

Abstract
Imaginal disc development in Drosophila requires coordinated cellular proliferation and tissue patterning. In our studies of TGFβ superfamily signaling components, we found that a protein null mutation of Smad2, the only Activin subfamily R-Smad in the fruit fly, produces overgrown wing discs that resemble gain of function for BMP subfamily signaling. The wing discs are expanded specifically along the anterior-posterior axis, with increased proliferation in lateral regions. The morphological defect is not observed in mutants for the TGFβ receptor baboon, and epistasis tests showed that baboon is epistatic to Smad2 for disc overgrowth. Rescue experiments indicate that Baboon binding, but not canonical transcription factor activity, of Smad2 is required for normal disc growth. Smad2 mutant discs generate a P-Mad stripe that is narrower and sharper than the normal gradient, and activation targets are correspondingly expressed in narrowed domains. Repression targets of P-Mad are profoundly mis-regulated, with brinker and pentagone reporter expression eliminated in Smad2 mutants. Loss of expression requires a silencer element previously shown to be controlled by BMP signaling. Epistasis experiments show that Baboon, Mad and Schnurri are required to mediate the ectopic silencer output in the absence of Smad2. Taken together, our results show that loss of Smad2 permits promiscuous Baboon activity, which represses genes subject to control by Mad-dependent silencer elements. The absence of Brinker and Pentagone in Smad2 mutants explains the compound wing disc phenotype. Our results highlight the physiological relevance of substrate inhibition of a kinase, and reveal a novel interplay between the Activin and BMP pathways.
Keywords: Smad2, Baboon, Mad, Drosophila
INTRODUCTION
Tissues and organs must coordinate cell proliferation and patterning to generate functional units. Insect imaginal tissues have offered a fruitful context in which to study these broad issues. In particular, extensive study of the wing imaginal disc of Drosophila melanogaster has revealed a system in which successive waves of patterning occur in the context of spatially uniform cell proliferation. The Dpp morphogen, an ortholog of vertebrate BMP2/4, is required for both proper disc size and patterning along the anterior-posterior (A/P) axis. How these phenomenon are coordinated at the molecular level remains an active area of research (Schwank and Basler, 2010; Wartlick et al., 2011).
The Activin/Transforming growth factor beta (TGFβ) signaling family plays diverse roles in growth and patterning in mammals. Several functions have been identified in Drosophila, including neuroblast proliferation (Zhu et al., 2008), axonal remodeling (Zheng et al., 2003; Ng, 2008), axon outgrowth (Serpe and O'Connor, 2006; Parker et al., 2006) and competence of the endocrine gland to regulate developmental timing (Gibbens et al., 2011). In the wing disc, loss of function of Baboon, the Activin/TGFβ type-I receptor, does not affect patterning but the disc is reduced in size (Brummel et al., 1999). One study described the application of RNA interference (RNAi) to reduce Smad2 (Smox – FlyBase) function in the developing wing and reported a patterning defect in the disc and venation defects in the adult wing (Sander et al., 2010). Smad2 opposes Mad function during both of these phases of wing development, but the mechanisms are unknown. There are several other examples of bone morphogenetic protein (BMP) and TGFβ signals limiting or competing with each other. The sub-branches share Type II receptors and a common Smad, and these are potential points of interplay. Indeed, it has been reported that competition for Smad4 mediates opposing actions of TGFβ and BMP signals in Xenopus mesoderm specification (Candia et al., 1997), and competition for ActIIR (Acvr2 – Mouse Genome Informatics) underpins the TGFβ and BMP antagonism that regulates mouse visceral endoderm development (Yamamoto et al., 2009).
Starting from the observation of larval patterning defects observed in a null mutant for Smad2, we describe here how mutation in the Activin pathway can impinge on the BMP patterning system to profoundly perturb wing disc growth and patterning. Our results uncover a surprising mode of regulation whereby a substrate limits the activity of a receptor. In the absence of its main substrate, aberrant Activin receptor signaling disrupts tissue patterning by the BMP signaling pathway.
MATERIALS AND METHODS
Fly stocks and crosses
dSmad2MB388, babofd4 (Zheng et al., 2003), dSmad2F4 and dSmad2C15 (Peterson et al., 2012) have been described. dSmad2C15 gives the same phenotypes as dSmad2F4 in terms of anal pad, CNS and wing disc size. The baboΔ chromosome is a deletion uncovering baboon (Bloomington Drosophila Stock Center, #5234). Mutant larvae were selected based on lack of GFP balancers, and sorted by gender when necessary. Some experiments used a y, dSmad2F4 chromosome stocked with y FM7GFP and {y+}Y to avoid confounding effects of XXY females.
GAL4 RNAi crosses were conducted at 25°C in vials of cornmeal-based food, and wandering stage larvae were examined when possible. In several cases where the desired larvae were impaired by RNAi, the largest surviving larvae were pulled from the food, or in the case of phm+C765 GAL4 drivers, mixed stage larvae were transferred to a yeast paste plate for several days. da-GAL4 >Smad2 RNAi was conducted in vial food and yeast paste with the same outcome. The RNAi lines used for Smad2 (insert 3B3) and Mad (insert 6C3) have been described (Peterson et al., 2012). The babo RNAi construct was made in a similar fashion and includes nucleotides 1416-1932 of NM_057652 (insert 58B3). The schnurri RNAi (KK series, stock 105643) and UAS-Dicer-2 stocks were purchased from the Vienna Drosophila RNAi Center. Recombinant chromosomes were used for epistasis tests.
The following GAL4 drivers are available from the Bloomington Drosophila Stock Center: da-GAL4 (#5460), sd (#8609), 32B (#1782), 71B (#1747), T80 (#1878), esg (#26816) and A9 (#8761). phm-GAL4 has been described (Ono et al., 2006). C765-GAL4 (Nellen et al., 1996) was provided by C. Mirth (Fundacao Calouste Gulbenkian, Portugal), and nub-GAL4 and tsh-GAL4 (Calleja et al., 1996) were provided by G. Morata (CSIC-UAM, Madrid, Spain). Recombinant chromosomes were made for phm+nub, C765+nub and nub+tsh, and additive expression was verified in GFP reporter test crosses. UAS-Babo-WT and -Babo-CA have been described (Brummel et al., 1999).
For rescue experiments, wild-type or mutated FLAG-tagged Smad2 proteins were expressed in a Smad2 mutant. Smad2-AAMA (alanine substitutions of C-terminal serines) and Smad2-RB4 (alanine substitutions of G450, Y455, R457 and Q458) were generated by site-directed mutagenesis and verified by sequencing. The RB4 variant does not bind to its receptor as judged by lack of phosphorylation by Baboon in S2 cells. pUASattB constructs were recombined into the VK31 site (injections carried out by Genetivision) and expressed as recombinants with da-Gal4. Expression of each protein was verified by immunohistochemistry for the FLAG epitope.
dpp-lacZ (dpp10638; Bloomington Drosophila Stock Center, #12379), Dad-lacZ (DadP1833) (Minami et al., 1999) and sal-lacZ (salm03602; Bloomington Drosophila Stock Center, #11340) reporters were assayed by crossing to Smad2 heterozygous females and comparing the β-galactosidase staining of female (control) and male (mutant) progeny. omb-lacZ (biP1) (Minami et al., 1999) was assayed as a recombinant chromosome with dSmad2C15. The B14-lacZ reporter has been described (Müller et al., 2003). E14, 8mT, BrkF brinker reporter (Müller et al., 2003; Pyrowolakis et al., 2004) and pent- and pentΔSE-lacZ reporter flies (Vuilleumier et al., 2010) were kindly provided by G. Pyrowolakis (Albert-Ludwigs-University of Freiburg, Freiburg, Germany).
GAL4 flp-out clones were induced by activating hs-flp by incubation for 30 minutes at 37°C. Larvae were assayed 48 hours after clone induction, or as indicated. Clone-bearing animals had the generalized genotype of hs-flp; B14-lacZ/+; actin[stop]GAL4, UAS-GFP/UAS-RNAi. Animals of the desired genotype were identified by the presence of GFP-marked clones, absence of balancers, and β-galactosidase staining.
