

Background: The vasoconstricting thromboxane A2 prostanoid receptor (TP) is physically coupled to the MaxiK channel α-subunit, down-regulating its activity, but the role of the MaxiK β1-subunit is unknown.
Results: The MaxiK β1-subunit can interact with TP independently of the MaxiK α-subunit and counteracts activated TP-induced current inhibition, and its ablation increases TP vasoconstricting potency.
Conclusion: The β1-subunit modifies TP actions.
Significance: This is the first demonstration that the MaxiK β1-subunit associates with a vasoconstricting receptor.
Keywords: G Protein-coupled Receptors (GPCRs), Membrane Proteins, Potassium Channels, Smooth Muscle, Vascular, BK Channel, MaxiK Channel, Thromboxane A2 Receptors, β1-Subunit
Abstract
The large conductance voltage- and Ca2+-activated K+ channel (MaxiK, BKCa, BK) is composed of four pore-forming α-subunits and can be associated with regulatory β-subunits. One of the functional roles of MaxiK is to regulate vascular tone. We recently found that the MaxiK channel from coronary smooth muscle is trans-inhibited by activation of the vasoconstricting thromboxane A2 prostanoid receptor (TP), a mechanism supported by MaxiK α-subunit (MaxiKα)-TP physical interaction. Here, we examined the role of the MaxiK β1-subunit in TP-MaxiK association. We found that the β1-subunit can by itself interact with TP and that this association can occur independently of MaxiKα. Subcellular localization analysis revealed that β1 and TP are closely associated at the cell periphery. The molecular mechanism of β1-TP interaction involves predominantly the β1 extracellular loop. As reported previously, TP activation by the thromboxane A2 analog U46619 caused inhibition of MaxiKα macroscopic conductance or fractional open probability (FPo) as a function of voltage. However, the positive shift of the FPo versus voltage curve by U46619 relative to the control was less prominent when β1 was coexpressed with TP and MaxiKα proteins (20 ± 6 mV, n = 7) than in cells expressing TP and MaxiKα alone (51 ± 7 mV, n = 7). Finally, β1 gene ablation reduced the EC50 of the U46619 agonist in mediating aortic contraction from 18 ± 1 nm (n = 12) to 9 ± 1 nm (n = 12). The results indicate that the β1-subunit can form a tripartite complex with TP and MaxiKα, has the ability to associate with each protein independently, and diminishes U46619-induced MaxiK channel trans-inhibition as well as vasoconstriction.
Introduction
The regulation of vascular tone is an integrative physiological process involving a broad range of vasoconstricting and vasorelaxing factors. The large conductance voltage- and Ca2+-activated K+ channel (MaxiK, BKCa, BK)6 is one of the key regulators of vascular tone depending on the particular level of activity achieved in response to vasoactive substances.
In smooth muscle, MaxiK is usually composed of pore-forming α- and regulatory β1-subunits. The β1-subunit increases the channel voltage/calcium sensitivity and slows its gating kinetics (1–4). Physiologically, β1 expression influences blood pressure and is down-regulated in hypertension and diabetes (5–9). Only a few studies have examined the role of β1 in vasorelaxant- or vasoconstrictor-mediated alterations of MaxiK channel activity. In this respect, β1 is essential for lithocholate-mediated MaxiK activation linked to cerebral vasodilation and for alcohol-induced MaxiK inhibition and associated vasoconstriction (10, 11), yet the β1 relationship to vasoconstricting G protein-coupled receptors such as the thromboxane A2 prostanoid receptor (TP) is unknown.
We have recently shown that activation of TP trans-inhibits MaxiK channel activity in native coronary arterial muscle; this G-protein independent coupling is supported by the ability of TP to physically interact with the MaxiK α-subunit (MaxiKα) (12). Because in coronary arterial muscle the majority of currents resemble channels accompanied by the β1-subunit (13), the question of whether (and how) β1 relates to vasoconstricting G protein-coupled receptors (in this case) to TP actions becomes relevant.
The results from this study reveal the following. (i) The MaxiK regulatory β1-subunit can by itself interact with vasoconstricting TP. (ii) β1 expression decreases activated TP-induced positive shift in MaxiKα voltage dependence of the fractional open probability. (iii) β1 expression is linked to a reduced potency of the thromboxane A2 analog U46619 in producing vasoconstriction.
EXPERIMENTAL PROCEDURES
Animals
Three-month-old wild-type (C57BL/6NCrL, Charles River Laboratories) and β1 knock-out (β1−/−) male mice were used. The β1−/− line was prepared using conventional homologous recombination by deleting exons II and III. Gene knock-out was confirmed by genotyping and the absence of β1 mRNA in the aorta, uterus, and bladder. All animal protocols received institutional approval.
Materials and Constructs
Human MaxiKα (amino acid sequence as in U11058) with or without an N-terminal c-Myc epitope (14), and human TPα (referred to here as TP; NM_001060, TBXA2R, transcript variant 2) with or without an N-terminal c-Myc epitope were in pcDNA3 (Invitrogen). Human β1 (NM_004137, KCNMB1), human β1(1–71), and human or mouse β1(1–102) (NM_031169, Kcnmb1; mouse and human β1 share 90.2% amino acid identity in this region) were subcloned into the p3XFLAG-CMV-14 (Sigma) vector, and thus expressed a C-terminal FLAG epitope. TP and N-terminally c-Myc-tagged MaxiKα were also subcloned in the pIRES vector (TP-pIRES-c-Myc-MaxiKα; Clontech) and used only in electrophysiological experiments. Anti-c-Myc polyclonal and monoclonal antibodies and anti-FLAG polyclonal and monoclonal antibodies were from Sigma. Anti-MaxiKα monoclonal and polyclonal antibodies were from NeuroMab (clone L6/60, catalogue no. 75-022; UC Davis/NIH NeuroMab Facility) and Alomone Labs (catalogue no. APC-021, lot N05), respectively. Anti-TP polyclonal antibody (catalog 10004452, lot 129010-12011) was from Cayman Chemical. Secondary antibodies for immunolabeling and immunoblotting were from Molecular Probes and LI-COR Biosciences, respectively. U46619 was from Cayman Chemical.
HEK293T Cell Culture and DNA Transfection
HEK293T cells were grown in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 2 units/ml penicillin, and 2 mg/ml streptomycin at 37 °C in a humidity-controlled incubator supplemented with 5% CO2. HEK293T cells at 80% confluence were transiently transfected using Lipofectamine 2000 (Invitrogen). Plasmid concentrations used for transfection are given in the figure legends.
