

Background: Cathepsin D has been implicated in Niemann-Pick type C (NPC) disease, which is associated with intracellular cholesterol accumulation.
Results: Increased cytosolic and extracellular levels of cathepsin D enhanced neuronal death via different mechanisms.
Conclusion: Leakage of cathepsin D within and outside the cell can cause cell death.
Significance: Cathepsin D may be involved in the degeneration of neurons in NPC pathology.
Keywords: Cell Death, Cell Signaling, Cholesterol Regulation, Lysosomes, Neurodegeneration, Neurodegenerative Diseases
Abstract
Cathepsin D is an aspartyl protease that plays a crucial role in normal cellular functions and in a variety of neurodegenerative disorders, including Niemann-Pick type C (NPC) disease, which is characterized by intracellular accumulation of cholesterol and glycosphingolipids in many tissues, including the brain. There is evidence that the level and activity of cathepsin D increased markedly in vulnerable neurons in NPC pathology, but its involvement in neurodegeneration remains unclear. In the present study, using mouse hippocampal cultured neurons, we evaluated the significance of cathepsin D in toxicity induced by U18666A, a class II amphiphile, which triggers cell death by impairing the trafficking of cholesterol, as observed in NPC pathology. Our results showed that U18666A-mediated toxicity is accompanied by an increase in cathepsin D mRNA and enzyme activity but a decrease in the total peptide content. The cytosolic level of cathepsin D, on the other hand, was increased along with cytochrome c and activated caspase-3 in U18666A-treated neurons. The cathepsin D inhibitor, pepstatin A, partially protected neurons against toxicity by attenuating these signaling mechanisms. Additionally, down-regulation of cathepsin D level prevented, whereas overexpression of the protease increased, vulnerability of cultured N2a cells to U18666A-induced toxicity. We also showed that extracellular cathepsin D from U18666A-treated neurons or application of exogenous enzyme can induce neurotoxicity by activating the autophagic pathway. These results suggest that increased release/activation of cathepsin D can trigger neurodegeneration and possibly development of NPC pathology. Thus, targeting cathepsin D level/activity may provide a new therapeutic opportunity for the treatment of NPC pathology.
Introduction
Cathepsin D is a soluble lysosomal aspartic protease of the pepsin superfamily, which is widely distributed in tissues, including the brain. This protease, after being synthesized in the rough endoplasmic reticulum as preprocathepsin D, undergoes post-translational modification to remove the signal peptide and is then transported to prelysosomes (also termed late endosomes) in clathrin-coated vesicles by mannose 6-phosphate receptors (1–3). The acidic milieu of the prelysosomes triggers the release of the enzymes from the receptors, which are then transported by capillary movement to the lysosomes. In certain physiological and pathological conditions, cathepsin D escapes normal targeting mechanisms and is secreted from the cell (2, 4). Functionally, the enzyme has been involved in a variety of biological activities, including metabolic degradation of intracellular proteins, activation of some hormones and growth factors, brain antigen processing, and regulation of cell death mechanisms (2, 3, 5–8).
A role for cathepsin D in cell death has been supported by experimental data that showed (i) activation or overexpression of the protease can mediate/sensitize cells to apoptosis induced by a variety of cytotoxic and stress agents (2, 3), (ii) cathepsin D-deficient fibroblasts are resistant to adriamycin- and etoposide-induced apoptosis (9, 10), and (iii) intracellular microinjection of cathepsin D can induce caspase-dependent apoptosis in human fibroblasts (11). There is evidence that partial lysosomal permeabilization with subsequent cytosolic release of cathepsin D can trigger apoptosis or apoptosis-like death, whereas generalized lysosomal rupture results in rapid cellular necrosis. In many instances, lysosomal leakage of cathepsin D is believed to precede release of cytochrome c, loss of mitochondrial membrane potential, and morphologic manifestation of apoptosis (2, 7, 8). Taken together, these results raise the possibility that cathepsin D may have an important role not only in normal cellular functioning but also in a variety of lysosomal storage disorders that are associated with extensive neurodegeneration and progressive cognitive decline. However, at present, the significance of cathepsin D in the degeneration of neurons and/or development of pathological features associated with any of these diseases remains unclear.
Niemann-Pick type C (NPC)4 disease is an autosomal recessive neurovisceral disorder characterized by abnormal accumulation of unesterified cholesterol and glycosphingolipids within the endosomal-lysosomal system in a number of tissues, including the brain. These defects trigger widespread neurological deficits, such as ataxia, dystonia, seizures, and dementia, that eventually lead to premature death (12–15). Interestingly, certain neuropathological features associated with NPC disease exhibit some striking similarity with Alzheimer disease (15–17). The overlaps between the two diseases include the presence of phospho-Tau-containing neurofibrillary tangles, increased levels of intracellular amyloid β (Aβ) peptide, and the loss of neurons in selected regions of the brain (18–23). There is also evidence that the endosomal-lysosomal system is altered in “at risk” neurons of both Alzheimer disease and NPC brains, which are reflected by an increased volume of early endosomes and lysosomes and enhanced synthesis of all classes of lysosomal hydrolases, including cathepsin D (20, 21). Some recent studies from NPC1-deficient mice recapitulating NPC pathology have also shown an up-regulation of cathepsin D levels in both neurons and microglia in selected brain regions (21, 24, 25), but their significance in the development of pathology and/or degeneration of neurons has yet to be established. Earlier reports have shown that the class II amphiphile U18666A can induce cell death under the in vitro paradigm by impairing the trafficking as well as the accumulation of cholesterol, as observed in NPC pathology (26–28). In the present study, we have demonstrated that cathepsin D plays a crucial role in the U18666A-induced degeneration of mouse primary cultured neurons by triggering lysosomal destabilization and enzyme leakage into the cytosol. Our results also revealed that fibroblasts from NPC patients are more susceptible to staurosporine-induced cell death, known to be mediated by cathepsin D (29), than fibroblasts from normal individuals. Additionally, we showed that extracellular cathepsin D released from U18666A-treated neurons or exogenous application of the enzyme can induce degeneration of neurons. These results, taken together, suggest that the increased level/activity of cathepsin D observed in NPC disease may be directly involved in the degeneration of neurons associated with the pathology.
EXPERIMENTAL PROCEDURES
Materials
Timed pregnant BALB/c mice purchased from Charles River (St. Constant, Canada) were maintained according to the Animal Care and Use Committee of the University of Alberta and the Canadian Council for Animal Care Committee guidelines. The U18666A was purchased from Biomol Research Laboratories (Plymouth, PA), whereas anti-glial fibrillary acidic protein antibody, the cathepsin D assay kit, and its inhibitor pepstatin A were from Sigma-Aldrich. Cathepsin D small interfering RNA (siRNA), scrambled cathepsin D siRNA, protein A/G-PLUS-agarose, agarose bead-tagged cathepsin D antibody, polyclonal anti-cathepsin D, anti-N-cadherin, anti-histone, anti-apoptosis-inducing factor (AIF), anti-microtubule-associated protein 2 (MAP2) antisera, monoclonal anti-Beclin-1, and all secondary antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), anti-caspase-9 and anti-cleaved caspase-3 antibodies were from Cell Signaling (Beverly, MA), anti-cytochrome c antibody was from BD Biosciences, anti-Atg5 and anti-p62 antibodies were from Millipore (Etobicoke, Canada), anti-Iba1 (ionized calcium-binding adaptor molecule 1) antibody was from Wako Chemicals (Richmond, VA), and anti-LC3 antibody was from MBL International (Woburn, MA). Cell culture reagents, such as Dulbecco's modified Eagle's medium (DMEM), neurobasal medium, Hanks' balanced salt solution, fetal bovine serum (FBS), B27, and Lipofectamine 2000 were from Invitrogen, whereas Hoechst 33258, filipin, 3-(4,5-dimethylthiozolyl)-2,5-diphenyltetrazolium bromide (MTT), 3-methyladenine (3-MA), the active form of human cathepsin D, and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as well as anti-β-actin antisera were from Sigma-Aldrich. The Qproteome cell compartment kit and RNeasy minikit were from Qiagen Inc. (Mississauga, Canada), reverse transcriptase was from Invitrogen, and SYBR Green real-time PCR master mix was from Bio-Rad, and the bicinchoninic acid (BCA) protein assay kit was from Pierce. The Live/Dead cell viability assay kit and LysoSensor Yellow/Blue DND-160 were from Molecular Probes, Inc. (Eugene, OR), whereas Cell Line Nucleofector® Solution V electroporation reagent was from Amaxa (Lnonza, Cologne, Germany). Polyacrylamide electrophoresis gels (4–20%) were from Invitrogen, and the enhanced chemiluminescence (ECL) kit was from Amersham Biosciences. All other reagents were from Sigma-Aldrich or Fisher.
Mouse Hippocampal Neuronal Cultures
Primary hippocampal cultures were prepared from 16- or 17-day-old embryos of timed pregnant BALB/c mice as described previously (30, 31). In brief, the pregnant mice were anesthetized with halothane and decapitated. The hippocampi from pup brains were dissected in Hanks' balanced salt solution supplemented with 15 mm HEPES, 10 units/ml penicillin, and 10 mg/ml streptomycin and digested with 0.25% trypsin-EDTA. The cell suspension was filtered through a cell strainer and then plated on 96-well plates (2 × 103 cells/well for survival/death assay), 6-well plates (2 × 104 cells/well for biochemical assays), or 12-mm glass coverslips (2 × 104 cells/coverslip for immunostaining). The cultures were grown at 37 °C in a 5% CO2 humidified atmosphere in Neurobasal medium supplemented with B27, 50 μm l-glutamine, 15 mm HEPES, 10 units/ml penicillin, 10 mg/ml streptomycin, and 1% FBS. The medium was replaced 1 day later without glutamine or FBS, and all experiments were performed on day 6/7 after plating.
