. L. (2003) Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 302, 1775–1779 [DOI] [PubMed] [Google Scholar]
- 17. Cheng P. L., Lu H., Shelly M., Gao H., Poo M. M. (2011) Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron 69, 231–243 [DOI] [PubMed] [Google Scholar]
- 18. Zhao L., Huang J., Guo R., Wang Y., Chen D., Xing L. (2010) Smurf1 inhibits mesenchymal stem cell proliferation and differentiation into osteoblasts through JunB degradation. J. Bone Miner. Res. 25, 1246–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang C., Rajfur Z., Yousefi N., Chen Z., Jacobson K., Ginsberg M. H. (2009) Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nat. Cell Biol. 11, 624–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Narimatsu M., Bose R., Pye M., Zhang L., Miller B., Ching P., Sakuma R., Luga V., Roncari L., Attisano L., Wrana J. L. (2009) Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell 137, 295–307 [DOI] [PubMed] [Google Scholar]
- 21. Li S., Lu K., Wang J., An L., Yang G., Chen H., Cui Y., Yin X., Xie P., Xing G., He F., Zhang L. (2010) Ubiquitin ligase Smurf1 targets TRAF family proteins for ubiquitination and degradation. Mol. Cell. Biochem. 338, 11–17 [DOI] [PubMed] [Google Scholar]
- 22. Yuan C., Qi J., Zhao X., Gao C. (2012) Smurf1 protein negatively regulates interferon-γ signaling through promoting STAT1 protein ubiquitination and degradation. J. Biol. Chem. 287, 17006–17015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ozdamar B., Bose R., Barrios-Rodiles M., Wang H. R., Zhang Y., Wrana J. L. (2005) Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science 307, 1603–1609 [DOI] [PubMed] [Google Scholar]
- 24. Orvedahl A., Sumpter R., Jr., Xiao G., Ng A., Zou Z., Tang Y., Narimatsu M., Gilpin C., Sun Q., Roth M., Forst C. V., Wrana J. L., Zhang Y. E., Luby-Phelps K., Xavier R. J., Xie Y., Levine B. (2011) Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fukunaga E., Inoue Y., Komiya S., Horiguchi K., Goto K., Saitoh M., Miyazawa K., Koinuma D., Hanyu A., Imamura T. (2008) Smurf2 induces ubiquitin-dependent degradation of Smurf1 to prevent migration of breast cancer cells. J. Biol. Chem. 283, 35660–35667 [DOI] [PubMed] [Google Scholar]
- 26. Cui Y., He S., Xing C., Lu K., Wang J., Xing G., Meng A., Jia S., He F., Zhang L. (2011) SCFFBXL regulates BMP signalling by directing the degradation of HECT-type ubiquitin ligase Smurf1. EMBO J. 30, 2675–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilkinson S. C., Higham S. M., Ingram G. S., Edgar W. M. (1997) Visualization of root caries lesions by means of a diazonium dye. Adv. Dent. Res. 11, 515–522 [DOI] [PubMed] [Google Scholar]
- 28. Fraile J. M., Quesada V., Rodríguez D., Freije J. M., López-Otín C. (2012) Deubiquitinases in cancer. New functions and therapeutic options. Oncogene 31, 2373–2388 [DOI] [PubMed] [Google Scholar]
- 29. Reyes-Turcu F. E., Ventii K. H., Wilkinson K. D. (2009) Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 78, 363–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Komander D., Clague M. J., Urbé S. (2009) Breaking the chains. Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 [DOI] [PubMed] [Google Scholar]
- 31. Taya S., Yamamoto T., Kanai-Azuma M., Wood S. A., Kaibuchi K. (1999) The deubiquitinating enzyme Fam interacts with and stabilizes β-catenin. Genes Cells 4, 757–767 [DOI] [PubMed] [Google Scholar]