Immunohistochemistry and microscopy
Rabbit anti-atypical Protein kinase C (aPKC) (Santa Cruz Biotechnology, #SC-216), mouse anti-FasIII (Fas3) (7G10, Developmental Studies Hybridoma Bank, University of Iowa, IA, USA) and DAPI were used as general stains for fixed larval tissues. Detection of lacZ reporter expression was carried out using anti-β-galactosidase (Promega, #Z378A). Sal was detected with rabbit α-Sal (Halachmi et al., 2007) (gift from A. Salzberg, Technion-Israel Institute of Technology, Haifa, Israel). Secondaries antibodies used for fluorescence microscopy were conjugated with AlexaFluor 488, 568 or 647 (Invitrogen). The Histone H3 P-Ser10 antibody was from Sigma (#H 0412). 5-Ethynyl-2′-deoxyuridine (EdU) incorporation and labeling was carried out using a Click-iT 555 Kit (Invitrogen, #C10338). Incorporation used freshly everted larvae incubated in M3 media with 10 μM EdU for 30 minutes at 21°C. Phosphorylated Mad (P-Mad) was detected as described (Peterson et al., 2012). To compare signals (dpp-lacZ and P-Mad in Fig. 5) accurately between wild-type (WT) and Smad2 null discs, fixation and staining was performed in one tube and the discs were subsequently genotyped by morphology. Whole larvae were heat-fixed at 50°C for 3 minutes prior to imaging.
Fig. 5.
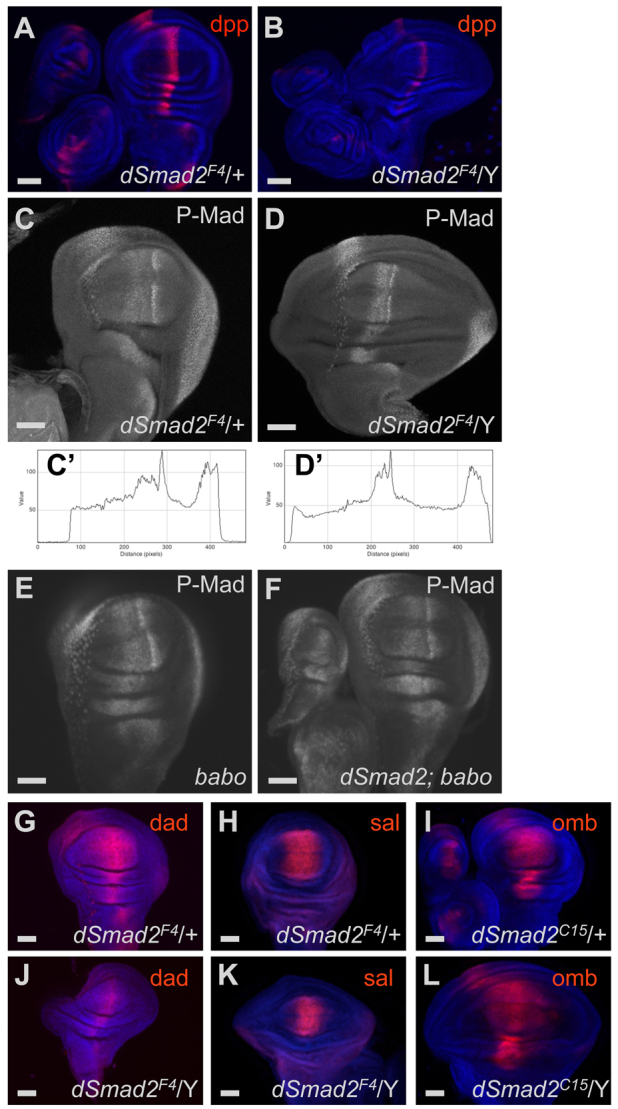
Moderate mis-expression of BMP activation targets in Smad2 mutant discs. (A,B) dpp-lacZ expression in wing discs. The normal stripe of dpp expression (A) was narrowed and weakened in Smad2 mutants (B). (C-F) P-Mad immunohistochemistry in wing discs. The canonical late L3 P-Mad profile (C) was narrowed and sharpened in Smad2 discs (D). Profiles running across the entire disc through the dorsal pouch are shown in C′ and D′. babo (E) and Smad2; babo double mutants (F) had nearly normal P-Mad patterns. (G-L) Dad-lacZ reporter expression was present but narrowed in Smad2 discs (J versus G). sal-lacZ expression was also narrowed (K versus H). omb-lacZ expression in dSmad2C15 occupied a smaller fraction of the enlarged discs (L versus I). Wing discs are oriented with the distal end up and the anterior to the left. Scale bars: 50 μm.
Wide-field images were captured using a 10× objective on a Zeiss Axiovert microscope, and confocal images were captured using a 20× objective using a CARV attachment, or using a 20× objective on a Zeiss LSM710 microscope. Single confocal sections or maximal intensity projections (MIP) are shown, as indicated in the figure legends. Projections were generated using Zeiss Axiovision or Zen software. P-Mad profiles were generated using ImageJ.
To estimate the number of cells in the wing disc, a circle of a fixed size (36 μm radius) was placed over wing blade images at the z-position at which the cross-section of the cells stained with membrane markers was most visible.
RESULTS
Phenotypic differences between Smad2 and baboon alleles expose non-canonical signaling
Drosophila has only one R-Smad of the Activin branch, encoded by Smad2 (also called Smox) (Brummel et al., 1999; Henderson and Andrew, 1998), and one R-Smad of the BMP branch, encoded by Mad (Newfeld et al., 1997). A mis-sense allele of Smad2 was identified in a screen for genes required for mushroom body neuronal remodeling (Zheng et al., 2003). This Smad2MB388 allele was categorized as a strong loss-of-function allele because it displays several phenotypes that resemble loss-of-function baboon alleles, including small central brain lobes and the namesake enlarged larval anal pads. The mutation causes a single amino acid substitution, and we recently found that the mutated protein can still interact with the Baboon receptor and be phosphorylated at the C-terminal SXSS motif (Peterson et al., 2012). We therefore examined morphological characteristics of larvae harboring a recently described null allele of Smad2 to catalog differences and similarities between baboon loss-of-function mutants and the two Smad2 mutants.
Visible inspection of larvae revealed a difference in the presentation of the ‘baboon’ anal pad phenotype for the two Smad2 alleles. Larvae completely lacking Smad2 maintained normal anal pad structures (Fig. 1A,C), whereas animals expressing a mis-sense protein displayed enlarged anal pads like baboon mutants (Fig. 1D,B). By contrast, the small central brain lobes first noted in baboon mutants were recapitulated in both the null and point mutation Smad2 alleles (Fig. 1E-H). This is consistent with the requirement of canonical Baboon to Smad2 signaling for neuronal proliferation (Zhu et al., 2008). We observed an additional morphological defect in Smad2-null larvae: the wing imaginal discs were drastically widened. This extreme overgrowth phenotype was seen only with the Smad2 null, and not the Smad2 point mutant or the baboon mutant (Fig. 1I-L).
Fig. 1.
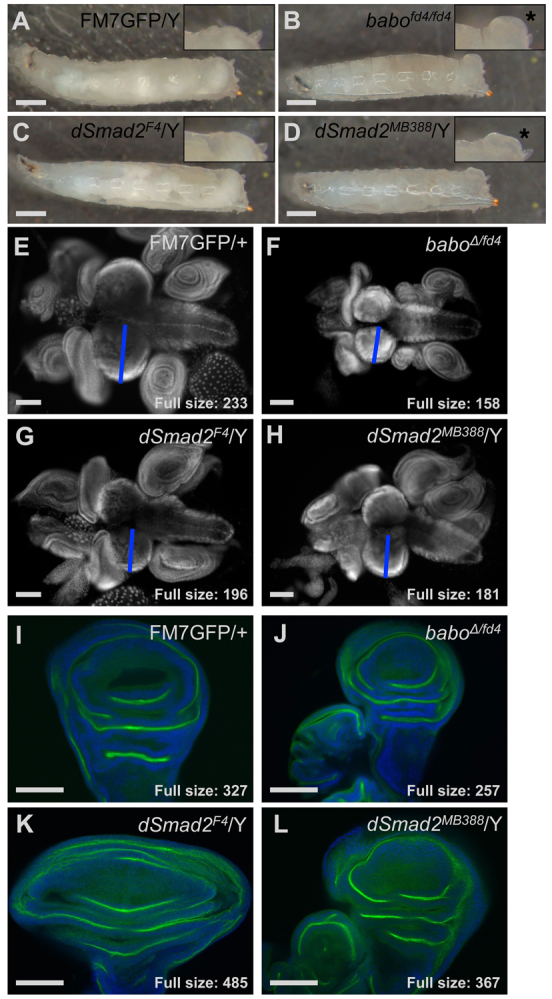
Phenotypes of a Smad2 null allele highlight non-canonical signaling. (A-D) Heat-fixed larvae of the indicated genotype were photographed to show overall morphology and insets (2× magnification) highlight anal pad region. Swollen anal pads (asterisks) were present in babo mutant (B) and dSmad2MB388 point mutant (D) larvae, but dSmad2F4 null larvae had normal anal pads (C versus A). The subtle change in larval shape and color observed for babo mutants was also seen for dSmad2MB388. (E-H) Wide-field images of DAPI-stained brain complexes from late L3 larvae of the indicated genotypes. Hypotrophy of the larval CNS is seen in babo mutant larvae and both dSmad2 alleles (compare F-H with E). Blue bar indicates width of one central brain lobe. Full size indicates the average central brain lobe width (μm) for the five largest samples of each genotype. (I-L) Late L3 wing discs of the indicated genotype were labeled with DAPI (blue) and aPKC (green). Single confocal sections are shown. Hypertrophy of the wing disc was seen in the Smad2 null (K versus I) but not babo (J) and was minimally observed with dSmad2MB388 (L). Full size indicates the average width (μm) of the ten largest discs of each genotype. Wing discs are oriented with the distal end up and the anterior to the left. Scale bars: in A-D, 500 μm; in E-L, 100 μm.
