

Background: Lem3p-Dnf1p and Lem3p-Dnf2p are phospholipid flippases that generate phospholipid asymmetry in yeast.
Results: The tryptophan permease Tat2p is missorted from the trans-Golgi network to the vacuole in the lem3Δ mutant.
Conclusion: Phospholipid asymmetry supports plasma membrane transport of Tat2p by inhibiting its improper ubiquitination at the trans-Golgi network.
Significance: Phospholipid asymmetry may be involved in proper sorting of membrane proteins.
Keywords: Amino Acid Transport, Membrane Bilayer, Phosphatidylserine, Protein Sorting, Ubiquitination, Yeast, Flippase, Phospholipid Asymmetry, Type 4 P-type ATPase
Abstract
The type 4 P-type ATPases are flippases that generate phospholipid asymmetry in membranes. In budding yeast, heteromeric flippases, including Lem3p-Dnf1p and Lem3p-Dnf2p, translocate phospholipids to the cytoplasmic leaflet of membranes. Here, we report that Lem3p-Dnf1/2p are involved in transport of the tryptophan permease Tat2p to the plasma membrane. The lem3Δ mutant exhibited a tryptophan requirement due to the mislocalization of Tat2p to intracellular membranes. Tat2p was relocalized to the plasma membrane when trans-Golgi network (TGN)-to-endosome transport was inhibited. Inhibition of ubiquitination by mutations in ubiquitination machinery also rerouted Tat2p to the plasma membrane. Lem3p-Dnf1/2p are localized to endosomal/TGN membranes in addition to the plasma membrane. Endocytosis mutants, in which Lem3p-Dnf1/2p are sequestered to the plasma membrane, also exhibited the ubiquitination-dependent missorting of Tat2p. These results suggest that Tat2p is ubiquitinated at the TGN and missorted to the vacuolar pathway in the lem3Δ mutant. The NH2-terminal cytoplasmic region of Tat2p containing ubiquitination acceptor lysines interacted with liposomes containing acidic phospholipids, including phosphatidylserine. This interaction was abrogated by alanine substitution mutations in the basic amino acids downstream of the ubiquitination sites. Interestingly, a mutant Tat2p containing these substitutions was missorted in a ubiquitination-dependent manner. We propose the following model based on these results; Tat2p is not ubiquitinated when the NH2-terminal region is bound to membrane phospholipids, but if it dissociates from the membrane due to a low level of phosphatidylserine caused by perturbation of phospholipid asymmetry in the lem3Δ mutant, Tat2p is ubiquitinated and then transported from the TGN to the vacuole.
Introduction
Phospholipid asymmetry of bilayer membranes is generally observed in the plasma membrane of eukaryotic organisms. In this phospholipid asymmetry, phosphatidylcholine (PC)2 is predominantly distributed in the outer leaflet facing extracellular space (exoplasmic leaflet), whereas phosphatidylethanolamine (PE) and phosphatidylserine (PS) are distributed in the inner leaflet facing the cytoplasm (cytoplasmic leaflet). The type 4 subfamily of P-type ATPase (P4-ATPase) seems to play an essential role to generate, maintain, and regulate phospholipid asymmetry by working as a “flippase,” which translocates aminophospholipids from the exoplasmic leaflet to the cytoplasmic one in an energy-dependent manner (1–5).
Five P4-ATPases, i.e. Drs2p, Dnf1p, Dnf2p, Dnf3p, and Neo1p, are identified in the budding yeast Saccharomyces cerevisiae, and these proteins, except Neo1p, require Cdc50 family proteins as noncatalytic subunits for their localization, function, and flippase activity (6–10). Drs2p, Dnf1p and Dnf2p, and Dnf3p are complexed with Cdc50p, Lem3p, and Crf1p, respectively. Drs2p is localized to endosomes, the trans-Golgi network (TGN), and post-Golgi secretory vesicles (11–13), whereas Dnf1p and Dnf2p are localized to the plasma membrane and early endosome/TGN membranes (13–15). Dnf1p and Dnf2p seem to be directly regulated by two serine/threonine kinases Fpk1p and Fpk2p (15). Functional studies revealed that flippases are involved in various vesicle transport pathways (5), and flippases in these functions are implicated in vesicle formation by inducing local membrane curvature (16, 17). Flippases also regulate functions of membrane proteins by changing transbilayer phospholipid composition; changes in PE and PS content in the cytoplasmic leaflet of the plasma membrane regulate Cdc42p localization in polarized cell growth (18, 19). However, other functions of flippases need to be investigated.
Tat2p is a high affinity tryptophan permease, which is the main machinery for tryptophan uptake in budding yeast. Similar to other permeases, localization of this permease is precisely regulated responding to extracellular tryptophan concentration; at low tryptophan, Tat2p is transported to the plasma membrane, but it is transported from the TGN to the vacuole at high tryptophan (20). Localization of yeast permeases is regulated by ubiquitination. In particular, monoubiquitination and lysine 63-linked polyubiquitin chain direct target permeases to the vacuole from the plasma membrane or from the sorting compartment without going through the plasma membrane (21). In the case of Tat2p, five lysine residues in the NH2-terminal cytoplasmic domain are identified as ubiquitin acceptor sites, which are recognized by ubiquitin ligase complexes Rsp5p-Bul1p and Rsp5p-Bul2p (20, 22).
Several reports have shown that changes in membrane lipid environment cause tryptophan requirement, which is probably due to mislocalization of Tat2p to the vacuole. Tat2p was inappropriately ubiquitinated in the erg6Δ mutant, which has a defect in the last step of ergosterol biosynthesis, and resulted in the missorting of Tat2p to the vacuole (20). The cho1Δ mutant, which is defective in phosphatidylserine synthesis, exhibited impaired tryptophan uptake (23). Fluidization of membrane lipids by increased unsaturation of fatty acids also caused tryptophan requirement (24). However, little is known about how Tat2p is missorted to the vacuole by changes in the lipid microenvironment.
Here, we show that Tat2p is also missorted to the vacuole by ubiquitination at the TGN in the lem3Δ mutant. Our results suggest that the destination of Tat2p is regulated by the interaction of its NH2-terminal region with phospholipids, including phosphatidylserine in the cytoplasmic leaflet of membranes.
EXPERIMENTAL PROCEDURES
Media and Genetic Methods
Cells were cultivated in YP-based rich medium, yeast extract/peptone/dextrose/adenine (YPDA) medium (1% Bacto-yeast extract (Difco), 2% Bacto-peptone (Difco), 2% glucose, and 0.01% adenine) supplemented with YPDA/tryptophan (YPDAW) or without 200 μg/ml tryptophan. Strains carrying plasmids were selected in synthetic medium (SD) containing the required nutritional supplements (25). Synthetic complete medium (SC) was SD medium containing all required nutritional supplements. When indicated, 0.5% casamino acids were added to SD medium with (SDA) or without (SDA-U) 20 μg/ml uracil. Tryptophan was supplemented to SDA medium at a concentration of 200 μg/ml (SDA200W) or 30 μg/ml (SDA30W), or it was not supplemented (SDA-W). Standard genetic manipulations of yeast were performed as described previously (26). Escherichia coli strains DH5α and XL1-Blue were used for construction and amplification of plasmids. The lithium acetate method was used for introduction of plasmids into yeast cells (27, 28).
Strains and Plasmids
Yeast strains used in this study are listed in Table 1. The vps27Δ and TAT2–3HA strains were constructed by transforming a BY4743 background strain (29) with the 3.6-kb EcoRI-SalI fragment from pKU65 and PmaCI-digested pKU51 (gifts from A. Nakano, Riken, Saitama, Japan), respectively. Other strains carrying complete gene deletions, GFP-tagged DNF1 and DNF2, and monomeric red fluorescence protein 1 (mRFP)-tagged SEC7 were constructed in the BY4743 background by PCR-based procedures as described (30, 31). All strains constructed by the PCR-based procedure were verified by colony-PCR amplification to confirm that the replacement had occurred at the expected locus. To construct the TAT23K>R mutant, tat2Δ::KanMX4 was transformed with the 2.9-kb PstI-EcoRI fragment of TAT23K>R from pKT2008, and the transformants were screened for tryptophan prototrophy on a YPDA plate. The transformant that lost the KanMX4 marker was further selected. The rsp5-1 mutant was constructed by two successive backcrosses to a BY4743 background strain.
TABLE 1.
Yeast strains used in this study
| Straina | Relevant genotype | Derivation/source |
|---|---|---|
| BY4743 | MATa/α LYS2/lys2Δ0 ura3Δ0/ura3Δ0 his3Δ1/his3Δ1 TRP1/TRP1 leu2Δ0/leu2Δ0 met15Δ0/MET15 | 29 |
| KKT33 | MATa lys2Δ0 ura3Δ0 his3Δ1 TRP1 leu2Δ0 met15Δ0 DNF1-GFP::HIS3MX6 | 15 |
| KKT61 | MATa LYS2 ura3Δ0 his3Δ1 TRP1 leu2Δ0 MET15 | 15 |
| KKT102 | MATa LYS2 ura3Δ0 his3Δ1 TRP1 leu2Δ0 MET15 lem3Δ::KanMX6 | 15 |
| KKT144 | MATα lys2Δ0 ura3Δ0 his3Δ1 TRP1 leu2Δ0 met15Δ0 vrp1Δ::LEU2 | This study |
| KKT268 | MATa LYS2 ura3Δ0 his3Δ1 TRP1 leu2Δ0 MET15 fpk1Δ::HphMX4 fpk2Δ::KanMX6 | 15 |
| KKT290 | MATα lys2Δ0 ura3Δ0 his3Δ1 TRP1 leu2Δ0 met15Δ0 end3Δ::HphMX4 | This study |
| KKT334 | MATa LYS2 ura3Δ0 his3Δ1 TRP1 leu2Δ0 MET15 DNF2-GFP::HIS3MX6 | 15 |
| KKT369 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 | This study |
| KKT372 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 lem3Δ::KanMX6 | This study |
| KKT381 | MATa/α LYS2/LYS2 ura3Δ0/ura3Δ0 his3Δ1/his3Δ1 trp1Δ-63/trp1Δ-63 leu2Δ0/leu2Δ0 MET15/MET15 | This study |
| KKT402 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 tat2Δ::KanMX4 | This study |
| KKT403 | MATα LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 dnf1Δ::HphMX4 | This study |
| KKT404 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 dnf2Δ::KanMX4 | This study |
| KKT405 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 dnf1Δ::HphMX4 dnf2Δ::KanMX4 | This study |
| KKT406 | MATα lys2Δ0 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 drs2Δ::KanMX4 | This study |
| KKT407 | MATα lys2Δ0 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 cho1Δ::HIS3MX6 | This study |
| KKT418 | MATa/α LYS2/LYS2 ura3Δ0/ura3Δ0 his3Δ1/his3Δ1 trp1Δ-63/trp1Δ-63 leu2Δ0/leu2Δ0 MET15/MET15 tat2Δ::KanMX4/tat2Δ::KanMX4 | This study |
| KKT419 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 met15Δ0 gga1Δ:: HIS3MX6 gga2Δ::KanMX4 | This study |
| KKT420 | MATα LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 met15Δ0 lem3Δ::KanMX6 gga1Δ::HIS3MX6 gga2Δ::KanMX4 | This study |
| KKT421 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 pep12Δ::KanMX4 | This study |
| KKT422 | MATα LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 lem3Δ::HIS3MX6 pep12Δ::KanMX4 | This study |
| KKT423 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 vps27Δ::HIS3 | This study |
| KKT424 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 lem3Δ::KanMX6 vps27Δ::HIS3 | This study |
| KKT425 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 bul1Δ::HIS3MX6 bul2Δ::KanMX6 | This study |