- 32. Taya S., Yamamoto T., Kano K., Kawano Y., Iwamatsu A., Tsuchiya T., Tanaka K., Kanai-Azuma M., Wood S. A., Mattick J. S., Kaibuchi K. (1998) The Ras target AF-6 is a substrate of the fam deubiquitinating enzyme. J. Cell Biol. 142, 1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Théard D., Labarrade F., Partisani M., Milanini J., Sakagami H., Fon E. A., Wood S. A., Franco M., Luton F. (2010) USP9x-mediated deubiquitination of EFA6 regulates de novo tight junction assembly. EMBO J. 29, 1499–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schwickart M., Huang X., Lill J. R., Liu J., Ferrando R., French D. M., Maecker H., O'Rourke K., Bazan F., Eastham-Anderson J., Yue P., Dornan D., Huang D. C., Dixit V. M. (2010) Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463, 103–107 [DOI] [PubMed] [Google Scholar]
- 35. Rott R., Szargel R., Haskin J., Bandopadhyay R., Lees A. J., Shani V., Engelender S. (2011) α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. U.S.A. 108, 18666–18671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Makarova O., Kamberov E., Margolis B. (2000) Generation of deletion and point mutations with one primer in a single cloning step. BioTechniques 29, 970–972 [DOI] [PubMed] [Google Scholar]
- 37. Huang S. M., Mishina Y. M., Liu S., Cheung A., Stegmeier F., Michaud G. A., Charlat O., Wiellette E., Zhang Y., Wiessner S., Hild M., Shi X., Wilson C. J., Mickanin C., Myer V., Fazal A., Tomlinson R., Serluca F., Shao W., Cheng H., Shultz M., Rau C., Schirle M., Schlegl J., Ghidelli S., Fawell S., Lu C., Curtis D., Kirschner M. W., Lengauer C., Finan P. M., Tallarico J. A., Bouwmeester T., Porter J. A., Bauer A., Cong F. (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620 [DOI] [PubMed] [Google Scholar]
- 38. Jin C., Yang Y. A., Anver M. R., Morris N., Wang X., Zhang Y. E. (2009) Smad ubiquitination regulatory factor 2 promotes metastasis of breast cancer cells by enhancing migration and invasiveness. Cancer Res. 69, 735–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang H. R., Ogunjimi A. A., Zhang Y., Ozdamar B., Bose R., Wrana J. L. (2006) Degradation of RhoA by Smurf1 ubiquitin ligase. Methods Enzymol. 406, 437–447 [DOI] [PubMed] [Google Scholar]
- 40. Wood S. A., Pascoe W. S., Ru K., Yamada T., Hirchenhain J., Kemler R., Mattick J. S. (1997) Cloning and expression analysis of a novel mouse gene with sequence similarity to the Drosophila fat facets gene. Mech. Dev. 63, 29–38 [DOI] [PubMed] [Google Scholar]
- 41. Diehl J. A., Zindy F., Sherr C. J. (1997) Inhibition of cyclin D1 phosphorylation on threonine 286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 11, 957–972 [DOI] [PubMed] [Google Scholar]
- 42. Thompson E. W., Reich R., Shima T. B., Albini A., Graf J., Martin G. R., Dickson R. B., Lippman M. E. (1988) Differential regulation of growth and invasiveness of MCF-7 breast cancer cells by antiestrogens. Cancer Res. 48, 6764–6768 [PubMed] [Google Scholar]
- 43. Sahai E., Garcia-Medina R., Pouysségur J., Vial E. (2007) Smurf1 regulates tumor cell plasticity and motility through degradation of RhoA leading to localized inhibition of contractility. J. Cell Biol. 176, 35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Macias M. J., Wiesner S., Sudol M. (2002) WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 513, 30–37 [DOI] [PubMed] [Google Scholar]
- 45. Shearwin-Whyatt L., Dalton H. E., Foot N., Kumar S. (2006) Regulation of functional diversity within the Nedd4 family by accessory and adaptor proteins. Bioessays 28, 617–628 [DOI] [PubMed] [Google Scholar]
- 46. Sangadala S., Boden S. D., Metpally R. P., Reddy B. V. (2007) Modeling and analysis of molecularinteraction between Smurf1-WW2 domain and various isoforms of LIM mineralization protein. Proteins 68, 690–701 [DOI] [PubMed] [Google Scholar]