Taken together, these observations indicate that (1) the protein null and mis-sense alleles of Smad2 are not equivalent, (2) non-canonical signaling by Baboon might be operative in multiple tissues, and (3) the relationship between Baboon and Smad2 varies with tissue. We were particularly intrigued by the wing disc phenotype because it resembles a gain of function for BMP signaling, exemplified by Dpp ligand overexpression (Nellen et al., 1996; Burke and Basler, 1996) or loss of the Brinker transcription factor (Campbell and Tomlinson, 1999). We conducted a series of experiments to examine interactions between components of the Activin/TGFβ and BMP signaling branches in wing disc growth.
Tissue-autonomous loss of Smad2 leads to wide wing discs
The wide wing disc phenotype has several notable features. One, the overgrowth is restricted to the anterior-posterior axis of the pouch and hinge portion of the disc. The long proximal-distal axis is neither shortened nor lengthened, and the notum portion of the disc is not expanded. To quantify the shape change, we determined the width to height (W/H) ratio, including the wing and hinge regions of the disc but excluding the notum. This W/H ratio did not change appreciably during normal L3 growth. The range of heights was unchanged between Smad2F4 and control discs, indicating that changes in width drives changes in the W/H ratio (supplementary material Fig. S1). Another characteristic is that the epithelial layers remained intact and the pattern of epithelial folding was similar to normal discs. These features suggest a specific growth or patterning defect along the A/P axis, as opposed to the general loss of organization reported for tumor suppressor mutants (Bryant and Levinson, 1985; Bilder et al., 2000).
Our first step in identifying the mechanism by which loss of Smad2 leads to patterning defects in the wing was to define the spatiotemporal requirement of Smad2 using RNAi knockdown. Expression of double-stranded RNA targeting the Smad2 message under the control of the constitutive da-GAL4 driver produced widening of wing discs similar to the null mutant (Fig. 2A). To determine whether the phenotype was caused by tissue-autonomous loss of Smad2 protein, we drove Smad2 RNAi with GAL4 lines expressed in the wing disc. Somewhat surprisingly, many wing disc drivers did not produce the wide wing phenotype (Fig. 2B,C; supplementary material Fig. S2). The lack of widening is unlikely to be caused by incomplete RNAi knockdown because co-expression of Dicer-2 had little effect. A handful of GAL4 drivers, however, did lead to various degrees of wing disc overgrowth (Fig. 2E,F; supplementary material Fig. S2). The drivers that reproduced the widened discs all have broad expression in the wing disc, but are also expressed in other tissues, complicating the interpretation of tissue-autonomous effects. Notably, they are all expressed in the prothoracic gland (PG), an endocrine tissue known to require Smad2 function (Gibbens et al., 2011). Simultaneous Smad2 knockdown in the PG (phm-GAL4) and the wing disc (C765 or nub-GAL4) did not induce widening, suggesting that wing overgrowth is independent of Smad2 in the PG (supplementary material Fig. S3). To define further the spatial requirements of Smad2 function in the disc, we combined the expression pattern of two partial wing drivers. nub and tsh are expressed in complementary domains of the wing disc beginning at L2 (Zirin and Mann, 2007) (Fig. 2G,H). Neither nub-GAL4 nor tsh-GAL4 on its own led to disc hypertrophy, but simultaneous expression did lead to wing disc widening (Fig. 2G-I), indicating that Smad2 must be removed from the entire disc. This requirement would explain the lack of effect with A9 and 71B, which are not expressed in the entire wing disc. The C765-GAL4 result (Fig. 2D) indicates a temporal parameter as it is expressed in the entire wing disc, but not strongly until late L3 (Wartlick et al., 2011). We conclude that Smad2 function must be removed prior to early L3 in nearly all of the wing disc cells to produce a morphological phenotype.
Fig. 2.
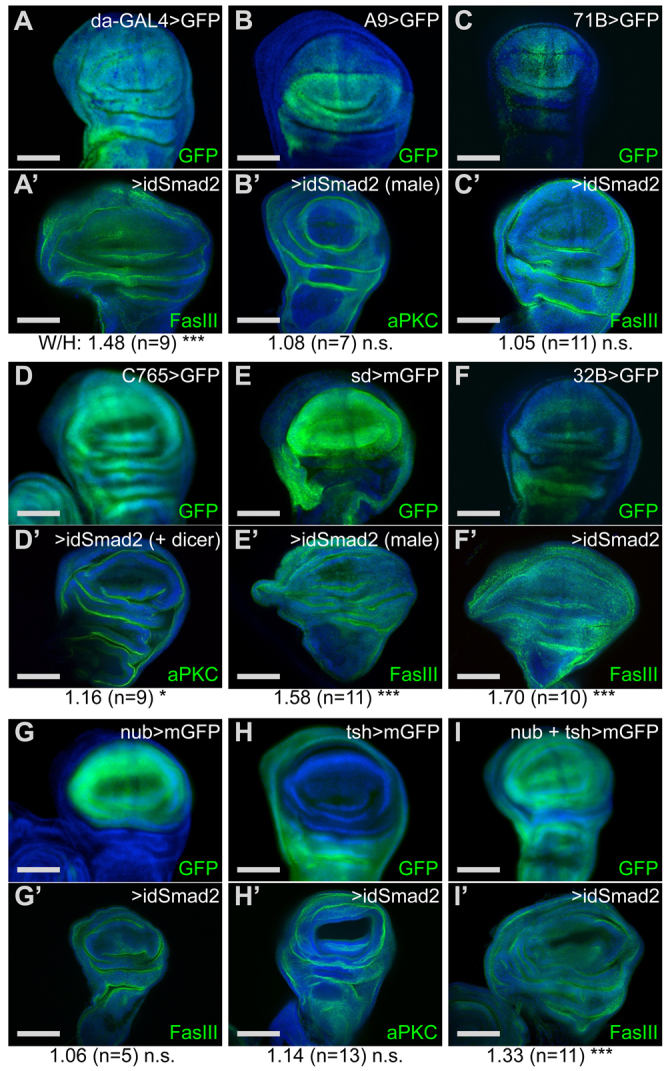
Smad2 RNAi produces tissue-autonomous wing disc widening. (A-I′) For each GAL4 driver, GFP reporter shows late L3 wing disc expression (A-I). mGFP indicates membrane-localized GFP. The effect of Smad2 RNAi is shown in the corresponding prime panels (A′-I′) with the W/H ratio presented as the mean for the indicated number of samples. Statistical significance of a one-tailed t-test for widening is indicated. *P<0.05, ***P<0.001. n.s., not significant. da-GAL4 (A) is a constitutive driver. A9 (B) and 71B (C) have restricted disc expression but caused no widening. C765 (D) is expressed in late L3 but caused minimal widening. sd (E) and 32B (F) are expressed in the entire disc and caused widening. sd-GAL4 is strongest in a central region including the wing blade with weaker expression in the remainder of the disc (see cells in upper left area in B and E for negative and weak GFP expression). tsh and nub expression domains produced widening only when used together (G-I). Wing discs are oriented with the distal end up and the anterior to the left. Confocal images shown except panels D, G, H and I, which show wide-field images. Scale bars: 100 μm.
Developmental time course of morphology and proliferation patterns in Smad2F4 discs
Change in tissue size can be due to changes in cell number, or changes in cell size or shape. To determine how cell number and size contribute to disc widening, we measured the cell density in normal and widened discs. Smad2 mutant discs had significantly smaller cells than did wild type (Fig. 3A-C), so cell enlargement is not the explanation for overgrown discs. Indeed, a larger tissue made up of smaller cells means that more cells are present. Taking into account the smaller cells and larger tissue area, we estimate that there are 1.8 times the normal number of cells in the Smad2 mutant wing blade.
Fig. 3.