Co-immunoprecipitation Assay
HEK293T cells expressing the different constructs were lysed in lysis buffer (150 mm NaCl, 50 mm Tris, 5 mm EDTA, 10 mm HEPES, 0.1% IGEPAL CA-630, 0.25% sodium deoxycholate (pH 7.4), and protease inhibitors (Complete protease inhibitor mixture tablets (one tablet/50 ml), Roche Applied Science). Lysates were centrifuged at 13,000 × g for 10 min at 4 °C, and the supernatants were precleared with 10 μl of protein A/G resin (Pierce)/mg of protein for 1 h at 4 °C with shaking and centrifuged at 2000 × g for 2 min. The precleared lysates (1 mg of protein) were incubated overnight at 4 °C with 10 μl of antibody-saturated protein A/G resin (2 μg of antibody/10 μl of resin were incubated for 2 h at 4 °C with shaking) in a final volume of 500 μl. Samples were centrifuged at 2000 × g for 2 min and washed five times with lysis buffer. The immunoprecipitated proteins were eluted from the beads with 30 μl of 3×Laemmli sample buffer at 37 °C for 1 h and centrifuged at 13,000 × g for 3 min at 4 °C. Immunoprecipitated proteins and input lysates were analyzed by SDS-PAGE and immunoblotting. Molecular weight markers were from LI-COR Biosciences (catalogue no. 928-40000) except those used for TP, which were low-range SDS-PAGE standards from Bio-Rad (catalogue no. 161-0305). The Odyssey® infrared imaging system (LI-COR Biosciences) was used to analyze single- or double-labeled immunoblots.
Immunolabeling
HEK293T cells were plated onto poly-d-lysine-coated coverslips 24 h after transfection and incubated at 37 °C for another 24 h. Live cells were incubated with 5 μg/ml anti-c-Myc polyclonal antibody for 1 h on ice in a 37 °C incubator supplemented with 95% air and 5% CO2 atmosphere; washed once with PBS (2.67 mm KCl, 138 mm NaCl, 1.47 mm KH2PO4, and 8.1 mm Na2HPO4 (pH 7.4)), and then fixed with 4% paraformaldehyde in phosphate buffer (0.1 m Na2HPO4, and 0.022 m NaH2PO4 (pH 7.4)). Cells were permeabilized and blocked with PBS containing 0.2% Triton X-100 and 10% normal goat serum for 30 min at room temperature, followed by incubation with 5 μg/ml anti-FLAG monoclonal antibody in PBS containing 0.2% Triton X-100 and 1% normal goat serum overnight at 4 °C. Cells were washed three times with PBS containing 0.2% Triton X-100 and labeled with 2 μg/ml secondary antibodies (Alexa Fluor 568 anti-mouse and Alexa Fluor 488 anti-rabbit) for 1 h at room temperature. Cells were rinsed twice with PBS containing 0.2% Triton X-100 and once with PBS alone. Cells were then mounted on slides using ProLong Gold (Invitrogen). Images were taken with an Olympus confocal microscope. All conditions, including optical sectioning and acquisition parameters, were identical for a given experiment.
Protein Proximity Index Analysis
To calculate the protein proximity index (PPI) (12, 15), pairs of digital images were acquired at 0.0288 μm/pixel and subjected to the following analysis using a custom-made program.
Median Filter
First, the nonspecific background value at each pixel was estimated by calculating the median value of a 32 × 32 pixel square centered at the target pixel. This value was then subtracted from the intensity of the target pixel.
Autocorrelation and Cross-correlation Analysis
Second, three-dimensional autocorrelation (of each TP and β1 image) and cross-correlation (of TP and β1 images) plots as a function of pixel shift in the x,y axis were constructed. Corresponding contour plots were generated, and line scans were obtained to plot the correlation intensity values as a function of pixel shift.
Fitting
Third, line scan plots were then fitted to the sum of two Gaussian functions: (A1exp(−(M1 − x)2/2B12)) + (A2exp(−(M2 − x)2/2B22)) − Base, where A1 and A2 are the amplitudes, B1 and B2 are the widths, M1 and M2 are the means of the position of the peaks in the x axis, x is the pixel shift, and Base is a constant value. The fits displayed sharp and shallow components. The sharp components (A1) correspond to specific labeling, and the shallow components (A2) correspond to antibody background and random colocalization.
PPI Calculation
Finally, the PPI values were obtained by dividing A1 from the cross-correlation analysis by A1 from each of the autocorrelation analyses.
HEK293T Cell Patch Recording
TP and c-Myc-MaxiKα were subcloned into the pIRES vector with TP under the control of the CMV promoter and with c-Myc-MaxiKα under the control of the internal ribosome entry site (TP-pIRES-c-Myc-MaxiKα). This was necessary to ensure coexpression of both proteins in the same cell. GFP was coexpressed to help monitor the transfected HEK293T cells. Twenty-four hours after transfection with TP-pIRES-c-Myc-MaxiKα with or without the β1-subunit, macroscopic currents were recorded in the inside-out patch configuration. The half-activation potential (V½) was measured to monitor the coexpression of β1. A negative shift of ∼90 mV by the β1-subunit indicated full coupling (2). The pipette and bath solution contained 105 mm potassium methanesulfonate, 5 mm KCl, 5 mm HEDTA, 3.9 mm CaCl2, and 10 mm HEPES (pH 7.4). The free calcium concentration ([Ca2+]i) of the bath solution measured with a Ca2+ electrode was 6.7 μm (facing the intracellular side of the channel). U46619 (500 nm) was dissolved in bath solution and perfused. Pipette resistances were 2.5–3.5 megohms. Currents were filtered at 1 kHz and digitized at 10 kHz. Instantaneous tail currents (I) measured at the beginning of a constant repolarizing pulse to −70 mV were used to fit a Boltzmann distribution of the following form. FPo = G/Gmax = 1/(1 + exp((V½ − V)zδF/RT)), where FPo is the fractional open probability; G is the macroscopic conductance; Gmax is the maximum macroscopic conductance; V is the voltage of the test pulse (preceding the repolarizing pulse); V½ is the half-activation potential; zδ is the effective valence; and F, R, and T have their usual thermodynamic meanings. G = I/(V − EK), where I is the instantaneous tail current, V is the voltage of the constant repolarizing pulse (in this case, −70 mV), and EK is the reversal potential for K+ (in this case, 0 mV). Gmax = Imax/(V − EK), where Imax is the maximum instantaneous tail current and the other parameters have been defined. Note that the voltage dependence of the macroscopic conductance (G) reflects the voltage dependence of channel Po (16), and thus, G/Gmax = FPo.