Culture of N2a Cells, Human Fibroblasts, and m5-7 Mouse Embryonic Fibroblasts
The initial stock of N2a mouse neuroblastoma cells were from the American Type Cell Collection. N2a cells were cultured in DMEM containing 10% FBS and penicillin/streptomycin as described earlier (32). Human fibroblasts from normal control (GM05659) and NPC1 patients (GM03123), purchased from the Coriell Cell Repositories (Camden, NJ), were cultured in DMEM containing 10% FBS, 2 mm l-glutamine, 100 units/ml penicillin, and 100 mg/ml streptomycin as described earlier (33). Both N2a and human fibroblast cells were grown at 37 °C with 5% CO2, and the medium was changed every second day. The cells were split every 3–4 days, and experiments were performed on day 2 after plating in 96-well plates. m5-7 mouse embryonic fibroblast cells (Atg5 Tet-off system by doxycycline), kindly provided by Dr. N. Mizushima (Tokyo Medical and Dental University), were cultured in DMEM containing 10% FBS, 100 units/ml penicillin, and 100 mg/ml streptomycin with or without 10 ng/ml doxycycline to regulate the expression of Atg5 as described earlier (34).
RNA Interference
A smart pool of siRNA containing a mixture of three target-specific 19–25-nucleotide siRNAs designed to knock down cathepsin D gene expression was obtained from Santa Cruz Biotechnology, Inc. N2a cells were transfected with cathepsin D or scrambled siRNA (100 nm) using Cell Line Nucleofector® Solution V electroporation reagent in an Amaxa Nucleofector system. Cathepsin D knockdown was analyzed 24 and 48 h following transfection by immunoblotting (35). After 24 h of transfection, cells were treated with or without 3 μg/ml U18666A, and the viability of N2a cells was measured using MTT assay.
Transient Transfection
A pCMV-SPORT6 plasmid containing the cathepsin D insert was obtained from Thermo Scientific (Ottawa, Canada). Cultured N2a cells were transfected with the cathepsin D plasmid or a control plasmid using Lipofectamine 2000 following the manufacturer's protocol. The overexpression of cathepsin D was analyzed 48 h past transfection by immunoblotting (35). The transfected cells were then treated with or without 3 μg/ml U18666A at 24 h post-transfection for a further 24 h, and cell viability was measured using MTT and Live/Dead assays.
Treatments
Six days after plating, mouse hippocampal neurons were treated either with 0.1–50 μg/ml U18666A for 24 h or with 5 μg/ml U18666A for 6–96 h time periods. In some experiments, hippocampal neurons were either co-treated with 1–50 μm pepstatin A, a cathepsin D inhibitor, or pretreated with the inhibitor for 24 h before being exposed to 5 μg/ml U18666A or 150 nm cathepsin D. Additionally, some experiments were performed where hippocampal neurons were exposed for 24 h with 25–150 nm active cathepsin D alone or with 100/150 nm cathepsin D in the presence or absence of 5 mm 3-MA or 200 mm sucrose. Human control and NPC1 fibroblast cells following platting were treated with 0.01–0.5 μm staurosporine for 24 h. The m5-7 fibroblast cells, on the other hand, were exposed to 100 or 150 nm exogenous cathepsin D for 24 h. Control and treated neuronal cultures from different experimental paradigms were then processed for cell viability/toxicity, quantitative RT-PCR, Western blotting, enzyme activity assays, subcellular fractionation, or confocal microscopy.
Cell Viability and Toxicity Assays
Viability of cells/neurons was determined using the colorimetric MTT assay (36, 37). Control and drug-treated culture plates were replaced with new medium containing 0.25% MTT and then incubated for 2 h at 37 °C. The reaction was terminated and measured spectrophotometrically at 570 nm. The experiment was repeated 3–5 times in triplicate. In parallel, neuronal apoptosis was assessed by using the nuclear marker Hoechst 33258 as described earlier (36). In brief, control and drug-treated cultures were fixed with 4% paraformaldehyde, washed in phosphate-buffered saline (PBS), and then stained with Hoechst 33258 (50 ng/ml). The chromatin staining pattern was analyzed for individual cells under a Zeiss Axioskop-2 epifluorescence microscope. The percentage of apoptotic cells was calculated by counting condensed and/or fragmented nuclei versus evenly stained nuclei of normal cells. Neuronal/cell viability was also assessed using the Live/Dead assay kit containing calcein AM and ethidium homodimer (EthD-1) as the fluorescent probes. Calcein AM is a cell-permeant dye that fluoresces in live cells with a functional intracellular esterase, whereas EthD-1 is a membrane-impermeable DNA-binding dye that is excluded from live cells. In this paradigm, control and treated cultures were incubated with medium containing 2 μm calcein AM and 4 μm EthD-1 for 30 min and then visualized under a Zeiss Axioskop-2 fluorescent microscope. The data, which are presented as mean ± S.E., were analyzed using one-way analysis of variance followed by Newman-Keuls post hoc analysis with significance set at p < 0.05.
Filipin Staining
Filipin labels unesterified cholesterol (38). To determine cholesterol accumulation, control and U18666A-treated hippocampal cultured neurons or N2a cells were incubated in the dark with 125 μg/ml filipin in PBS for 1 h. Stained sections were examined using a Zeiss Axioskop-2 microscope.
Western Blotting
For Western blotting, control and drug-treated cells from different experimental paradigms were rinsed with cold TBS and then harvested in radioimmunoprecipitation assay buffer (TBS containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and 10% glycerol with inhibitors 50 mm NaF, 1 mm NaVO3, 10 μg/ml aprotinin, and 10 μg/ml leupeptin). Samples were then denatured in modified Laemmli sample buffer (40 mm Tris-HCl, pH 6.8, 1% SDS, 4% 2-mercaptoethanol, 10% glycerol, and 0.002% bromphenol blue) and boiled for 2–5 min, and equal amounts of proteins (20 μg) were separated by 4–20% polyacrylamide gel electrophoresis (30). The proteins were subsequently transferred to nitrocellulose membranes, blocked with 5% nonfat milk, and incubated overnight at 4 °C with anti-cathepsin D (1:200), anti-cleaved caspase-9 (1:200), anti-cleaved caspase-3 (1:1000), anti-AIF (1:300), anti-LC3 (1:1000), anti-Beclin-1 (1:200), anti-p62 (1:1000) or anti-Atg5 (1:1000) antibodies. Membranes were then incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (1:5000) and visualized using an ECL detection kit. To determine the extracellular cathepsin D level following treatment with U18666A, equal volumes of culture medium collected at different times (i.e. 12, 24, 48, and 72 h) from control and treated cells were centrifuged, and then proteins were precipitated by treating the samples with ice-cold acetone overnight. The proteins were subsequently recovered by centrifugation at 10,000 × g at 4 °C for 1 h, solubilized in the sample buffer, and then processed for Western blotting with anti-cathepsin D (1:200) antiserum. All blots were reprobed with anti-β-actin (1:1000) antiserum and quantified using an Microcomputer Imaging Device (MCID) image analysis system as described earlier (39). The data, which are presented as mean ± S.E., were analyzed using one-way analysis of variance followed by Newman-Keuls post hoc analysis with significance set at p < 0.05.
Real-time Polymerase Chain Reaction
Quantitative real-time polymerase chain reaction (PCR) was performed as described elsewhere (40). In brief, cellular RNA was first extracted with the RNeasy minikit and then reverse-transcribed using reverse transcriptase as described earlier (41). Quantitative PCR was then carried out using SYBR Green real-time PCR master mix according to the manufacturer's instructions. Mouse primer sequences used in the study were as follows: cathepsin D (sense, 5′-CGCAGTGTTTCACAGTCGT-3′; antisense, 5′-TGAGCCGTAGTGGATGTCAA-3′); GAPDH (sense, 5′-TGAAGCAGGCATCTGAGGG-3′; antisense, 5′-CGAAGGTGGAAGAGTGGGAG-3′); and β-actin (sense, 5′-GGGAAATCGGTGACATT-3′; antisense, 5′-GCGGCAGTGGCCATCTC-3′). All primers were purchased from IDT (San Jose, CA). PCR was performed with the MyiQ single-color real-time PCR detection system (Bio-Rad), and conditions used were as follows: 95 °C for 10 min, 40 cycles at 95 °C for 30 s, and 60 °C for 1 min. The relative quantitative values of cathepsin D expression were normalized by the expression levels of β-actin as well as GAPDH genes. The expression levels of cathepsin D mRNA are presented as fold increase to the mean value of the control.
Activity Assay of Cathepsin D
Control, U18666A- and cathepsin D-treated cultured neurons from various experiments were homogenized in assay buffer (0.5 m Tris, 1 μm MgCl2, 0.1% BSA, pH 7.4) on ice and then centrifuged (12,000 × g, 4 °C, 10 min) to yield the supernatant. The protein amount was equalized after a protein assay with the BCA assay kit, and then cathepsin D activity was measured using the fluorogenic immunocapture activity assay kit as described earlier (30).
Confocal Microscopy with LysoSensor
To evaluate endosomal/lysosomal changes after a 24-h treatment with 5 μg/ml U18666A, control and treated hippocampal cultured neurons were exposed to the pH-sensitive endosomal dye, LysoSensor Yellow/Blue DND-160, at 5 μm for 10 min (42). The fluorescent signal was measured with excitation at 360 nm and emission at 420 nm and then visualized under a Zeiss confocal microscope (LSM510). The endosome/lysosome volumes were calculated using Nikon NIS-3.0 (NIS-Element Advanced Research) software.
Subcellular Fractionation
Hippocampal cultured neurons from various experimental paradigms were homogenized, fractionated using the Qproteome cell compartment kit, and then processed for immunoblotting with anti-cathepsin D (1:200) or anti-cytochrome c (1:1000) antibodies. Membranes were then washed with TBST, incubated with appropriate secondary antibodies (1:5000), and visualized using an ECL detection kit. Blots were subsequently reprobed with anti-N cadherin (1:200), anti-GAPDH (1:1000), or anti-histone (1:1000) antisera as described earlier (30).