| KKT426 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 lem3Δ::KanMX6 bul1Δ::HIS3MX6 bul2Δ::KanMX6 | This study |
| KKT427 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 TAT23K>R | This study |
| KKT428 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 lem3Δ::HIS3MX6 TAT23K>R | This study |
| KKT429 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 met15Δ0 vps1Δ::KanMX4 | This study |
| KKT430 | MATα lys2Δ0 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 lem3Δ::HIS3MX6 vps1Δ::KanMX4 | This study |
| KKT433 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 cdc50Δ::HphMX4 | This study |
| KKT434 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 fpk1Δ::HphMX4 fpk2Δ::KanMX6 | This study |
| KKT435 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 met15Δ0 psd1Δ::KanMX4 psd2Δ::HIS3MX6 | This study |
| KKT436 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 tat1Δ::HIS3MX6 | This study |
| KKT437 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 lem3Δ::KanMX6 tat1Δ::HIS3MX6 | This study |
| KKT438 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 lem3Δ::HIS3MX6 tat2Δ::KanMX4 | This study |
| KKT439 | MATa lys2Δ0 ura3Δ0 his3Δ1 TRP1 leu2Δ0 met15Δ0 cho1Δ::KanMX6 | This study |
| KKT440 | MATα lys2Δ0 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 end3Δ::HphMX4 | This study |
| KKT441 | MATα lys2Δ0 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 met15Δ0 vrp1Δ::LEU2 | This study |
| KKT442 | MATa lys2Δ0 ura3Δ0 his3Δ1 TRP1 leu2Δ0 met15Δ0 end3Δ::HphMX4 DNF1-GFP::HIS3MX6 | This study |
| KKT443 | MATa lys2Δ0 ura3Δ0 his3Δ1 TRP1 leu2Δ0 met15Δ0 end3Δ::HphMX4 DNF2-GFP::HIS3MX6 | This study |
| KKT444 | MATa lys2Δ0 ura3Δ0 his3Δ1 TRP1 leu2Δ0 met15Δ0 DNF1-GFP::HIS3MX6 SEC7-mRFP1::HIS3MX6 | This study |
| KKT445 | MATa LYS2 ura3Δ0 his3Δ1 TRP1 leu2Δ0 met15Δ0 DNF2-GFP::HIS3MX6 SEC7-mRFP1::HIS3MX6 | This study |
| KKT446 | MATα LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 rsp5-1 | This study |
| KKT447 | MATα LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 lem3Δ::KanMX6 rsp5-1 | This study |
| KKT448 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 end3Δ::HphMX4 TAT23K>R | This study |
| KKT449 | MATa lys2Δ0 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 met15Δ0 end3Δ::HphMX4 bul1Δ::HIS3MX6 | This study |
| KKT450 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 vrp1Δ::LEU2 TAT23K>R | This study |
| KKT451 | MATα LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 met15Δ0 vrp1Δ::LEU2 bul1Δ::HIS3MX6 | This study |
| KKT452 | MATa LYS2 ura3Δ0 his3Δ1 TRP1 leu2Δ0 MET15 pep4Δ::KanMX4 prb1Δ::NatMX4 | This study |
| KKT453 | MATa lys2Δ0 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 met15Δ0 ypt6Δ::KanMX4 | This study |
| KKT454 | MATa lys2Δ0 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 met15Δ0 lem3Δ::HIS3MX6 ypt6Δ::KanMX4 | This study |
| KKT455 | MATa LYS2 ura3Δ0 his3Δ1 TRP1 leu2Δ0 MET15 lem3Δ::HIS3MX6 pep4Δ::KanMX4 prb1Δ::NatMX4 | This study |
| KKT456 | MATa/α LYS2/LYS2 ura3Δ0/ura3Δ0 his3Δ1/his3Δ1 trp1Δ-63/trp1Δ-63 leu2Δ0/leu2Δ0 MET15/MET15 lem3Δ::HIS3MX6/lem3Δ::HIS3MX6 tat2Δ::KanMX4/tat2Δ::KanMX4 | This study |
| KKT457 | MATα LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 tat2Δ::KanMX4 bul1Δ::HIS3MX6 bul2Δ::KanMX6 | This study |
| KKT458 | MATa LYS2 ura3Δ0 his3Δ1 TRP1 leu2Δ0 met15Δ0 tat2Δ::KanMX4 | This study |
| KKT459 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 TAT2-GFP::HIS3MX6 lem3Δ::KanMX6 | This study |
| KKT460 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 TAT2-GST::HIS3MX6 lem3Δ::KanMX6 | This study |
| KKT461 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 TAT2–13myc::HIS3MX6 lem3Δ::KanMX6 | This study |
| KKT462 | MATa LYS2 ura3Δ0 his3Δ1 trp1Δ-63 leu2Δ0 MET15 TAT2–3HA::URA3 | This study |
| KKT465 | MATa LYS2 ura3Δ0 his3Δ1 TRP1 leu2Δ0 MET15 lem3Δ::HIS3MX6 tat2Δ::KanMX4 | This study |
a KKT strains are isogenic derivatives of BY4743.
The plasmids used in this study are listed in Table 2. Schemes detailing the construction of plasmids and DNA sequences of nucleotide primers are available on request. Site-directed mutations were introduced into the NH2-terminal region of Tat2p by the overlap extension PCR method using pKT1747 as a template (32). The PCR-amplified region in each construct was sequenced to verify that only the desired mutations were introduced.
TABLE 2.
Plasmids used in this study
| Plasmid | Characteristics | Derivation/source |
|---|---|---|
| YCplac33 | URA3 CEN4 | 76 |
| YEplac195 | URA3 2 μm | 76 |
| pKO10 | PGAL1-HA URA3 2 μm | 77 |
| pGEX | GST AmpR | GE Healthcare |
| pKU51 [YIplac211-TAT2–3HA] | TAT2–3HA URA3 AmpR | 20 |
| pKU65 [pUC18-vps27Δ] | vps27Δ::HIS3 AmpR | 20 |
| pKT1742 [YCplac33–3HA-TAT2] | 3HA-TAT2 URA3 CEN4 | 78 |
| pKT1747 [YEplac195-TAT2] | TAT2 URA3 2 μm | 79 |
| pKT1763 [YCplac33–2HA-TAT2] | 2HA-TAT2 URA3 CEN4 | 79 |
| pKT1765 [YCplac33-TAT2] | TAT2 URA3 CEN4 | This study |
| pKT1855 [pGEX-GST-TAT2NT] | GST-TAT2 (residues 1–85) AmpR | This study |
| pKT2008 [pBSII-TAT23K>R] | TAT23K>R AmpR | This study |
| pKT2019 [YCplac33-TAT25K>A] | TAT25K>A URA3 CEN4 | This study |
| pKT2020 [YCplac33-TAT22R>A] | TAT22R>A URA3 CEN4 | This study |
| pKT2021 [YCplac33-tat22K2R>A] | tat22K2R>A URA3 CEN4 | This study |
| pKT2026 [pKO10-GST-TAT2NT] | PGAL1-HA-GST-TAT2 (residues 1–85) URA3 2 μm | This study |
| pKT2027 [pKO10-GST-TAT2NT5K>A] | PGAL1-HA-GST-TAT25K>A (residues 1–85) URA3 2 μm | This study |
| pKT2028 [pKO10-GST-TAT2NT2R>A] | PGAL1-HA-GST-TAT22R>A (residues 1–85) URA3 2 μm | This study |
| pKT2029 [pKO10-GST-TAT2NT2K2R>A] | PGAL1-HA-GST-TAT22K2R>A (residues 1–85) URA3 2 μm | This study |
| pKT2045 [pRS316-PACT1-3myc-TAT2] | PACT1-3myc-TAT2 URA3 CEN6 | This study |
| pKT2046 [pRS316-PACT1-GFP-TAT2] | PACT1-GFP-TAT2 URA3 CEN6 | This study |
Sucrose Gradient Fractionation
Fractionation of subcellular organelles based on sedimentation through a sucrose step gradient was performed as described previously (33) with a slight modification. In brief, 50 A600 units of early log phase cells were harvested and washed with ice-cold 10 mm NaN3. The cells were collected by centrifugation and resuspended in 0.5 ml of STE10 buffer (10% sucrose (w/w), 10 mm Tris-HCl, pH 7.5, and 10 mm EDTA) with protease inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mm PMSF), followed by agitation with glass beads. After addition of 1 ml of STE10 buffer with protease inhibitors, unlysed cells and debris were removed by centrifugation (500 × g for 3 min). 0.2 ml of the total cell lysate was subjected to centrifugation on a three-step sucrose gradient (0.2 ml of 55%, 0.5 ml of 45%, and 0.4 ml of 30% sucrose (w/w) in 10 mm Tris-HCl, pH 7.5, and 10 mm EDTA) at 55,000 rpm in a TLS55 rotor (Beckman, Brea, CA) for 2.5 h. Fractions (0.2 ml) were collected manually from the top and analyzed by immunoblotting, which was performed as described previously (34). For membrane proteins, SDS-PAGE samples were heated at 37 °C for 30 min before loading. Protein bands were visualized by chemiluminescence using ECL or ECL Advance (GE Healthcare). Where indicated, only the results of plasma membrane-rich fractions were shown.
Microscopy
GFP-tagged Dnf1p and Dnf2p were observed in living cells, which were grown to early log phase, collected, and resuspended in SC medium. Cells were mounted on microslide glass and immediately observed. Co-localization of Dnf1p-GFP or Dnf2p-GFP with Sec7-mRFP was examined in fixed cells. Fixation was performed by addition of a commercial 37% formaldehyde stock (Wako Pure Chemicals, Osaka, Japan) to a final concentration of 3.7% in the medium, followed by a 10-min incubation at 30 °C. After fixation, cells were washed twice with phosphate-buffered saline and examined.
Cells were observed using a Nikon ECLIPSE E800 microscope (Nikon Instec, Tokyo, Japan) equipped with an HB-10103AF super high pressure mercury lamp and a 1.4 NA 100× plan Apo oil immersion objective with the appropriate fluorescence filter sets and differential interference contrast optics. Images were acquired with a digital cooled charge-coupled device camera (C4742-95-12NR; Hamamatsu Photonics, Hamamatsu, Japan) using AQUACOSMOS software (Hamamatsu Photonics). Observations were compiled from the examination of at least 200 cells.
Liposome Flotation Experiments
Binding of the Tat2p NH2-terminal fragment to liposomes was assayed by liposome flotation experiments (35). The NH2-terminal cytoplasmic region (residues 1–85) of Tat2p (Tat2pNT) and its mutant proteins were expressed and purified as a GST fusion protein from the cells of yeast KKT452 (pep4Δ prb1Δ TRP1) containing pKT2026, pKT2027, pKT2028, or pKT2029 as described (15) with the following modifications. In ammonium sulfate precipitation, the fraction that precipitated between 15 and 35% (w/v) salt saturation was used. In affinity purification with glutathione-Sepharose 4B (GE Healthcare), the column was washed three times with 3.3 bed volumes of 20 mm HEPES buffer, pH 8.0, after loading the sample, and GST fusion proteins were successively eluted five times with 1 bed volume of the same buffer containing 10 mm reduced glutathione. Liposomes were prepared as follows. A dried film, prepared by evaporation of a mixture of defined lipids in chloroform, was resuspended in 20 mm HEPES buffer, pH 7.2, followed by brief sonication. After five steps of freezing and thawing in liquid nitrogen, the liposome suspension was extruded through a polycarbonate filter (pore size 0.1 μm, GE Healthcare) using LiposoFast (Avestin, Ottawa, Canada). Liposomes were stored at room temperature and used within 2 days after preparation. Lipids used were dioleoylphosphatidylcholine (DOPC), dioleoylphosphatidylethanolamine (DOPE), dioleoylphosphatidylserine (DOPS), l-α-phosphatidic acid (PA) (Avanti Polar Lipids, Alabaster, AL), and l-α-phosphatidylinositol (PI) (Nacalai Tesque, Kyoto, Japan). A fluorescence 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD)-labeled PE, 1-palmitoyl-2-(6-NBD-aminocaproyl)-PE (Avanti Polar Lipids), was added to every liposome at 0.2 mol % to visualize flotation of liposomes. Liposome flotation assay was modified as follows. GST-Tat2pNT or its mutant protein (1 μm) was incubated at 30 °C for 5 min with liposomes (1 mm lipids) in 20 mm HEPES buffer, pH 7.2. The suspension was then adjusted to 30% (w/v) sucrose and sequentially overlaid with 25 and 0% sucrose in 20 mm HEPES buffer, pH 7.2, followed by centrifugation at 20 °C for 1 h. The top (0.1 ml), middle (0.2 ml), and bottom (0.25 ml) fractions were manually collected, and proteins were precipitated with 10% TCA. After washing twice with 1 ml of cold acetone, the pellet was air-dried and resuspended in SDS-PAGE sample buffer. After SDS-PAGE, proteins were stained with SYPRO Red (Lonza, Rockland, ME), followed by quantification with a FLA-3000 fluorescence imaging system (Fuji Photo Film, Tokyo, Japan).
