

Abstract
Mammalian mitochondrial DNA (mtDNA) is replicated by the heterotrimeric Pol γ comprised of a single catalytic subunit, encoded by Polg, and a homodimeric accessory subunit encoded by the Polg2 gene. While the catalytic subunit has been shown to be essential for embryo development, genetic data regarding the accessory subunit are lacking in mammalian systems. Here, we describe the generation of heterozygous (Polg2+/−) and homozygous (Polg2−/−) knockout (KO) mice. Polg2+/− mice are haplosufficient and develop normally with no discernable difference in mitochondrial function through 2 years of age. In contrast, the Polg2−/− is embryonic lethal at day 8.0–8.5 p.c. with concomitant loss of mtDNA and mtDNA gene products. Electron microscopy shows severe ultra-structural defects and loss of organized cristae in mitochondria of the Polg2−/− embryos as well as an increase in lipid accumulation compared with both wild-type (WT) and Polg2+/− embryos. Our data indicate that Polg2 function is critical to mammalian embryogenesis and mtDNA replication, and that a single copy of Polg2 is sufficient to sustain life.
INTRODUCTION
In animal cells, mitochondrial DNA (mtDNA) is replicated by the DNA polymerase γ (pol γ) complex consisting of a large catalytic subunit of 140 kDa (p140) and a smaller accessory subunit of 55 kDa (p55). The human p140 catalytic subunit, encoded by the POLG gene on chromosome 15, contains the DNA polymerase activity, a 3′–5′ exonuclease required for proofreading and a 5′-dRP lyase activity required for single-nucleotide base excision repair (1). The fidelity of DNA synthesis by the catalytic subunit is highly faithful, making less than one error per 250 000 nucleotides incorporated (2). The human p55 accessory subunit is encoded by the POLG2 gene on chromosome 17 and promotes highly processive DNA synthesis and enhanced DNA binding for pol γ (3). One consequence of increased processivity offered by the accessory subunit is a 25% increase in single-nucleotide misincorporation events (2). The accessory subunit is not required for in vitro polymerase activity by the catalytic subunit (3).
The crystal structure of the mouse p55 accessory subunit revealed a tight homodimer complex of 110 kDa (4). Physical and functional experiments have demonstrated that the human p55 homodimer binds with one p140 catalytic subunit to form the heterotrimeric holoenzyme (5). The crystal structure of the human Pol γ heterotrimer revealed that the p55 dimer binds the catalytic subunit in an asymmetric fashion. Multiple contacts are made between the p140 and the proximal p55 subunit, while only a single salt bridge is made between a residue of the distal p55 monomer and p140 (6). The proximal p55 subunit appears to strengthen the interaction of the complex with DNA, while the distal p55 subunit accelerates nucleotide incorporation (7).
The accessory subunit gene is not conserved throughout eukaryotes as evident by its absence in some single-celled eukaryotes and the multicellular nematode, Caenorhabditis elegans (8). While Polg2 is conserved in metazoans, its presence is sporadic among other eukaryotes, thus raising the question of its relative importance regarding mtDNA replication. The dispensable characteristic of p55 for in vitro polymerase activity, the reduced fidelity associated with Pol γ harboring the p55 dimer and the lack of evolutionary conservation suggest that p55 may not be essential for mtDNA replication. On the other hand, studies in a Drosophila Polg2 knockout (KO) revealed a failure to develop beyond the pupal stage, suggesting an essential role of Polg2 in animal cell mtDNA replication (9). In further support of POLG2 being essential in mtDNA replication, patients with mutations in POLG2 are heterozygous, and these mutations are associated with mitochondrial disease (8).
Defects in mtDNA replication lead to a number of debilitating and life-threatening diseases (10–12). Mutations in the POLG gene can cause depletion of mtDNA, such as in myocerebrohepatopathy spectrum disorder and Alpers syndrome, or cause deletions in mtDNA associated with progressive external ophthalmoplegia (PEO), ataxia neuropathy spectrum disorders or myoclonus epilepsy myopathy sensory ataxia (13). To date, nearly 200 different types of pathogenic POLG mutations have been described in the literature (http://tools.niehs.nih.gov/polg/), and mutations in POLG represent one of the most common genetic causes of mitochondrial diseases. It has been calculated that 2% of the general population are heterozygous carriers of POLG pathological mutations (14).
Diseases associated with mutations in POLG2 have not been investigated as extensively as those in POLG. The first described POLG2 mutation was documented in a patient with late-onset autosomal dominant PEO (adPEO) with multiple mtDNA deletions caused by a single heterozygous G451E (c.1352G>A) variant that compromised the ability of the accessory subunit to associate with the catalytic subunit, and hence, failed to stimulate processive DNA synthesis (15). The second identified POLG2 mutation also involved a late-onset adPEO patient with mtDNA deletions and harbored a c.1207-1208ins24 mutation, causing mis-splicing and skipping of exon 7 (16). More recently, in a cohort of 112 patients with mitochondrial disease with no POLG mutations, eight heterozygous mutations in POLG2 were identified of which seven were novel (17). Biochemical analysis of these seven p55 variants revealed that four were similar to wild-type (WT), but two variants, P205R and R369G p55, had reduced stimulation of processivity and decreased affinity for the catalytic subunit (17). One variant, L475DfsX2, resulted in a premature stop codon that truncates 10-carboxyl-terminal amino acid residues and was generally unstable and unable to bind either the catalytic subunit or double-stranded DNA. Patients harboring P205R and L475DfsX2 variants had early age-of-onset disease, suggesting POLG2-related disorders include both early- and late-onset (PEO) mitochondrial disorders. The failure to enhance processivity in the catalytic subunit by these mutant variants presumably causes the complex to stall during mtDNA replication. Stalling during replication is consistent with the accumulation of mtDNA deletions found in PEO patients harboring POLG2 variants G451E and R369G (15,18). In order to determine the requirements of the Polg2 gene in mammalian systems, we generated a Polg2 KO mouse, characterized mitochondrial ultrastructure, determined mtDNA content, evaluated for mtDNA deletions, assessed cytochrome c oxidase I (COXI) activity, conducted extensive pathology and measured transcript levels of genes associated with mtDNA replication in the Polg+/+, Polg+/− and Polg−/− animals.