Immunostaining
To assess the purity of our neuronal cultures, primary cultured neurons after 6 days of plating were fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100, and then incubated overnight with anti-MAP2 (1:500), anti-glial fibrillary acidic protein (1:1000), and anti-Iba1 (1:5000) antisera. In a parallel series of experiments, hippocampal neurons following treatment with or without 150 nm cathepsin D for 24 h were incubated overnight with an anti-AIF antibody (1:750). The cells were then exposed to appropriate fluorescein isothiocyanate or Texas Red-conjugated secondary antibody, washed, and stained with or without DAPI. The presence of neurons, astrocytes, and microglia in our cultured conditions was evaluated using a Zeiss Axioskop-2 microscope. Additionally, the localization of AIF in the cytoplasm versus nucleus was evaluated in 700–750 cells from control and cathepsin D-treated cultured neurons using a Zeiss Axioskop-2 epifluorescence microscope. This experiment was repeated in two separate cultures, and the data, which are presented as mean ± S.E., were analyzed using Student's t test followed by Mann-Whitman post hoc analysis with significance set at p < 0.05.
RESULTS
U18666A-induced Toxicity in Primary Neuronal Cultures
Mouse primary hippocampal neuronal cultures that contain <10% glial cells (such as astrocytes and microglia; see supplemental Fig. 1) are found to be vulnerable to U18666A-induced toxicity, as evident by a reduction in MTT values and concurrent increase in apoptotic nuclei following Hoechst 33258 nuclear staining and Live/Dead assays (Fig. 1, A–H). A concentration-dependent (0.1–50 μg/ml) effect of U18666A over a 24-h treatment revealed a significant decrease in MTT values from a dose of 1 μg/ml upward. Exposure of neuronal cultures to 5 μg/ml U18666A decreased MTT values in a time-dependent (6–96 h) manner, with a marked reduction in cell viability observed after 24 h of treatment with the drug (Fig. 1, A and B). The toxicity of U18666A toward neuronal cells was supported by a concentration- and time-dependent increased number of pycnotic nuclei as evidenced by Hoechst 33258 staining (Fig. 1, C–F). The Live/Dead assay also revealed that exposure to 5 μg/ml U18666A over 24 h can induce a marked increase in the number of dead neuronal cells (Fig. 1, G and H). Accompanying the toxicity, filipin-labeled cholesterol accumulation was increased in cultured neurons following a 24-h treatment with 5 μg/ml U18666A (Fig. 1, I and J).
FIGURE 1.

Increased U18666A-induced neurotoxicity and cholesterol accumulation in mouse primary hippocampal neurons. After 5 days of plating, cultured neurons were treated with 0.1–50 μg/ml U18666A for 24 h (A) or with 5 μg/ml U18666A for 6–96 h (B). MTT values were significantly attenuated in concentration-dependent (A) and time-dependent (B) manners in U18666A-treated cultures compared with mock-treated cultures. C and D, increase in the number of Hoechst 33258-labeled pycnotic nuclei following treatment with 0.1–50 μg/ml U18666A for 24 h (C) or with 5 μg/ml U18666A for 6–96 h (D). E and F, presence of condensed and/or fragmented nuclei (arrows) in control and U18666A-treated neuronal cultures. G and H, Live/Dead assay showing calcein AM staining in living neurons (green, G; arrowheads) and EthD-1-labeled dead neurons following treatment with 5 μg/ml U18666A for 24 h (red, H; arrows). I and J represent cholesterol accumulation, as evident by filipin staining in control and U18666A-treated neurons. All results, which are presented as means ± S.E. (error bars), were obtained from three separate experiments, each performed in triplicate. UA, U18666A; Ctrl, control. Scale bar, 25 μm. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Cathepsin D Level/Activity in U18666A-treated Neuronal Cultures
To examine the possible involvement of cathepsin D in U18666A-induced toxicity, we evaluated peptide and mRNA levels as well as activity of the enzyme in control and U18666A-treated cultured neurons. Our results showed that the ratio of mature to immature cathepsin D increased until 24 h and then declined significantly by 72 h post-treatment (Fig. 2, A and B). Intriguingly, cathepsin D mRNA levels were found to be markedly increased at 24, 48, and 72 h following treatment with 5 μg/ml U18666A (Fig. 2C). These results raise the possibility of either an increased turnover or a release of the enzyme in U18666A-treated primary neurons. Indeed, the activity of cathepsin D in these neurons was increased dramatically compared with untreated cultures. Following a 24- and 48-h treatment with 5 μg/ml U18666A, enzyme activity was increased 2.5- and 4-fold over control levels, respectively (Fig. 2D). Moreover, labeling with the LysoSensor dye DND-160 revealed that U18666A-treated cultured neurons have a larger lysosomal/endosomal vesicles compared with the control neurons (Fig. 2, E–G).
FIGURE 2.

Cathepsin D regulation in U18666A-treated neuronal cultures. A–D, cathepsin D immunoblot and quantification (A and B), mRNA levels (C), and enzyme activity (D) in hippocampal neuronal cultures treated with 5 μg/ml U18666A for different periods of time. These results showed a decrease in the ratio of mature to immature cathepsin D and increased activity and mRNA levels of cathepsin D compared with control cultures. E–G, photomicrographs (E and F) and the histogram (G) showing larger endosomal/lysosomal vesicles labeled with LysoSensor dye DND-160 in hippocampal neurons treated for 24 h with U18666A (F, arrows) compared with control cultures (E). UA, UA18666A; Ctrl, control. Scale bar, 40 μm. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.E.
A number of earlier studies have shown that an altered level and/or activity of cathepsin D may represent an adaptive response to overcome abnormal protein accumulation, or it may lead to loss of cell viability. In general, an increased enzyme activity within lysosomes or limited release of enzymes into the cytosol can prevent sublethal damage (5, 39, 43), whereas lysosomal leakage, leading to sustained release of the enzymes into the cytosol, can induce cell death directly and/or indirectly via cytochrome c release from mitochondria (6, 7, 44). Once in the cytosol, cytochrome c associates with Apaf-1, forming an apoptosome complex that, in the presence of dATP/ATP, is capable of activating caspase-9 followed by caspase-3, leading to cell death (5, 26, 45). Lysosomal enzymes can induce mitochondria permeability either by activating phospholipase A2 (46) or by cleaving the Bcl-2 family member Bid, which in its truncated form translocates to mitochondria, resulting in Bax/Bak activation (9, 47). There is also evidence that damage to mitochondria may cause release of other factors, such as AIF, which can trigger cell death in a caspase-independent manner following its translocation to the nucleus (48). To determine the role of cathepsin D in U18666A-induced toxicity, cultured neurons were treated with 5 μg/ml U18666A for different time periods (i.e. 6, 24, and 48 h) and then submitted to a subcellular fractionation followed by Western blotting. Our results clearly revealed that cytosolic cathepsin D levels were not altered at 6 h but increased at 24 and 48 h following treatment with U18666A compared with untreated control cultures (Fig. 3, A–E). In parallel, cytosolic levels of cytochrome c were also enhanced in 24- and 48-h U18666A-treated neurons compared with untreated neurons (Fig. 3, A–E). Additionally, the active forms of caspase-9 and caspase-3 (17 kDa), the downstream effectors, were significantly increased time-dependently from 6 and 12 h onward, respectively, in cultured cells treated with 5 μg/ml U18666A (Fig. 3F).
FIGURE 3.

Cathepsin D, cytochrome c, and cleaved caspase-3 in U18666A-treated neuronal cultures. A and B, immunoblots (A) and quantification (B) depicting subcellular distribution of cathepsin D and cytochrome c and their relative alterations (B) in the mouse primary neurons treated with 5 μg/ml U18666A for 24 h. Note the higher cytosolic levels of cathepsin D and cytochrome c in the treated hippocampal neurons compared with controls. C–E, immunoblots (C) and quantification (D and E) depicting subcellular levels of cathepsin D and cytochrome c in the mouse primary neurons treated with 5 μg/ml U18666A for 6 and 48 h. Note the higher cytosolic levels of cathepsin D and cytochrome c at 48 h but not at 6 h following treatment with U18666A. F, immunoblots showing the relative increase in caspase-9 and cleaved caspase-3 levels following treatment with 5 μg/ml U18666A for 6–72 h. UA, UA18666A; Ctrl, control; Cyto, cytoplasmic; Memb, membrane; Nuc, nuclear. All results, which are presented as means ± S.E. (error bars), were obtained from three separate experiments. *, p < 0.05; **, p < 0.01.
Cathepsin D Inhibitor- and U18666A-treated Neuronal Cultures
To establish whether increased cathepsin D activity was a cause or a consequence of cell death, primary neurons were treated with various concentrations (1–50 μm) of the cathepsin D inhibitor pepstatin A either concurrently or 24 h prior to exposure with 5 μg/ml U18666A, and cell viability was assessed using an MTT assay, Hoechst 33258 staining, or a Live/Dead assay. The concentrations of pepstatin A used were based on earlier data (49). Our results showed that only 10 and 20 μm pepstatin A, and not lower or higher concentrations, can significantly protect neuronal cultures against U18666A-induced toxicity (Fig. 4, A–E) and can also attenuate the corresponding enzyme activity (Fig. 4F). Furthermore, the protective effect of pepstatin A was equivalent when the cells were co- or pretreated with the inhibitor (Fig. 4A). It is also evident that pepstatin A treatment partially reversed the increase in cytosolic cathepsin D and cytochrome c levels (Fig. 4G) as well as activation of caspase-9 and caspase-3 in U18666A-treated neuronal cultures (Fig. 4, H and I).
FIGURE 4.