Tryptophan Uptake Assay
Tryptophan uptake was assayed as follows based on a previous study (36). Cells were grown to early log phase in SDA-W medium at 30 °C. 0.9 A600 units of the cells were washed twice with wash buffer (10 mm sodium citrate, pH 4.5, and 20 mm (NH4)2SO4). The cells were then resuspended in 2.7 ml of incubation medium (10 mm sodium citrate, pH 4.5, 20 mm (NH4)2SO4, and 2% glucose), and the absorbance at 600 nm was measured. The assay was initiated by the addition of 300 μl of radiolabeled tryptophan solution (297 μl of the incubation medium and 3 μl of l-[5-3H]tryptophan, 20 Ci/mmol; American Radiolabeled Chemicals, St. Louis) at a final tryptophan concentration of ∼10 μg/ml. An aliquot (500 μl) was withdrawn at each time point and chilled by the addition of 1 ml of the ice-cold incubation medium. The cells were collected by filtration through a nitrocellulose filter (pore size 0.45 μm, Schleicher & Schuell) presoaked in the wash buffer and washed three times with chilled water. The filters were completely dried at 50 °C for more than 2 h, and intracellular radiolabeled tryptophan was quantified by scintillation counting.
When tryptophan import activity of Tat2p was estimated (Fig. 7B), the Tat2p content in the plasma membrane was determined. Cells were grown to early log phase in SDA-UW medium at 30 °C. 50 A600 units of the cells were subjected to the sucrose gradient fractionation as described above to obtain the plasma membrane-rich fraction, which was analyzed by immunoblotting. The Tat2p content was quantified by densitometric scanning with LAS-1000 (Fuji Photo Film). Other 3.5 A600 units of the cells instead of 0.9 A600 units were subjected to tryptophan uptake assay as described above. The data (104 dpm/A600) were corrected for the difference of Tat2p content in the plasma membrane-rich fraction.
FIGURE 7.
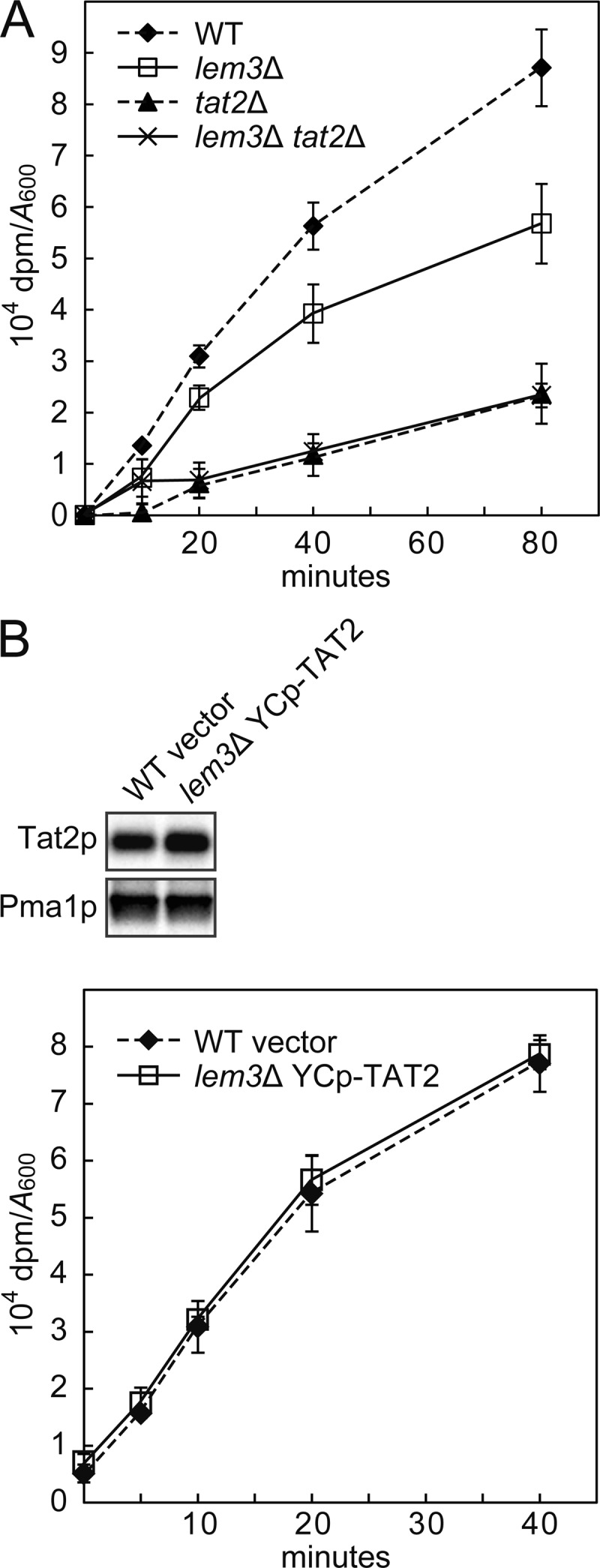
Tryptophan uptake activity of Tat2p in the lem3Δ mutant. A, tryptophan uptake in the lem3Δ and tat2Δ mutants. Tryptophan import activity was assayed with l-[5-3H]tryptophan as described under “Experimental Procedures.” The data represent means ± S.D. of three independent experiments. Strains were KKT61 (wild-type TRP1), KKT102 (lem3Δ), KKT458 (tat2Δ), and KKT465 (lem3Δ tat2Δ). These were TRP1 background strains. B, tryptophan uptake activity of Tat2p was not affected by the lem3Δ mutation. Upper panel, Tat2p content in the PM-rich fraction. PM-rich fractions were isolated by sucrose gradient fractionation from KKT61 (wild-type TRP1) harboring YCplac33 (vector) and KKT102 (lem3Δ TRP1) harboring pKT1765 (YCp-TAT2), followed by immunoblotting with anti-Tat2pN2 or anti-Pma1p (a loading control) antibodies. The plasma membrane Tat2p level in the lem3Δ mutant was 129 ± 15% of the wild type. Lower panel, tryptophan uptake activity of Tat2p. Tryptophan import activity (104 dpm/A600), which was assayed as described in A, was corrected for the difference of Tat2p content in the PM-rich fraction.
Antibodies
Rabbit anti-Tat2p polyclonal antibodies were generated against two synthetic peptides of 17 amino acids, N1 (residues 5–21) and N2 (residues 66–82), from the NH2-terminal cytoplasmic tail of Tat2p. Peptide synthesis and rabbit immunizations were performed in MBL (Nagoya, Japan). Antibodies were affinity-purified with a protein column in which 5 mg of GST-Tat2pNT (residues 1–85) was coupled to 0.5 ml of CNBr-activated Sepharose 4B beads (GE Healthcare). GST-Tat2pNT was expressed in E. coli and purified as described previously (37). The antiserum was incubated with the protein beads for 1 h at room temperature with gentle rotation. The beads were then successively washed five times with 1 ml of high salt wash buffer (50 mm Tris-HCl, pH 7.5, and 500 mm NaCl) and five times with 1 ml of low salt wash buffer (50 mm Tris-HCl, pH 7.5, and 150 mm NaCl). The antibodies were successively eluted five times with 0.5 ml of 0.1 m glycine-HCl, pH 3.5, and five times with 0.5 ml of 0.1 m glycine-HCl, pH 2.5, and the eluates were immediately equilibrated to pH 7.5 by mixing with 1 m Tris-HCl, pH 8.5. Fractions were divided into aliquots and stored at −20 °C. The antibodies against the N1 and N2 peptides were named anti-Tat2pN1 and anti-Tat2pN2 antibodies, respectively. Mouse anti-Pep12p monoclonal antibody was purchased from Invitrogen. Rabbit anti-Kex2p and anti-Pma1p polyclonal antibodies were gifts from S. Nothwehr (University of Missouri, Columbia, MO) and R. Serrano (Polytechnic University of Valencia, Valencia, Spain), respectively. For immunoblot analyses, these antibodies were used at the following dilutions: anti-Pep12p, 1:1000; anti-Kex2p, 1:5000; anti-Pma1p, 1:50,000; anti-Tat2pN1, 1:10,000; anti-Tat2pN2, 1:5000. Horseradish peroxidase-conjugated secondary antibodies (sheep anti-mouse IgG and donkey anti-rabbit IgG) used for immunoblotting were purchased from GE Healthcare.
RESULTS
Flippase Mutants Are Defective in Tryptophan Uptake Because of Dysregulation of Tat2p
The TRP1 gene product (phosphoribosylanthranilate isomerase) catalyzes the third step in the tryptophan biosynthesis pathway in budding yeast (38). Thus, trp1Δ cells require extracellular tryptophan for growth. As shown in Fig. 1A, trp1Δ cells grew normally in YPDA-rich medium containing tryptophan at a standard concentration (∼100 μg/ml), which was estimated by amino acid compositional analysis.3 Tryptophan uptake at this tryptophan concentration is thought to mainly depend on a high affinity tryptophan permease, Tat2p (39). Thus, tat2Δ trp1Δ cells did not grow on YPDA but grew on YPDAW, which contained a high concentration of tryptophan (∼300 μg/ml), because tryptophan could be taken up by a low affinity tryptophan permease, Tat1p (Fig. 1A) (39). We previously identified Fpk1p and Fpk2p as kinases that phosphorylated Dnf1p and Dnf2p flippases for activation (15). We noticed that fpk1Δ fpk2Δ trp1Δ cells did not grow on YPDA but rather on YPDAW (Fig. 1A). Consistently, dnf1Δ dnf2Δ trp1Δ as well as lem3Δ trp1Δ cells also exhibited tryptophan requirement, albeit weakly (Fig. 1A). Similar tryptophan requirement was also observed in drs2Δ trp1Δ and cdc50Δ trp1Δ mutants, but the phenotype seems to be milder compared with the dnf1Δ dnf2Δ/lem3Δ trp1Δ mutants, because the drs2Δ/cdc50Δ trp1Δ mutants exhibited slower growth even in YPDAW medium. Tryptophan requirement was not seen in the dnf3Δ/crf1Δ trp1Δ mutants.3 These results suggest that the tryptophan requirement of lem3Δ and dnf1Δ dnf2Δ mutants reflects a unique function of Lem3p-Dnf1/2p. The severest tryptophan requirement in the fpk1Δ fpk2Δ trp1Δ mutant could be due to reduced phosphorylation of Drs2p, Ypk1p, or other substrates in addition to Dnf1p/Dnf2p (15, 40). The dnf1Δ dnf2Δ trp1Δ and lem3Δ trp1Δ mutants exhibited clearer tryptophan-dependent growth at 18 °C (Fig. 1B). Interestingly, the tat2Δ trp1Δ mutant did not grow even in YPDAW, suggesting that the function or localization of Tat1p may be impaired at 18 °C. The cho1Δ trp1Δ mutant also exhibited tryptophan requirement for growth as reported previously (23), but it grew slowly even in YPDAW (Fig. 1A).
FIGURE 1.

lem3Δ trp1Δ mutant exhibits tryptophan-dependent growth. A and B, tryptophan requirement in flippase and flippase-related mutants. Cells were grown to early log phase in YPDAW, washed, and adjusted at a concentration of 2.5 × 107 cells/ml. 4-μl drops of 5-fold serial dilutions were spotted on YPDAW or YPDA, followed by incubation at 30 °C for 20 h (A) or at 18 °C for 3 days (B). Strains were KKT61 (wild-type TRP1), KKT369 (wild-type trp1Δ), KKT402 (tat2Δ), KKT403 (dnf1Δ), KKT404 (dnf2Δ), KKT405 (dnf1Δ dnf2Δ), KKT406 (drs2Δ), KKT372 (lem3Δ), KKT433 (cdc50Δ), KKT434 (fpk1Δ fpk2Δ), and KKT407 (cho1Δ). These were trp1Δ background strains except for KKT61. C, suppression of tryptophan requirement of flippase and flippase-related mutants by overproduction of Tat2p. Cell growth was examined as in A. Strains were KKT372 (lem3Δ), KKT434 (fpk1Δ fpk2Δ), and KKT407 (cho1Δ) harboring YEplac195 (vector) or pKT1747 (YEp-TAT2). KKT369 was used as a WT control. D, lem3Δ tat1Δ trp1Δ mutant exhibits severe tryptophan requirement. Cells were spotted as in A, followed by incubation at 25 °C for 1.5 days. Strains were KKT369 (wild-type trp1Δ), KKT372 (lem3Δ), KKT436 (tat1Δ), KKT437 (lem3Δ tat1Δ), KKT402 (tat2Δ), and KKT438 (lem3Δ tat2Δ). These were also trp1Δ background strains.