RESULTS
Generation of Polg2 knockout mice
The C-terminus of the accessory subunit includes the domains responsible for dimerization, DNA binding, the ability to bind to the catalytic subunit (6), and is highly conserved between human and mouse (Supplementary Material, Fig. S1) (4). In our model, the Polg2 gene was disrupted in a C57/B6 mouse using the cre-loxp recombination system (Fig. 1A) (19). The embryonic stem (ES) cells were positively selected for homologous recombination via neomycin selection. Homologous recombination was further confirmed in ES cells by Southern blots. The mouse genomic DNA was digested with the restriction enzyme SpeI. The 5′ probe detected the WT fragment at 26.0 kb and the Polg2 KO fragment at 9.6 kb. The 3′ probe detected the WT fragment at 26.0 kb, and the KO fragment at 15.0 kb (Fig. 1B). The resulting Polg2+/loxp mice were crossed to generate a homozygous floxed mouse (Polg2loxp/loxp). The Polg2loxp/loxp male mice were then crossed with ubiquitously expressing cre female mice (Sox2-cre) resulting in a cre-mediated excision of exons 5–7 of the Polg2 gene in the pups generated from this cross. The resulting genotypes were confirmed via PCR. Further evidence of gene disruption was obtained by extracting total RNA from the kidneys of adult Polg2 heterozygous mice, and using traditional reverse transcriptase PCR methods to identify the expected 324 bp deletion in Polg2 transcript (Fig. 1C). The internal deletion of the mRNA results in the loss of functional p55 domains, and we predict that this will inactivate the protein.
Figure 1.

Generation of the Polg2 KO mouse. (A) The Polg2+/− were derived from parental floxed mice harboring homologous arms for recombination (blue), the loxP sequences (purple triangles) flanking exons 5–7 (exons designated by orange bars) at ∼2.4 kb in size (green) and loxP sequences flanking the Neo expression cassette (black and white). (B) Mouse genomic DNA was digested with SpeI (Sp) following neomycin selection. Southern blots confirmed homologous recombination in ES cells using a 5′ arm probe (panel 1) with the WT band located at 26 kb and the recombinant 9.6 kb allele, and a 3′ arm probe (panel 2) to identify the WT band at 26.0 kb and the recombinant allele at 15.0 kb. (C) The identification of the internally deleted transcript in adult Polg2+/− mice further confirms the KO of the three critical exons. Using RT-PCR, we amplified a fragment that spanned exons 4–8 of the Polg2 gene from total RNA isolated from kidney. The WT cDNA yielded a 660 bp band, and the genetically altered transcript with the deletions of exons 5–7 yielded a 336 bp amplicon.
Characterization of adult Polg2 heterozygous mice
The Polg2 heterozygous mice were monitored until 2 years of age and no distinguishable differences were found from the WT controls in life span or health. Body weights of the Polg2+/− mice were measured at 40 weeks of age and 2 years of age with no significant differences from the age-matched WT mice (Supplementary Material, Table S1A). Relative liver weights were determined at 2 years of age again with no difference from the WT controls (Supplementary Material, Table S1B). Extensive histopathology and clinical pathology (Supplementary Material, Tables S2–S5) were conducted on 2-year-old Polg2 heterozygous mice with individual differences noted, but they were not consistent nor attributable to the loss of one copy of the Polg2 gene (Supplementary Material, data). In addition, electron microscopy (EM) was used to examine the ultra-structural features of heart tissue of 1-year-old Polg2+/− mice, and again no differences were found from the WT (Supplementary Material, Fig. S2).
To investigate any effects on mtDNA replication due the loss of half of the WT transcript level of Polg2, we used quantitative PCR (qPCR) to determine the mtDNA content in brain, heart, liver and muscle tissues of adult heterozygous animals. Whole tissue lysates were used to measure the copy number of mtDNA using primers and probes that target a mtDNA gene outside of the arc of common insertions and deletions, Nd1. We first measured mtDNA in each of the four tissues at 40 weeks of age in three animals of each genotype and found no change in the mtDNA content of Polg2+/− tissues from their age-matched WT counterparts (Fig. 2). We repeated this analysis at 1 year of age with an increased sample size of 10 animals for each genotype, and again found no statistical difference in the mtDNA content from the WT levels in any of the tissues (Fig. 2). At 2 years of age, mtDNA measurements were repeated in brain, heart, liver and muscle tissues with an even greater number of samples (n = 21), and the results were similar to the earlier time points (Fig. 2). Although, there were large individual differences in the mtDNA content measured, these differences existed in both the WT and the Polg2+/− tissues. In summary, there was no significant difference (P > 0.05) found in the mtDNA content of brain, heart, liver and muscle tissues compared with the corresponding tissues in age-matched WT mice.