Prevention of U18666A-induced neuronal toxicity by cathepsin D inhibitor. A–E, protective effects of cathepsin D inhibitor pepstatin A against U18666A-mediated toxicity in primary hippocampal neurons as measured using the MTT assay (A), Hoechst 33258 labeling (B), and the Live/Dead assay (C–E; living neurons are marked by arrowheads, and dead neurons are marked by arrows). Note that pre- or co-treatment of hippocampal cultures with 10 and 20 μm pepstatin A can significantly protect the neurons against 5 μg/ml UA18666A-mediated toxicity (A). F, histogram showing that treatment of hippocampal neurons with 20 μm pepstatin A can attenuate UA18666A-induced activation of cathepsin D enzyme activity. G, immunoblots depicting pepstatin A treatment can partially reverse the relative increase in the cytosolic cathepsin D and cytochrome c levels in UA18666A-treated cultured neurons. H and I, immunoblots showing pepstatin A treatment can partially reverse the levels of caspase-9 (H) and cleaved caspase-3 (I) in U18666A-treated cultured neurons. All results, which are presented as means ± S.E. (error bars), were obtained from three separate experiments. Ctrl, control; UA, U18666A; Pep A, pepstatin A; Cyto, cytoplasmic; Memb, membrane; Nuc, nuclear. Scale bar, 25 μm. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Effects of Experimental Modulation of Cathepsin D on Cell Viability
To further validate the role of cathepsin D in U18666A-mediated apoptosis, we evaluated whether knocking down its expression using siRNA could protect neuronal cells against U18666A toxicity. Because transfection of the hippocampal neuronal culture by electroporation or Lipofectamine 2000 did not yield significant siRNA incorporation to observe knockdown (data not shown), we used N2a cells, which are known to be vulnerable to U18666A treatment (28, 50). Moreover, these cells showed similar cholesterol accumulation and cell death when treated with 3 μg/ml U18666A for 24 h as that observed in primary neuronal cultures (supplemental Fig. 2).
In N2a cells, cathepsin D-targeting siRNAs efficiently decreased the levels of immature cathepsin D at 24 h and both immature and mature cathepsin D at 48 h following U18666A treatment compared with non-targeting scrambled siRNAs (Fig. 5, A and B). After 24 h of transfection, N2a cells were treated with or without 3 μg/ml U18666A for an additional 24 h before cell viability was evaluated using the MTT assay. Cathepsin D knockdown significantly prevented U18666A-induced toxicity compared with cells transfected with the scrambled siRNA (Fig. 5C), thus suggesting a critical role for the lysosomal enzyme in cell death induction. To complement these data, N2a cells were transfected with plasmid containing cathepsin D (Fig. 5D), and cell viability was assessed following treatment with or without 3 μg/ml U18666A (Fig. 5E). Our results indicate that both cathepsin D overexpression and U18666A treatment of empty vector-transfected cells significantly reduce (i.e. by ∼25%) cell viability. When cathepsin D-overexpressing cells were treated with U18666A, there was a further, although not significant, decrease in cell viability compared with cells transfected with an empty vector. This is also evident in the Live/Dead assay, which showed that cathepsin D-overexpressing cells are more vulnerable to U18666A treatment than cells transfected with an empty vector (Fig. 5, F–I).
FIGURE 5.

Cathepsin D and cell toxicity in cultured N2a and human fibroblasts cells. A and B, immunoblots (A) and quantifications (B) showing the decreased levels of both immature (Imm) and mature (Mat) cathepsin D after transfection of N2a cells with cathepsin D siRNA. C, cathepsin D siRNA prevents toxicity induced by 3 μg/ml U16888A in N2a cells compared with cells treated with scrambled siRNA, as detected using the MTT assay. D, immunoblot showing the increased cathepsin D levels after N2a cells were transiently transfected with a cathepsin D-expressing vector compared with an empty vector. E–I, enhanced cathepsin D levels following transfection increased cell death when treated with 3 μg/ml U18666A as evident by the MTT assay (E) and Live/Dead assay (F–I), which showed reduced cell density and increased EthD-1-labeled dead cells (red-colored cells). J, MTT assay showing fibroblasts from NPC patients were significantly more vulnerable to staurosporine-induced toxicity than normal control fibroblasts. All results, which are presented as means ± S.E. (error bars), were obtained from at least three separate experiments. Scale bar, 25 μm. Ctrl, control; Scram, scrambled siRNA. **, p < 0.01; ***, p < 0.001.
Effects of Staurosporine on Human NPC Fibroblasts
Staurosporine, a broad-spectrum protein kinase inhibitor, has been used widely to induce apoptosis in a variety of cells, including human fibroblasts (51). It has been demonstrated that cathepsin D acts as a positive mediator of staurosporine-induced apoptosis, acting upstream of cytochrome c release and caspase activation in human fibroblasts (29). Our results revealed that viability of NPC fibroblasts did not differ from control fibroblasts under normal conditions, but they were more susceptible to staurosporine-induced toxicity than control fibroblasts (Fig. 5J). This is consistent with earlier reports, which showed that a lack of functional NPC1 protein may not directly influence cell viability (52, 53) but can render them more vulnerable to toxic insults than control cells (52). The present results, together with the evidence of altered distribution of cathepsin D in the cerebellar Purkinje cells of the brain from NPC patients (21), suggest a potential role for this aspartic protease in disease pathology.
Extracellular Release of Cathepsin D by U18666A-treated Neuronal Cultures
Unlike cytosolic levels, very little is currently known about whether extracellular or released cathepsin D can trigger degeneration of neurons. Our results from primary hippocampal neurons revealed an increase in the steady state cathepsin D levels in the supernatants of the U18666A-treated neurons compared with untreated cultures (Fig. 6, A and B). The immature cathepsin D, however, was not evident in the conditioned medium of treated cultured neurons. To define the role of extracellular cathepsin D in the loss of neurons, hippocampal neurons were cultured for 24 h with or without 5 μg/ml U18666A before replacing with the fresh medium. Conditioned media, collected after an additional 24 h in culture, were applied to untreated neurons for 12 or 24 h, and cell viability was measured using the MTT assay (Fig. 6C). It is apparent from our results that conditioned media from U18666A-treated neuronal cultures, but not from untreated cultures, were toxic to neurons (Fig. 6D). To substantiate the role of extracellular cathepsin D in the death of neurons, cathepsin D levels were depleted from the media using cathepsin D antibody-coupled beads, which markedly attenuated toxicity induced by U18666A-treated conditioned media (Fig. 6E). Taken together, these results suggest that extracellular cathepsin D from U18666A-treated cultured neurons can induce toxicity.
FIGURE 6.

U18666A treatment enhanced extracellular levels of cathepsin D in hippocampal neuronal cultures. A and B, immunoblots (A) and respective quantification (B) showing elevated cathepsin D levels in the conditioned medium of the neurons treated with 5 μg/ml U18666A for 12–72 h compared with respective control cultures. C, illustration of the experimental paradigm followed to study the influence of the conditioned medium containing extracellular cathepsin D on the viability of hippocampal neurons. D, toxicity induced by conditioned medium obtained from neurons treated for 24 h with or without 5 μg/ml U18666A. As evident from histograms, conditioned medium from UA18666A-treated neurons, but not from untreated neurons, can time-dependently reduce viability of hippocampal neurons as detected by the MTT assay. E, MTT assay revealed that toxicity induced by UA18666A-treated conditioned medium was partially reversed after depletion of cathepsin D in the media using cathepsin D antibody-coupled beads. All results, which are presented as means ± S.E. (error bars), were obtained from three separate experiments. Cat D, cathepsin D; Ctrl, control; sup, supernatant; UA, U18666A. **, p < 0.01; ***, p < 0.001.
To further validate the toxic potential of extracellular cathepsin D, hippocampal cultured neurons were exposed to 25–150 nm exogenous cathepsin D, which was found to induce toxicity in a concentration-dependent manner over a 24-h period (Fig. 7A). This is supported by a parallel increase in the number of EthD-1-positive dead neurons in the cathepsin D-treated cultures (Fig. 7, B and C). We observed that activity of the exogenous cathepsin D in the conditioned media decreased with time possibly due to higher pH of the media (Fig. 7D). Additionally, treatment with 200 mm sucrose, a potent inhibitor of clathrin-mediated endocytosis (54), was found to attenuate cathepsin D-induced toxicity, thus suggesting that uptake of exogenous cathepsin D is required to trigger death of cultured neurons (Fig. 7E). As a first step to understand the mechanisms by which exogenous cathepsin D can trigger cell death, we measured the activity of the enzyme in treated cultures, but no alteration was evident compared with untreated cultures, suggesting that cell death is not accompanied by an increase of cathepsin D activity (data not shown). This is supported by the evidence that pepstatin A was unable to protect the neurons against toxicity (Fig. 7F). Additionally, we did not observe any changes in the levels of caspase-9, cleaved caspase-3, or AIF in cathepsin D-treated cultured neurons (Fig. 7G). The nuclear distribution of AIF showed an increasing trend in cathepsin D-treated neurons, but this was not found to be significant compared with control neurons (Fig. 7, H–J). Interestingly, we observed a significant increase in the levels of LC3-II, p62, Atg5, and Beclin-1, which are known to be associated with autophagy-induced cell death (Fig. 8, A–E) (55–58). The involvement of the autophagic pathway is partly substantiated by our data, which showed that treatment of cultured neurons with 3-MA, a selective inhibitor of LC3-II (59, 60), not only attenuated the increased levels of LC3-II, p62, Atg5, and Beclin-1 (Fig. 8, A–E) but also markedly protected the hippocampal neurons against cathepsin D-induced toxicity (Fig. 8F). The effects of exogenous cathepsin D were further validated using mouse m5-7 fibroblast cells that are regulated by the Tet-off system (34). In the presence of doxycycline, these cells exhibit reduced expression of Atg5 and are resistant to autophagy-induced cell death (61). Our results clearly showed that doxycyline treatment not only decreased Atg5 expression (Fig. 9A) but also rendered these cells partially resistant to cathepsin D-induced toxicity compared with control cells (Fig. 9B). Taken together, these results suggest that exogenous cathepsin D may induce cell death via autophagy.
FIGURE 7.

Exogenous cathepsin D triggered apoptosis-independent cell death. A–C, hippocampal cultured neurons showing a reduction in MTT value (A) and an increase in EthD-1-positive dead cells (arrows represent dead cells, and the arrowhead shows a live cell) following treatment with cathepsin D (C) compared with untreated control cultures (B). D, histogram showing decreased in exogenous cathepsin D enzyme activity in cultured medium. E, MTT assay showing that 200 mm sucrose treatment protected cultured hippocampal neurons against cathepsin D-induced toxicity. F, MTT assay showing that 10 μm pepstatin A was unable to protect cultured neurons against exogenous cathepsin D-induced toxicity. G, unaltered levels of caspase-9, cleaved caspase-3, and AIF after exposure of hippocampal cultured neurons to 25–150 nm cathepsin D for 24 h. H and I, photomicrographs (H and I) and histograms (J) showing that cathepsin D treatment did not alter the number of neurons exhibiting nuclear localization of AIF (arrowheads) compared with untreated control cultures. Cat D, cathepsin D; Ctrl, control; Pep A, pepstatin A. Scale bar, 25 μm. *, p < 0.05; **, p < 0.01. Error bars, S.E.