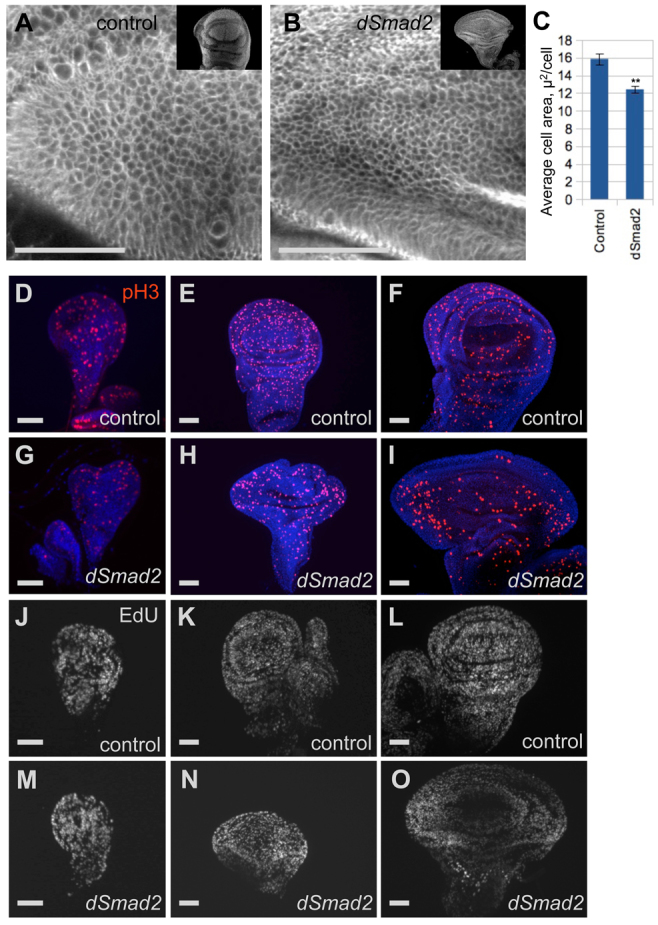
Reduced cell size and uneven proliferation are associated with altered wing disc morphology. (A-C) Membrane FasIII staining showed reduced size of wing blade cells for Smad2 mutant discs (B) relative to heterozygous controls (A). Example single confocal sections from the dorsal-anterior quadrant are shown along with DAPI-stained discs (inset). Cells in a fixed area were counted (three circles per disc, three discs per genotype) and the cross-sectional cell area is plotted in C as mean cell area ± s.e.m. **P<0.01 for two-tailed t-test. (D-I) Histone H3 PSer10 (pH3) staining to mark M phase cells. Representative wing discs from early, mid and late L3 are shown for WT (D-F) and dSmad2F4 (G-I) animals. Note that WT discs have uniform A/P distribution, but mutant discs have increased numbers of M phase cells in lateral regions at mid and late stages (H,I) when the overgrowth is visible. (J-O) EdU incorporation to label S phase cells. The normally uniform distribution (J-L) is altered in Smad2 mutants at mid and late L3 (N,O). Images in several panels include associated haltere and leg discs. Wing discs are oriented with the distal end up and the anterior to the left. Scale bars: in A,B, 100 μm; in D-O, 50 μm.
The wing disc undergoes extensive growth and patterning during the third larval instar. To define the temporal progression of the morphological abnormality, we compared wing discs from early, mid and late L3 larvae. The mutant wing discs displayed normal size and shape at the beginning of L3 (Fig. 3G), but by mid L3 the widening had already taken place (Fig. 3H), giving these discs a similar W/H ratio as later mutant larvae. The discs grew during the remaining 24 hours of larval growth, but the abnormal W/H ratio persisted (Fig. 3I).
Apoptosis is not prevalent in third instar larval wing discs and is not thought to play a major role in normal growth during this phase (Milán et al., 1997), so the observation of increased cell number implicates an increase in proliferation in the Smad2 mutant. We examined cells at two cell cycle stages to characterize how proliferation correlates with atypical morphogenesis. Mitotic cells in the wing disc were identified by staining for Histone H3 P-Ser10. As has been reported (Schwank et al., 2008), a remarkable feature of wing disc proliferation is that it is normally uniform, despite mitogenic inputs from several morphogen gradients. In fully developed Smad2 wing discs, the total number of mitotic cells was reduced (Fig. 3I compared with 3F). There was also a marked asymmetry along the A/P axis, with the lateral regions containing far more mitotic cells than the medial region. We examined earlier developmental stages to correlate mitotic patterns with the progression of the abnormal morphology. At early L3, when the wing discs were normal in size and shape, the mitotic cell patterns appeared normal (Fig. 3D,G). At mid L3, when the widening was apparent, the asymmetry was observed and the total number of mitotic cells was modestly reduced relative to wild-type discs (Fig. 3E,H). Labeling of S phase cells by staining for EdU incorporation led to similar findings (Fig. 3J-O), suggesting a general increase in proliferation rates in the lateral regions. We conclude that asymmetry in proliferation along the A/P axis produces the wide wing discs in the Smad2 mutant.
Baboon is epistatic to Smad2 function in the wing disc
One curious aspect of the Smad2 wing disc phenotype is that it is markedly different from the baboon mutant wing disc phenotype. We considered several molecular explanations for the difference between components of the same signaling pathway. First, Smad2 might have Baboon-independent roles in limiting proliferation in discs. Alternatively, Baboon might be controlling proliferation in a manner influenced by the presence of Smad2 protein. We carried out an epistasis test to distinguish between these possibilities and found that the wing discs of Smad2; baboon double mutants resembled baboon single mutants. The widening caused by loss of Smad2 was completely suppressed by concomitant loss of Baboon (Fig. 4C,D). The receptor kinase is thus epistatic to the Smad substrate in this assay. We also observed this Baboon dependence when the disc widening was generated by Smad2 RNAi. Single Smad2 knockdown in the esg-GAL4 domain led to widening (Fig. 4G compared with 4E). Single Baboon knockdown had no significant effect (Fig. 4F), but simultaneous knockdown of Smad2 and Baboon produced discs with normal W/H ratios (Fig. 4H). These parallel results between the whole animal mutants and the disc RNAi conditions (Fig. 4I) strengthen the conclusion that tissue-autonomous perturbation of the Activin/TGFβ pathway can lead to wing disc hypertrophy. Expression of constitutively active Baboon also caused disc widening (supplementary material Fig. S4), implicating excessive Baboon activity as the cause of widening in Smad2 mutants.
Fig. 4.

The Baboon receptor is epistatic to the Smad2 substrate. (A-H) L3 wing discs stained with DAPI and anti-FasIII. Lateral overgrowth of wing discs did not occur in babo mutants (B versus A) but did in Smad2 null mutants (C versus A). Double mutants had normal W/H ratios (D). esg-GAL4 driving babo RNAi did not affect growth parameters (F versus E), but Smad2 RNAi led to dramatic widening (G). Simultaneous RNAi restored the normal W/H ratio (H). (I-L) L3 wing discs stained with DAPI and anti-aPKC. Expression of transgenic WT Smad2 protein with da-GAL4 in a Smad2 null restored normal disc shape (J versus I). Smad2-AAMA mutant protein also normalized the W/H ratio (K), but the RB4 mutant did not (L). (M) Plot of mean ratios ± s.e.m. for each condition. Statistical significance relative to control is indicated. ***P<0.001. n.s., not significant (P>0.05). For rescue tests, comparison is with control lacking da-GAL4. Wing discs are oriented with the distal end up and the anterior to the left. Scale bars: 100 μm.
The finding that the wide disc phenotype requires both the absence of Smad2 and the presence of Baboon suggests a role for Smad2 acting not as a transcription factor, but as an inhibitor of Baboon. We tested several mutated versions of Smad2 for the ability to restore normal wing development to ascertain the contributions of canonical transcription factor activity and receptor binding. Constitutive expression of Smad2-WT in a Smad2 mutant led to normal wing disc growth (Fig. 4I,J). Expression of Smad2-AAMA, which can bind to Baboon (Brummel et al., 1999) but lacks the C-terminal serine phosphorylation sites, also restored disc morphology (Fig. 4K,M). By contrast, Smad2-RB4, which cannot bind Baboon owing to mutations in conserved receptor binding residues (Lo et al., 1998), did not rescue the phenotype (Fig. 4L). These data show that Smad2 inhibition of its receptor is crucial for wing patterning, but canonical TGFβ signaling is dispensable.
BMP pathway activity is altered in Smad2 mutants
As mentioned above, the shape and size of Smad2 loss-of-function wing discs resembles BMP signaling gain-of-function situations. This similarity is especially intriguing in light of the reported ability of mammalian and fly TGFβ type I receptors to directly phosphorylate BMP R-Smads in addition to TGFβ R-Smads (Liu et al., 2009; Wrighton et al., 2009; Peterson et al., 2012). We therefore looked for Dpp pathway alterations upon Smad2 loss. dpp expression was monitored by a lacZ enhancer trap. A stripe of expression was observed at the normal A/P boundary, but it was narrower (average width of 39 μm compared with 53 μm for control) and the reporter expression was weaker (average peak intensity of 1173 units compared with 2582 units for control) in Smad2 mutant discs (Fig. 5A,B). This indicates that any gain-of-function alteration of Dpp signaling is downstream of ligand production.