These results suggest that the tryptophan requirement in the dnf1Δ dnf2Δ/lem3Δ trp1Δ mutant is caused by dysregulation of Tat2p, and this was supported by the following results. (i) The tryptophan requirement of lem3Δ trp1Δ, fpk1Δ fpk2Δ trp1Δ, and cho1Δ trp1Δ mutants was suppressed by the overexpression of TAT2 (Fig. 1C). (ii) The tat2Δ mutation is synthetically lethal with the tat1Δ mutation in the trp1Δ background due to the severe uptake defect of tryptophan (39). The lem3Δ trp1Δ mutation was synthetically lethal with tat1Δ, but not with tat2Δ (Fig. 1D). Our strain background contains not only trp1Δ but also leu2Δ, his3Δ, met15Δ, and lys2Δ mutations, but the lem3Δ mutant did not require a higher level of these amino acids for growth,3 suggesting that dysregulation of an amino acid transporter is specific to Tat2p.
Plasma Membrane Tat2p Is Decreased in the lem3Δ Mutant
Our previous microarray analysis suggested that the lem3Δ mutation did not affect transcription of TAT2.3 To investigate intracellular localization of Tat2p, we constructed tagged versions of Tat2p, in which GFP, GST, 13 copies of the Myc epitope (13myc), and three copies of the HA epitope (3HA) were appended at the COOH terminus of TAT2 in the genomic locus. However, TAT2-GFP, TAT2-GST, and TAT2–13myc resulted in suppression of the tryptophan requirement in the lem3Δ trp1Δ mutant, whereas TAT2–3HA was nonfunctional in the tat2Δ mutant (Fig. 2, A and B). TAT2-GFP also suppressed the tryptophan requirement in the fpk1Δ fpk2Δ trp1Δ and cho1Δ trp1Δ mutants.3 We also constructed the NH2-terminally tagged versions of Tat2p, but again they did not complement the tat2Δ mutation (Fig. 2C). Therefore, we decided to generate antibodies against Tat2p.
FIGURE 2.

Tagged versions of Tat2p are abnormally regulated or are not fully functional. 10-Fold serial dilutions of cell suspension were prepared as described in Fig. 1A and spotted on YPDAW and YPDA plates, followed by incubation at 30 °C for 22 h. A, strains were KKT372 (lem3Δ), KKT459 (TAT2-GFP lem3Δ), KKT460 (TAT2-GST lem3Δ), and KKT461 (TAT2–13myc lem3Δ). B, strains were KKT369 (wild-type trp1Δ) and KKT462 (TAT2–3HA). C, strains were KKT381 (wild-type trp1Δ/trp1Δ) and KKT418 (tat2Δ/tat2Δ trp1Δ/trp1Δ) harboring YCplac33 (vector), pKT1765 (YCp-TAT2), pKT1763 (YCp-2HA-TAT2), pKT1742 (YCp-3HA-TAT2), pKT2045 (YCp-PACT1-3myc-TAT2), and pKT2046 (YCp-PACT1-GFP-TAT2). These were trp1Δ background strains.
Anti-Tat2p polyclonal antibodies were generated in rabbits against two Tat2p peptides (N1, residues 5–21, and N2, residues 66–82) from the NH2-terminal cytoplasmic region of Tat2p. As shown in Fig. 3A, Tat2p was detected as a band of about 50 kDa in wild-type cells, but not in tat2Δ cells, by immunoblot assay with affinity-purified antibodies, although some cross-reactive bands were also detected especially with the anti-N2 antibodies. In the following experiments, the anti-N1 antibodies were used if not otherwise specified.
FIGURE 3.

Mislocalization of Tat2p in the lem3Δ mutant. A, detection of Tat2p by anti-Tat2p antibodies. Protein extracts from bulk membrane fraction after centrifugation at 100,000 × g were subjected to immunoblotting with antibodies against the NH2-terminal cytoplasmic region of Tat2p. α-N1 and α-N2 indicate anti-Tat2pN1 and anti-Tat2pN2 antibodies, respectively. Strains were KKT369 (wild-type trp1Δ) and KKT402 (tat2Δ trp1Δ). B, effect of extracellular tryptophan concentration on the expression level of Tat2p. Total cell lysates prepared from cells of KKT61 (wild-type TRP1) were subjected to immunoblotting with the anti-Tat2pN1 or anti-Kex2p (a loading control) antibodies. The cells were grown to early log phase in SDA medium containing 200 (200W), 30 (30W), or 0 (-W) μg/ml of tryptophan at 30 °C. C, effect of extracellular tryptophan concentration on the subcellular distribution of Tat2p. Total cell lysates prepared from the KKT61 cells grown as described in B were subjected to sucrose gradient fractionation as described under “Experimental Procedures,” followed by immunoblotting with antibodies against Tat2p, Pma1p, Kex2p, and Pep12p. PM and IMs indicate the plasma membrane-rich fraction and internal membranes-rich fractions, respectively. Distribution of Pma1p, Kex2p, and Pep12p was not affected by extracellular tryptophan concentration (data not shown). D, mislocalization of Tat2p in the lem3Δ mutant. Subcellular distribution of Tat2p was examined by sucrose gradient fractionation as in C. Cells were grown in SDA-W medium at 30 or 18 °C. Strains were KKT61 (wild-type TRP1), KKT102 (lem3Δ), KKT268 (fpk1Δ fpk2Δ), and KKT439 (cho1Δ). For the cho1Δ mutant, 1 mm ethanolamine was supplemented to the culture medium. E, mislocalization of Tat2p in the lem3Δ trp1Δ mutant. Subcellular distribution of Tat2p was examined as in C. Cells were grown in YPDA medium at 30 °C. Strains were KKT369 (wild-type trp1Δ) and KKT372 (lem3Δ trp1Δ). F, vacuole-dependent degradation of Tat2p in the lem3Δ mutant. Total cell lysates prepared from cells grown in SDA-W medium at 18 °C were subjected to immunoblotting with the anti-Tat2pN1 or anti-Kex2p (a loading control) antibodies. Strains were KKT61 (wild-type TRP1), KKT102 (lem3Δ), KKT452 (pep4Δ prb1Δ), and KKT455 (lem3Δ pep4Δ prb1Δ). These were TRP1 background strains.
The plasma membrane Tat2p level is regulated by extracellular tryptophan in the trp1Δ mutant as well as wild type, and this is accomplished by rerouting intracellular transport of Tat2p (20). Because previous reports used strains expressing tagged Tat2 proteins, we reassessed the endogenous Tat2p level in cells grown in a different tryptophan concentration. In these assays, we first used TRP1 strains rather than trp1Δ mutants to examine the effect of tryptophan depletion. Tat2p was most expressed when tryptophan was depleted from the medium (SDA-W) compared with the SDA medium with a low (30 μg/ml) or high (200 μg/ml) concentration of tryptophan (Fig. 3B). We next examined intracellular localization of Tat2p by separating organelles on a sucrose density gradient. As shown in Fig. 3C, the plasma membrane marker Pma1p fractionated in a denser fraction (lane 6), whereas the TGN marker Kex2p and the endosome marker Pep12p fractionated in lighter fractions (lanes 3 and 4). Hereafter, fraction 6 is referred to as the plasma membrane (PM)-rich fraction, whereas fractions 3 and 4 are referred to as the internal membrane (IM)-rich fractions. Tat2p was clearly increased in the PM-rich fraction by tryptophan depletion, whereas Tat2p in the IM-rich fraction was little affected by extracellular tryptophan concentration (Fig. 3C).
The fractionation profile of Tat2p was next examined in the lem3Δ and other mutants grown at 30 °C. Tat2p in the PM-rich fraction was slightly decreased in the lem3Δ mutant, although it was clearly decreased in the cho1Δ mutant (Fig. 3D). These results paralleled weak and strong tryptophan requirements in the lem3Δ trp1Δ and cho1Δ trp1Δ mutants, respectively (Fig. 1A). In contrast, although the fpk1Δ fpk2Δ mutant exhibited a strong tryptophan requirement (Fig. 1A), Tat2p was only slightly decreased in this mutant, suggesting that the plasma membrane Tat2p might be less functional in the fpk1Δ fpk2Δ mutant. Tat2p in the PM-rich fraction was more decreased at 18 °C in the lem3Δ and fpk1Δ fpk2Δ mutants (Fig. 3D), again consistent with the growth phenotype (Fig. 1B). The fractionation profile of Tat2p was also examined in the lem3Δ trp1Δ mutant grown in YPDA at 30 °C, and it was confirmed that Tat2p was decreased in the PM-rich fraction (Fig. 3E). These results suggest that the tryptophan requirement in the lem3Δ mutant is due to the decreased Tat2p in the plasma membrane. Tat2p that was not delivered to the plasma membrane seemed to be degraded, because the total cellular Tat2p level was decreased in the lem3Δ mutant (Fig. 3F). This reduction was suppressed by mutations in PEP4 and PRB1, encoding vacuolar proteases that are required for maturation and activation of most vacuolar hydrolases (41, 42). These results suggest that Tat2p is missorted to vacuoles in the lem3Δ mutant.
Tat2p May be Missorted to the Vacuole from the TGN in the lem3Δ Mutant
Lem3p-Dnf1p and Lem3p-Dnf2p are recycled through the endocytic recycling pathway to maintain their primary localization to the plasma membrane (6, 43, 44). Thus, Lem3p-Dnf1/2p could regulate phospholipid asymmetry of endosomal/TGN membranes in addition to the plasma membrane. Two mechanisms can be envisioned to account for the decreased plasma membrane localization of Tat2p as follows: (i) Tat2p is missorted to the vacuole from the TGN in the lem3Δ mutant, and (ii) Tat2p is rapidly endocytosed in the lem3Δ mutant. We first examined whether a mutation in genes involved in the vacuolar sorting pathway suppressed the tryptophan requirement in the lem3Δ mutant. The VPS1 and GGA1/GGA2 genes encode a dynamin-like GTPase (45, 46) and Golgi-associated coat proteins with homology to γ-adaptin (47, 48), respectively. Both proteins are implicated in vesicle formation from the TGN for transport to the vacuole via late endosomes. Thus, mutations in these genes would redeliver cargo proteins destined for the vacuole to the plasma membrane (46, 49–51). As shown in Fig. 4A, both vps1Δ and gga1Δ gga2Δ mutations suppressed growth defects of the lem3Δ mutant on YPDA medium. If Tat2p is missorted to the vacuole through the vacuolar sorting pathway, mutations in late endosome-to-vacuole transport would also suppress the lem3Δ mutation. The PEP12 and VPS27 genes encode a t-SNARE required for vesicle fusion with late endosomes (52, 53) and a subunit of the ESCRT-0 complex involved in the sorting of ubiquitinated cargo into intraluminal budding of the endosomal membrane (54–56), respectively. pep12Δ and vps27Δ mutations also suppressed growth defects of the lem3Δ mutant (Fig. 4A).
FIGURE 4.

Tat2p is redelivered to the plasma membrane by inhibition of the vacuolar sorting pathway in the lem3Δ mutant. A, suppression of tryptophan requirement in the lem3Δ mutant. 10-Fold serial dilutions of cell suspension were prepared as described in Fig. 1A and spotted on YPDAW and YPDA plates, followed by incubation at 30 °C for 22 h. Strains were KKT369 (wild-type), KKT372 (lem3Δ), KKT429 (vps1Δ), KKT430 (lem3Δ vps1Δ), KKT419 (gga1Δ gga2Δ), KKT420 (lem3Δ gga1Δ gga2Δ), KKT421 (pep12Δ), KKT422 (lem3Δ pep12Δ), KKT423 (vps27Δ), and KKT424 (lem3Δ vps27Δ). These were trp1Δ background strains. B, restoration of the plasma membrane localization of Tat2p in the lem3Δ mutant. The cells of strains in A were grown in YPDA at 30 °C and the PM-rich fractions were isolated, followed by immunoblotting with anti-Tat2pN1 (upper panel) or anti-Pma1p (lower panel, a loading control) antibodies as described in Fig. 3C. C, co-localization of Dnf1p-GFP and Dnf2p-GFP with Sec7p-mRFP. Cells were grown to early log phase in YPDA at 30 °C, followed by fluorescence microscopic observation after fixation in 3.7% formaldehyde. Strains used were KKT444 (DNF1-GFP SEC7-mRFP, upper panel) and KKT445 (DNF2-GFP SEC7-mRFP, lower panel). Images were merged to compare two signal patterns. Arrowheads indicate co-localization between Dnf1p-GFP or Dnf2p-GFP and Sec7p-mRFP. Bars, 5 μm.