Isometric Contraction
Aortic rings (endothelium denuded with a cotton thread) were mounted in an organ bath kept at 37 °C, filled with modified Krebs solution (119 mm NaCl, 4.7 mm KCl, 1.6 mm CaCl2, 1.2 mm MgSO4, 1.2 mm KH2PO4, 22 mm NaHCO3, 8 mm HEPES, 5 mm creatine, 20 mm taurine, 5 mm pyruvate, and 5 mm glucose (pH 7.4); gassed with 95% O2 and 5% CO2), and connected to an isometric force transducer (World Precision Instruments). Tension was recorded with WinDaq (DATAQ Instruments). After equilibration for 60 min at the optimum resting tension (∼7.8 millinewtons), rings were contracted with 80 mm KCl for ∼15 min and then washed out with Krebs solution. After equilibration for another 30 min, cumulative concentrations of U46619 (0.01 nm to 3 μm) were applied. Contraction was normalized to KCl-induced contraction and expressed as a percentage. The calculation was as follows: % contraction = 100 × (U46619-induced aortic tension − basal tension)/(80 mm KCl-induced aortic tension − basal tension). Half-maximum effective concentration values (EC50) were calculated using a Hill function: % contraction = Emax/(1 + EC50/[U46619])H, where Emax is the maximum contraction and H is the Hill coefficient. To compare KCl contraction in wild-type (β1+/+) versus knock-out (β1−/−) aortic rings, maximum contraction was normalized to the dry weight of each ring.
Statistical Analysis
Data are presented as means ± S.E. Statistical comparisons between groups were made using Student's t test. A p value <0.05 was considered statistically significant.
RESULTS
β1 Does Not Prevent MaxiKα-TP Interaction and Is Able to Independently Associate with TP
To investigate whether (and how) β1 modifies TP actions, we first determined if β1 can alter MaxiKα-TP association. To this end, HEK293T cells were transiently cotransfected with human β1 (C-terminally tagged with a FLAG epitope), TP (N-terminally tagged with a c-Myc epitope), and MaxiKα (Fig. 1A) or with any protein pair to perform parallel co-immunoprecipitation experiments (n = 3 each). Anti-TP antibody readily co-immunoprecipitated MaxiKα in the absence of β1 (Fig. 1B, lane 2) or in its presence (lane 4). Negative controls in which TP (Fig. 1B, lane 1) and MaxiKα (lane 3) were not cotransfected showed no signal. Used as a positive control, β1 pulled down MaxiKα in the absence of TP (Fig. 1C, lane 1) but also in its presence (lane 4). The appropriate expression of TP in the input lysates is shown in Fig. 1D; for proper immunoprecipitation of β1, see Fig. 1G. Quantification of TP efficiency in pulling down MaxiKα yielded similar results in the absence (control) and presence of β1 (Fig. 1E, white and black bars); co-immunoprecipitation efficiency was calculated by normalizing the band intensities of co-immunoprecipitated MaxiKα (Fig. 1B, lanes 2 and 4) to those of immunoprecipitated TP (Fig. 1F, lanes 2 and 4) and to MaxiKα expression in the corresponding input lysates (Fig. 1H, lanes 2 and 4). Overall, the experiments demonstrate that β1 is unable to disrupt MaxiKα-TP association but, at the same time, can interact with MaxiKα.
FIGURE 1.

Association of the MaxiK β1-subunit with TP is independent of MaxiKα. A, scheme of constructs used: MaxiKα, c-Myc-TP, and β1-FLAG. B, TP co-immunoprecipitates MaxiKα in the presence (lane 4) and absence (lane 2) of β1. C, β1 pulls down MaxiKα in the presence (lane 4) and absence of TP (lane 1). D, proper expression of TP in lysates from transfected cells. E, TP co-immunoprecipitates (Co-IP) MaxiKα with similar efficiency regardless of the presence (black bar) or absence (white bar; control) of β1. Similarly, TP co-immunoprecipitates β1 regardless of the presence (checkered bar) or absence (dotted bar; control) of MaxiKα. F, proper immunoprecipitation of TP. G, proper immunoprecipitation of β1. H, proper expression of MaxiKα in cell lysates. I, β1 immunoprecipitates TP independently of the presence (lane 4) or absence (lane 3) of MaxiKα. J, reverse co-immunoprecipitation showing that TP is also able to immunoprecipitate β1 in the presence (lane 4) and absence (lane 3) of MaxiKα. K, proper expression of β1 in input cell lysates. D, H, and K, 20 μg of protein/lane; H and K, double-immunolabeled. The double dots mark the position corresponding to the molecular mass of the TP dimer. HEK293T cells were cotransfected with 8 μg of each plasmid/4 ml of medium in a 10-cm dish; pcDNA3 was used for mock transfection (−). In this and the following figures, numbers on the left of each blot indicate mass in kilodaltons. IP, immunoprecipitation; IB, immunoblot.
Unexpectedly, we also found that β1 could readily immunoprecipitate TP without MaxiKα being present (Fig. 1I, lane 3 versus lane 4). This novel interaction was verified by reverse co-immunoprecipitation, where TP pulled down β1 in the absence of MaxiKα (Fig. 1J, lane 3). Moreover, the presence of MaxiKα did not modify the degree of interaction between TP and β1, as co-immunoprecipitation efficiency was the same in the absence (control) and presence of MaxiKα (Fig. 1E, dotted and checkered bars). Efficiency was obtained by normalizing co-immunoprecipitated β1 (Fig. 1J, lanes 3 and 4) to immunoprecipitated TP (Fig. 1F, lanes 3 and 4) and to β1 expression in the corresponding cell lysates (Fig. 1K, lanes 3 and 4). The proper expression of β1, TP, and MaxiKα is shown in immunoblots of input lysates in Fig. 1 (D, H, and K). In summary, β1 is able to associate with both MaxiKα and TP, forming a tripartite complex, but its association with TP is independent of its interaction with MaxiKα.