We examined Mad to see whether it was ectopically stimulated in Smad2 mutants. We found that Smad2 mutant discs displayed a stripe of P-Mad along the A/P boundary, but the pattern was abnormal (Fig. 5C,D). In late L3 discs, the P-Mad stripe was clearly condensed. Staining profiles across the breadth of the disc show that the normal two-humped gradient was condensed into a narrower stripe with rapid drop off in lateral regions (Fig. 5C′,D′). A similar narrowing of the P-Mad stripe was observed for discs during mid L3, but the difference between genotypes was less dramatic because the WT pattern is less elaborate at earlier time points (not shown). The P-Mad pattern was close to normal in baboon single mutants and in Smad2; baboon double mutants (Fig. 5E,F). These results demonstrate that neither Smad2 nor Baboon is absolutely required for the formation of a P-Mad gradient, rather that mis-regulation of the TGFβ components can perturb the distribution of P-Mad.
We examined several downstream transcriptional targets of Mad to define the coupling between P-Mad and targets in Smad2 mutants. sal (salm – FlyBase), Dad and omb (bi – FlyBase) have Dpp- and P-Mad-dependent expression domains centered on the A/P boundary (Minami et al., 1999). Reporters for each of these three genes were expressed along the A/P boundary in the wing discs of Smad2 mutant animals, but the absolute widths of the Dad and sal stripes were reduced and the omb reporter was expressed in a narrower fraction of the mutant disc (Fig. 5G-L). baboon mutants do not show narrowing of activation targets and double mutants resemble baboon (supplementary material Fig. S5). By contrast, we found that a P-Mad repression target was profoundly mis-regulated. brinker is normally expressed in a complementary pattern to P-Mad, but in Smad2 mutants a brinker expression reporter was essentially shut down throughout the entire disc (Fig. 6B). This result is consistent with the morphological phenotype because loss of Brinker leads to expanded discs (Campbell and Tomlinson, 1999). The interesting twist in the Smad2 mutant is that brinker expression is uncoupled from the P-Mad pattern.
Fig. 6.
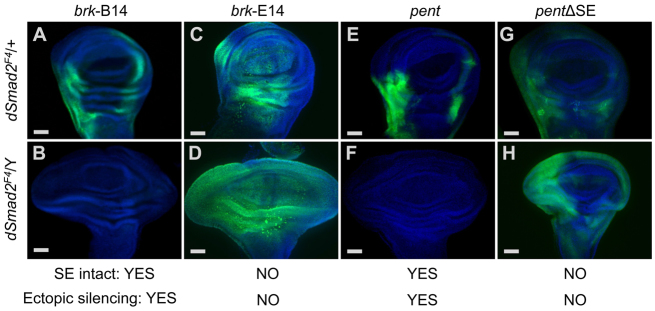
Ectopic repression mediated by SEs. (A-D) Brinker expression was monitored by lacZ reporters (green) and is shown with DAPI staining (light blue). The B14 reporter was Smad2 dependent (A,B) and the E14 brinker reporter lacking the SE was not Smad2 dependent (C,D). (E-H) A pentagone reporter with intact SEs lost expression in a Smad2 mutant (E,F) but a reporter with mutation of the SEs (pentΔSE) was no longer Smad2 dependent (G,H). Relationship between SE and ectopic silencing in the absence of Smad2 is summarized for each reporter. Wing discs are oriented with the distal end up and the anterior to the left. The green channel was captured and is displayed with matching settings for panels A and B and for E and F. Scale bars: 50 μm.
We next determined brinker expression at several developmental time points. brinker reporters are already expressed at the flanks of the early L3 wing discs, and expression was normal in Smad2 mutants at this time point, indicating that brinker expression initiated properly. By mid L3, brinker expression was severely reduced and remained off for the duration of larval development (supplementary material Fig. S6). The loss of brinker thus roughly parallels the perturbed cell proliferation and the appearance of abnormal disc morphology, consistent with the role of Brinker in regulating wing disc proliferation (Martín et al., 2004; Schwank et al., 2008).
Ectopic repression through Dpp silencing elements
The transcriptional regulation of brinker in the wing has been extensively studied (Müller et al., 2003, Yao et al., 2008). Mad-responsive silencer elements repress brinker expression that is otherwise found in the entire wing disc. We considered two general possibilities for loss of brinker expression in Smad2 mutants. One possibility is that Smad2 actively promotes brinker expression, and the other is that in the absence of Smad2 brinker is ectopically silenced. To differentiate between these models, we assayed the expression of a panel of reporter constructs in Smad2 mutant discs.
The B14 enhancer region contains a strong silencer element (SE) adjacent to activation elements (Müller et al., 2003). A smaller E14 element containing the activation sequences but lacking the SE was not affected by the absence of Smad2 (Fig. 6C,D). This means that Smad2 is not required for the pan-disc active expression of brinker. These results implicate the SE in the Smad2-dependent effect. To test whether Smad2 generally regulates SE-containing genes, we tested a reporter from another gene. Among a handful of characterized Dpp target genes, pentagone (pent; now known as magu) was recently identified as an SE-containing Mad target that is required for wing disc patterning (Vuilleumier et al., 2010). The intact pent reporter recapitulates the normal pent expression pattern in wild-type discs and, like the B14 brinker reporter, its expression is abolished in Smad2 mutant discs (Fig. 6E,F). To assess the role of the SE directly, we tested a reporter construct with its two SEs disabled by mutation (Vuilleumier et al., 2010). The pentΔSE reporter was expressed in a similar pattern regardless of the Smad2 genotype (Fig. 6G,H). We also found that brk-B14 and pent reporters were expressed normally when Baboon or Smad2 and Baboon were removed (supplementary material Fig. S7). Taken together, these data show that ectopic silencing requires the absence of Smad2, the presence of Baboon, and intact target gene SEs.
What is acting through the silencer element?
Our results indicate that loss of Smad2 leads to aberrant repression of genes under control of regulatory elements previously characterized as Mad-dependent silencers, and that the Baboon receptor is required for this behavior. Wing disc clones subject to Smad2 RNAi did not express the B14 brinker reporter, showing that clones lacking Smad2 mimic this behavior of disc cells in the null mutant (Fig. 7A). The effect is spatially robust because it was observed at many positions within the normal brinker expression domains (not shown). Next, we generated clones that simultaneously knocked down Smad2 and another protein of interest. Smad2; baboon double RNAi clones maintained B14 expression 48 hours after clone induction (Fig. 7C), again in line with the whole animal double mutants in which loss of Baboon suppressed loss of Smad2. Some baboon RNAi clones had reduced B14 levels relative to their neighbors, and this was also seen with Smad2; baboon double RNAi clones (Fig. 7B,C).
Fig. 7.