- 47. Sangadala S., Boden S. D., Viggeswarapu M., Liu Y., Titus L. (2006) LIM mineralization protein-1 potentiates bone morphogenetic protein responsiveness via a novel interaction with Smurf1 resulting in decreased ubiquitination of Smads. J. Biol. Chem. 281, 17212–17219 [DOI] [PubMed] [Google Scholar]
- 48. Lu K., Yin X., Weng T., Xi S., Li L., Xing G., Cheng X., Yang X., Zhang L., He F. (2008) Targeting WW domains linker of HECT-type ubiquitin ligase Smurf1 for activation by CKIP-1. Nat. Cell Biol. 10, 994–1002 [DOI] [PubMed] [Google Scholar]
- 49. Wan L., Zou W., Gao D., Inuzuka H., Fukushima H., Berg A. H., Drapp R., Shaik S., Hu D., Lester C., Eguren M., Malumbres M., Glimcher L. H., Wei W. (2011) Cdh1 regulates osteoblast function through an APC/C-independent modulation of Smurf1. Mol. Cell 44, 721–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Crose L. E., Hilder T. L., Sciaky N., Johnson G. L. (2009) Cerebral cavernous malformation 2 protein promotes smad ubiquitin regulatory factor 1-mediated RhoA degradation in endothelial cells. J. Biol. Chem. 284, 13301–13305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu X., Yen L., Irwin L., Sweeney C., Carraway K. L., 3rd (2004) Stabilization of the E3 ubiquitin ligase Nrdp1 by the deubiquitinating enzyme USP8. Mol. Cell Biol. 24, 7748–7757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lu Y., Adegoke O. A., Nepveu A., Nakayama K. I., Bedard N., Cheng D., Peng J., Wing S. S. (2009) USP19 deubiquitinating enzyme supports cell proliferation by stabilizing KPC1, a ubiquitin ligase for p27Kip1. Mol. Cell. Biol. 29, 547–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stegmeier F., Rape M., Draviam V. M., Nalepa G., Sowa M. E., Ang X. L., McDonald E. R., 3rd, Li M. Z., Hannon G. J., Sorger P. K., Kirschner M. W., Harper J. W., Elledge S. J. (2007) Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature 446, 876–881 [DOI] [PubMed] [Google Scholar]
- 54. Sowa M. E., Bennett E. J., Gygi S. P., Harper J. W. (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kee Y., Lyon N., Huibregtse J. M. (2005) The Rsp5 ubiquitin ligase is coupled to and antagonized by the Ubp2 deubiquitinating enzyme. EMBO J. 24, 2414–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kee Y., Muñoz W., Lyon N., Huibregtse J. M. (2006) The deubiquitinating enzyme Ubp2 modulates Rsp5-dependent Lys63-linked polyubiquitin conjugates in Saccharomyces cerevisiae. J. Biol. Chem. 281, 36724–36731 [DOI] [PubMed] [Google Scholar]
- 57. Mouchantaf R., Azakir B. A., McPherson P. S., Millard S. M., Wood S. A., Angers A. (2006) The ubiquitin ligase itch is auto-ubiquitylated in vivo and in vitro but is protected from degradation by interacting with the deubiquitylating enzyme FAM/USP9X. J. Biol. Chem. 281, 38738–38747 [DOI] [PubMed] [Google Scholar]
- 58. Pérez-Mancera P. A., Rust A. G., van der Weyden L., Kristiansen G., Li A., Sarver A. L., Silverstein K. A., Grützmann R., Aust D., Rümmele P., Knösel T., Herd C., Stemple D. L., Kettleborough R., Brosnan J. A., Li A., Morgan R., Knight S., Yu J., Stegeman S., Collier L. S., ten Hoeve J. J., de Ridder J., Klein A. P., Goggins M., Hruban R. H., Chang D. K., Biankin A. V., Grimmond S. M., Australian Pancreatic Cancer Genome Initiative, Wessels L. F., Wood S. A., Iacobuzio-Donahue C. A., Pilarsky C., Largaespada D. A., Adams D. J., Tuveson D. A. (2012) The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486, 266–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kwei K. A., Shain A. H., Bair R., Montgomery K., Karikari C. A., van de Rijn M., Hidalgo M., Maitra A., Bashyam M. D., Pollack J. R. (2011) SMURF1 amplification promotes invasiveness in pancreatic cancer. PLoS One 6, e23924. [DOI] [PMC free article] [PubMed] [Google Scholar]