Figure 2.
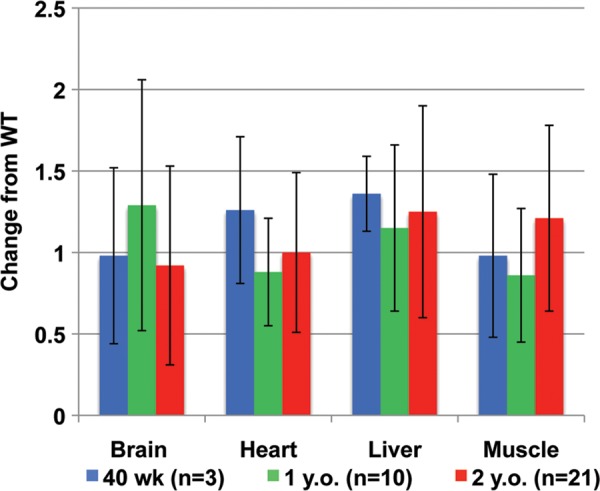
Determination of the mtDNA content in tissues. qPCR analysis was used to determine mtDNA levels in brain, heart, liver and muscle tissues of adult Polg2+/− animals. Data are expressed as fold change from the mean levels of WT tissues. Tissue samples were obtained at 40 weeks (n = 3, blue), 1 year (n = 10, green) and 2 years of age (n = 21, red). No significant changes (P > 0.05) in the mtDNA content were found in any of the tissues at any ages. Error bars indicate ± standard deviation.
Additional molecular mtDNA parameters were measured such as transcript levels of genes associated with mtDNA replication (Supplementary Material, Fig. S3) and mtDNA deletions (Supplementary Material, Figs S4 and S5). Again, there was no statistically significant difference seen in any of these studies between the Polg2+/− and age-matched WT control mice (Supplementary Material, data). In conclusion, the heterozygous Polg2 mice are haplosufficient with no differences in histology, clinical pathology, mitochondrial ultrastructure, mtDNA content, mtDNA deletions or gene expression associated with mtDNA replication.
Polg2 homozygous knockout is embryonic lethal
Interbreeding Polg2+/− mice generated Polg2 homozygous KO mice. The analysis of 251 pups from a total of 40 litters resulted in a genotype ratio for the Polg2 gene of 68% heterozygous (Polg2+/−), 32% WT and a complete absence of homozygous KOs (Polg2−/−). The absence of Polg2−/− pups indicated a possible embryonic lethal phenotype. Working back through the various stages of development, beginning with embryonic day (E) 18.5, structurally intact albeit non-viable Polg2−/− embryos were first observed at E9.5 (Fig. 3). Somite counts for viable, turned WT littermates ranged from 13 to 19; both the somite counts and the turned status corresponded with a gestational age of E9.5 (20,21). Developmental arrest of non-viable Polg2−/− embryos was observed in these litters as evidenced by the presence of unturned embryos with eight somites, this provided sufficient evidence to conclude that lethality occurred at approximately gestation day E8.0–E8.5. (Fig. 3). In order to confirm that the non-viable embryos were Polg2−/−, serial frozen sections of implanted, intact E8.5 embryos (Supplementary Material, Fig. S6) were extracted from the deciduum by laser capture microdissection (LCM) and genotyped by PCR. The H&E-stained slide was evaluated for histopathological changes. The WT and Polg2+/− embryos were morphologically similar, with nucleated red blood cells (RBCs) present in the E8.5 unturned conceptus indicating continued embryo viability. Most of the Polg2−/− embryos lacked nucleated RBCs, again denoting a nonviable conceptus in which developmental arrest had occurred at approximately E8.0–E8.5 (20). Tissue necrosis was extensive in the non-viable embryos, which correlated with their grossly pale and friable appearances.
Figure 3.

Morphology of embryos 9.5 days post coitis. (A) A sagittal view of a turned viable WT embryo depicting all 16 somites at E9.5 (×20 magnification), (B) an unturned non-viable Polg2−/− embryo littermate at E9.5 depicting only 8 somites (×40 magnification). This embryo is phenotypically similar to a WT embryo 8.5 days p.c., suggesting arrested development around that time. Arrows indicate somites, NF: neural fold, AS: amniotic sac and H: heart.
Mitochondrial dysfunction in Polg2−/− embryos
To assess mitochondrial function in E8.5 Polg2−/− embryos, a histochemical stain was employed on serial frozen sections to detect the presence or absence of the COXI enzyme, a subunit of cytochrome c oxidase, uniquely encoded by the mtDNA. COXI staining of intact embryos enveloped in the maternally derived deciduum revealed a complete lack of COXI activity in the Polg2−/− embryos, in contrast to the widespread staining of WT conceptuses (Fig. 4). The complete absence of COX1 activity in the Polg2−/− embryos implied respiratory-chain failure. Homogenous positive staining was observed in the maternal decidual tissue of Polg2−/−, which was identical to the pattern in the WT control decidua.
Figure 4.