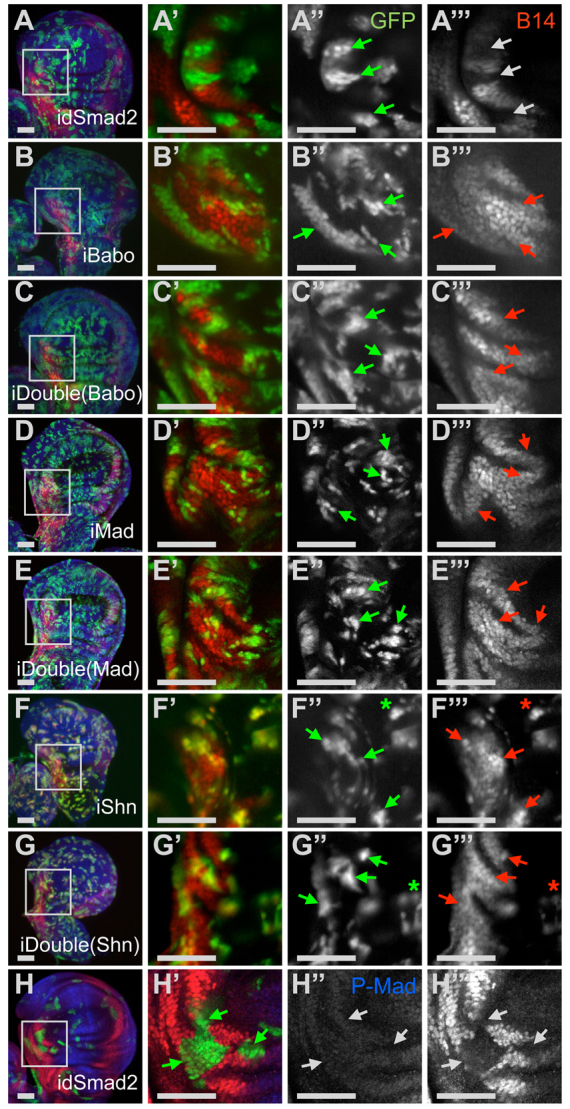
Loss of Smad2 causes cell-autonomous brinker silencing and requires Baboon, Mad and Schnurri. Clones expressing RNAi elements were generated and monitored 48 hours after clone induction. All clones are marked with GFP (green). (A-G) Maximum intensity projections of clone-containing disc showing B14-lacZ expression (red) and stained with DAPI (blue). Single confocal sections (A′-G′) show higher magnification views of boxed areas in A-G, including clones in a B14-expressing area near the anterior-dorsal pouch, with GFP shown in A″-G″ and lacZ expression shown in A‴-G‴. Condensed labels indicate genes targeted by RNAi, with double always including Smad2. B14 expression was lost in idSmad2 clones (A-A‴). Arrows mark selected clone positions. White arrows indicate loss of B14 and red arrows indicate retention of B14. iBabo clones retained B14 expression (B-B‴) as did iBabo + idSmad2 together (C-C‴). iMad single (D-D‴) and iMad + idSmad2 double clones (E-E‴) also retained proper B14 expression. A similar result was observed for iShn (F-F‴) and iShn + idSmad2 double (G-G‴) RNAi clones, and ectopic B14 expression (asterisks) was readily observed in these clones. (H-H‴) P-Mad detection (blue in H, gray in H″) was not elevated in idSmad2 clones with ectopic B14 repression (red in H′, gray in H‴). White arrows in H″ and H‴ point to GFP-positive RNAi clone areas shown by green arrows in H′. Wing discs are oriented with the distal end up and the anterior to the left. Scale bars: 50 μm.
Reporter assays pinpointed the SE in mediating the Baboon-dependent repression. The SE can bind Mad, Medea and the Schnurri repressor in vitro (Müller et al., 2003), so we tested several of these factors using the RNAi epistasis assay. Single Mad RNAi clones lacked P-Mad, had normal B14 expression in the lateral domains, and had weak ectopic B14 expression near the center of the discs (Fig. 7D; data not shown). In Mad and Smad2 double RNAi clones, B14 was still expressed in lateral regions (Fig. 7E). Lack of Mad thus reverses the effect on brinker seen when Smad2 is absent. A similar finding was observed for Schnurri. schnurri RNAi clones showed increased B14 expression (Fig. 7F), as expected from published reports of schnurri mutant clone behavior (Marty et al., 2000). Smad2 and schnurri double RNAi clones maintained B14 expression in the normal lateral regions (Fig. 7G). Taken together, these data indicate that the Smad2-Baboon module influences the activity of a Mad-Medea-Schnurri complex at silencing elements of target genes.
By what mechanism does Smad2 influence the output of the canonical Mad-dependent silencer? In principle, Smad2 could be acting as a transcription factor, but the reversal of the phenotype upon removal of Baboon strongly suggests that Baboon itself can regulate the SE, and that Smad2 limits Baboon activity. Trans-branch cross-over, in which stimulation of a TGFβ Type I receptor can lead to phosphorylation of a BMP R-Smad, has been reported, and we have observed this reaction in vivo in Drosophila (Peterson et al., 2012). To test whether Baboon phosphorylates Mad in the absence of Smad2, we examined P-Mad in Smad2 RNAi clones. We found that P-Mad was not increased in such clones at late L3 (Fig. 7H). To test whether the behavior is different at earlier stages, we examined clones throughout the two days of L3 development, and we did not observe ectopic P-Mad staining (supplementary material Fig. S8). To test whether brinker mis-regulation can occur without detectable P-Mad increase, we assayed a brinker reporter and P-Mad simultaneously and confirmed that clones with ectopic silencing did not have increased P-Mad (Fig. 7H). Thus, despite the demonstrated requirement for baboon and Mad in the silencing activity, we find no evidence for the direct involvement of Baboon cross-talk in producing ectopic P-Mad.
DISCUSSION
The relationship between Baboon and Smad2 varies by tissue
Based on phenotypic comparison of baboon loss of function and two Smad2 alleles, we can model three ways that Smad2 mutation alters development in various tissues. For canonical signaling events, exemplified by neuroblast proliferation and axonal pruning, loss of function for baboon or Smad2 is equivalent. In cases in which the Smad2 null phenotype differs from the point mutant phenotype, we invoke non-canonical signaling. For the development of enlarged anal pads, the point mutant acts as a neomorphic allele that blocks Baboon function, whereas the null allele does not produce the phenotype, indicating that Baboon is able to function properly in a Smad-independent manner. For the wing disc overgrowth phenotype, the point mutant protein is produced and can bind to Baboon to prevent the promiscuous activity of Baboon that occurs when Smad2 is completely removed. We focus here on the wing growth process, and one key finding from our experiments is that Smad2 restricts the output of Baboon. Together with our earlier report on the competition between Smad2 and Mad for activation by Baboon, this is another example of a substrate regulating receptor activity. This wiring may be relevant to some situations in which antagonism is observed between TGFβ and BMP signaling, and might also be an under-appreciated feature of signal pathway regulation in general.
Anti-signaling from Smad2 to Baboon
Smads are direct phosphorylation targets of TGFβ superfamily Type I receptors, and are thus typically considered to be downstream of the receptor. Our finding that Baboon is clearly epistatic to Smad2 for the wing disc overgrowth inverts this relationship to generate a configuration we term ‘anti-signaling’. Other proteins are known to bind and negatively regulate TGFβ receptors, such as I-Smads (Hayashi et al., 1997). Anti-signaling by Smad2 resembles this activity, but offers additional regulatory possibilities because the same molecule can transduce the canonical signal and inhibit non-canonical signals. One interpretation of our data is that the function of Smad2 during wing development is to inhibit Baboon, thus permitting BMP signaling to control growth and patterning. Alternatively, the Baboon receptor could normally play a supporting role in controlling these processes, with inhibition by Smad2 as one of several inputs into Baboon activity. baboon mutants have small wing discs, but it is difficult to attribute this to a wing-autonomous function because of pleiotropic defects. Indeed, RNAi of baboon using several GAL4 drivers expressed in the wing disc had no observable effect on the shape or size of larval wing discs or adult wings (data not shown). If Baboon normally plays a role, then its absence can be compensated for by other growth control mechanisms, or it might only be required under certain developmental conditions. Furthermore, the finding that Smad2; baboon double mutant wing disc phenotypes resemble those of baboon mutants indicates that the major function of Baboon must be non-canonical (Fig. 4; supplementary material Figs S5, S7). We cannot rule out a minor role of canonical TGFβ signaling in wing disc patterning or growth, but our data clearly indicate that canonical transcription factor activity is dispensable for proper spatial proliferation.
Silencing of multiple genes leads to a composite wing phenotype
Our second principal finding is that unrestrained Baboon activity leads to ectopic repression through the SEs found in a subset of known BMP target genes. The phenotype we observe in the Smad2-null mutant can be explained as a compound loss of expression of BMP targets that would otherwise have lateral expression (Fig. 8A,B). Loss of Brinker on its own leads to widened discs, which neatly explains the lateral overgrowth of Smad2 mutant discs. Loss of Pentagone on its own leads to constriction of the P-Mad gradient and narrow wing discs. The narrow P-Mad stripe in Smad2-null discs is consistent with a loss of Pentagone, and manifests as narrowed expression of positive targets of Dpp signaling. The brinker morphological effect dominates over the pentagone morphological effect because brinker expression is uncoupled from the P-Mad pattern. Pentagone is a secreted molecule, which might explain why Smad2 function must be removed in nearly all cells of the wing blade to see the widening. Note that these two defects are separable, as brinker mutant discs elaborate a normal P-Mad gradient (Moser and Campbell, 2005) and pentagone mutants express brinker laterally (Vuilleumier et al., 2010). The Smad2 phenotype is thus a compound phenotype caused by loss of multiple SE-controlled genes (Fig. 8B). Additional genes contain predicted SEs (Pyrowolakis et al., 2004), so we expect that other targets are also silenced in the Smad2 mutant but any resulting phenotype is probably obscured by the strong effects of Brinker and Pentagone loss.
Fig. 8.