β1 and TP Colocalize at the Plasma Membrane
We next examined whether β1 and TP can colocalize at the plasma membrane without the assistance of MaxiKα. HEK293T cells cotransfected with β1-FLAG (tagged at its intracellular C terminus) and c-Myc-TP (tagged at its extracellular N terminus) were first labeled live to assess the plasma membrane localization of TP. This was followed by fixation, permeabilization, and labeling of β1. Confocal images of immunolabeled cells show a high degree of correlation between TP signals at the plasma membrane (Fig. 2A) and β1 labeling (Fig. 2B), which is emphasized in the overlay (Fig. 2C). PPI (12, 15) was calculated to quantify β1 and TP “colocalization” as described under “Experimental Procedures.” Panels D and E in Fig. 2 show the autocorrelation three-dimensional plots as a function of pixel shift of TP (panel A) and β1 (panel B) images, respectively. The cross-correlation three-dimensional plot of both TP and β1 images is shown in Fig. 2F. At zero pixel shift, the surface plots have a peak that decays abruptly by shifting the image a few pixels, indicating specific TP and β1 signals (Fig. 2, D and E) and specific colocalization (Fig. 2F). Fig. 2G is the contour plot of Fig. 2D, where the dashed white line was used to construct the line scan plot (autocorrelation intensity versus pixel shift plot) in Fig. 2H. Similar contour plots were constructed for panels E and F to obtain the line scan plots in panels I and J. Experimental data (black dashed lines) in panels H–J were individually fitted to the sum of two Gaussian functions (red lines). All fittings showed two components, a sharp and a shallow component. The sharp component corresponds to the specific labeling (panels H and I) and specific colocalization (panel J), whereas the shallow component corresponds to antibody background (panels H and I) and random colocalization (panel J). The blue dashed lines in panels H–J are the calculated sharp components using the amplitude (A1) and width (M1) of each fit. Amplitudes were then used to calculate the fraction of TP in proximity (colocalized) with β1 and vice versa. In this specific case, PPI for TP → β1 = 35.4/48.9 = 0.72 and that for β1 → TP = 35.4/66.6 = 0.53. The mean values of PPI for 18 cells were 0.58 ± 0.03 for TP → β1 and 0.58 ± 0.04 for β1 → TP (Fig. 2K) indicating that at least 58% of labeled TP is in close proximity to β1 and vice versa. These results map β1-TP interaction to the plasma membrane, although other trafficking locations are not excluded, and confirm that β1-TP association does not require MaxiKα expression.
FIGURE 2.

TP and β1 are expressed in close proximity. A and B, HEK293T cells expressing c-Myc-TP and β1-FLAG were immunolabeled live for TP with anti-c-Myc polyclonal antibody (green). After permeabilization, cells were immunolabeled for β1 with anti-FLAG monoclonal antibody (red). C, overlay of images in A and B. D–F, three-dimensional plots of c-Myc-TP or β1-FLAG autocorrelation or c-Myc-TP + β1-FLAG cross-correlation versus pixel shift on the x,y axis. G, contour plot of D. H, line scan plot of the white dashed line in G. I and J, line scan plots of contour plots from E and F, respectively. In H–J, the black dashed lines are the experimental data, the red lines are the fit to a double Gaussian distribution, and the blue dashed lines are the sharp component. The amplitude of the sharp components (A1) was used to calculate PPI. In this example, PPI values, which quantify the degree of TP clusters interacting with β1 (TP → β1) and vice versa (β1 → TP) were as follows: TP → β1 = 35.4/48.9 = 0.72 and β1 → TP = 35.4/66.6 = 0.53. K, mean PPI values were near 0.6 (on a scale of 0–1) in both instances (n = 18). Cells were cotransfected with 3.5 μg of each plasmid/2 ml of medium in a 60-mm dish.
β1 Extracellular Loop Contributes to β1-TP Interaction
Which domain of β1 is required for association with TP? Fig. 3A illustrates the topology of wild-type β1 (β1(1–191)) and deletion constructs (β1(1–102) and β1(1–71)) designed to circumscribe the region relevant for TP association. Black circles represent residues encoded by exon II, gray circles represent those encoded by exon III, and open circles represent those encoded by exon IV. All three constructs were tagged with a FLAG epitope at the C terminus. The first deletion construct tested, β1(1–102), lacked all residues encoded by exon IV (residues 103–191, open circles). The idea behind this was that sequences in exons may have been evolutionarily selected as functional cassettes and thus could be a good starting point to look for residues that interact with TP. Fig. 3B shows that similar to wild-type β1 (β1(1–191)), β1(1–102) readily interacted with TP (lanes 1 and 2), discarding residues 103–191 as relevant for β1-TP association. We next deleted amino acids 72–102 (encoded within exon III), making construct β1(1–71). The β1(1–71) construct lost its ability to interact with TP (Fig. 1B, lane 3). Because the β1(1–102) construct contained residues 72–102 and associated with TP in contrast to β1(1–71), which did not, it is safe to suggest that the sequence 72PQYPCLWVNVSAAGRWAVLYHTEDTRDQNQ102 plays an important role in determining β1-TP association.
FIGURE 3.

β1 domain involved in β1-TP interaction. A, topological schemes of wild-type β1 (β1(1–191)) and deletion constructs β1(1–102) and β1(1–71). Black circles represent amino acids 1–45 encoded by exon II, gray circles represent amino acids 46–102 encoded by exon III, and open circles represent amino acids 103–191 encoded by exon IV. β1 constructs were tagged with a FLAG epitope at the C terminus. B–F, HEK293T cells were transfected with c-Myc-TP and β1 constructs (4 μg of each plasmid/2 ml of medium in a 60-mm dish) for co-immunoprecipitation analysis. B, TP effectively pulls down β1(1–191) (wild-type) and the β1(1–102) deletion construct. β1(1–102) from mouse or human yielded similar results. In contrast, TP associated poorly with β1(1–71). TP immunoprecipitation (IP) was performed with anti-c-Myc polyclonal antibody, and immunoblotting (IB) was carried out with anti-FLAG monoclonal antibody. C, the control shows similar TP immunoprecipitation in all instances, confirming the poor association of TP with β1(1–71). Anti-c-Myc polyclonal antibody was used for immunoprecipitation, and anti-c-Myc monoclonal antibody was used for immunoblotting. D and E, double-labeled immunoblot of the corresponding input lysates (18 μg of protein/lane) showing the expression levels of β1 constructs and TP, respectively. The dashed boxes mark β1-subunit dimers. The double dots mark the position corresponding to the putative TP dimer. F, mean values of co-immunoprecipitation (Co-IP) efficiency (n = 3) calculated as follows: Co-IP efficiency (%) = 100 × (density of co-immunoprecipitated β1 construct)/((density of immunoprecipitated TP) × (density of β1 construct in input cell lysate)). *, significantly different from TP + β1(1–191) (p < 0.01).