We next confirmed that the suppression was caused by increased Tat2p at the plasma membrane. The PM-rich fraction was isolated by sucrose gradient centrifugation as described in Fig. 3C, and Tat2p content was examined (Fig. 4B). The vps1Δ mutation slightly increased Tat2p at the plasma membrane in the lem3Δ mutant, and the gga1Δ gga2Δ mutations substantially increased it, but to a lesser extent than the pep12Δ and vps27Δ mutations. In the vps1Δ and gga1Δ gga2Δ mutants, Tat2p might be transported to the vacuole via the TGN-to-early endosome pathway, which would not be affected by these mutations. As shown in Fig. 4C, Dnf1p-GFP and Dnf2p-GFP were partially co-localized with a TGN marker Sec7p-mRFP (arrowheads) (57). Taken together, these results suggest that Tat2p is missorted to the vacuole from the TGN membrane whose phospholipid asymmetry is abnormally regulated in the lem3Δ mutant.
As shown in Fig. 4B, the plasma membrane Tat2p level in pep12Δ and vps27Δ mutants was not clearly decreased by the lem3Δ mutation, suggesting that Tat2p was not rapidly cleared from the plasma membrane by endocytosis in the lem3Δ mutant. In addition, Pomorski et al. (14) reported that flippase mutants are rather defective in endocytosis; the uptake of a lipophilic dye FM4-64 was delayed in the dnf1Δ dnf2Δ mutant, and the internalization of the mating factor receptor is inhibited in the dnf1Δ dnf2Δ drs2Δ mutant. If endocytosis of Tat2p is enhanced in the lem3Δ mutant, inhibition of endocytosis would recover Tat2p on the plasma membrane. END3 and VRP1 encode a factor of the endocytic coat/adaptor complex (58) and a regulator of cortical actin patch assembly (59), respectively, and removal of them causes a defect in receptor-mediated and fluid-phase endocytosis (60, 61). However, to our great surprise, the end3Δ trp1Δ and vrp1Δ trp1Δ mutants exhibited tryptophan-dependent slow growth, which was suppressed by the overproduction of Tat2p (Fig. 5A). Consistently, the plasma membrane Tat2p was decreased in these mutants as estimated by sucrose density gradient fractionation (Fig. 5B). These results suggest that Tat2p is not properly delivered to the plasma membrane due to some indirect defect in the TGN-to-plasma membrane transport of Tat2p.
FIGURE 5.
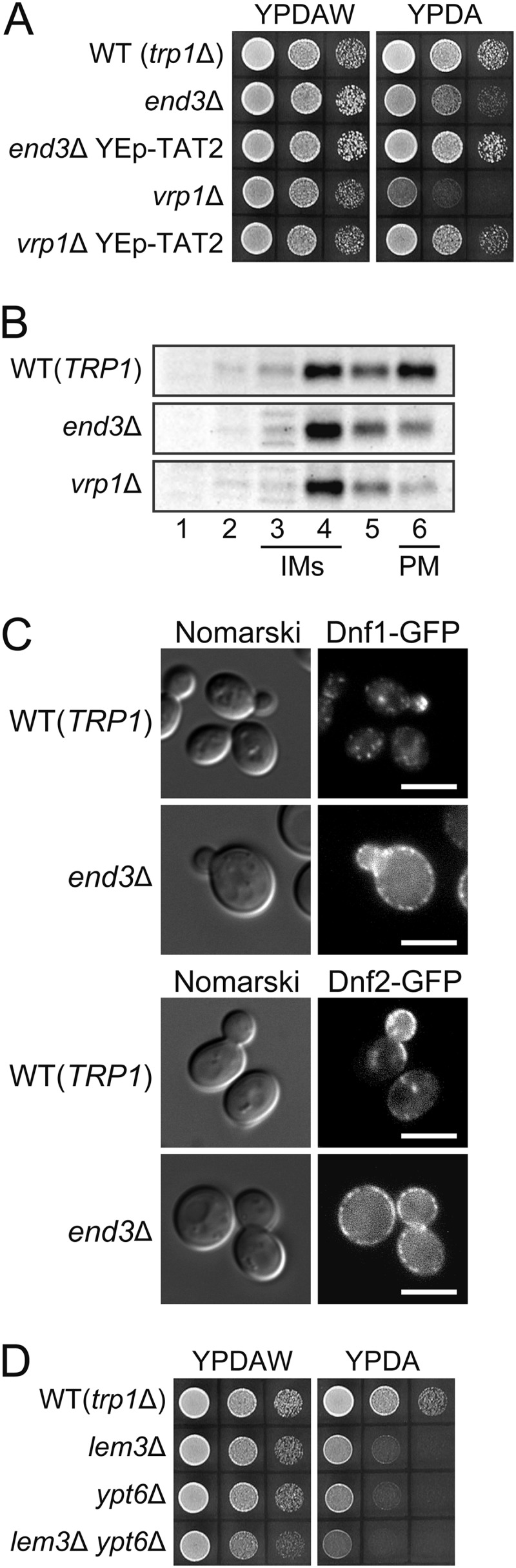
Mislocalization of Tat2p in endocytosis-defective mutants. A, tryptophan requirement in endocytosis-defective mutants. 10-Fold serial dilutions of cell suspension were prepared and spotted on YPDAW and YPDA plates as in Fig. 1A, followed by incubation at 25 °C for 1.5 days. Stains were KKT369 (wild-type trp1Δ), KKT440 (end3Δ), KKT441 (vrp1Δ), and KKT440 and KKT441 containing pKT1747 (YEp-TAT2). These were trp1Δ background strains. B, subcellular distribution of Tat2p in endocytosis-defective mutants. The cells of KKT61 (wild-type TRP1), KKT290 (end3Δ TRP1), and KKT144 (vrp1Δ TRP1) were grown in SDA-W medium at 25 °C, and the cell lysates were subjected to sucrose gradient fractionation, followed by immunoblotting with anti-Tat2pN1 antibodies. C, accumulation of Dnf1p-GFP and Dnf2p-GFP at the plasma membrane in endocytosis-defective mutants. The cells of wild-type and end3Δ strains expressing either Dnf1p-GFP or Dnf2p-GFP were grown to early log phase in YPDA at 25 °C, followed by fluorescence microscopic observation. Strains were KKT33 (DNF1-GFP), KKT334 (DNF2-GFP), KKT442 (end3Δ DNF1-GFP), and KKT443 (end3Δ DNF2-GFP). Bars, 5 μm. D, tryptophan requirement in the ypt6Δ mutant. Cell growth was examined as in Fig. 1A at 30 °C for 22 h. Strains were KKT369 (wild-type trp1Δ), KKT372 (lem3Δ), KKT453 (ypt6Δ), and KKT454 (lem3Δ ypt6Δ). These were trp1Δ background strains.
Lem3p-Dnf1p is recycled from the plasma membrane through early endosomes to the TGN and back to the plasma membrane (6, 43). We previously showed that Dnf1p-GFP was exclusively localized to the plasma membrane in the vrp1Δ mutant (6). Similarly, Dnf1p-GFP and Dnf2p-GFP were localized to the plasma membrane in end3Δ cells (Fig. 5C). Thus, in end mutants, Lem3p-Dnf1/2p could not be localized to the TGN, resulting in deregulated phospholipid asymmetry of this membrane. This would account for the possible missorting of Tat2p from the TGN to the vacuole in end mutants. Consistently, the ypt6Δ trp1Δ mutant, in which Dnf2p was mislocalized to endocytic recycling vesicles (62), also exhibited tryptophan requirement that was not strongly exacerbated by the additional lem3Δ mutation (Fig. 5D). However, because various membrane proteins are recycled through the endocytic pathway, mislocalization of other proteins than Lem3p-Dnf1/2p may be also involved in the missorting of Tat2p.
Inhibition of Ubiquitination Restores Plasma Membrane Localization of Tat2p
How does deregulated phospholipid asymmetry drive Tat2p to the vacuolar sorting pathway? Ubiquitination of yeast permeases is involved in their transport from the TGN to endosomes, endocytosis from the plasma membrane, and invagination into late endosomes (21). It was reported that ubiquitination at the TGN redirected Gap1p general amino acid permease to vacuoles from the exocytosis pathway (63). Thus, we examined whether inhibition of Tat2p ubiquitination restored plasma membrane localization of Tat2p in the lem3Δ mutant. Beck et al. (22) identified five lysine residues in the NH2-terminal domain of Tat2p as ubiquitin acceptor sites during starvation, and Umebayashi and Nakano (20) reported that three (residues 10, 17, and 20) of them were mainly ubiquitinated. TAT23K>R, in which these three lysine residues were replaced by arginine, was constructed and the genomic TAT2 was replaced with this allele. TAT23K>R suppressed the tryptophan requirement in the lem3Δ trp1Δ mutant (Fig. 6A). Rsp5p is an E3 ubiquitin ligase of the NEDD4 family, and Bul1/2p are Rsp5p adaptors required for the substrate recognition (21). Both bul1Δ bul2Δ and rsp5-1 mutations suppressed the tryptophan-dependent growth in the lem3Δ trp1Δ mutant (Fig. 6A). To confirm that the plasma membrane localization of Tat2p was restored by inhibition of ubiquitination, the PM-rich fraction was isolated by sucrose gradient fractionation, and the Tat2p content was examined by immunoblotting. In these experiments, we used the anti-Tat2pN2 antibodies instead of the anti-Tat2pN1 antibodies, which were raised against the NH2-terminal peptide containing the substitution sites of Tat2p3K>R. All mutations increased the plasma membrane Tat2p in the lem3Δ mutant (Fig. 6B). However, the increase by TAT23K>R and rsp5-1 mutations was not high, probably due to ubiquitination on other ubiquitin acceptor sites in Tat2p3K>R (e.g. residues 29 and 31) (22) and leakiness of the temperature-sensitive rsp5-1 mutation at 30 °C, respectively.
FIGURE 6.

Inhibition of ubiquitination restores the plasma membrane localization of Tat2p in the lem3Δ mutant. A, suppression of tryptophan requirement in the lem3Δ mutant by mutations in ubiquitination machinery. 10-Fold serial dilutions of cell suspension were prepared and spotted on YPDAW and YPDA plates as in Fig. 1A, followed by incubation at 30 °C for 22 h. Strains were KKT369 (wild-type trp1Δ), KKT372 (lem3Δ), KKT427 (TAT23K>R), KKT428 (lem3Δ TAT23K>R), KKT425 (bul1Δ bul2Δ), KKT426 (lem3Δ bul1Δ bul2Δ), KKT446 (rsp5-1), and KKT447 (lem3Δ rsp5-1). These were trp1Δ background strains. B, restoration of the plasma membrane localization of Tat2p in the lem3Δ mutant. The cells of strains in A were grown in YPDA at 30 °C, and the PM-rich fractions were isolated by sucrose gradient fractionation, followed by immunoblotting with anti-Tat2pN2 (upper panel) or anti-Pma1p (lower panel, a loading control) antibodies as described in Fig. 3C. C, suppression of tryptophan requirement in end mutants by mutations in ubiquitination machinery. 10-Fold serial dilutions of cell suspension were prepared and spotted on YPDAW and YPDA plates as in Fig. 1A, followed by incubation at 25 °C for 1.5 days. Strains were KKT369 (wild-type trp1Δ), KKT440 (end3Δ), KKT448 (end3Δ TAT23K>R), KKT449 (end3Δ bul1Δ), KKT441 (vrp1Δ), KKT450 (vrp1Δ TAT23K>R), and KKT451 (vrp1Δ bul1Δ). These were trp1Δ background strains.