Models of wing widening due to cavalier Baboon signaling in the absence of Smad2. (A) Functions of BMP and Activin/TGFβ pathways in larval wing disc development are illustrated. Normally, Dpp and other BMP branch proteins generate a P-Mad gradient about the A/P axis, and the activated and repressed targets control proliferation (Brinker) and the P-Mad gradient itself (Pentagone). (B) In the absence of Smad2, promiscuous Baboon activity leads to ectopic silencing through SE-containing Dpp target genes. The absence of Brinker causes lateral overgrowth and the absence of Pentagone leads to a contracted P-Mad stripe. (C,D) Models of non-canonical Baboon functions at the membrane. (C) An unknown phosphorylation target (T) of Baboon could lead to ectopic repression at SEs. Based on the genetic results, Smad2 (S) restrains Baboon activity, and removal of Baboon removes the corrupting influence on the SE. (D) Alternatively, the role of Baboon could be to bind one or more membrane proteins the availability of which affects repression at SEs. To conform to the epistasis of baboon over Smad2, the function of this hypothetical molecule (J) would be altered by Baboon only in the absence of Smad2.
We show that Baboon, Mad and Schnurri are all required for the ectopic silencing through SE. Sander et al. reported that Mad and Medea are required for Smad2 RNAi-induced disc widening (Sander et al., 2010). Because Medea is a common partner for Smad2 and Mad, it is not straightforward to determine which one of these roles is relevant. An alternative role for Medea would be to limit Smad2 or Mad action by way of competition for a limited amount of Medea, but we found that Medea overexpression did not phenocopy Smad2 loss in the wing disc (data not shown). Instead, our data suggest that the cell-autonomous requirement for Medea is due to its inclusion in the Mad-Medea-Schnurri repressive complex that binds SEs.
Models of Baboon-SE coupling
It is unknown how Baboon influences events at the SE. As a transmembrane receptor with kinase activity, the two ways Baboon can function are by post-translational modification of substrates, or by binding and regulating the activity of other proteins (Fig. 8C,D). The only known substrates for Baboon are Smad2 and Mad. In the wing disc, an active requirement for Smad2 in transmitting the signal is ruled out by the double mutant phenotype. We found no direct evidence for the simple hypothesis that unrestrained Baboon phosphorylates Mad at the canonical C-terminal serine residues. Mad is regulated by other modifications that control its stability and nuclear localization (Zeng et al., 2007; Eivers et al., 2009), so an intriguing possibility is that the increased access to Baboon afforded Mad in the absence of Smad2 may alter its modification state. Other potential substrates for the kinase Baboon could mediate the gain-of-function Baboon activity uncovered by lack of Smad2 (T in Fig. 8C). No non-Smad substrate is known for any Drosophila receptor, but a growing list in mammals includes endoglin, mps1, ShcA and PAR6 (Ray et al., 2010; Zhu et al., 2007; Lee et al., 2007; Ozdamar et al., 2005). Alternatively, Baboon could regulate other factors merely by binding to them. Baboon has been proposed to act independently of R-Smads in axon projection in the mushroom body (Ng, 2008), and there are examples from the broader TGFβ superfamily in which receptors regulate the availability of other factors by binding, such as regulation of LIMK activity by binding to BMPRII (Lee-Hoeflich et al., 2004). Figure 8D models the conjectured Baboon binding partner as a transmembrane protein, but a cytoplasmic protein could also fill this role. Epistasis experiments showed that aberrant output is observed only in the situation in which Baboon is present and Smad2 is absent. We therefore model a complex of Baboon and another protein as the agent that leads to silencing, either by promoting SE repression, or preventing the other molecule from carrying out an anti-repression role (Fig. 8D).
At the level of the SE DNA sequence, we assume that the Mad-Medea-Schnurri complex is the only complex that can repress transcription from the SE. This is consistent with the requirement for the proteins of this complex even when Baboon is ‘trying’ to engage. Unrestrained Baboon might alter the sensitivity to the repressive complex, such that in all cells the basal level of P-Mad is sufficient to engage the SE. To engender this state, Baboon could be promoting the formation, availability or activity of the silencing complex. Alternatively, Baboon could be removing an SE input that normally opposes the action of the silencing complex. The action of additional factors binding at or near the SE is speculative, but it is common for regulatory DNA elements to bind competing complexes to permit graded activities. An example is the competitive binding of Brinker and Mad to the activation element of Dpp target genes in the wing (Weiss et al., 2010). Based on the available data, it is not known whether Baboon action at the membrane directly regulates a component of a DNA-associated complex or if such regulation is mediated by intermediate factors.
Future work will provide molecular details of how the Activin and BMP branches of the ancient TGFβ superfamily pathway are wired to coordinately regulate growth in the wing disc. It will be interesting to learn whether the mechanism is tailored to the specific system or represents a common mechanism. More generally, it will be important to gauge the significance of substrate inhibition in TGFβ superfamily signaling. TGFβ signaling networks are present in all metazoan cells and control many cell behaviors in development and disease. Mutations in Smad proteins have long been known to be associated with various aspects of cancer (Padua and Massagué, 2009), and Smad3 mutations were recently reported to be associated with a dominant vascular syndrome (van de Laar et al., 2011). If Smad2/3 proteins control the activity of Activin or TGFβ receptors, then additional effects beyond loss of function of the canonical signaling pathway need to be considered. As exemplified by loss of Baboon function caused by a mutant Smad2 protein, Smad proteins can cause loss of non-canonical receptor functions. As illustrated by Baboon-dependent patterning defects when Smad2 is completely absent, lack of Smads can lead to gain of function for TGFβ superfamily receptors. Beyond the appreciated cell-type dependent roles of Smad proteins as transcription factors, these new possibilities add further complexity to the TGFβ signaling network that need to be studied to predict the effects of therapeutic intervention to regulate TGFβ signaling.
Supplementary Material
Acknowledgements
MaryJane Shimell made and characterized the cell culture behavior of the Smad2-RB4 quadruple point mutant. We are grateful to the Pyrowolakis, Affolter, Neufeld, Morata, Nakato, Selleck, Truman and Haerry groups for sharing fly lines; to Melissa Ritter for characterization of RNAi lines; and to Phil Jensen for discussions.
Footnotes
Funding
This work was supported by the National Institutes of Health [grant R01 GM95746 to M.B.O.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.085605/-/DC1
References
- Bilder D., Li M., Perrimon N. (2000). Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 289, 113-116 [DOI] [PubMed] [Google Scholar]
- Brummel T., Abdollah S., Haerry T. E., Shimell M. J., Merriam J., Raftery L., Wrana J. L., O'Connor M. B. (1999). The Drosophila activin receptor baboon signals through dSmad2 and controls cell proliferation but not patterning during larval development. Genes Dev. 13, 98-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant P. J., Levinson P. (1985). Intrinsic growth control in the imaginal primordia of Drosophila, and the autonomous action of a lethal mutation causing overgrowth. Dev. Biol. 107, 355-363 [DOI] [PubMed] [Google Scholar]
- Burke R., Basler K. (1996). Dpp receptors are autonomously required for cell proliferation in the entire developing Drosophila wing. Development 122, 2261-2269 [DOI] [PubMed] [Google Scholar]
- Calleja M., Moreno E., Pelaz S., Morata G. (1996). Visualization of gene expression in living adult Drosophila. Science 274, 252-255 [DOI] [PubMed] [Google Scholar]
- Campbell G., Tomlinson A. (1999). Transducing the Dpp morphogen gradient in the wing of Drosophila: regulation of Dpp targets by brinker. Cell 96, 553-562 [DOI] [PubMed] [Google Scholar]
- Candia A. F., Watabe T., Hawley S. H., Onichtchouk D., Zhang Y., Derynck R., Niehrs C., Cho K. W. (1997). Cellular interpretation of multiple TGF-β signals: intracellular antagonism between activin/BVg1 and BMP-2/4 signaling mediated by Smads. Development 124, 4467-4480 [DOI] [PubMed] [Google Scholar]