COXI staining was used to determine mitochondrial function. Ten micrometer cryostat sections of embryos at E8.5 post coitis still enveloped in the maternally derived, deciduum at ×10 magnification and the inset panel at ×2.5 magnification. (A) A Polg2+/+ embryo and the maternal deciduum both stained positive for COXI activity. (B) A Polg2−/− embryo lacking COXI activity. (C) A Polg2+/+ embryo counterstained with hematoxylin showing a dual staining with COXI-positive cells. (D) A Polg2−/− embryo counter stained with hematoxylin showing COXI-negative cells stained in purple revealing the presence of an embryo in the deciduum.
To further investigate mitochondrial dysfunction in the Polg2−/− embryo, we used EM to study the ultrastructures of the mitochondria in the E8.5 embryonic tissues. Using EM (at a magnification of ×11 500), there was a noticeable increase in fatty droplets and swollen cristae in the Polg2−/− compared with the WT (Fig. 5A and C), which may indicate a disruption in the oxidation of long-chain fatty acids. At an even greater magnification (×20 500) of the KO, the overall morphology of the mitochondria was altered with a noticeable formation of inner-compartmentalized structures and the lack of organized cristae (Fig. 5B and D). At ×43 000 (Fig. 5E), a disruption of the mitochondrial outer and inner membranes was revealed, and the aberrant mitochondria morphology of the KO was further pronounced.
Figure 5.

EM was used to analyze the morphology of the mitochondria in E8.5 embryos. (A) Mitochondria from a WT embryo seen here at ×11 500 and at ×20 500 magnifications (B), both the panels are evidence of normal morphology. In the Polg2−/− embryos, many mitochondria have an aberrant morphology. (C) At ×11 500 magnification, there is an increase in fatty droplets and evidence of swollen intra-cristal spaces in the KO, and at ×20 500 (D) many mitochondria have the formation of inner-compartmentalized structures, and lack organized cristae. (E) At ×43 000, the aberrant morphology of the KO is further pronounced, and evidence of outer and inner-mitochondrial membrane disruption is observed.
To determine the effect of the Polg2 KO on mtDNA replication, we measured the mtDNA content using qPCR on Polg2−/− embryo samples. LCM was used to discern embryonic from maternal tissues, and whole tissue lysates were used in a qPCR assay. The mtDNA content was normalized to the nuclear gene, Actin, and expressed as the mean relative copy number. The WT (n = 4) and Polg2+/− (n = 4) embryos at day E8.5 were very similar in mtDNA content ranging from 131 to 236 copies per cell relative to Actin (Fig. 6). The Polg2−/− KO embryos (n = 7) had significantly less mtDNA (P ≤ 0.001) relative to the WT ranging from 0.5 to 2 copies (Fig. 6). The few residual copies in the KO may be maternal mtDNA that originated from the oocyte.
Figure 6.
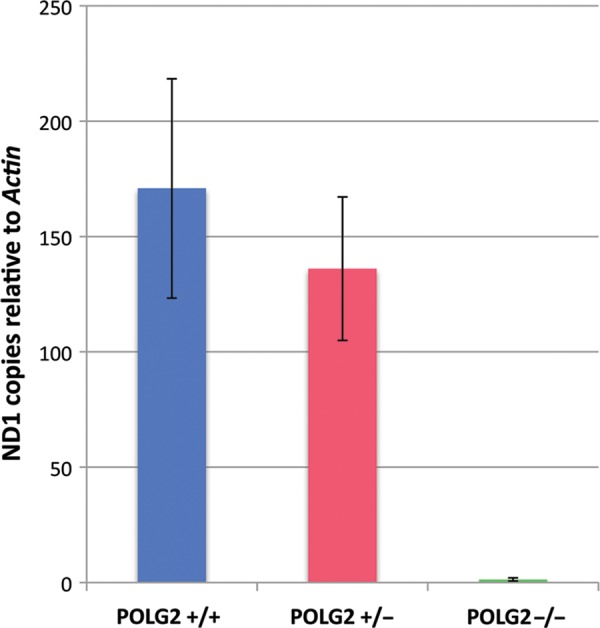
Evaluating the mtDNA content of Polg2 KOs. The mtDNA content of embryonic tissues was determined by using qPCR. The mtDNA target, Nd1, was normalized to nuclear target, Actin. Data are expressed as mtDNA copies relative to Actin copies per cell. There was no significant difference (P > 0.05) between the Polg2+/+ (blue, n = 4) and the Polg2+/− (red, n = 4), ranging from 131 to 236 copies. The homozygous KO mice (n = 7) showed a dramatic decrease of mtDNA copies ranging from 0.5 to 2 (P < 0.01). Error bars indicate ± standard deviation.
DISCUSSION
Abundant evidence exists to show that pol γ is essential for mtDNA replication. Deletion of the yeast pol γ gene (MIP1) results in 100% petites and concomitant loss of mtDNA (rho0 cells) (22). Chemically induced mutations in the pol γ catalytic subunit in Drosophila disrupt larval behavior and cell growth, and result in reduced mitochondrial mass (9). A mouse KO of the catalytic subunit, Polg, resulted in embryonic lethality at day 7.5–8.5 post coitis (p.c.) with subsequent loss of mtDNA (23). Moreover, there are numerous citations involving clinical patients with severe mitochondrial diseases, such as Alpers and PEO, which can be attributed to mutations in the catalytic subunit (10–12). Conversely, there has been only one animal model, Drosophila, that has described the importance of the gene Polg2 encoding the accessory subunit of polγ. In addition, there have been ∼19 mutations and single nucleotide polymorphisms in POLG2 identified in clinical patients.