The results described above imply that Tat2p is ubiquitinated at the TGN and missorted to the vacuole in the lem3Δ mutant. However, we cannot exclude a possibility that the plasma membrane Tat2p was increased due to blockade of endocytosis. Interestingly, TAT23K>R and bul1Δ also suppressed the tryptophan requirement in the end3Δ trp1Δ and vrp1Δ trp1Δ mutants (Fig. 6C). These results suggest that ubiquitination of Tat2p occurs at the TGN, not at the plasma membrane, at least in end mutants. We also examined increased ubiquitination of Tat2p in the lem3Δ mutant. Tat2p was immunoprecipitated with the anti-Tat2p antibodies from cells expressing Myc-tagged ubiquitin, followed by immunoblotting with the anti-Myc antibody. However, we could not reproducibly detect an increase of ubiquitination in the lem3Δ mutant, because of abundant ubiquitinated Tat2p in wild-type cells, possibly originating from Tat2p localized in intracellular membranes (Fig. 3, C and D).3
Tryptophan Import Activity of Tat2p Was Not Affected by the lem3Δ Mutation
Because Lem3p-Dnf1/2p are implicated in phospholipid asymmetry in the plasma membrane (14, 64), it is possible that tryptophan import activity of Tat2p is decreased by perturbed phospholipid asymmetry in the lem3Δ mutant. Tryptophan uptake was examined in the lem3Δ mutant using radiolabeled tryptophan. As shown in Fig. 7A, tryptophan uptake was decreased in the lem3Δ mutant in accordance with the decreased Tat2 protein level in the PM-rich fraction (Fig. 3D). Tat2p was mainly responsible for this tryptophan uptake in the lem3Δ mutant as well as in the wild type. To compare the Tat2p activity more precisely between the wild type and lem3Δ mutant, we constructed a lem3Δ mutant that expressed Tat2p at the plasma membrane in a level comparable with that in the wild type by transforming the lem3Δ mutant with YCp-TAT2. The plasma membrane Tat2p level in this strain was 129 ± 15% of the wild type. As shown in Fig. 7B, tryptophan uptake was not affected by the lem3Δ mutation when the Tat2p level in the lem3Δ mutant was normalized to the wild type. These results suggest that tryptophan requirement in the lem3Δ mutant was not caused by decreased Tat2p activity.
Interaction of the Tat2p NH2-terminal Cytoplasmic Region with Phosphatidylserine May be Involved in the Ubiquitination of Tat2p
We next investigated a mechanism for sensing perturbed phospholipid asymmetry by Tat2p. If Tat2p is directly involved in the sensing, the sensing site might be located in the vicinity of the ubiquitination sites in the NH2-terminal region. Thus, we examined whether the Tat2p NH2-terminal region (residues 1–85, Tat2pNT) directly interacted with phospholipids by the liposome flotation assay (35). In these experiments, liposomes and their bound proteins move to the top fraction from the bottom fraction after centrifugation, whereas unbound proteins are left in the bottom fraction. Interestingly, GST-Tat2pNT bound to liposomes composed of 80% DOPC and 20% DOPS, or 80% DOPC and 20% PA (mol/mol) (Fig. 8A). GST-Tat2pNT very weakly bound to 80% DOPC and 20% PI liposomes but did not bind to DOPC-only liposomes or 80% DOPC and 20% DOPE liposomes, suggesting that GST-Tat2pNT specifically bound to acidic phospholipid-containing liposomes in a preferable manner for PS and PA. These results are interesting, because Lem3p-Dnf1/2p are suggested to translocate PS but not PA to the cytoplasmic leaflet (see under “Discussion”) (14).
FIGURE 8.

Mutational analysis of basic residues in the NH2-terminal region of Tat2p for interaction with liposomes and in vivo functions. A, NH2-terminal region of Tat2p preferentially binds to PS- or PA-containing liposomes. Liposome flotation experiments were performed with GST-Tat2p(residues 1–85) (GST-Tat2pNT) as described under the “Experimental Procedures.” T, M, and B indicate top, middle, and bottom fractions, respectively. Percent Tat2p in the top fraction is shown (mean ± S.D. of three independent experiments). The liposome composition was 100 mol % DOPC (PC), 80% DOPC, and 20% DOPS (20% PS), 80% DOPC and 20% DOPE (20% PE), 80% DOPC and 20% PA (20% PA), or 80% DOPC and 20% PI (20% PI). Arrowheads indicate GST-Tat2pNT. B, alanine substitution mutations in the NH2-terminal region of Tat2p. Partial amino acid sequence of the NH2-terminal region (residues 1–85) and three alanine substitution mutants are shown. Asterisks indicate ubiquitin (Ub) acceptor lysines. C, interaction of Tat2pNT mutant proteins with PS liposomes. Binding of GST-fused mutant Tat2pNT to PS liposomes (80% DOPC and 20% DOPS) was examined by liposome flotation experiments as in A. D, tryptophan-dependent growth phenotypes of the tat2 mutants. 10-Fold serial dilutions of cell suspension were prepared and spotted on YPDAW and YPDA plates as in Fig. 1A, followed by incubation at 30 °C for 22 h. Strains were KKT381 (wild-type trp1Δ/trp1Δ) and KKT418 (tat2Δ/tat2Δ) and KKT456 (lem3Δ/lem3Δ tat2Δ/tat2Δ) harboring YCplac33 (vector), pKT1765 (TAT2), pKT2019 (TAT25K>A), pKT2020 (TAT22R>A), or pKT2021 (tat22K2R>A). These were trp1Δ background strains. E, subcellular distribution of mutant Tat2 proteins. Cells were grown to early log phase in SDA-UW medium at 30 °C, and the cell lysates were subjected to sucrose gradient fractionation, followed by immunoblotting with anti-Tat2pN1 (α-N1) and -Tat2pN2 (α-N2) antibodies. Tat2p5K>A exhibited mobility shifts possibly because of post-translational modifications or structural alteration by substitutions. An arrowhead indicates the original Tat2p position. Strains were KKT458 (tat2Δ TRP1) harboring pKT1765 (Tat2p), pKT2021 (Tat2p2K2R>A), pKT2019 (Tat2p5K>A), or pKT2020 (Tat2p2R>A). F, suppression of tryptophan-dependent growth in the tat22K2R>A mutant by bul1Δ bul2Δ mutations. Cell growth was examined as described in D. Strains were KKT369 (wild-type trp1Δ) and KKT457 (bul1Δ bul2Δ tat2Δ trp1Δ) harboring YCplac33 (vector), pKT1765 (TAT2), or pKT2021 (tat22K2R>A).
To evaluate the physiological significance of the above findings, we isolated mutant Tat2pNT proteins that were impaired in the binding to PS-containing liposomes. Given that the interaction was sensitive to high salt concentrations (e.g. 200 mm KCl),3 we speculated that positively charged amino acids may be involved in the lipid binding. The partial amino acid sequence of Tat2pNT is shown in Fig. 8B. This region is highly variable among yeast amino acid permeases except for the conserved sequence (residues 78–85) that precedes the first transmembrane domain. Two lysine- and arginine-containing sequences, regions 1 and 2, which are separated by a serine-rich sequence (residues 34–47) were found. In region 1, five lysine residues (10, 17, 20, 29, and 31) had been identified as ubiquitin acceptor lysines (22), and these lysines were replaced with alanines to construct Tat2pNT5K>A. There are also two arginine residues (11 and 19) in this region, and they were replaced with alanines to construct Tat2pNT2R>A. In region 2, there are two arginine residues (55 and 60) and lysine residues (54 and 66), and these four residues were replaced with alanines to construct Tat2pNT2K2R>A. These three mutant proteins fused to GST were expressed, purified, and examined for binding to the PC liposomes containing 20% PS (Fig. 8C). Interestingly, the 2K2R>A substitutions greatly impaired the interaction as follows: only 14.8 ± 1.1% of GST-Tat2pNT2K2R>A was found in the top fraction compared with 71.2 ± 4.5% of the wild type. The binding of GST-Tat2pNT5K>A was slightly impaired to 42.4 ± 9.6% but that of GST-Tat2pNT2R>A was not affected. These results suggest that the four basic residues in region 2 are mainly involved in the PS-liposome binding.
We next constructed mutant tat2 genes containing these substitutions and expressed them in the tat2Δ trp1Δ mutant. Most interestingly, the tat22K2R>A gene failed to complement the tat2Δ mutation (Fig. 8D). This was because the plasma membrane Tat2p2K2R>A was decreased as estimated by sucrose gradient fractionation (Fig. 8E). We next expressed the tat22K2R>A gene in the bul1Δ bul2Δ tat2Δ trp1Δ mutant. As shown in Fig. 8F, bul1Δ bul2Δ mutations clearly suppressed the tryptophan requirement of the tat22K2R>A mutant. Taken together, these results suggest that Tat2p2K2R>A is ubiquitinated and missorted to the vacuole, possibly because its NH2-terminal region does not interact with membranes.
We similarly examined the functionality of TAT25K>A and TAT22R>A mutant genes. The TAT25K>A mutation restored growth in the lem3Δ tat2Δ trp1Δ mutant as well as in the tat2Δ trp1Δ mutant (Fig. 8D), consistent with the previous observation that the mutated lysine residues are ubiquitin acceptor sites; this protein would be continuously transported to the plasma membrane from the TGN and would be defective in its endocytosis (22, 65). In fact, Tat2p5K>A was exclusively found in the PM-rich fraction (Fig. 8E). As shown in Fig. 8C, GST-Tat2pNT5K>A exhibited an ∼30% reduction in liposome binding. It is an interesting possibility that these lysine residues are also involved in sensing the PS level for ubiquitination of Tat2p. The TAT22R>A mutant behaved like wild type in tryptophan requirement for growth and subcellular localization (Fig. 8, D and E). Consistently, this mutation did not affect the binding to PS-containing liposomes (Fig. 8C).
DISCUSSION
Here, we report that phospholipid flippases Lem3p-Dnf1/2p are involved in the plasma membrane localization of Tat2p. Considering the primary localization site of Lem3p-Dnf1/2p at the plasma membrane, we first speculated that the missorting of Tat2p was due to the perturbation of phospholipid asymmetry at the plasma membrane. However, the missorting seems to occur at the TGN, because the inhibition of TGN-to-late endosome transport rerouted Tat2p to the plasma membrane. Interestingly, endocytosis-defective mutants also exhibited the defects in the plasma membrane localization of Tat2p, suggesting that, in end mutants, Tat2p was also missorted from the TGN to the vacuole. In end mutants, various membrane proteins that are recycled through the endocytic pathway are trapped at the plasma membrane. Thus, the Tat2p missorting seems to be caused by sequestering some proteins, including Lem3p-Dnf1/2p, from the TGN. Consistently, the end3Δ mutation did not exhibit a synthetic tryptophan requirement with the lem3Δ trp1Δ mutations at 25 °C.3 Although we cannot exclude the possibility that Tat2p is endocytosed at an increased rate in the lem3Δ mutant, it is not plausible, because of the following: (i) the plasma membrane-associated Tat2p level was not very different between pep12Δ and lem3Δ pep12Δ mutants (Fig. 4B), and (ii) Lem3p-Dnf1/2p were rather required for endocytosis (14).
If the phospholipid flip at the TGN is generally required for the plasma membrane transport of Tat2p, the cdc50Δ/drs2Δ trp1Δ mutant should exhibit a stronger tryptophan requirement, because Cdc50p-Drs2p is mainly localized to TGN/endosomal membranes (13). However, the cdc50Δ/drs2Δ mutants exhibited a weaker tryptophan requirement than the lem3Δ/dnf1/2Δ mutants, taking into account the slower growth rates of the cdc50Δ/drs2Δ mutants in a tryptophan-rich medium (Fig. 1A). One possibility is that Lem3p-Dnf1/2p and Tat2p are present in a similar membrane microenvironment; both proteins have been reported to be present in detergent-resistant membrane domains, the so-called lipid rafts (20, 64). In yeast, most plasma membrane proteins, including Lem3p and Tat2p, were found to be associated with lipid rafts. Accumulating evidence suggests that lipid rafts function as a sorting platform at the TGN for cell surface delivery of plasma membrane proteins (66). Quantitative analysis of isolated secretory vesicles by mass spectrometry revealed that they are highly enriched in ergosterol and sphingolipids compared with the donor TGN membranes (67). Thus, Lem3p-Dnf1/2p and Tat2p may be segregated at the TGN from Cdc50p-Drs2p, which is normally transported to the early endosome. It is unknown how phospholipid asymmetry is organized in these raft domains, but Lem3p-Dnf1/2p may affect the destination of Tat2p by flipping phospholipids in raft domains.