Used as a positive control, TP was effectively immunoprecipitated in all instances (Fig. 3C); both monomeric and dimeric TP species were detected at ∼40 and ∼80 kDa (arrow and double dots, respectively). Proper expression of β1 constructs (Fig. 3D) and TP (Fig. 3E) was tested by immunoblotting cell lysates used for immunoprecipitation. Wild-type β1 and β1(1–102) deletion constructs showed clear bands at higher molecular masses, which would correspond to dimers of the protein (Fig. 3D, lanes 1 and 2, dashed boxes). To compare the efficiency of TP in pulling down the β1 constructs, the density of co-immunoprecipitated β1 bands (Fig. 3B) was quantified in each case and normalized to immunoprecipitated TP signals (Fig. 3C) and to the signal of β1 bands in cell lysates (Fig. 3D). The bands corresponding to the protein monomer and dimer were taken into account when analyzing the data. Mean values of normalized density are shown in Fig. 3F. The efficiency of TP in co-immunoprecipitating β1(1–102) was 94 ± 14% (n = 3), which was not significantly different from that of TP in pulling down wild-type β1(1–191) (set to 100%). These data demonstrate no dramatic change in β1-TP interaction after deletion of residues encoded by exon IV. On the other hand, further deletion of residues 72–102 of the β1 extracellular loop (β1(1–71)) yielded a protein that was no longer able to interact well with TP. The co-immunoprecipitation efficiency dropped nearly 90% to an efficiency of 13 ± 6% (n = 3), indicative of the role of the β1 extracellular loop, likely of residues 72–102, in β1-TP association.
β1 Decreases Activated TP Reduction in MaxiKα Fractional Open Probability
The results so far indicate that β1 can independently interact with either TP or MaxiKα. Therefore, we next asked the question whether β1 could modify TP and MaxiKα functional coupling, whereby activated TP trans-inhibits MaxiKα (12). To address this question, we compared the effect of a stable analog of thromboxane A2 (U46619) on MaxiKα macroscopic currents coexpressed with TP in the presence and absence of β1 in HEK293T cells (Fig. 4). In all experiments, the same patch expressing TP and MaxiKα alone (TP-pIRES-c-Myc-MaxiKα) or in conjunction with the β1-subunit was used to measure the current before (control) and after U46619 treatment. The holding potential was 0 mV, [K+] was the same in the bath and patch pipette, and [Ca2+] facing the intracellular side of the membrane was 6.7 μm. Under these conditions, at 0 mV, there was no net current flux (Fig. 4, A–D), as the K+ concentration was equal at both sides of the membrane, yet under control conditions, channels had a significant FPo of >0.6 at this voltage (Fig. 4, E and F, black circles). As a consequence of the latter, when patches were stimulated by a series of test pulses, control currents displayed very fast activation kinetics (unresolved by the acquisition system, yielding traces that were almost square) (Fig. 4, A, C, and D), and at very negative potentials, it was possible to observe “tail” currents (arrowheads) that deactivated to reach the steady state for the particular voltage.
FIGURE 4.

β1 reduces TP-mediated positive shift in MaxiK channel voltage dependence of FPo. A and B, MaxiK currents from an inside-out HEK293T cell patch expressing TP and MaxiKα. Current traces are from the same patch upon excision (A; control) and after U46619 (500 nm) application (B). C and D, MaxiK currents from an inside-out patch of a cell coexpressing TP, MaxiKα, and β1. Current traces are from the same cell patch before (C; control) and after (D) 500 nm U46619 treatment. Insets in A–D are corresponding I-V curves. E, mean FPo versus membrane potential plot before (●; control) and after (○) 500 nm U46619 treatment from cells coexpressing TP and MaxiKα. As reported earlier, U46619 induced a rightward shift of the plot, consistent with significant MaxiK channel trans-inhibition (12). F, mean FPo versus membrane potential plot from cells coexpressing TP, MaxiKα, and β1. Data were fitted to Boltzmann distributions (continuous lines; see “Experimental Procedures”). U46619-containing solution was perfused to the bath (intracellular side of the patch), and its effect was recorded after 15 min of exposure. MaxiK currents were elicited by step depolarizations (test pulses) from −140 to 140 mV for A and B and from −180 to 100 mV for C and D. The repolarizing potential was −70 mV. The arrowheads mark tail currents elicited at the beginning of test pulses, and the arrows indicate the time point of instantaneous tail current measurements. [Ca2+]i was maintained constant at 6.7 μm. The holding potential was set at 0 mV. The TP-pIRES-c-Myc-MaxiKα plasmid was transfected at 0.6 or 0.9 μg, and β1 was at 1.6 μg, both in 1 ml of medium in a 35-mm dish.
Confirming our previous findings (12), in patches expressing TP and MaxiKα alone, application of 500 nm U46619 to the same cell patch dramatically slowed down MaxiKα current activation kinetics at positive potentials (Fig. 4, B versus A) and reduced tail currents at the beginning of negative test pulses (arrowheads), both indicative of a TP-mediated decrease in channel Po or channel inhibition. This inference is based on the well established concepts that the time course of channel average Po corresponds to the macroscopic current (16) and that the instantaneous tail current is proportional to channel Po at the end of the preceding pulse (17).
The macroscopic current (I) at the end of test pulses was measured to construct current-voltage relationships (I-V curves). The I-V curves show that TP activation by U46619 produced a decrease in macroscopic current amplitude at several potentials (Fig. 4, B versus A, insets). Because I = NiPo, where N is the number of channels, i is the unitary conductance, and Po is the channel open probability, a reduction in I could be due to an inhibition of any of the three parameters. However, we showed previously that activated TP inhibits channel Po but does not decrease i or N in inside-out patches (12). To confirm an inhibition of MaxiK Po by activated TP, we evaluated the voltage dependence of the macroscopic conductance or FPo before (control) and after U46619 treatment (Fig. 4E) by measuring instantaneous tail currents at the beginning of the repolarizing pulse (Fig. 4, A and B, arrows) (see “Experimental Procedures”). It is clear from these measurements that U46619 induced a 51 ± 7 mV shift of the voltage dependence of FPo from a half-activation potential (V½) of −15 ± 5 mV (control; n = 7) to a V½ of 37 ± 7 mV (n = 7). This positive shift by U46619 can be interpreted as an inhibition of MaxiKα channel Po by activated TP.