In our mouse model, the Polg2 gene is haplosufficient. While the homozygous mice were embryonic lethal, the heterozygous Polg2 mice appeared normal at birth and progressed to 2 years of life with no noticeable symptoms. To evaluate the biochemical and physiological effects of gene dose, WT and heterozygous mice were sacrificed at 40 weeks, 1 and 2 years followed by extensive analysis of pathology, clinical pathology and molecular genetics. MtDNA levels, ultrastructure of the mitochondria, pathology, hematology and blood chemistry were similar to age-matched WT mice. Analysis of Polg2 transcripts in these aged heterozygotes revealed the expected 50% reduction of Polg2 mRNA levels in all tissues examined. To explore the consequence and/or compensatory mechanisms in tissues with only 50% Polg2 message levels, we measured the expression levels of key nuclear encoded mtDNA maintenance proteins. No difference in expression was seen in any of the following transcripts: Polg, mtRnap, Tfam, Twinkle, mtSsb, Tfb2or Tfb1.
A homozygous KO of Polg2 in our mouse model was embryonic lethal at approximately E8.0–E8.5. MtDNA depletion was monitored by a corresponding loss of the mitochondrial gene, Nd1. Enzymatic staining for COXI activity confirmed that mtDNA gene products were reduced in our KOs, while the corresponding WT embryonic littermates were positive for COXI activity. The loss of COXI activity has been observed in muscle biopsies of mitochondrial disease patients, such as the patients harboring the G451E and the R369G variants of POLG2 (15,18). A loss of mtDNA has also been reported in several patients diagnosed with Alpers (10–12).
Electron microscopy showed severe ultra-structural defects with the loss of cristae organization, swollen mitochondria and formation of intra-compartmental structures within the mitochondria at day E8.5 of homozygous Polg2 KO mice. These aberrant morphologies in mitochondria were similar to those described in a DNA ligase III conditional KO (24) and Death-associated protein-3 (dap3) KO mouse models (25). DNA ligase III has a role in mtDNA replication and repair. Similar to what we observed in our Polg2−/− embryos, the mitochondria of the DNA ligase III KO also exhibited the formation of inner-compartmental structures, swelling and disorganized cristae (24). The Ligase III conditional KO mice were born normal, but within 20 days developed mitochondrial dysfunction and ultimately died as a result. A second example of aberrant mitochondrial morphology similar to that of the Polg2−/− is in the homozygous KO of the gene that encodes dap-3, which is a GTP-binding protein that has been implicated in the synthesis of intra-mitochondrial proteins and in apoptosis (25). The dap3 KO was embryonic lethal at day 9.5 p.c., with decreased levels of COXI protein, and the EM analysis of the mitochondria revealed an abnormal morphology with swollen cristae (25). Additionally, the EM analysis of the homozygous Polg2 KO revealed an accumulation of lipid droplets in developing embryonic tissue that were not present in the corresponding WT littermates. This phenotype may be indicative of disruption of mitochondrial fatty acid oxidation (β-oxidation). Disruption of mitochondrial function has been associated with lipid accumulation in the liver of some HIV patients taking nucleoside reverse-transcriptase inhibitors (26).
The POLG2 protein has not been found in single-cell eukaryotes such as Saccharomyces cerevisiae, nor is it present in the multicellular eukaryote C. elegans, which precludes genetic studies in these systems (8). The lack of Polg2 in some lower eukaryotes, and its existence as a monomer in some insects has hindered the development of research models to study Polg2. In animal cells, the catalytic subunit forms a complex with the accessory subunit protein. In Drosophila, where the accessory subunit exists as a monomer (27,28), the disruption of the accessory subunit gene results in normal embryo and larval development, but causes lethality during early pupation with a loss of mtDNA (9). In mammalian cells, the accessory subunit exists as a dimer to form a heterotrimer complex with the catalytic subunit. Our novel Polg2 mouse model provides an excellent system to study the accessory subunit.
Mature oocytes in mice contain 100 000–250 000 copies of mtDNA per oocyte (29). Upon fertilization and cell division, this number is progressively diluted to <200 copies of mtDNA per cell until mtDNA replication is initiated post implantation and in concert with cell differentiation around embryonic day 7.0 (30). The Polg, Tfam and Ligase III KO mice models demonstrate the importance of the onset of mtDNA transcription and replication for embryogenesis. The Polg and the Tfam KO mice were embryonic lethal at days 8.5 and 11.5, respectively, and both demonstrated severe mtDNA depletion and respiratory-chain defects (23,31). DNA ligase III plays a role in mtDNA replication and repair in mitochondria, and the ablation of this gene in mouse resulted in cell death at day E9.5 p.c. The conditional KO of Ligase III in the central nervous system of mice resulted in death at 2 weeks of age with associated loss of mtDNA and abnormal mitochondria (24). Interestingly, the heterozygous mice of the Polg, Tfam and Ligase III models were haplosufficient, as is the case with the Polg2+/− model. Our Polg2 KO model is similar to the aforementioned KO models in embryonic lethality and aberrant mitochondrial ultra-structural defects.
