


Abstract
Existence of some extra-genetic (epigenetic) codes has been postulated since the discovery of the primary genetic code. Evident effects of histone post-translational modifications or DNA methylation over the efficiency and the regulation of DNA processes are supporting this postulation. EMdeCODE is an original algorithm that approximate the genomic distribution of given DNA features (e.g. promoter, enhancer, viral integration) by identifying relevant ChIPSeq profiles of post-translational histone marks or DNA binding proteins and combining them in a supermark. EMdeCODE kernel is essentially a two-step procedure: (i) an expectation-maximization process calculates the mixture of epigenetic factors that maximize the Sensitivity (recall) of the association with the feature under study; (ii) the approximated density is then recursively trimmed with respect to a control dataset to increase the precision by reducing the number of false positives. EMdeCODE densities improve significantly the prediction of enhancer loci and retroviral integration sites with respect to previous methods. Importantly, it can also be used to extract distinctive factors between two arbitrary conditions. Indeed EMdeCODE identifies unexpected epigenetic profiles specific for coding versus non-coding RNA, pointing towards a new role for H3R2me1 in coding regions.
INTRODUCTION
The role of epigenetic mechanisms as modulators of transcriptional activation and repression, DNA methylation, cell memory maintenance, stem cell differentiation, cancerogenesis and other major genomic processes have been widely demonstrated [reviewed in (1–3)]. Moreover, it has been already observed that epigenetic features such as histone post-translational modifications and DNA methylation can act in concert to promote or silence specific cellular functions (4). Accordingly, I have recently shown that retroviral insertional site selection into the host DNA could be strongly influenced by a set of specific histone modifications acting as a sort of beacon for incoming retroviruses (5). This information could be taken as the prelude to postulate the existence of a not-yet-well-defined epigenetic code (6).
Here I propose a statistical approach to identify patterns of epigenetic signatures associated with specific genomic functions. Relatively new technologies, notably ChIPSeq, are able to track the position of DNA binding proteins as transcription factors or histones carrying specific modifications and yield a genome-wide density profile. When searching for association to a given genomic process, this information might be probabilistically interpreted and combined to generate a new virtual mark of greater statistical power with respect to the ‘real’ mark alone. Several approaches have been developed. Some (7–10) are based on unsupervised genome segmentation where the aim is to find clusters of epigenetic factors repeated over the genome (transcription factor binding sites, histone marks, DNase profile etc.). These patterns are then associated ‘a posteriori’ with known annotations as promoter or enhancer regions. As another example, (11) uses Bayesian networks to explores causal relationships among clusters of DNA binding proteins in Drosophila and infers direct or indirect interactions among them. In general, unsupervised methods are useful to label large portions of the genome but they lack a false-positive correction resulting in poor performance especially in terms of positive predicted value (PPV or Precision <0.5) (10). On the other hand, supervised methods use training subsets of specific functional regions (enhancers and promoters in most cases) to predict the genomic position of other unknown functional sites (12,13), and they use a control (random) dataset to increase the Precision. Additionally these methods yield better Sensitivity (Recall) than unsupervised methods (12). Previously I implemented a supervised heuristic methodology based on F score statistics to create a ‘supermarker’ out of ChIPSeq profiles (5). This methodology requires the pre-calculation of individual F scores for each mark, the selection of a subset of significant marks and the eventual addition or deletion of some marks to the subset. EMdeCODE is a general extension of this idea, based on mixture modeling Expectation and Maximization (EM) and statistical selection. It is specifically designed to deal with post-translational histone modifications and it presents some peculiar features. The input data is a set of peaks calculated by an independent peak caller from row ChIPSeq data [similarly to (9)]. The rationale behind this design is to decouple the peak calling task that can be accomplished by several continuously improved algorithms [see (14) for a review] from the statistical combination of histone marks. The advantage is 2-fold: (i) EMdeCODE is independent from current and likely soon obsolete treatments of ChIPSeq data; (ii) EMdeCODE can work with peaks called from different algorithms and from different experiments. The peak caller can be chosen accordingly with the expected ChIPSeq profile and the experimental conditions, for example HOMER (15) for narrow peaked marks as H3K4me3, and RSEG (16) for wider distribution as H3K27me3. Differently from all other approaches, for each input peak set, EMdeCODE recreates a new histone mark distribution by interpreting each peak as a single or stretch of nucleosomes (accordingly to the genomic size of the peak) and modeling the nucleosome occupancy by Gaussian functions of equal amplitude and variance (=200 bp). The idea here is to drastically reduce the experimental noise and facilitate the interaction of data obtained by different experiments. These new distributions are then combined during the training phase to generate a genome-wide probability mass distribution that approximate the unknown functional distribution. Here, another peculiarity of EMdeCODE is the maximization of the F Score, a weighted combination of precision (positive predictive value) and recall (Sensitivity), to reduce the influence of false negatives similarly to what presented in (5). The main difference is the addition of the EM procedure that combines a set of factors and selects the relevant subset without supervision. As shown in the Results section, a consequence of this approach is a gain of precision with respect to other methods. EMdeCODE can therefore be used to discover new associations between marks and genomic functions. Applications to enhancer identification in comparison with previous supervised methods, retroviral integration site prediction and to discriminate between coding versus non-coding genes are discussed.