Parallel experiments were performed in cells coexpressing TP and MaxiKα (in the pIRES vector) together with the β1-subunit (Fig. 4, C and D). To saturate the effect of β1, the molar ratio of transfected β1 to TP + MaxiKα was 3:1. Under these conditions and consistent with previous studies (2), in the presence of β1, the V½ of MaxiKα was −99 ± 2 mV at [Ca2+]i = 6.7 μm (Fig. 4F, Control).
The presence of β1 modified the U46619 effect on MaxiKα macroscopic currents. The kinetics of current activation at positive test potentials and tail currents at the beginning of negative test pulses induced by U46619 remained practically the same as under control conditions (Fig. 4, D versus C), whereas I-V curves measured at the end of test pulses showed a modest decrease (Fig. 4, D versus C, insets). The voltage dependence of FPo was also calculated from tail currents at the beginning of the repolarizing pulse (Fig. 4, C and D, arrows). In this case, U46619 caused a smaller but significant shift of 20 ± 6 mV in the voltage dependence of FPo from a V½ of −99 ± 2 mV (n = 7) in the control to −79 ± 8 mV (n = 7) after treatment (Fig. 4F).
To rule out the remote possibility that the decreased effect of U46619 in cells coexpressing TP + MaxiKα + β1 was due to a selective inhibition of TP expression compared with MaxiKα when coexpressed with β1, we measured protein expression levels in these cells and compared them with the levels of expression in cells transfected with TP and MaxiKα alone. Immunoblot analysis showed that when the β1-subunit was expressed, both TP and MaxiKα relative expression levels were proportionally decreased, TP by 65 ± 1% (n = 3) and MaxiKα by 59 ± 7% (n = 3) (p = 0.37), ruling out selective decreased TP expression by β1 cotransfection. Together, the results support the view that β1 modifies the way activated TP modulates MaxiKα activity.
β1 and U46619-induced Aortic Vasoconstriction
In addition to causing MaxiKα channel inhibition, activation of TP by the thromboxane A2 analog U46619 induces vasoconstriction as an end point of its signaling cascade. Because β1 decreased the TP-mediated shift in the MaxiKα voltage dependence of FPo, we questioned whether β1 could also reduce U46619-induced vasoconstriction. Aortic rings isolated from wild-type β1 (β1+/+) and β1 knock-out (β1−/−) mice were contracted by cumulative concentrations of U46619 (0.01–3000 nm) (Fig. 5A). Prior to U46619 treatment, aortic rings were contracted with 80 mm KCl and then washed with Krebs solution. The mean maximum contractile responses to 80 mm KCl in β1+/+ and β1−/− mice were similar after being normalized to the dry weight of the aortic rings (Fig. 5B, inset). Likewise, the maximum contraction elicited by U46619 was similar in β1+/+ and β1−/− mice (Fig. 5B). In contrast, the U46619 potency in eliciting contractile responses was increased in β1−/− aortic rings with respect to those from β1+/+ animals. The mean dose-response curves in Fig. 5B (normalized to 80 mm KCl contraction) demonstrate that the U46619 dose needed to achieve 50% of the contraction (EC50) in β1−/− mice was only half (9 ± 1 nm, n = 12) of that needed in β1+/+ mice (18 ± 1 nm, n = 12). Fig. 5C also shows that the percent contraction (normalized to KCl contraction) at 18 nm U46619 (EC50 in β1+/+ mice) was lower in mice expressing the β1-subunit (β1+/+) than in its absence. These results indicate that the presence of β1 plays an important role in controlling TP agonist-induced contraction as well.
FIGURE 5.

β1 buffers U46619-induced aortic contraction. A, typical contractile responses of aortic rings from β1+/+ (upper) and β1−/− (lower) mice elicited by 80 mm KCl and by cumulative concentrations of U46619 (0.01–3000 nm). The arrows mark the times of addition of U46619, and w indicates washout. B, mean dose-response curves of U46619 in aortic rings isolated from β1+/+ (○) and β1−/− (●) mice. Contraction was normalized to KCl response and is expressed as a percentage. A significant leftward shift was observed in the β1−/− mice (change in potency) without a change in maximum contraction (efficacy). The EC50 (a measure of drug potency) was 9 ± 1 nm (n = 12) for β1−/− mice versus 18 ± 1 nm (n = 12) for β1+/+ mice (p < 0.001), whereas the maximum contraction was 284 ± 69% and 315 ± 98% (p = 0.5) with respect to KCl contraction, respectively. The inset shows similar KCl-induced contraction in β1+/+ and β1−/− mice when normalized to the dry weight of the rings. This normalization was applied because relying on the approximate width of aortic rings (∼3–4 mm) can yield significant errors. C, the calculated percent contraction at 18 nm U46619 (EC50 in β1+/+ mice) was higher in β1−/− mice than in β1+/+ mice. *, statistically different with respect to β1−/− animals (p < 0.01).
DISCUSSION
In this study, we present the first demonstration that the MaxiK regulatory β1-subunit can by itself interact with TP and at the same time can assemble in a tripartite complex with MaxiKα and TP. Furthermore, our study provides evidence that β1 is able to reduce TP agonist thromboxane A2-induced functional effects, i.e. thromboxane A2-induced MaxiKα inhibition as well as vasoconstriction.