In summary, we have described the construction and characterization of heterozygous and homozygous KOs of the Polg2 gene in mice. Homozygous Polg2 KOs are embryonic lethal at days 8.0–8.5 p.c., while the heterozygous mouse was asymptomatic through 2 years of age. The inability to replicate mtDNA in a Polg2−/− embryo resulted in abnormal mitochondrial morphology, the loss of COXI activity, delayed development and ultimately embryonic death. Although not a true mitochondrial disease model, these data demonstrate the essential role of Polg2 in mtDNA replication, mitochondrial function and mammalian embryonic development, and suggests a more fundamental role of Polg2 not only in mtDNA elongation, but also in mtDNA replication initiation and distribution. This suggestion is consistent with the idea that POLG2 may play a role in the recruitment of other proteins to the D-loop for initiation of mtDNA replication and the distribution of mtDNA within nucleoids (32). Our novel KO mouse has demonstrated the importance of the Polg2 gene in vertebrates, and now affords us the opportunity to construct conditional Polg2 KO mice with tissue specificity at various stages of development, as well as to explore environmental factors that compromise mitochondria in the heterozygous Polg2 mice. The evidence that Polg2 is essential in mammalian systems may also lead to the development of a true mitochondrial disease model harboring a heterozygous missense mutation in the Polg2 gene.
MATERIALS AND METHODS
Animals
The generation of a cre-mediated Polg2 KO mouse on a C57/B6 background strain was contracted out to Xenogen (now Caliper Life Sciences). The loxP sites were introduced into the genome of ES cells, as previously described (19). Neomycin selection was used for the identification of ES cells that had undergone homologous recombination. Southern blots were used to confirm the homologous recombination in ES cells. The resulting loxP-flanked Polg2 heterozygous mice were shipped and held in the NIEHS animal facility in accordance with an approved animal study and NIH guidelines for the care and maintenance of experimental animals. Mice were provided food and water ad libitum and held under a 12 h light–dark cycle.
Disruption of Polg2 via cre-loxP recombination
The loxP-flanked Polg2 heterozygous mice (Polg2+/loxp) were crossed to generate a homozygous floxed (Polg2loxp/loxp) mouse. The Polg2+/loxp and Polg2loxp/loxp mice were healthy and fertile, and had a normal life span, which indicated that the insertion of the loxP sites had no adverse effects on the animals. The Polg2loxp/loxp male mouse was crossed with a heterozygous ubiquitously expressing cre-recombinase female mouse (Sox2-cre, Jackson Laboratory) allowing for the cre-mediated excision of Polg2, exons 5 through 7.
Whole tissue lysates for genotyping and qPCR
Whole tissue lysates generated from ear punch tissue were used for genotyping. In addition, whole tissue lysates generated from 15 mg of brain, liver, muscle and heart tissues were used in a qPCR assay for mtDNA content determination. All tissues were incubated at 98°C for 1 h in 200 µl of 25 mm NaOH (Sigma) followed by a neutralization step with the addition of 20 µl of 1 m Tris (Invitrogen). Lysates were diluted 1:15 with PCR grade water (Ambion) for qPCR reactions.
mtDNA content determination
The mtDNA content was determined using qPCR. The primers and the FAM/TAMRA-labeled probe targeting the mitochondrial gene, Nd1, were designed by Applied Biosystems specifically for our needs. The endogenous control, Actin, was purchased from Applied Biosystems (catalog No. Mm00607939_s1). qPCR amplifications were carried out on an ABI PRISM 7900HT Sequence Detector (Applied Biosystems) with the following cycling protocol: 95°C for 10 min, and 40 cycles at 95°C for 15 s and 60°C for 1 min. All reactions were done in triplicate, and 1 µl of a diluted (1:15) tissue lysate was used in each 25 µl reaction using TaqMan 2X Universal Mix (Applied Biosystems). Standard curves for both Nd1 and Actin were created by serially diluting known quantities of plasmid DNA (102–107 copies) containing the targeted amplicon and the values were quantitated. The copy number of Nd1 relative to the copy number of Actin was employed to determine the mtDNA content. The relative standard curve method (http://www3.appliedbiosystems.com/cms/groups/mcb_support/documents/generaldocuments/cms_042380.pdf) was used to quantify mtDNA in tissues.
Laser capture microdissection
LCM was used to isolate E8.5 embryos from the maternally derived deciduum for genotyping. Frozen samples and formalin-fixed paraffin-embedded sample blocks were serially sectioned at 8 µm and adhered to PET foil slides. The frozen sections were fixed with 75% ethanol in diethylpyrocarbonate-treated water, and stained with fresh/filtered 1% Cresyl Violet acetate (Sigma), then desiccated to harvest embryonic tissue for genotyping. The MMI Cellcut® Plus (Molecular Machined & Industries, Inc.) was used for microdissection of all samples (33). DNA was isolated using an Arcturus PicoPure DNA extraction kit (Invitrogen).
Histopathology
E8.5 embryos were used for COXI staining. Embryos were collected intact within the deciduum, embedded in an Optimal Cutting Temperature compound (Tissue-Tek) and frozen in the vapor phase of liquid nitrogen. Ten micrometer cryostat sections were applied to charged slides (Erie Scientific, LLC), and were stained for COXI activity, washed in water, counterstained with hematoxylin and mounted in Advantage Permanent mounting media (Axell). Staining protocols were adapted from Chen et al. (34), and the Washington University Neuromuscular Disease Center website (http://www.neuro.wustl.edu/neuromuscular/index.html).
Electron microscopy
For EM analysis of each E8.5 embryo and heart sample, the sections were fixed in modified Karnovsky's fixative, processed, and embedded in resin blocks routinely for transmission EM. The resin blocks were sectioned at ∼70–90 nm, placed on Formvar copper grids, and then stained with uranyl acetate and lead citrate (35). The grids were then examined on a FEI Tecnai 110KV TEM.