FIGURE 8.

Exogenous cathepsin D increased autophagy markers in cultured neurons. A–E, immunoblots (A) and quantifications (B–E) depicting increased levels of LC3-II, p62, Atg5, and Beclin-1 following treatment with cathepsin D and their reductions with 3-MA treatment in hippocampal cultured neurons. F, MTT assay showing that 3-MA treatment protected cultured hippocampal neurons against cathepsin D-induced toxicity. All results, which are presented as means ± S.E. (error bars), were obtained from three separate experiments. Cat D, cathepsin D; Ctrl, control. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 9.
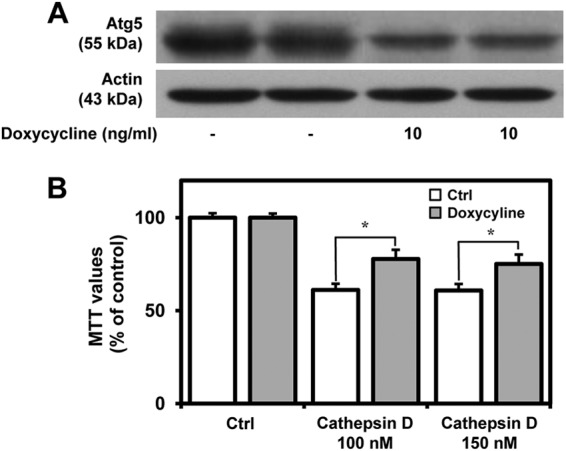
Depletion of Atg5 level partially protected the cells from exogenous cathepsin D-induced toxicity. A, immunoblot depicting Atg5 levels in mouse m5-7 fibroblast cells with or without doxycycline treatment. Note the reduction in Atg5 levels following doxycycline treatment. B, MTT assay showing that doxycycline-treated m5-7 cells are partially resistant to cathepsin D-induced toxicity compared with untreated m5-7 fibroblast cells. All results, which are presented as means ± S.E. (error bars), were obtained from three separate experiments. Ctrl, control. *, p < 0.05.
DISCUSSION
Using a variety of experimental approaches, the present study showed that increased intracellular and extracellular levels of cathepsin D following treatment with U18666A can trigger degeneration of neurons. Our results reveal that (i) U18666A-mediated toxicity in hippocampal neuronal cultures is accompanied by increased levels of cathepsin D mRNA, enzyme activity, and cytosolic levels of the peptide along with activation of caspase-9 and -3; (ii) cathepsin D inhibitor pepstatin A can protect neurons against toxicity by attenuating activation of a caspase-dependent pathway; (iii) down-regulation of cathepsin D levels prevented U18666A-induced cell death in N2a cells, whereas transient overexpression of the enzyme was sufficient to induce cell death; (iv) fibroblasts from NPC disease are more vulnerable to staurosporine-induced toxicity than normal, control fibroblasts; and (v) extracellular cathepsin D from U18666A-treated cultured neurons or application of exogenous enzyme can induce toxicity to hippocampal neurons by activating autophagy. Taken together, these results suggest that increased levels of cathepsin D in the cytosol or in the extracellular medium can be toxic to neurons. The evidence that pepstatin A can prevent U18666A-induced toxicity raises the possibility that cathepsin D inhibitors may be of therapeutic relevance in the treatment of NPC pathology.
The main biochemical manifestation of NPC pathology is the abnormal accumulation of free cholesterol and glycosphingolipids within the endosomal-lysosomal system in various brain regions, including cortex, hippocampus, and cerebellum (13, 14, 62, 63). The hydrophobic amine, U18666A (3-β-[2-(diethylamino) ethoxy]androst-5-en-17-one), is a well known class II amphiphile, which has been shown to trigger accumulation of cholesterol by inhibiting its intracellular transport, as observed in NPC pathology (22). It has been reported that U18666A treatment can decrease the cholesterol content of the endoplasmic reticulum and impede the movement of cholesterol between the lysosomal compartment and the plasma membrane and from the plasma membrane to other intracellular compartments (64, 65). Additionally, U18666A may act on the activity and/or synthesis of other proteins or lipids that facilitate cholesterol movement or alter cellular distribution of the NPC1 protein, thus causing accumulation of intracellular cholesterol (66–68). U18666A is therefore considered to be one of the best-characterized drugs to mimic the cellular effects of NPC in normal cells through dysfunction of lipid storage and inhibition of cholesterol movement (22, 64, 65).
Earlier studies have shown that U18666A can induce degeneration of cortical cultured neurons by triggering mitochondrial depolarization, increase production of reactive oxygen species and the occurrence of other oxidative events, such as lipid peroxidation, DNA oxidation, and protein oxidation, along with the activation of caspases and calpains (27, 28). Cotreatment of U18666A with cyclodextrin, which depletes cellular cholesterol, was able to rescue cortical neurons from cell death, suggesting that accumulation of cholesterol leads to U18666A-mediated neuronal apoptosis (28). More recently, fibroblasts exposed to a lower concentration of U18666A (0.5 μg/ml) have been somewhat protected against staurosporine- and cisplatin-induced toxicity, thus raising the possibility that class II amphilic amine, depending on the dose and time of exposure, can have a biphasic effect on cell viability, but the underlying mechanisms remain to be established (69).
In our study, we showed that U18666A treatment can cause degeneration of primary hippocampal neurons as well as N2a cells in a concentration-dependent manner (0.1–50 μg/ml) and was accompanied by intracellular accumulation of cholesterol and increased endosomal/lysosomal volumes, as seen in NPC pathology (20, 21). There was also an increase in cathepsin D mRNA levels and enzymatic activity, but the overall level of the cellular peptide was found to be decreased. Earlier studies reported that cathepsin D can have a survival or apoptotic effect depending on the subcellular localization and activity of the enzyme. If the levels and activity of the enzyme are increased within the lysosomes, it may represent an adaptive response to protect cells against toxic insults (39, 43, 70, 71), whereas an enhanced activity and cytosolic levels of the enzyme can lead to cell death either directly or indirectly via cytochrome c release from mitochondria (6–8). This phenomenon has been described under in vitro paradigms using a variety of toxic insults (29, 44, 73–77) and in some animal models of neurodegenerative disorders (78–80). The present study clearly showed that degeneration of neurons following U18666A treatment is accompanied by an increased activity and cytosolic levels of cathepsin D, as observed in toxicity induced by oxidative stress (81). This is supported by three distinct lines of evidence: (i) U18666A-induced toxicity was associated with increased levels of cytochrome c and activation of caspase-9 and caspase-3; (ii) cathepsin D inhibitor pepstatin A significantly protected hippocampal neurons against U18666A-induced toxicity by attenuating the aforesaid signaling mechanisms; and (iii) down-regulation of the cathepsin D level by siRNA treatment was found to protect cultured N2a cells against U18666A-mediated toxicity, whereas overexpression of the enzyme was sufficient to induce cell death. Collectively, these results suggest an important role for cathepsin D in U18666A-induced toxicity in cultured hippocampal neurons.
Increasing evidence now indicates that cathepsin D can be secreted in a variety of pathological conditions, and its effect can be mediated by an unidentified cell surface receptor (82). Aberrant secretion of cathepsin D is believed to be the consequence of defective acidification of intracellular organelles (83), but its implication in cell death mechanisms, if any, remains unclear. The present study showed that U18666A treatment, apart from increasing cellular mRNA and activity of cathepsin D, can enhance the level of the enzyme secreted into the conditioned media of hippocampal cultures. Additionally, we revealed that extracellular cathepsin D in the conditioned medium of U18666A-treated cultures can trigger degeneration in naive neuronal cultures and that removing the enzyme from the conditioned medium using agarose beads coupled to cathepsin D antibodies prevented neuronal death. Thus, extracellular cathepsin D secreted from U18666A-treated neurons can induce toxicity in a manner similar to that reported for cathepsin B, which is secreted following Aβ-mediated activation of microglia (84). This is further validated by the observation that exogenous cathepsin D can trigger degeneration of hippocampal cultured neurons in a concentration-dependent manner. Inhibition of endocytosis was found to attenuate toxicity, thus suggesting that internalization of cathepsin D may be involved in mediating death of cultured neurons. This effect, however, appears to be independent of intracellular cathepsin D because (i) the proteolytic activity of the enzyme was unaltered in the presence of exogenous cathepsin D, and (ii) cell viability was unaffected following treatment with pepstatin A. Additionally, exogenous cathepsin D did not activate the caspase-dependent pathway, as observed with U18666A-treated cultured neurons. This suggests that the underlying mechanisms of cell death may differ from that triggered by intracellular cathepsin D following treatment with U18666A. This led us to investigate the role of the caspase-independent pathway, where the involvement of the mitochondrial flavoprotein AIF has been well established (85, 86). Under normal conditions the protein is located in the inner mitochondrial membrane space (87), but upon activation, AIF is cleaved and translocated to the nucleus to participate in chromatin condensation and DNA fragmentation, leading to cell death (86, 88). Cultured neurons exposed to exogenous cathepsin D for 24 h exhibited neither altered level nor distribution of AIF, thus excluding the possible involvement of a caspase-independent pathway in the death of neurons. Interestingly, autophagy has been shown to promote cell death via a caspase-independent pathway under certain conditions (89, 90). Our results clearly showed that neurotoxicity induced by exogenous cathepsin D is accompanied by increased levels of autophagic markers, including LC3-II, p62, Atg5, and Beclin-1, in cultured hippocampal neurons. Because elevated LC3-II level can lead to cell death via a defective autophagosome clearance in some cases (57, 91, 92), it is possible that the loss of neurons observed following cathepsin D treatment is mediated via the autophagic pathway. This is supported by two distinct lines of evidence: (i) inhibition of autophagy by 3-MA treatment not only protected neurons against cathepsin D-induced toxicity but also attenuated the levels of LC3-II, p62, Atg5, and Beclin-1 in treated cultured neurons, and (ii) doxycycline-regulated m5-7 fibroblast cells are somewhat more resistant to cathepsin D-induced toxicity than control fibroblast cells. Thus, it appears that increased levels of intracellular and extracellular cathepsin D following U18666A treatment can trigger degeneration of neurons by activating different cell death mechanisms.