The binding of Tat2pNT to liposomes required positively charged amino acid residues and PS or PA on liposomes, suggesting that the NH2-terminal region of Tat2p electrostatically interacts with negatively charged membranes. It was reported that the trp1Δ mutant exhibited growth sensitivity to weak organic acid stress, which was suppressed by tryptophan supplementation or overexpression of Tat2p (68). Acid stress might disrupt the electrostatic interaction of Tat2p with membranes, resulting in mislocalization of Tat2p. Electrostatic interaction between a cytoplasmic region and membranes is involved in the regulation of various membrane proteins. The cytoplasmic COOH-terminal domain of the epithelial Na+/H+ exchanger NHE3 interacted with negatively charged membranes through basic residues, and this association seemed to be involved in the regulation of NHE3 activity in vivo (69). In rhodopsin, a cytoplasmic helical segment (H8) extending from the transmembrane domain seven could act as a membrane-dependent conformational switch by interacting with PS (70). In addition, it has been proposed that a cytoplasmic juxtamembrane domain of epidermal growth factor receptor binds electrostatically to acidic phospholipids in the plasma membrane, resulting in autoinhibition of the tyrosine kinase activity (71).
It has not been clearly demonstrated that Lem3p-Dnf1/2p flip PS, because NBD-labeled PS was still flipped in the lem3Δ mutant probably due to an unidentified protein on the plasma membrane (72), and NBD-PS was a less preferred substrate of Dnf1p compared with NBD-PC and NBD-PE (73). However, growth of the lem3Δ mutant was clearly sensitive to papuamide B, a cyclic lipopeptide that shows cytotoxicity by binding to PS in biological membranes (74), and this sensitivity was suppressed by the cho1Δ mutation,3 indicating that PS is exposed on the cell surface in this mutant. These results may suggest that Lem3p-Dnf1/2p flips PS more efficiently than NBD-PS. The results that Tat2p was missorted in the cho1Δ mutant (Fig. 3D) also support that PS is involved in Tat2p transport. Involvement of PS was also demonstrated for the ferrichrome-induced plasma membrane transport of the siderophore transporter Arn1p; Arn1p-GFP was mislocalized to intracellular structures in the cho1Δ mutant (75). Interestingly, in the drs2Δ mutant, the ferrichrome-induced plasma membrane transport of Arn1p-GFP was not affected, whereas Arn1p-GFP was mislocalized to the plasma membrane in the absence of ferrichrome. It seems that phospholipid asymmetry is involved in sorting of various membrane proteins in a different manner.
Because it was suggested that Lem3p-Dnf1/2p flip PE and PC (14, 64), these phospholipids may be also involved in Tat2p missorting. Interaction with the Tat2p NH2-terminal region was not detected with PE or PC in the liposome flotation experiments, but it is possible that changes in asymmetric distribution of these lipids are sensed through other regions of Tat2p or by other proteins that regulate Tat2p ubiquitination. Further studies are required to examine these possibilities.
Inhibition of ubiquitination restored plasma membrane localization of Tat2p in the lem3Δ mutant, suggesting that perturbation of phospholipid asymmetry induces ubiquitination of Tat2p. Thus, an important question is how changes in phospholipid asymmetry are sensed and ultimately result in ubiquitination of Tat2p. Identification of the liposome binding activity in the Tat2p NH2-terminal region shed light on this mechanism. Amino acid substitutions that reduced interaction with liposomes (2K2R>A: K54A, R55A, R60A, and K66A) caused ubiquitination-dependent missorting of Tat2p. These results suggest that Tat2p2K2R>A mimics Tat2p in the lem3Δ mutant, although we cannot exclude a possibility that the substitutions cause other defects such as structural change or the inability to interact with an interacting protein. Because these residues are close to the ubiquitin acceptor lysines (residues 10, 17, 20, 29, and 31), we propose that the interaction of the Tat2p NH2-terminal region with PS/PA-rich membranes through the basic residues plays an important role for whether Tat2p is ubiquitinated or not. When the Tat2p NH2-terminal region is bound to membrane phospholipids, it is not ubiquitinated, but when it dissociates from the membrane, Tat2p would be ubiquitinated and then transported to the vacuole. To test this hypothesis, ubiquitination of Tat2p should be reconstituted in vitro with liposomes. PA, which seems not to be flipped by Lem3p-Dnf1/2p (14), is likely to exist in TGN/endosomal membranes at a level comparable with that of PS (67). This might account for the mild missorting of Tat2p in the lem3Δ mutant.
Plasma membrane localization of Tat2p is also sensitive to perturbation of other lipids, including ergosterol; Tat2p was missorted to the vacuole by ubiquitination in the erg6Δ mutant (20). Thus, Tat2p is suitable for the study of effects of lipid microenvironment on protein sorting.
Acknowledgments
We thank Drs. Akihiko Nakano, Fumiyoshi Abe, Ramon Serrano, and Steven Nothwehr for yeast strains, plasmids, and antibodies. We thank Tomohiro Hirose (Instrumental Analysis Division, Equipment Management Center, Creative Research Institution, Hokkaido University) for amino acid compositional analysis. We thank our colleagues in the Tanaka laboratory for valuable discussions and Eriko Itoh for technical assistance.
This work was supported by Japan Society for the Promotion of Science KAKENHI Grants 21570192 and 21370085.
T. Hachiro, T. Yamamoto, and K. Tanaka, unpublished results.
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PS
- phosphatidylserine
- P4-ATPase
- type 4 P-type ATPase
- TGN
- trans-Golgi network
- mRFP
- monomeric red fluorescent protein 1
- Tat2pNT
- NH2-terminal cytoplasmic region (residues 1–85) of Tat2p
- DOPC
- dioleoylphosphatidylcholine
- DOPE
- dioleoylphosphatidylethanolamine
- DOPS
- dioleoylphosphatidylserine
- PA
- phosphatidic acid
- NBD
- 7-nitrobenz-2-oxa-1,3-diazol-4-yl
- PM
- plasma membrane
- IM
- internal membrane
- PI
- phosphatidylinositol.
REFERENCES
- 1. Devaux P. F., López-Montero I., Bryde S. (2006) Proteins involved in lipid translocation in eukaryotic cells. Chem. Phys. Lipids 141, 119–132 [DOI] [PubMed] [Google Scholar]
- 2. Daleke D. L. (2007) Phospholipid flippases. J. Biol. Chem. 282, 821–825 [DOI] [PubMed] [Google Scholar]
- 3. Lenoir G., Williamson P., Holthuis J. C. (2007) On the origin of lipid asymmetry. The flip side of ion transport. Curr. Opin. Chem. Biol. 11, 654–661 [DOI] [PubMed] [Google Scholar]
- 4. Tanaka K., Fujimura-Kamada K., Yamamoto T. (2011) Functions of phospholipid flippases. J. Biochem. 149, 131–143 [DOI] [PubMed] [Google Scholar]
- 5. Sebastian T. T., Baldridge R. D., Xu P., Graham T. R. (2012) Phospholipid flippases. Building asymmetric membranes and transport vesicles. Biochim. Biophys. Acta 1821, 1068–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Saito K., Fujimura-Kamada K., Furuta N., Kato U., Umeda M., Tanaka K. (2004) Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol. Biol. Cell 15, 3418–3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Furuta N., Fujimura-Kamada K., Saito K., Yamamoto T., Tanaka K. (2007) Endocytic recycling in yeast is regulated by putative phospholipid translocases and the Ypt31p/32p-Rcy1p pathway. Mol. Biol. Cell 18, 295–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lenoir G., Williamson P., Puts C. F., Holthuis J. C. (2009) Cdc50p plays a vital role in the ATPase reaction cycle of the putative aminophospholipid transporter Drs2p. J. Biol. Chem. 284, 17956–17967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takahashi Y., Fujimura-Kamada K., Kondo S., Tanaka K. (2011) Isolation and characterization of novel mutations in CDC50, the noncatalytic subunit of the Drs2p phospholipid flippase. J. Biochem. 149, 423–432 [DOI] [PubMed] [Google Scholar]
- 10. Jacquot A., Montigny C., Hennrich H., Barry R., le Maire M., Jaxel C., Holthuis J., Champeil P., Lenoir G. (2012) Phosphatidylserine stimulation of Drs2p·Cdc50p lipid translocase dephosphorylation is controlled by phosphatidylinositol 4-phosphate. J. Biol. Chem. 287, 13249–13261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Natarajan P., Wang J., Hua Z., Graham T. R. (2004) Drs2p-coupled aminophospholipid translocase activity in yeast Golgi membranes and relationship to in vivo function. Proc. Natl. Acad. Sci. U.S.A. 101, 10614–10619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alder-Baerens N., Lisman Q., Luong L., Pomorski T., Holthuis J. C. (2006) Loss of P4 ATPases Drs2p and Dnf3p disrupts aminophospholipid transport and asymmetry in yeast post-Golgi secretory vesicles. Mol. Biol. Cell 17, 1632–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hua Z., Fatheddin P., Graham T. R. (2002) An essential subfamily of Drs2p-related P-type ATPases is required for protein trafficking between Golgi complex and endosomal/vacuolar system. Mol. Biol. Cell 13, 3162–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pomorski T., Lombardi R., Riezman H., Devaux P. F., van Meer G., Holthuis J. C. (2003) Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol. Biol. Cell 14, 1240–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakano K., Yamamoto T., Kishimoto T., Noji T., Tanaka K. (2008) Protein kinases Fpk1p and Fpk2p are novel regulators of phospholipid asymmetry. Mol. Biol. Cell 19, 1783–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Devaux P. F., Herrmann A., Ohlwein N., Kozlov M. M. (2008) How lipid flippases can modulate membrane structure. Biochim. Biophys. Acta 1778, 1591–1600 [DOI] [PubMed] [Google Scholar]
- 17. Graham T. R., Kozlov M. M. (2010) Interplay of proteins and lipids in generating membrane curvature. Curr. Opin. Cell Biol. 22, 430–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saito K., Fujimura-Kamada K., Hanamatsu H., Kato U., Umeda M., Kozminski K. G., Tanaka K. (2007) Transbilayer phospholipid flipping regulates Cdc42p signaling during polarized cell growth via Rga GTPase-activating proteins. Dev. Cell 13, 743–751 [DOI] [PubMed] [Google Scholar]
- 19. Das A., Slaughter B. D., Unruh J. R., Bradford W. D., Alexander R., Rubinstein B., Li R. (2012) Flippase-mediated phospholipid asymmetry promotes fast Cdc42 recycling in dynamic maintenance of cell polarity. Nat. Cell Biol. 14, 304–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Umebayashi K., Nakano A. (2003) Ergosterol is required for targeting of tryptophan permease to the yeast plasma membrane. J. Cell Biol. 161, 1117–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lauwers E., Erpapazoglou Z., Haguenauer-Tsapis R., André B. (2010) The ubiquitin code of yeast permease trafficking. Trends Cell Biol. 20, 196–204 [DOI] [PubMed] [Google Scholar]
- 22. Beck T., Schmidt A., Hall M. (1999) Starvation induces vacuolar targeting and degradation of the tryptophan permease in yeast. J. Cell Biol. 146, 1227–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakamura H., Miura K., Fukuda Y., Shibuya I., Ohta A., Takagi M. (2000) Phosphatidylserine synthesis required for the maximal tryptophan transport activity in Saccharomyces cerevisiae. Biosci. Biotechnol. Biochem. 64, 167–172 [DOI] [PubMed] [Google Scholar]
- 24. Rodríguez-Vargas S., Sánchez-García A., Martínez-Rivas J. M., Prieto J. A., Randez-Gil F. (2007) Fluidization of membrane lipids enhances the tolerance of Saccharomyces cerevisiae to freezing and salt stress. Appl. Environ. Microbiol. 73, 110–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rose M. D., Winston F., Hieter P. (1990) Methods in Yeast Genetics: A Laboratory Course Manual, pp. 177–186, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 26. Guthrie C., Fink G. R. (eds) (1991) Guide to Yeast Genetics and Molecular Biology, Vol 194, Academic Press, San Diego [Google Scholar]
- 27. Elble R. (1992) A simple and efficient procedure for transformation of yeasts. BioTechniques 13, 18–20 [PubMed] [Google Scholar]
- 28. Gietz R. D., Woods R. A. (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350, 87–96 [DOI] [PubMed] [Google Scholar]
- 29. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C. A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- 30. Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 31. Goldstein A. L., McCusker J. H. (1999) Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15, 1541–1553 [DOI] [PubMed] [Google Scholar]