SUPPLEMENTARY MATERIAL
FUNDING
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (ES 065078 and ES 065080).
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to the efforts of Mr Scotty Dowdy for animal care, Dr Janssen Daly for animal breeding advice, Dr Dave Malarkey for analysis of fatty droplets, Dr Connie Cummings and Ms Deloris Sutton for EM analysis, Ms Pat Stockton for laser capture microscopy, Dr Grace Kissling for statistical analysis of blood chemistry and Dr Gordon Flake and the NIEHS Histology Core for histological services. We also thank Drs Matthew J. Longley and Katarzyna Bebenek for the critical reading of this manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Graziewicz M.A., Longley M.J., Copeland W.C. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem. Rev. 2006;106:383–405. doi: 10.1021/cr040463d. doi:10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- 2.Longley M.J., Nguyen D., Kunkel T.A., Copeland W.C. The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J. Biol. Chem. 2001;276:38555–38562. doi: 10.1074/jbc.M105230200. doi:10.1074/jbc.M105230200. [DOI] [PubMed] [Google Scholar]
- 3.Lim S.E., Longley M.J., Copeland W.C. The mitochondrial p55 accessory subunit of human DNA polymerase gamma enhances DNA binding, promotes processive DNA synthesis, and confers N-ethylmaleimide resistance. J. Biol. Chem. 1999;274:38197–38203. doi: 10.1074/jbc.274.53.38197. doi:10.1074/jbc.274.53.38197. [DOI] [PubMed] [Google Scholar]
- 4.Carrodeguas J.A., Theis K., Bogenhagen D.F., Kisker C. Crystal structure and deletion analysis show that the accessory subunit of mammalian DNA polymerase gamma, Pol gamma B, functions as a homodimer. Mol. Cell. 2001;7:43–54. doi: 10.1016/s1097-2765(01)00153-8. doi:10.1016/S1097-2765(01)00153-8. [DOI] [PubMed] [Google Scholar]
- 5.Yakubovshaya E., Chen Z., Carrodeguas J.A., Kisker C., Bogenhagen D.F. Functional human mitochondrial DNA polymerase gamma forms a heterotrimer. J. Biol. Chem. 2006;281:374–382. doi: 10.1074/jbc.M509730200. doi:10.1074/jbc.M509730200. [DOI] [PubMed] [Google Scholar]
- 6.Lee Y.S., Kennedy W.D., Yin Y.W. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell. 2009;139:312–324. doi: 10.1016/j.cell.2009.07.050. doi:10.1016/j.cell.2009.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee Y.S., Lee S., Demeler B., Molineux I.J., Johnson K.A., Yin Y.W. Each monomer of the dimeric accessory protein for human mitochondrial DNA polymerase has a distinct role in conferring processivity. J. Biol. Chem. 2010;285:1490–1499. doi: 10.1074/jbc.M109.062752. doi:10.1074/jbc.M109.062752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young M.J., Copeland W.C. In: Mitochondrial Disorders Caused by Nuclear Genes. Wong L.J.C., editor. Springer Science+Business Media, New York, NY; 2013. pp. 49–72. [Google Scholar]
- 9.Iyengar B., Luo N., Farr C.L., Kaguni L.S., Campos A.R. The accessory subunit of DNA polymerase gamma is essential for mitochondrial DNA maintenance and development in Drosophila melanogaster. Proc. Natl Acad. Sci. USA. 2002;99:4483–4488. doi: 10.1073/pnas.072664899. doi:10.1073/pnas.072664899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Copeland W.C. Inherited mitochondrial diseases of DNA replication. Annu. Rev. Med. 2008;59:131–146. doi: 10.1146/annurev.med.59.053006.104646. doi:10.1146/annurev.med.59.053006.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Copeland W.C. The mitochondrial DNA polymerase in health and disease. Subcell. Biochem. 2010;50:211–222. doi: 10.1007/978-90-481-3471-7_11. doi:10.1007/978-90-481-3471-7_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stumpf J.D., Copeland W.C. Mitochondrial DNA replication and disease: insights from DNA polymerase gamma mutations. Cell. Mol. Life Sci. 2011;68:219–233. doi: 10.1007/s00018-010-0530-4. doi:10.1007/s00018-010-0530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong L.J., Naviaux R.K., Brunetti-Pierri N., Zhang Q., Schmitt E.S., Truong C., Milone M., Cohen B.H., Wical B., Ganesh J., et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum. Mutat. 2008;29:E150–E172. doi: 10.1002/humu.20824. doi:10.1002/humu.20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen B.H., Naviaux R.K. The clinical diagnosis of POLG disease and other mitochondrial DNA depletion disorders. Methods. 2010;51:364–373. doi: 10.1016/j.ymeth.2010.05.008. doi:10.1016/j.ymeth.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 15.Longley M.J., Clark S., Yu Wai Man C., Hudson G., Durham S.E., Taylor R.W., Nightingale S., Turnbull D.M., Copeland W.C., Chinnery P.F. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am. J. Hum. Genet. 2006;78:1026–1034. doi: 10.1086/504303. doi:10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walter M.C., Czermin B., Muller-Ziermann S., Bulst S., Stewart J.D., Hudson G., Schneiderat P., Abicht A., Holinski-Feder E., Lochmuller H., et al. Late-onset ptosis and myopathy in a patient with a heterozygous insertion in POLG2. J. Neurol. 2010;257:1517–1523. doi: 10.1007/s00415-010-5565-9. doi:10.1007/s00415-010-5565-9. [DOI] [PubMed] [Google Scholar]