A number of studies have shown that dysfunction of the NPC1 protein can lead to intracellular accumulation of cholesterol and degeneration of neurons in selected regions of the brain, but the underlying mechanisms remain unclear (13, 62, 63). Some recent studies have indicated that deregulation of the phosphatidylinositol 3-kinase pathway (93), Aβ-mediated signaling cascades (20, 21), or increased levels/activity of cathepsins (24, 30) may contribute to the loss of neurons in NPC1 knock-out mouse brains. It has been shown that cathepsin D level/expression is increased not only in neurons but also in activated microglia located in close proximity to neurons in vulnerable regions of NPC1 knock-out mouse brains (21, 24–26, 30). Because microglia can play an active role in the loss of neurons (94, 95), it is possible that enhanced levels of cathepsin D in microglia following its release into the extracellular milieu may contribute to the degeneration of neurons, as shown in our study. Alternatively, increased levels/activity of cathepsin D in the microglia may be involved in the removal of neuronal debris because this enzyme is capable of degrading microtubule-associated proteins (96) and myelin basic proteins (97). On the other hand, given the evidence that cathepsin D can have a direct role in the degeneration of cultured neurons in U18666A-induced toxicity, which mimics NPC pathology under in vitro conditions, it is likely that the enzyme may also be involved in the loss of neurons in NPC1 knock-out mouse brains. This is partly supported by two lines of evidence: (i) attenuation of NPC pathology by pharmacological treatments (i.e. β-cyclodextrin or allopregnanolone) is accompanied by reduced expression/levels of the enzyme (98, 99), and (ii) NPC fibroblasts, as demonstrated in the present study, were more vulnerable than normal fibroblasts to toxicity induced by staurosporine, which is known to trigger cell death via a cathepsin D-mediated caspase-dependent pathway (29). However, it is of interest to note that NPC1 deficiency, unlike U18666A treatment, does not induce cell autonomous toxicity (52, 53) but can render cells more susceptible to toxic insults than normal control cells (52), as observed in the present study. Whether this relates to the differential effects of U18666A treatment versus lack of functional NPC1 protein remains to be established. Nevertheless, it would be of therapeutic relevance to determine whether attenuation of cathepsin D activity can ameliorate degeneration of neurons and associated pathology in NPC1 knock-out mice. This, however, may depend on two important factors: (i) ability of the inhibitor to cross the blood-brain barrier and (ii) reduction of cathepsin D activity to a degree sufficient to block degeneration of neurons without causing generalized disturbances of lysosomal operations. Although pepstatin A acts as a general inhibitor of aspartic peptidases, it remains the most potent inhibitor of cathepsin D activity known so far (100). Intraperitoneal administration of pepstatin A has been shown to be beneficial for experimental colitis and bone mineralization (101, 102), but it needs to be determined whether intracerebroventricular administration of the inhibitor can mitigate NPC pathology. Given the evidence that selective cathepsin B inhibitor can reduce brain Aβ levels/deposition and improve cognitive behavioral deficits in animal models of Alzheimer disease (72, 103), it is likely that the availability of rather selective and potent cathepsin D inhibitors that can cross the blood-brain barrier may provide a new therapeutic opportunity in the treatment of NPC pathology.
This work was supported by grants from the Canadian Institutes of Health Research (MOP-94375 and MOP-97837).

This article contains supplemental Figs. 1 and 2.
- NPC
- Niemann-Pick type C
- AIF
- apoptosis-inducing factor
- MTT
- 3-(4,5-dimethylthiozolyl)-2,5-diphenyltetrazolium bromide
- 3-MA
- 3-methyladenine.
REFERENCES
- 1. Turk B., Turk D., Turk V. (2000) Lysosomal cysteine proteases. More than scavengers. Biochim. Biophys. Acta 1477, 98–111 [DOI] [PubMed] [Google Scholar]
- 2. Benes P., Vetvicka V., Fusek M. (2008) Cathepsin D. Many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 68, 12–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zaidi N., Maurer A., Nieke S., Kalbacher H. (2008) Cathepsin D. A cellular roadmap. Biochem. Biophys. Res. Commun. 376, 5–9 [DOI] [PubMed] [Google Scholar]
- 4. Mullins C., Bonifacino J. S. (2001) The molecular machinery for lysosome biogenesis. BioEssays 23, 333–343 [DOI] [PubMed] [Google Scholar]
- 5. Bursch W. (2001) The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 8, 569–581 [DOI] [PubMed] [Google Scholar]
- 6. Turk B., Stoka V., Rozman-Pungercar J., Cirman T., Droga-Mazovec G., Oresić K., Turk V. (2002) Apoptotic pathways. Involvement of lysosomal proteases. Biol. Chem. 383, 1035–1044 [DOI] [PubMed] [Google Scholar]
- 7. Chwieralski C. E., Welte T., Bühling F. (2006) Cathepsin-regulated apoptosis. Apoptosis 11, 143–149 [DOI] [PubMed] [Google Scholar]
- 8. Boya P., Kroemer G. (2008) Lysosomal membrane permeabilization in cell death. Oncogene 27, 6434–6451 [DOI] [PubMed] [Google Scholar]
- 9. Heinrich M., Neumeyer J., Jakob M., Hallas C., Tchikov V., Winoto-Morbach S., Wickel M., Schneider-Brachert W., Trauzold A., Hethke A., Schütze S. (2004) Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ. 11, 550–563 [DOI] [PubMed] [Google Scholar]
- 10. Wu G. S., Saftig P., Peters C., El-Deiry W. S. (1998) Potential role for cathepsin D in p53-dependent tumor suppression and chemosensitivity. Oncogene 16, 2177–2183 [DOI] [PubMed] [Google Scholar]
- 11. Roberg K., Kågedal K., Ollinger K. (2002) Microinjection of cathepsin D induces caspase-dependent apoptosis in fibroblasts. Am. J. Pathol. 161, 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mukherjee S., Maxfield F. R. (2004) Lipid and cholesterol trafficking in NPC. Biochim. Biophys. Acta 1685, 28–37 [DOI] [PubMed] [Google Scholar]
- 13. Vanier M. T., Millat G. (2003) Niemann-Pick disease type C. Clin. Genet. 64, 269–281 [DOI] [PubMed] [Google Scholar]
- 14. Vance J. E. (2006) Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett. 580, 5518–5524 [DOI] [PubMed] [Google Scholar]
- 15. Pacheco C. D., Lieberman A. P. (2008) The pathogenesis of Niemann-Pick type C disease. A role for autophagy? Expert Rev. Mol. Med. 10, e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. López O. L., DeKosky S. T. (2003) [Neuropathology of Alzheimer's disease and mild cognitive impairment]. Rev. Neurol. 37, 155–163 [PubMed] [Google Scholar]
- 17. Selkoe D. J. (2008) Biochemistry and molecular biology of amyloid β-protein and the mechanism of Alzheimer's disease. Handb. Clin. Neurol. 89, 245–260 [DOI] [PubMed] [Google Scholar]
- 18. Auer I. A., Schmidt M. L., Lee V. M., Curry B., Suzuki K., Shin R. W., Pentchev P. G., Carstea E. D., Trojanowski J. Q. (1995) Paired helical filament τ (PHFτ) in Niemann-Pick type C disease is similar to PHFτ in Alzheimer's disease. Acta Neuropathol. 90, 547–551 [DOI] [PubMed] [Google Scholar]
- 19. Saito Y., Suzuki K., Nanba E., Yamamoto T., Ohno K., Murayama S. (2002) Niemann-Pick type C disease. Accelerated neurofibrillary tangle formation and amyloid β deposition associated with apolipoprotein E ϵ4 homozygosity. Ann. Neurol. 52, 351–355 [DOI] [PubMed] [Google Scholar]
- 20. Nixon R. A. (2004) Niemann-Pick type C disease and Alzheimer's disease. The APP-endosome connection fattens up. Am. J. Pathol. 164, 757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jin L. W., Shie F. S., Maezawa I., Vincent I., Bird T. (2004) Intracellular accumulation of amyloidogenic fragments of amyloid-β precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am. J. Pathol. 164, 975–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koh C. H., Cheung N. S. (2006) Cellular mechanism of U18666A-mediated apoptosis in cultured murine cortical neurons. Bridging Niemann-Pick disease type C and Alzheimer's disease. Cell. Signal. 18, 1844–1853 [DOI] [PubMed] [Google Scholar]
- 23. Tang Y., Li H., Liu J. P. (2010) Niemann-Pick disease type C. From molecule to clinic. Clin. Exp. Pharmacol. Physiol. 37, 132–140 [DOI] [PubMed] [Google Scholar]
- 24. Liao G., Yao Y., Liu J., Yu Z., Cheung S., Xie A., Liang X., Bi X. (2007) Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1−/− mouse brain. Am. J. Pathol. 171, 962–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. German D. C., Liang C. L., Song T., Yazdani U., Xie C., Dietschy J. M. (2002) Neurodegeneration in the Niemann-Pick C mouse. Glial involvement. Neuroscience 109, 437–450 [DOI] [PubMed] [Google Scholar]
- 26. Cheung N. S., Koh C. H., Bay B. H., Qi R. Z., Choy M. S., Li Q. T., Wong K. P., Whiteman M. (2004) Chronic exposure to U18666A induces apoptosis in cultured murine cortical neurons. Biochem. Biophys. Res. Commun. 315, 408–417 [DOI] [PubMed] [Google Scholar]
- 27. Huang Z., Hou Q., Cheung N. S., Li Q. T. (2006) Neuronal cell death caused by inhibition of intracellular cholesterol trafficking is caspase dependent and associated with activation of the mitochondrial apoptosis pathway. J. Neurochem. 97, 280–291 [DOI] [PubMed] [Google Scholar]