- Eivers E., Fuentealba L. C., Sander V., Clemens J. C., Hartnett L., De Robertis E. M. (2009). Mad is required for wingless signaling in wing development and segment patterning in Drosophila. PLoS ONE 4, e6543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbens Y. Y., Warren J. T., Gilbert L. I., O'Connor M. B. (2011). Neuroendocrine regulation of Drosophila metamorphosis requires TGFbeta/Activin signaling. Development 138, 2693-2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halachmi N., Schulze K. L., Inbal A., Salzberg A. (2007). Additional sex combs affects antennal development by means of spatially restricted repression of Antp and wg. Dev. Dyn. 236, 2118-2130 [DOI] [PubMed] [Google Scholar]
- Hayashi H., Abdollah S., Qiu Y., Cai J., Xu Y. Y., Grinnell B. W., Richardson M. A., Topper J. N., Gimbrone M. A., Jr, Wrana J. L., et al. (1997). The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 89, 1165-1173 [DOI] [PubMed] [Google Scholar]
- Henderson K. D., Andrew D. J. (1998). Identification of a novel Drosophila SMAD on the X chromosome. Biochem. Biophys. Res. Commun. 252, 195-201 [DOI] [PubMed] [Google Scholar]
- Lee M. K., Pardoux C., Hall M. C., Lee P. S., Warburton D., Qing J., Smith S. M., Derynck R. (2007). TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 26, 3957-3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Hoeflich S. T., Causing C. G., Podkowa M., Zhao X., Wrana J. L., Attisano L. (2004). Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J. 23, 4792-4801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu I. M., Schilling S. H., Knouse K. A., Choy L., Derynck R., Wang X. F. (2009). TGFbeta-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGFbeta switch. EMBO J. 28, 88-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo R. S., Chen Y. G., Shi Y., Pavletich N. P., Massagué J. (1998). The L3 loop: a structural motif determining specific interactions between SMAD proteins and TGF-β receptors. EMBO J. 17, 996-1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín F. A., Pérez-Garijo A., Moreno E., Morata G. (2004). The brinker gradient controls wing growth in Drosophila. Development 131, 4921-4930 [DOI] [PubMed] [Google Scholar]
- Marty T., Müller B., Basler K., Affolter M. (2000). Schnurri mediates Dpp-dependent repression of brinker transcription. Nat. Cell Biol. 2, 745-749 [DOI] [PubMed] [Google Scholar]
- Milán M., Campuzano S., García-Bellido A. (1997). Developmental parameters of cell death in the wing disc of Drosophila. Proc. Natl. Acad. Sci. USA 94, 5691-5696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami M., Kinoshita N., Kamoshida Y., Tanimoto H., Tabata T. (1999). brinker is a target of Dpp in Drosophila that negatively regulates Dpp-dependent genes. Nature 398, 242-246 [DOI] [PubMed] [Google Scholar]
- Moser M., Campbell G. (2005). Generating and interpreting the Brinker gradient in the Drosophila wing. Dev. Biol. 286, 647-658 [DOI] [PubMed] [Google Scholar]
- Müller B., Hartmann B., Pyrowolakis G., Affolter M., Basler K. (2003). Conversion of an extracellular Dpp/BMP morphogen gradient into an inverse transcriptional gradient. Cell 113, 221-233 [DOI] [PubMed] [Google Scholar]
- Nellen D., Burke R., Struhl G., Basler K. (1996). Direct and long-range action of a DPP morphogen gradient. Cell 85, 357-368 [DOI] [PubMed] [Google Scholar]
- Newfeld S. J., Mehra A., Singer M. A., Wrana J. L., Attisano L., Gelbart W. M. (1997). Mothers against dpp participates in a DDP/TGF-β responsive serine-threonine kinase signal transduction cascade. Development 124, 3167-3176 [DOI] [PubMed] [Google Scholar]
- Ng J. (2008). TGF-β signals regulate axonal development through distinct Smad-independent mechanisms. Development 135, 4025-4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono H., Rewitz K. F., Shinoda T., Itoyama K., Petryk A., Rybczynski R., Jarcho M., Warren J. T., Marqués G., Shimell M. J., et al. (2006). Spook and Spookier code for stage-specific components of the ecdysone biosynthetic pathway in Diptera. Dev. Biol. 298, 555-570 [DOI] [PubMed] [Google Scholar]
- Ozdamar B., Bose R., Barrios-Rodiles M., Wang H. R., Zhang Y., Wrana J. L. (2005). Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 307, 1603-1609 [DOI] [PubMed] [Google Scholar]
- Padua D., Massagué J. (2009). Roles of TGFbeta in metastasis. Cell Res. 19, 89-102 [DOI] [PubMed] [Google Scholar]
- Parker L., Ellis J. E., Nguyen M. Q., Arora K. (2006). The divergent TGF-β ligand Dawdle utilizes an activin pathway to influence axon guidance in Drosophila. Development 133, 4981-4991 [DOI] [PubMed] [Google Scholar]
- Peterson A. J., Jensen P. A., Shimell M., Stefancsik R., Wijayatonge R., Herder R., Raftery L. A., O'Connor M. B. (2012). R-Smad competition controls activin receptor output in Drosophila. PLoS ONE 7, e36548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrowolakis G., Hartmann B., Müller B., Basler K., Affolter M. (2004). A simple molecular complex mediates widespread BMP-induced repression during Drosophila development. Dev. Cell 7, 229-240 [DOI] [PubMed] [Google Scholar]
- Ray B. N., Lee N. Y., How T., Blobe G. C. (2010). ALK5 phosphorylation of the endoglin cytoplasmic domain regulates Smad1/5/8 signaling and endothelial cell migration. Carcinogenesis 31, 435-441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander V., Eivers E., Choi R. H., De Robertis E. M. (2010). Drosophila Smad2 opposes Mad signaling during wing vein development. PLoS ONE 5, e10383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwank G., Basler K. (2010). Regulation of organ growth by morphogen gradients. Cold Spring Harb. Perspect. Biol. 2, a001669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwank G., Restrepo S., Basler K. (2008). Growth regulation by Dpp: an essential role for Brinker and a non-essential role for graded signaling levels. Development 135, 4003-4013 [DOI] [PubMed] [Google Scholar]
- Serpe M., O'Connor M. B. (2006). The metalloprotease tolloid-related and its TGF-β-like substrate Dawdle regulate Drosophila motoneuron axon guidance. Development 133, 4969-4979 [DOI] [PubMed] [Google Scholar]
- van de Laar I. M., Oldenburg R. A., Pals G., Roos-Hesselink J. W., de Graaf B. M., Verhagen J. M., Hoedemaekers Y. M., Willemsen R., Severijnen L. A., Venselaar H., et al. (2011). Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 43, 121-126 [DOI] [PubMed] [Google Scholar]
- Vuilleumier R., Springhorn A., Patterson L., Koidl S., Hammerschmidt M., Affolter M., Pyrowolakis G. (2010). Control of Dpp morphogen signalling by a secreted feedback regulator. Nat. Cell Biol. 12, 611-617 [DOI] [PubMed] [Google Scholar]
- Wartlick O., Mumcu P., Kicheva A., Bittig T., Seum C., Jülicher F., González-Gaitán M. (2011). Dynamics of Dpp signaling and proliferation control. Science 331, 1154-1159 [DOI] [PubMed] [Google Scholar]
- Weiss A., Charbonnier E., Ellertsdóttir E., Tsirigos A., Wolf C., Schuh R., Pyrowolakis G., Affolter M. (2010). A conserved activation element in BMP signaling during Drosophila development. Nat. Struct. Mol. Biol. 17, 69-76 [DOI] [PubMed] [Google Scholar]
- Wrighton K. H., Lin X., Yu P. B., Feng X. H. (2009). Transforming growth factor β can stimulate Smad1 phosphorylation independently of bone morphogenic protein receptors. J. Biol. Chem. 284, 9755-9763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M., Beppu H., Takaoka K., Meno C., Li E., Miyazono K., Hamada H. (2009). Antagonism between Smad1 and Smad2 signaling determines the site of distal visceral endoderm formation in the mouse embryo. J. Cell Biol. 184, 323-334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L. C., Phin S., Cho J., Rushlow C., Arora K., Warrior R. (2008). Multiple modular promoter elements drive graded brinker expression in response to the Dpp morphogen gradient. Development 135, 2183-2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y. A., Rahnama M., Wang S., Sosu-Sedzorme W., Verheyen E. M. (2007). Drosophila Nemo antagonizes BMP signaling by phosphorylation of Mad and inhibition of its nuclear accumulation. Development 134, 2061-2071 [DOI] [PubMed] [Google Scholar]
- Zheng X., Wang J., Haerry T. E., Wu A. Y., Martin J., O'Connor M. B., Lee C. H., Lee T. (2003). TGF-β signaling activates steroid hormone receptor expression during neuronal remodeling in the Drosophila brain. Cell 112, 303-315 [DOI] [PubMed] [Google Scholar]
- Zhu S., Wang W., Clarke D. C., Liu X. (2007). Activation of Mps1 promotes transforming growth factor-β-independent Smad signaling. J. Biol. Chem. 282, 18327-18338 [DOI] [PubMed] [Google Scholar]
- Zhu C. C., Boone J. Q., Jensen P. A., Hanna S., Podemski L., Locke J., Doe C. Q., O'Connor M. B. (2008). Drosophila Activin- and the Activin-like product Dawdle function redundantly to regulate proliferation in the larval brain. Development 135, 513-521 [DOI] [PubMed] [Google Scholar]
- Zirin J. D., Mann R. S. (2007). Nubbin and Teashirt mark barriers to clonal growth along the proximal-distal axis of the Drosophila wing. Dev. Biol. 304, 745-758 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.