TP, β1, and MaxiKα in a Macromolecular Complex
MaxiKα and β1 are known to associate with each other without the need of an intermediary protein, e.g. TP. Expressed β1 is known to be in close contact with MaxiKα, enabling covalent cross-linking of engineered cysteines in both proteins (18). Our experiments also confirmed this physical association by co-immunoprecipitation without coexpression of TP (Fig. 1C). On the other hand, our previous studies have shown a close proximity of MaxiKα with TP, enabling Förster resonance energy transfer without the need of β1 (12). Based on these premises, it was possible that β1 and TP might associate via MaxiKα (Fig. 6A) or that β1 could disrupt MaxiKα-TP association (Fig. 6B). Unexpectedly, we found that β1 could co-immunoprecipitate TP without the assistance of MaxiKα (Fig. 1I) and vice versa (Fig. 1J) and that β1 did not disrupt MaxiKα-TP association (Fig. 1, B and E, white and black bars). Moreover, co-immunoprecipitation efficiency between β1 and TP was not modified by the additional coexpression of MaxiKα (Fig. 1, E (dotted and checkered bars), I, and J). Together, these data demonstrate that β1-TP association can occur independently of MaxiKα (Fig. 1B), reminiscent of MaxiKα-TP interaction that occurs independently of β1 (12). In this scenario, one can picture that in the tripartite complex, β1 interacts with both MaxiKα and TP via different regions (Fig. 6C). At present, however, we cannot discard the possibility that β1-TP interaction involves an intermediary protein.
FIGURE 6.
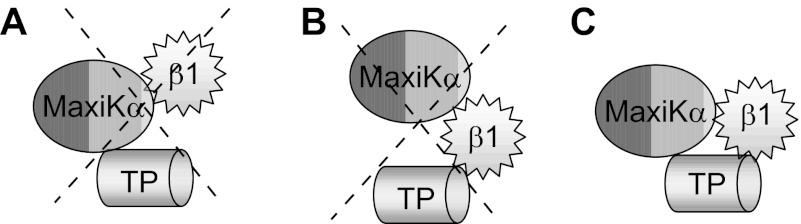
Model of β1, TP, and MaxiKα tripartite complex. A, model in which MaxiKα mediates β1-TP association. B, model in which β1 disrupts the previously demonstrated MaxiKα-TP physical interaction (12). These possibilities are not supported by the experiments in Fig. 1. C, model in which β1 interacts with MaxiKα (18), MaxiKα interacts with TP (12), and β1 associates with both via independent mechanisms. This model is supported by the experiments in Fig. 1. Whether β1 interacts directly with TP or via an intermediary protein is unknown.
Molecularity of β1-TP Interaction
β1 has a membrane topology with two membrane-spanning segments (TM1 and TM2), short intracellular N and C termini, and a large extracellular loop (Fig. 3A). Our results point to residues 72–102 within the β1 extracellular loop as relevant for the association between β1 and TP. On the other hand, it is known that β1 can interact with MaxiKα via multiple segments. For example, the β1 intracellular N and C termini are engaged in functional regulation of MaxiKα (19), whereas extracellular loop residues proximal to both TM1 and TM2 (positions 40–45 and 152–155) are close to MaxiKα, as interprotein disulfide bridges can be formed after appropriate cysteine engineering (18). In conjunction, these studies point to the view that the interacting sites between β1 and TP are distinct from those between β1 and MaxiKα and are consistent with the observation that the association efficiency of TP and β1 was not modified by coexpression of MaxiKα. In the scenario that TP interacts with β1 through its extracellular loop via residues within positions 72–102, as suggested by our studies, MaxiKα could still bind/interact with β1 through residues proximal to TM1 and TM2 without interrupting β1-TP interaction.
β1 and TP-mediated Function
β1 is an important regulatory subunit of MaxiK channels in the vasculature (7). In human coronary smooth muscle cells, single-channel voltage activation curves could be sorted in five groups, suggesting different degrees of MaxiK channel association with the β1-subunit, with ∼70% of the channels likely in complex with 3–4 β1-subunits, ∼25% of the channels in complex with 1–2 β1-subunits, and ∼5% of the channels not in apparent association with the β1-subunit (13). Moreover, reduced expression of β1 is related to vascular dysfunction in hypertension (20, 21), supporting a key role of this MaxiK channel subunit in vasoregulation.
Our present results indicate that the β1-subunit regulates TP-mediated MaxiK channel trans-inhibition in a way that the activated TP effect would be exerted most potently on channels formed by the α-subunit alone than on channels formed by α/β1-subunits (Fig. 4). Still, TP activation induces a significant inhibition of channels expressing saturating levels of the β1-subunit (confirmed by the very negative V½ value of approximately −100 mV at 6.7 μm Ca2+i under control conditions) as evident from a 20 mV rightward shift of the voltage dependence of the FPo curve. By comparison, in human coronary cells, TP-mediated MaxiK inhibition yielded an ∼35 mV rightward shift of the channel voltage activation curve (12), which is consistent with the majority of MaxiK channels formed by α- and β1-subunits in this smooth muscle cell type.
TP-mediated channel inhibition of cells expressing the α-subunit alone showed an ∼50 mV rightward shift (Fig. 4E) of the FPo versus voltage curve, which is larger than the ∼30 mV shift reported in our previous studies under similar experimental conditions (12). At present, we do not have a clear explanation for this difference, but we have noted that distinct batches of the commercially available TP agonist U46619 may display different potencies. Nevertheless, the present studies performed in parallel clearly show that channels formed by the α-subunit alone respond more effectively to U46619-induced inhibition than those formed by α/β1-subunits.
We also analyzed the potential effect of β1-subunit expression on the end result of TP activation, vasoconstriction. Utilizing aortas from β1 knock-out and wild-type mice, we found a significant EC50 reduction in U46619-induced aortic contraction (without a significant change in maximum contraction) in the absence of β1 expression. This result reveals a protective effect of β1 against thromboxane A2-induced vasoconstriction and also provides evidence for a physiological coupling between β1 and TP. A lack of change in the maximum response in β1−/− versus β1+/+ mice indicates no change in the number of activated TP receptors after knocking out β1 expression. However, the change in EC50 after normalization to KCl contraction reflects an intrinsic change in the TP-triggered contractile system in β1−/− mice, which could comprise (i) a change in cellular signaling pathways, including decreased MaxiK activity due to loss of β1, and/or (ii) a change in TP agonist affinity due to loss of β1-TP interaction. Interestingly, norepinephrine-induced aortic contraction normalized to KCl contractile response has been shown to be identical in β1−/− and β1+/+ animals (8). It would be interesting to determine whether β1 associates or not with α-adrenergic receptors.
In summary, β1 not only modifies MaxiK channel voltage/calcium sensitivity but also regulates TP-mediated channel inhibition and vasoconstriction. β1-TP association might be a force to dampen the inhibitory signal from TP to MaxiKα and to modify TP vasoconstricting potency.