- 28. Koh C. H., Whiteman M., Li Q. X., Halliwell B., Jenner A. M., Wong B. S., Laughton K. M., Wenk M., Masters C. L., Beart P. M., Bernard O., Cheung N. S. (2006) Chronic exposure to U18666A is associated with oxidative stress in cultured murine cortical neurons. J. Neurochem. 98, 1278–1289 [DOI] [PubMed] [Google Scholar]
- 29. Johansson A. C., Steen H., Ollinger K., Roberg K. (2003) Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ. 10, 1253–1259 [DOI] [PubMed] [Google Scholar]
- 30. Amritraj A., Peake K., Kodam A., Salio C., Merighi A., Vance J. E., Kar S. (2009) Increased activity and altered subcellular distribution of lysosomal enzymes determine neuronal vulnerability in Niemann-Pick type C1-deficient mice. Am. J. Pathol. 175, 2540–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zheng W. H., Bastianetto S., Mennicken F., Ma W., Kar S. (2002) Amyloid β peptide induces τ phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 115, 201–211 [DOI] [PubMed] [Google Scholar]
- 32. Vetrivel K. S., Gong P., Bowen J. W., Cheng H., Chen Y., Carter M., Nguyen P. D., Placanica L., Wieland F. T., Li Y. M., Kounnas M. Z., Thinakaran G. (2007) Dual roles of the transmembrane protein p23/TMP21 in the modulation of amyloid precursor protein metabolism. Mol. Neurodegener. 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Umeda A., Fujita H., Kuronita T., Hirosako K., Himeno M., Tanaka Y. (2003) Distribution and trafficking of MPR300 is normal in cells with cholesterol accumulated in late endocytic compartments. Evidence for early endosome-to-TGN trafficking of MPR300. J. Lipid Res. 44, 1821–1832 [DOI] [PubMed] [Google Scholar]
- 34. Hosokawa N., Hara Y., Mizushima N. (2006) Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett. 580, 2623–2629 [DOI] [PubMed] [Google Scholar]
- 35. Kodam A., Vetrivel K. S., Thinakaran G., Kar S. (2008) Cellular distribution of γ-secretase subunit nicastrin in the developing and adult rat brains. Neurobiol. Aging 29, 724–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Song M. S., Rauw G., Baker G. B., Kar S. (2008) Memantine protects rat cortical cultured neurons against β-amyloid-induced toxicity by attenuating τ phosphorylation. Eur. J. Neurosci. 28, 1989–2002 [DOI] [PubMed] [Google Scholar]
- 37. Wei Z., Song M. S., MacTavish D., Jhamandas J. H., Kar S. (2008) Role of calpain and caspase in β-amyloid-induced cell death in rat primary septal cultured neurons. Neuropharmacology 54, 721–733 [DOI] [PubMed] [Google Scholar]
- 38. Börnig H., Geyer G. (1974) Staining of cholesterol with the fluorescent antibiotic “filipin”. Acta Histochem. 50, 110–115 [PubMed] [Google Scholar]
- 39. Hawkes C., Kabogo D., Amritraj A., Kar S. (2006) Up-regulation of cation-independent mannose 6-phosphate receptor and endosomal-lysosomal markers in surviving neurons after 192-IgG-saporin administrations into the adult rat brain. Am. J. Pathol. 169, 1140–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arikketh D., Nelson R., Vance J. E. (2008) Defining the importance of phosphatidylserine synthase-1 (PSS1). Unexpected viability of PSS1-deficient mice. J. Biol. Chem. 283, 12888–12897 [DOI] [PubMed] [Google Scholar]
- 41. Iliev A. I., Stringaris A. K., Nau R., Neumann H. (2004) Neuronal injury mediated via stimulation of microglial Toll-like receptor-9 (TLR9). FASEB J. 18, 412–414 [DOI] [PubMed] [Google Scholar]
- 42. Hurwitz S. J., Terashima M., Mizunuma N., Slapak C. A. (1997) Vesicular anthracycline accumulation in doxorubicin-selected U-937 cells. Participation of lysosomes. Blood 89, 3745–3754 [PubMed] [Google Scholar]
- 43. Bendiske J., Bahr B. A. (2003) Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis. An approach for slowing Alzheimer disease? J. Neuropathol. Exp. Neurol. 62, 451–463 [DOI] [PubMed] [Google Scholar]
- 44. Roberg K., Ollinger K. (1998) Oxidative stress causes relocation of the lysosomal enzyme cathepsin D with ensuing apoptosis in neonatal rat cardiomyocytes. Am. J. Pathol. 152, 1151–1156 [PMC free article] [PubMed] [Google Scholar]
- 45. Oberst A., Bender C., Green D. R. (2008) Living with death. The evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ. 15, 1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao M., Antunes F., Eaton J. W., Brunk U. T. (2003) Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur. J. Biochem. 270, 3778–3786 [DOI] [PubMed] [Google Scholar]
- 47. Droga-Mazovec G., Bojic L., Petelin A., Ivanova S., Romih R., Repnik U., Salvesen G. S., Stoka V., Turk V., Turk B. (2008) Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 283, 19140–19150 [DOI] [PubMed] [Google Scholar]
- 48. Cande C., Vahsen N., Métivier D., Tourrière H., Chebli K., Garrido C., Tazi J., Kroemer G. (2004) Regulation of cytoplasmic stress granules by apoptosis-inducing factor. J. Cell Sci. 117, 4461–4468 [DOI] [PubMed] [Google Scholar]
- 49. Figueiredo C., Pais T. F., Gomes J. R., Chatterjee S. (2008) Neuron-microglia cross-talk up-regulates neuronal FGF-2 expression which mediates neuroprotection against excitotoxicity via JNK1/2. J. Neurochem. 107, 73–85 [DOI] [PubMed] [Google Scholar]
- 50. Davis W., Jr. (2008) The cholesterol transport inhibitor U18666a regulates amyloid precursor protein metabolism and trafficking in N2aAPP “Swedish” cells. Curr. Alzheimer Res. 5, 448–456 [DOI] [PubMed] [Google Scholar]
- 51. Koh J. Y., Wie M. B., Gwag B. J., Sensi S. L., Canzoniero L. M., Demaro J., Csernansky C., Choi D. W. (1995) Staurosporine-induced neuronal apoptosis. Exp. Neurol. 135, 153–159 [DOI] [PubMed] [Google Scholar]
- 52. Zampieri S., Mellon S. H., Butters T. D., Nevyjel M., Covey D. F., Bembi B., Dardis A. (2009) Oxidative stress in NPC1 deficient cells. Protective effect of allopregnanolone. J. Cell Mol. Med. 13, 3786–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Peake K. B., Campenot R. B., Vance D. E., Vance J. E. (2011) Niemann-Pick Type C1 deficiency in microglia does not cause neuron death in vitro. Biochim. Biophys. Acta 1812, 1121–1129 [DOI] [PubMed] [Google Scholar]
- 54. Hibbert A. P., Kramer B. M., Miller F. D., Kaplan D. R. (2006) The localization, trafficking and retrograde transport of BDNF bound to p75NTR in sympathetic neurons. Mol. Cell Neurosci. 32, 387–402 [DOI] [PubMed] [Google Scholar]
- 55. Marino G., López-Otín C. (2004) Autophagy. Molecular mechanisms, physiological functions and relevance in human pathology. Cell Mol. Life Sci. 61, 1439–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tanida I., Ueno T., Kominami E. (2004) LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xilouri M., Stefanis L. (2010) Autophagy in the central nervous system. Implications for neurodegenerative disorders. CNS Neurol. Disord. Drug Targets 9, 701–719 [DOI] [PubMed] [Google Scholar]
- 58. Son J. H., Shim J. H., Kim K. H., Ha J. Y., Han J. Y. (2012) Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 44, 89–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rideout H. J., Lang-Rollin I., Stefanis L. (2004) Involvement of macroautophagy in the dissolution of neuronal inclusions. Int. J. Biochem. Cell Biol. 36, 2551–2562 [DOI] [PubMed] [Google Scholar]
- 60. Zheng X., Chu F., Mirkin B. L., Sudha T., Mousa S. A., Rebbaa A. (2008) Role of the proteolytic hierarchy between cathepsin L, cathepsin D, and caspase-3 in regulation of cellular susceptibility to apoptosis and autophagy. Biochim. Biophys. Acta 1783, 2294–2300 [DOI] [PubMed] [Google Scholar]
- 61. Shin J. H., Park S. J., Kim E. S., Jo Y. K., Hong J., Cho D. H. (2012) Sertindole, a potent antagonist at dopamine D2 receptors, induces autophagy by increasing reactive oxygen species in SH-SY5Y neuroblastoma cells. Biol. Pharm. Bull. 35, 1069–1075 [DOI] [PubMed] [Google Scholar]
- 62. Li H., Repa J. J., Valasek M. A., Beltroy E. P., Turley S. D., German D. C., Dietschy J. M. (2005) Molecular, anatomical, and biochemical events associated with neurodegeneration in mice with Niemann-Pick type C disease. J. Neuropathol. Exp. Neurol. 64, 323–333 [DOI] [PubMed] [Google Scholar]
- 63. German D. C., Quintero E. M., Liang C. L., Ng B., Punia S., Xie C., Dietschy J. M. (2001) Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J. Comp. Neurol. 433, 415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lange Y., Ye J., Rigney M., Steck T. (2000) Cholesterol movement in Niemann-Pick type C cells and in cells treated with amphiphiles. J. Biol. Chem. 275, 17468–17475 [DOI] [PubMed] [Google Scholar]
- 65. Liscum L., Sturley S. L. (2004) Intracellular trafficking of Niemann-Pick C proteins 1 and 2. Obligate components of subcellular lipid transport. Biochim. Biophys. Acta 1685, 22–27 [DOI] [PubMed] [Google Scholar]
- 66. Härmälä A. S., Pörn M. I., Mattjus P., Slotte J. P. (1994) Cholesterol transport from plasma membranes to intracellular membranes is inhibited by 3β-[2-(diethylamino)ethoxy]androst-5-en-17-one. Biochim. Biophys. Acta 1211, 317–325 [DOI] [PubMed] [Google Scholar]
- 67. Underwood K. W., Jacobs N. L., Howley A., Liscum L. (1998) Evidence for a cholesterol transport pathway from lysosomes to endoplasmic reticulum that is independent of the plasma membrane. J. Biol. Chem. 273, 4266–4274 [DOI] [PubMed] [Google Scholar]