- 32. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 33. Valdivia R. H., Schekman R. (2003) The yeasts Rho1p and Pkc1p regulate the transport of chitin synthase III (Chs3p) from internal stores to the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 100, 10287–10292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Misu K., Fujimura-Kamada K., Ueda T., Nakano A., Katoh H., Tanaka K. (2003) Cdc50p, a conserved endosomal membrane protein, controls polarized growth in Saccharomyces cerevisiae. Mol. Biol. Cell 14, 730–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bigay J., Casella J. F., Drin G., Mesmin B., Antonny B. (2005) ArfGAP1 responds to membrane curvature through the folding of a lipid packing sensor motif. EMBO J. 24, 2244–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heitman J., Koller A., Kunz J., Henriquez R., Schmidt A., Movva N. R., Hall M. N. (1993) The immunosuppressant FK506 inhibits amino acid import in Saccharomyces cerevisiae. Mol. Cell. Biol. 13, 5010–5019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mochida J., Yamamoto T., Fujimura-Kamada K., Tanaka K. (2002) The novel adaptor protein, Mti1p, and Vrp1p, a homolog of Wiskott-Aldrich syndrome protein-interacting protein (WIP), may antagonistically regulate type I myosins in Saccharomyces cerevisiae. Genetics 160, 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Braus G. H. (1991) Aromatic amino acid biosynthesis in the yeast Saccharomyces cerevisiae. A model system for the regulation of a eukaryotic biosynthetic pathway. Microbiol. Rev. 55, 349–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schmidt A., Hall M. N., Koller A. (1994) Two FK506 resistance-conferring genes in Saccharomyces cerevisiae, TAT1 and TAT2, encode amino acid permeases mediating tyrosine and tryptophan uptake. Mol. Cell. Biol. 14, 6597–6606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roelants F. M., Baltz A. G., Trott A. E., Fereres S., Thorner J. (2010) A protein kinase network regulates the function of aminophospholipid flippases. Proc. Natl. Acad. Sci. U.S.A. 107, 34–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zubenko G. S., Park F. J., Jones E. W. (1982) Genetic properties of mutations at the PEP4 locus in Saccharomyces cerevisiae. Genetics 102, 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones E. W., Zubenko G. S., Parker R. R. (1982) PEP4 gene function is required for expression of several vacuolar hydrolases in Saccharomyces cerevisiae. Genetics 102, 665–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu K., Hua Z., Nepute J. A., Graham T. R. (2007) Yeast P4-ATPases Drs2p and Dnf1p are essential cargos of the NPFXD/Sla1p endocytic pathway. Mol. Biol. Cell 18, 487–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takagi K., Iwamoto K., Kobayashi S., Horiuchi H., Fukuda R., Ohta A. (2012) Involvement of Golgi-associated retrograde protein complex in the recycling of the putative Dnf aminophospholipid flippases in yeast. Biochem. Biophys. Res. Commun. 417, 490–494 [DOI] [PubMed] [Google Scholar]
- 45. Vater C. A., Raymond C. K., Ekena K., Howald-Stevenson I., Stevens T. H. (1992) The VPS1 protein, a homolog of dynamin required for vacuolar protein sorting in Saccharomyces cerevisiae, is a GTPase with two functionally separable domains. J. Cell Biol. 119, 773–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bensen E. S., Costaguta G., Payne G. S. (2000) Synthetic genetic interactions with temperature-sensitive clathrin in Saccharomyces cerevisiae. Roles for synaptojanin-like Inp53p and dynamin-related Vps1p in clathrin-dependent protein sorting at the trans-Golgi network. Genetics 154, 83–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dell'Angelica E. C., Puertollano R., Mullins C., Aguilar R. C., Vargas J. D., Hartnell L. M., Bonifacino J. S. (2000) GGAs. A family of ADP ribosylation factor-binding proteins related to adaptors and associated with the Golgi complex. J. Cell Biol. 149, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hirst J., Lui W. W., Bright N. A., Totty N., Seaman M. N., Robinson M. S. (2000) A family of proteins with γ-adaptin and VHS domains that facilitate trafficking between the trans-Golgi network and the vacuole/lysosome. J. Cell Biol. 149, 67–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nothwehr S. F., Conibear E., Stevens T. H. (1995) Golgi and vacuolar membrane proteins reach the vacuole in vps1 mutant yeast cells via the plasma membrane. J. Cell Biol. 129, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Costaguta G., Stefan C. J., Bensen E. S., Emr S. D., Payne G. S. (2001) Yeast Gga coat proteins function with clathrin in Golgi to endosome transport. Mol. Biol. Cell 12, 1885–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Scott P. M., Bilodeau P. S., Zhdankina O., Winistorfer S. C., Hauglund M. J., Allaman M. M., Kearney W. R., Robertson A. D., Boman A. L., Piper R. C. (2004) GGA proteins bind ubiquitin to facilitate sorting at the trans-Golgi network. Nat. Cell Biol. 6, 252–259 [DOI] [PubMed] [Google Scholar]
- 52. Gerrard S. R., Levi B. P., Stevens T. H. (2000) Pep12p is a multifunctional yeast syntaxin that controls entry of biosynthetic, endocytic and retrograde traffic into the prevacuolar compartment. Traffic 1, 259–269 [DOI] [PubMed] [Google Scholar]
- 53. Becherer K. A., Rieder S. E., Emr S. D., Jones E. W. (1996) Novel syntaxin homologue, Pep12p, required for the sorting of lumenal hydrolases to the lysosome-like vacuole in yeast. Mol. Biol. Cell 7, 579–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bilodeau P. S., Winistorfer S. C., Kearney W. R., Robertson A. D., Piper R. C. (2003) Vps27-Hse1 and ESCRT-I complexes cooperate to increase efficiency of sorting ubiquitinated proteins at the endosome. J. Cell Biol. 163, 237–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Piper R. C., Cooper A. A., Yang H., Stevens T. H. (1995) VPS27 controls vacuolar and endocytic traffic through a prevacuolar compartment in Saccharomyces cerevisiae. J. Cell Biol. 131, 603–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Katzmann D. J., Stefan C. J., Babst M., Emr S. D. (2003) Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J. Cell Biol. 162, 413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Franzusoff A., Redding K., Crosby J., Fuller R. S., Schekman R. (1991) Localization of components involved in protein transport and processing through the yeast Golgi apparatus. J. Cell Biol. 112, 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tang H. Y., Munn A., Cai M. (1997) EH domain proteins Pan1p and End3p are components of a complex that plays a dual role in organization of the cortical actin cytoskeleton and endocytosis in Saccharomyces cerevisiae. Mol. Cell. Biol. 17, 4294–4304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vaduva G., Martin N. C., Hopper A. K. (1997) Actin-binding verprolin is a polarity development protein required for the morphogenesis and function of the yeast actin cytoskeleton. J. Cell Biol. 139, 1821–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Raths S., Rohrer J., Crausaz F., Riezman H. (1993) end3 and end4. Two mutants defective in receptor-mediated and fluid-phase endocytosis in Saccharomyces cerevisiae. J. Cell Biol. 120, 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Munn A. L., Stevenson B. J., Geli M. I., Riezman H. (1995) end5, end6, and end7. Mutations that cause actin delocalization and block the internalization step of endocytosis in Saccharomyces cerevisiae. Mol. Biol. Cell 6, 1721–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Takagi K., Iwamoto K., Kobayashi S., Horiuchi H., Fukuda R., Ohta A. (2012) Involvement of Golgi-associated retrograde protein complex in the recycling of the putative Dnf aminophospholipid flippases in yeast. Biochem. Biophys. Res. Commun. 417, 490–494 [DOI] [PubMed] [Google Scholar]
- 63. Risinger A. L., Kaiser C. A. (2008) Different ubiquitin signals act at the Golgi and plasma membrane to direct GAP1 trafficking. Mol. Biol. Cell 19, 2962–2972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kato U., Emoto K., Fredriksson C., Nakamura H., Ohta A., Kobayashi T., Murakami-Murofushi K., Kobayashi T., Umeda M. (2002) A novel membrane protein, Ros3p, is required for phospholipid translocation across the plasma membrane in Saccharomyces cerevisiae. J. Biol. Chem. 277, 37855–37862 [DOI] [PubMed] [Google Scholar]
- 65. Nagayama A., Kato C., Abe F. (2004) The N- and C-terminal mutations in tryptophan permease Tat2 confer cell growth in Saccharomyces cerevisiae under high pressure and low-temperature conditions. Extremophiles 8, 143–149 [DOI] [PubMed] [Google Scholar]
- 66. Surma M. A., Klose C., Simons K. (2012) Lipid-dependent protein sorting at the trans-Golgi network. Biochim. Biophys. Acta 1821, 1059–1067 [DOI] [PubMed] [Google Scholar]
- 67. Klemm R. W., Ejsing C. S., Surma M. A., Kaiser H. J., Gerl M. J., Sampaio J. L., de Robillard Q., Ferguson C., Proszynski T. J., Shevchenko A., Simons K. (2009) Segregation of sphingolipids and sterols during formation of secretory vesicles at the trans-Golgi network. J. Cell Biol. 185, 601–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bauer B. E., Rossington D., Mollapour M., Mamnun Y., Kuchler K., Piper P. W. (2003) Weak organic acid stress inhibits aromatic amino acid uptake by yeast, causing a strong influence of amino acid auxotrophies on the phenotypes of membrane transporter mutants. Eur. J. Biochem. 270, 3189–3195 [DOI] [PubMed] [Google Scholar]
- 69. Alexander R. T., Jaumouillé V., Yeung T., Furuya W., Peltekova I., Boucher A., Zasloff M., Orlowski J., Grinstein S. (2011) Membrane surface charge dictates the structure and function of the epithelial Na+/H+ exchanger. EMBO J. 30, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Krishna A. G., Menon S. T., Terry T. J., Sakmar T. P. (2002) Evidence that helix 8 of rhodopsin acts as a membrane-dependent conformational switch. Biochemistry 41, 8298–8309 [DOI] [PubMed] [Google Scholar]
- 71. McLaughlin S., Smith S. O., Hayman M. J., Murray D. (2005) An electrostatic engine model for autoinhibition and activation of the epidermal growth factor receptor (EGFR/ErbB) family. J. Gen. Physiol. 126, 41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Stevens H. C., Malone L., Nichols J. W. (2008) The putative aminophospholipid translocases, DNF1 and DNF2, are not required for 7-nitrobenz-2-oxa-1,3-diazol-4-yl-phosphatidylserine flip across the plasma membrane of Saccharomyces cerevisiae. J. Biol. Chem. 283, 35060–35069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Baldridge R. D., Graham T. R. (2012) Identification of residues defining phospholipid flippase substrate specificity of type IV P-type ATPases. Proc. Natl. Acad. Sci. U.S.A. 109, E290–E298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Parsons A. B., Lopez A., Givoni I. E., Williams D. E., Gray C. A., Porter J., Chua G., Sopko R., Brost R. L., Ho C. H., Wang J., Ketela T., Brenner C., Brill J. A., Fernandez G. E., Lorenz T. C., Payne G. S., Ishihara S., Ohya Y., Andrews B., Hughes T. R., Frey B. J., Graham T. R., Andersen R. J., Boone C. (2006) Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126, 611–625 [DOI] [PubMed] [Google Scholar]
- 75. Guo Y., Au W. C., Shakoury-Elizeh M., Protchenko O., Basrai M., Prinz W. A., Philpott C. C. (2010) Phosphatidylserine is involved in the ferrichrome-induced plasma membrane trafficking of Arn1 in Saccharomyces cerevisiae. J. Biol. Chem. 285, 39564–39573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gietz R. D., Sugino A. (1988) New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534 [DOI] [PubMed] [Google Scholar]
- 77. Kikyo M., Tanaka K., Kamei T., Ozaki K., Fujiwara T., Inoue E., Takita Y., Ohya Y., Takai Y. (1999) An FH domain-containing Bnr1p is a multifunctional protein interacting with a variety of cytoskeletal proteins in Saccharomyces cerevisiae. Oncogene 18, 7046–7054 [DOI] [PubMed] [Google Scholar]
- 78. Abe F., Iida H. (2003) Pressure-induced differential regulation of the two tryptophan permeases Tat1 and Tat2 by ubiquitin ligase Rsp5 and its binding proteins, Bul1 and Bul2. Mol. Cell. Biol. 23, 7566–7584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Abe F., Horikoshi K. (2000) Tryptophan permease gene TAT2 confers high pressure growth in Saccharomyces cerevisiae. Mol. Cell. Biol. 20, 8093–8102 [DOI] [PMC free article] [PubMed] [Google Scholar]