This work was supported, in whole or in part, by National Institutes of Health Grants HL096740 and HL107418 (to E. S. and L. T.) and HL088640 (to E. S.). This work was also supported by American Heart Association Postdoctoral Fellowship 0825273F (to M. L.).
- MaxiK
- large conductance voltage- and Ca2+-activated K+ channel
- TP
- thromboxane A2 prostanoid receptor
- MaxiKα
- MaxiK α-subunit
- PPI
- protein proximity index
- HEDTA
- N-(2-hydroxyethyl)ethylenediaminetriacetic acid
- TM
- transmembrane domain.
REFERENCES
- 1. McManus O. B., Helms L. M., Pallanck L., Ganetzky B., Swanson R., Leonard R. J. (1995) Functional role of the β subunit of high conductance calcium-activated potassium channels. Neuron 14, 645–650 [DOI] [PubMed] [Google Scholar]
- 2. Meera P., Wallner M., Jiang Z., Toro L. (1996) A calcium switch for the functional coupling between α (hslo) and β subunits (KV,Caβ) of maxi K channels. FEBS Lett. 382, 84–88 [DOI] [PubMed] [Google Scholar]
- 3. Cox D. H., Aldrich R. W. (2000) Role of the β1 subunit in large-conductance Ca2+-activated K+ channel gating energetics. Mechanisms of enhanced Ca2+ sensitivity. J. Gen. Physiol. 116, 411–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nimigean C. M., Magleby K. L. (2000) Functional coupling of the β1 subunit to the large conductance Ca2+-activated K+ channel in the absence of Ca2+. Increased Ca2+ sensitivity from a Ca2+-independent mechanism. J. Gen. Physiol. 115, 719–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang D. M., He T., Katusic Z. S., Lee H. C., Lu T. (2010) Muscle-specific F-box only proteins facilitate BK channel β1 subunit downregulation in vascular smooth muscle cells of diabetes mellitus. Circ. Res. 107, 1454–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McGahon M. K., Dash D. P., Arora A., Wall N., Dawicki J., Simpson D. A., Scholfield C. N., McGeown J. G., Curtis T. M. (2007) Diabetes downregulates large-conductance Ca2+-activated potassium β1 channel subunit in retinal arteriolar smooth muscle. Circ. Res. 100, 703–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brenner R., Peréz G. J., Bonev A. D., Eckman D. M., Kosek J. C., Wiler S. W., Patterson A. J., Nelson M. T., Aldrich R. W. (2000) Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature 407, 870–876 [DOI] [PubMed] [Google Scholar]
- 8. Plüger S., Faulhaber J., Fürstenau M., Löhn M., Waldschütz R., Gollasch M., Haller H., Luft F. C., Ehmke H., Pongs O. (2000) Mice with disrupted BK channel β1 subunit gene feature abnormal Ca2+ spark/STOC coupling and elevated blood pressure. Circ. Res. 87, E53–E60 [DOI] [PubMed] [Google Scholar]
- 9. Nieves-Cintrón M., Amberg G. C., Nichols C. B., Molkentin J. D., Santana L. F. (2007) Activation of NFATc3 down-regulates the β1 subunit of large conductance, calcium-activated K+ channels in arterial smooth muscle and contributes to hypertension. J. Biol. Chem. 282, 3231–3240 [DOI] [PubMed] [Google Scholar]
- 10. Bukiya A. N., Liu J., Dopico A. M. (2009) The BK channel accessory β1 subunit determines alcohol-induced cerebrovascular constriction. FEBS Lett. 583, 2779–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bukiya A. N., Liu J., Toro L., Dopico A. M. (2007) β1 (KCNMB1) subunits mediate lithocholate activation of large-conductance Ca2+-activated K+ channels and dilation in small, resistance-size arteries. Mol. Pharmacol. 72, 359–369 [DOI] [PubMed] [Google Scholar]
- 12. Li M., Tanaka Y., Alioua A., Wu Y., Lu R., Kundu P., Sanchez-Pastor E., Marijic J., Stefani E., Toro L. (2010) Thromboxane A2 receptor and MaxiK-channel intimate interaction supports channel trans-inhibition independent of G-protein activation. Proc. Natl. Acad. Sci. U.S.A. 107, 19096–19101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tanaka Y., Meera P., Song M., Knaus H. G., Toro L. (1997) Molecular constituents of maxi KCa channels in human coronary smooth muscle: predominant α + β subunit complexes. J. Physiol. 502, 545–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meera P., Wallner M., Song M., Toro L. (1997) Large conductance voltage- and calcium-dependent K+ channel, a distinct member of voltage-dependent ion channels with seven N-terminal transmembrane segments (S0–S6), an extracellular N terminus, and an intracellular (S9-S10) C terminus. Proc. Natl. Acad. Sci. U.S.A. 94, 14066–14071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu Y., Eghbali M., Ou J., Lu R., Toro L., Stefani E. (2010) Quantitative determination of spatial protein-protein correlations in fluorescence confocal microscopy. Biophys. J. 98, 493–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bezanilla F., Stefani E. (1994) Voltage-dependent gating of ionic channels. Annu. Rev. Biophys. Biomol. Struct. 23, 819–846 [DOI] [PubMed] [Google Scholar]
- 17. Islas L. D., Sigworth F. J. (1999) Voltage sensitivity and gating charge in Shaker and Shab family potassium channels. J. Gen. Physiol. 114, 723–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu G., Zakharov S. I., Yang L., Wu R. S., Deng S. X., Landry D. W., Karlin A., Marx S. O. (2008) Locations of the β1 transmembrane helices in the BK potassium channel. Proc. Natl. Acad. Sci. U.S.A. 105, 10727–10732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Orio P., Torres Y., Rojas P., Carvacho I., Garcia M. L., Toro L., Valverde M. A., Latorre R. (2006) Structural determinants for functional coupling between the β and α subunits in the Ca2+-activated K+ (BK) channel. J. Gen. Physiol. 127, 191–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amberg G. C., Bonev A. D., Rossow C. F., Nelson M. T., Santana L. F. (2003) Modulation of the molecular composition of large conductance, Ca2+-activated K+ channels in vascular smooth muscle during hypertension. J. Clin. Invest. 112, 717–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amberg G. C., Santana L. F. (2003) Downregulation of the BK channel β1 subunit in genetic hypertension. Circ. Res. 93, 965–971 [DOI] [PubMed] [Google Scholar]