- 68. Lange Y., Ye J., Rigney M., Steck T. L. (2002) Dynamics of lysosomal cholesterol in Niemann-Pick type C and normal human fibroblasts. J. Lipid Res. 43, 198–204 [PubMed] [Google Scholar]
- 69. Appelqvist H., Nilsson C., Garner B., Brown A. J., Kågedal K., Ollinger K. (2011) Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. Am. J. Pathol. 178, 629–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Barlow C., Ribaut-Barassin C., Zwingman T. A., Pope A. J., Brown K. D., Owens J. W., Larson D., Harrington E. A., Haeberle A. M., Mariani J., Eckhaus M., Herrup K., Bailly Y., Wynshaw-Boris A. (2000) ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc. Natl. Acad. Sci. U.S.A. 97, 871–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Butler D., Hwang J., Estick C., Nishiyama A., Kumar S. S., Baveghems C., Young-Oxendine H. B., Wisniewski M. L., Charalambides A., Bahr B. A. (2011) Protective effects of positive lysosomal modulation in Alzheimer's disease transgenic mouse models. PLoS One 6, e20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hook V. Y., Kindy M., Hook G. (2008) Inhibitors of cathepsin B improve memory and reduce β-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, β-secretase site of the amyloid precursor protein. J. Biol. Chem. 283, 7745–7753 [DOI] [PubMed] [Google Scholar]
- 73. Venè R., Arena G., Poggi A., D'Arrigo C., Mormino M., Noonan D. M., Albini A., Tosetti F. (2007) Novel cell death pathways induced by N-(4-hydroxyphenyl)retinamide. Therapeutic implications. Mol. Cancer Ther. 6, 286–298 [DOI] [PubMed] [Google Scholar]
- 74. Gowran A., Campbell V. A. (2008) A role for p53 in the regulation of lysosomal permeability by δ9-tetrahydrocannabinol in rat cortical neurones. Implications for neurodegeneration. J. Neurochem. 105, 1513–1524 [DOI] [PubMed] [Google Scholar]
- 75. Tofighi R., Johansson C., Goldoni M., Ibrahim W. N., Gogvadze V., Mutti A., Ceccatelli S. (2011) Hippocampal neurons exposed to the environmental contaminants methylmercury and polychlorinated biphenyls undergo cell death via parallel activation of calpains and lysosomal proteases. Neurotox. Res. 19, 183–194 [DOI] [PubMed] [Google Scholar]
- 76. Ditaranto K., Tekirian T. L., Yang A. J. (2001) Lysosomal membrane damage in soluble Aβ-mediated cell death in Alzheimer's disease. Neurobiol. Dis. 8, 19–31 [DOI] [PubMed] [Google Scholar]
- 77. Yang A. J., Chandswangbhuvana D., Margol L., Glabe C. G. (1998) Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Aβ1–42 pathogenesis. J. Neurosci. Res. 52, 691–698 [DOI] [PubMed] [Google Scholar]
- 78. Yamashima T., Kohda Y., Tsuchiya K., Ueno T., Yamashita J., Yoshioka T., Kominami E. (1998) Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074. A novel strategy for neuroprotection based on “calpain-cathepsin hypothesis”. Eur. J. Neurosci. 10, 1723–1733 [DOI] [PubMed] [Google Scholar]
- 79. Vitner E. B., Dekel H., Zigdon H., Shachar T., Farfel-Becker T., Eilam R., Karlsson S., Futerman A. H. (2010) Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Hum. Mol. Genet. 19, 3583–3590 [DOI] [PubMed] [Google Scholar]
- 80. Ceccariglia S., D'Altocolle A., Del Fa' A., Pizzolante F., Caccia E., Michetti F., Gangitano C. (2011) Cathepsin D plays a crucial role in the trimethyltin-induced hippocampal neurodegeneration process. Neuroscience 174, 160–170 [DOI] [PubMed] [Google Scholar]
- 81. Castino R., Bellio N., Nicotra G., Follo C., Trincheri N. F., Isidoro C. (2007) Cathepsin D-Bax death pathway in oxidative stressed neuroblastoma cells. Free Radic. Biol. Med. 42, 1305–1316 [DOI] [PubMed] [Google Scholar]
- 82. Fusek M., Vetvicka V. (2005) Dual role of cathepsin D. Ligand and protease. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech. Repub. 149, 43–50 [DOI] [PubMed] [Google Scholar]
- 83. Kokkonen N., Rivinoja A., Kauppila A., Suokas M., Kellokumpu I., Kellokumpu S. (2004) Defective acidification of intracellular organelles results in aberrant secretion of cathepsin D in cancer cells. J. Biol. Chem. 279, 39982–39988 [DOI] [PubMed] [Google Scholar]
- 84. Gan L., Ye S., Chu A., Anton K., Yi S., Vincent V. A., von Schack D., Chin D., Murray J., Lohr S., Patthy L., Gonzalez-Zulueta M., Nikolich K., Urfer R. (2004) Identification of cathepsin B as a mediator of neuronal death induced by Aβ-activated microglial cells using a functional genomics approach. J. Biol. Chem. 279, 5565–5572 [DOI] [PubMed] [Google Scholar]
- 85. Joza N., Susin S. A., Daugas E., Stanford W. L., Cho S. K., Li C. Y., Sasaki T., Elia A. J., Cheng H. Y., Ravagnan L., Ferri K. F., Zamzami N., Wakeham A., Hakem R., Yoshida H., Kong Y. Y., Mak T. W., Zúñiga-Pflücker J. C., Kroemer G., Penninger J. M. (2001) Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410, 549–554 [DOI] [PubMed] [Google Scholar]
- 86. Krantic S., Mechawar N., Reix S., Quirion R. (2007) Apoptosis-inducing factor. A matter of neuron life and death. Prog. Neurobiol. 81, 179–196 [DOI] [PubMed] [Google Scholar]
- 87. Otera H., Ohsakaya S., Nagaura Z., Ishihara N., Mihara K. (2005) Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 24, 1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Susin S. A., Daugas E., Ravagnan L., Samejima K., Zamzami N., Loeffler M., Costantini P., Ferri K. F., Irinopoulou T., Prévost M. C., Brothers G., Mak T. W., Penninger J., Earnshaw W. C., Kroemer G. (2000) Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 192, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Denton D., Shravage B., Simin R., Mills K., Berry D. L., Baehrecke E. H., Kumar S. (2009) Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr. Biol. 19, 1741–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zaidi A. U., McDonough J. S., Klocke B. J., Latham C. B., Korsmeyer S. J., Flavell R. A., Schmidt R. E., Roth K. A. (2001) Chloroquine-induced neuronal cell death is p53 and Bcl-2 family-dependent but caspase-independent. J. Neuropathol. Exp. Neurol. 60, 937–945 [DOI] [PubMed] [Google Scholar]
- 91. Chen Y., Klionsky D. J. (2011) The regulation of autophagy. Unanswered questions. J. Cell Sci. 124, 161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ferguson C. J., Lenk G. M., Meisler M. H. (2009) Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum. Mol. Genet. 18, 4868–4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bi X., Liu J., Yao Y., Baudry M., Lynch G. (2005) Deregulation of the phosphatidylinositol-3 kinase signaling cascade is associated with neurodegeneration in Npc1−/− mouse brain. Am. J. Pathol. 167, 1081–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wada R., Tifft C. J., Proia R. L. (2000) Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. U.S.A. 97, 10954–10959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Flavin M. P., Coughlin K., Ho L. T. (1997) Soluble macrophage factors trigger apoptosis in cultured hippocampal neurons. Neuroscience 80, 437–448 [DOI] [PubMed] [Google Scholar]
- 96. Matus A., Green G. D. (1987) Age-related increase in a cathepsin D like protease that degrades brain microtubule-associated proteins. Biochemistry 26, 8083–8086 [DOI] [PubMed] [Google Scholar]
- 97. Williams K. R., Williams N. D., Konigsberg W., Yu R. K. (1986) Acidic lipids enhance cathepsin D cleavage of the myelin basic protein. J. Neurosci. Res. 15, 137–145 [DOI] [PubMed] [Google Scholar]
- 98. Liao G., Cheung S., Galeano J., Ji A. X., Qin Q., Bi X. (2009) Allopregnanolone treatment delays cholesterol accumulation and reduces autophagic/lysosomal dysfunction and inflammation in Npc1−/− mouse brain. Brain Res. 1270, 140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Xie X., Brown M. S., Shelton J. M., Richardson J. A., Goldstein J. L., Liang G. (2011) Amino acid substitution in NPC1 that abolishes cholesterol binding reproduces phenotype of complete NPC1 deficiency in mice. Proc. Natl. Acad. Sci. U.S.A. 108, 15330–15335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Dash C., Kulkarni A., Dunn B., Rao M. (2003) Aspartic peptidase inhibitors. Implications in drug development. Crit. Rev. Biochem. Mol. Biol. 38, 89–119 [DOI] [PubMed] [Google Scholar]
- 101. Rowe P. S., Matsumoto N., Jo O. D., Shih R. N., Oconnor J., Roudier M. P., Bain S., Liu S., Harrison J., Yanagawa N. (2006) Correction of the mineralization defect in hyp mice treated with protease inhibitors CA074 and pepstatin. Bone 39, 773–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Menzel K., Hausmann M., Obermeier F., Schreiter K., Dunger N., Bataille F., Falk W., Scholmerich J., Herfarth H., Rogler G. (2006) Cathepsins B, L and D in inflammatory bowel disease macrophages and potential therapeutic effects of cathepsin inhibition in vivo. Clin. Exp. Immunol. 146, 169–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Butler D., Hwang J., Estick C., Nishiyama A., Kumar S. S., Baveghems C., Young-Oxendine H. B., Wisniewski M. L., Charalambides A., Bahr B. A. (2011) Protective effects of positive lysosomal modulation in Alzheimer's disease transgenic mouse models. PLoS One 6, e20501. [DOI] [PMC free article] [PubMed] [Google Scholar]