- 17.Young M.J., Longley M.J., Li F.Y., Kasiviswanathan R., Wong L.J., Copeland W.C. Biochemical analysis of human POLG2 variants associated with mitochondrial disease. Hum. Mol. Genet. 2011;20:3052–3066. doi: 10.1093/hmg/ddr209. doi:10.1093/hmg/ddr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Craig K., Young M.J., Blakely E.L., Longley M.J., Turnbull D.M., Copeland W.C., Taylor R.W. A p.R369G POLG2 mutation associated with adPEO and multiple mtDNA deletions causes decreased affinity between polymerase gamma subunits. Mitochondrion. 2011;12:313–319. doi: 10.1016/j.mito.2011.11.006. doi:10.1016/j.mito.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu H., Marth J.D., Orban P.C., Mossmann H., Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. doi:10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 20.Kaufman M.H.e.a. The Atlas of Mouse Development. Academic Press, San Diego: 1992. [Google Scholar]
- 21.Theiler K.e.a. Atlas of Embryonic Development. New York: Springer-Verlag; 1989. [Google Scholar]
- 22.Foury F. Cloning and sequencing of the nuclear gene MIP1 encoding the catalytic subunit of the yeast mitochondrial DNA polymerase. J. Biol. Chem. 1989;264:20552–20560. [PubMed] [Google Scholar]
- 23.Hance N., Ekstrand M.I., Trifunovic A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum. Mol. Genet. 2005;14:1775–1783. doi: 10.1093/hmg/ddi184. doi:10.1093/hmg/ddi184. [DOI] [PubMed] [Google Scholar]
- 24.Gao Y., Katyal S., Lee Y., Zhao J., Rehg J.E., Russell H.R., McKinnon P.J. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature. 2011;471:240–244. doi: 10.1038/nature09773. doi:10.1038/nature09773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim H.R., Chae H.J., Thomas M., Miyazaki T., Monosov A., Monosov E., Krajewska M., Krajewski S., Reed J.C. Mammalian dap3 is an essential gene required for mitochondrial homeostasis in vivo and contributing to the extrinsic pathway for apoptosis. FASEB J. 2007;21:188–196. doi: 10.1096/fj.06-6283com. doi:10.1096/fj.06-6283com. [DOI] [PubMed] [Google Scholar]
- 26.Nunnari J., Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. doi:10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olson M.W., Wang Y., Elder R.H., Kaguni L.S. Subunit structure of mitochondrial DNA polymerase from Drosophila embryos. Physical and immunological studies. J. Biol. Chem. 1995;270:28932–28937. doi: 10.1074/jbc.270.48.28932. doi:10.1074/jbc.270.48.28932. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y., Farr C.L., Kaguni L.S. Accessory subunit of mitochondrial DNA polymerase from Drosophila embryos. Cloning, molecular analysis, and association in the native enzyme. J. Biol. Chem. 1997;272:13640–13646. doi: 10.1074/jbc.272.21.13640. doi:10.1074/jbc.272.21.13640. [DOI] [PubMed] [Google Scholar]
- 29.Aiken C.E., Cindrova-Davies T., Johnson M.H. Variations in mouse mitochondrial DNA copy number from fertilization to birth are associated with oxidative stress. Reprod. Biomed. online. 2008;17:806–813. doi: 10.1016/s1472-6483(10)60409-9. doi:10.1016/S1472-6483(10)60409-9. [DOI] [PubMed] [Google Scholar]
- 30.Cree L.M., Samuels D.C., de Sousa Lopes S.C., Rajasimha H.K., Wonnapinij P., Mann J.R., Dahl H.H., Chinnery P.F. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat. Genet. 2008;40:249–254. doi: 10.1038/ng.2007.63. doi:10.1038/ng.2007.63. [DOI] [PubMed] [Google Scholar]
- 31.Larsson N.G., Wang J., Wilhelmsson H., Oldfors A., Rustin P., Lewandoski M., Barsh G.S., Clayton D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. doi:10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 32.Di Re M., Sembongi H., He J., Reyes A., Yasukawa T., Martinsson P., Bailey L.J., Goffart S., Boyd-Kirkup J.D., Wong T.S., et al. The accessory subunit of mitochondrial DNA polymerase gamma determines the DNA content of mitochondrial nucleoids in human cultured cells. Nucleic Acids Res. 2009;37:5701–5713. doi: 10.1093/nar/gkp614. doi:10.1093/nar/gkp614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flake G.P., Rivera M.P., Funkhouser W.K., Maygarden S.J., Meadows K.L., Long E.H., Stockton P.S., Jones T.C., Yim H.W., Slebos R.J., et al. Detection of pre-invasive lung cancer: technical aspects of the LIFE project. Toxicol. Pathol. 2007;35:65–74. doi: 10.1080/01926230601052659. doi:10.1080/01926230601052659. [DOI] [PubMed] [Google Scholar]
- 34.Chen H., McCaffery J.M., Chan D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. doi:10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 35.Burghardt R.C., Droleskey R. Transmission electron microscopy. Curr. Protoc. Microbial. 2006 doi: 10.1002/9780471729259.mc02b01s03. Chapter 2, Unit 2B, 1.1–1.39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.