MATERIALS AND METHODS
Input data
High-Throughput Sequencing–based applications usually require large amount of computational power and software tools. The minimal computational ChIPSeq pipeline consists of a sequence aligner that maps millions of reads produced by the sequencer to the proper reference genome and a peak finder to identify enriched regions with respect to a control background according to some user pre-defined parameters [see (17) for a review about ChIPSeq and other similar technologies]. EMdeCODE takes as input the final peak set and assumes that these steps are correctly performed with proper controls and sufficient profile quality. In particular, all ChIPSeq histone profiles used in this study have been pre-processed with Fseq (18) for it produces good-quality peak set from both narrow and broad read distributions (14).
The algorithm
Here M DNA-binding factors are considered. Formally, each ChIPSeq profile for factor 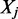 could be interpreted as the probability to find
could be interpreted as the probability to find 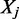 bound to a specific genomic region of size w centered on i = (chr, pos):
bound to a specific genomic region of size w centered on i = (chr, pos): 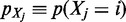 . This mass density is modeled by EMdeCODE with a sum of Gaussian functions, each one centered on one nucleosome. Nucleosome positions are extracted from ChIPSeq peaks by considering a genomic occupancy of 200 bp. This reduces the potential bias that can arise by combining ChIPSeq densities obtained over different experimental conditions. Briefly, each marker probability density function is written as
. This mass density is modeled by EMdeCODE with a sum of Gaussian functions, each one centered on one nucleosome. Nucleosome positions are extracted from ChIPSeq peaks by considering a genomic occupancy of 200 bp. This reduces the potential bias that can arise by combining ChIPSeq densities obtained over different experimental conditions. Briefly, each marker probability density function is written as
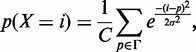 |
(1) |
where Γ is the peak set of factor X, and C is a normalization factor.
Similarly, the biological event could be modeled as a random variable B taking values on all chromosomal loci. In other words, the event modeled by B has an unknown (mass) probability to occur in the region centered on i:
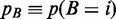 .
.
The aim is to find a new mass probability functional that can approximate 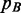 by means of the
by means of the 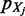 ’s:
’s: 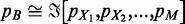 .
.
Assuming that B has been observed to occur on N loci 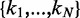 ,
, 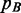 can be written as
can be written as
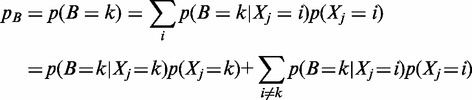 |
(2) |
where i spans across all possible loci.
The first right term in (2) can be interpreted as how well 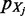 fits
fits 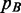 . The second term is the related error expressing the spreading of
. The second term is the related error expressing the spreading of 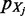 over loci unrelated to B.
over loci unrelated to B.
Summing (2) over j:
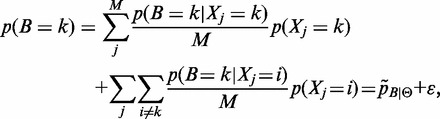 |
(3) |
where 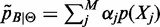 ,
, 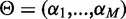 and
and 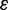 is the aggregate of all M approximation errors.
is the aggregate of all M approximation errors.
 can be estimated by a classical Maximum Likelihood approach. Indeed, maximizing the (log)likelihood of
can be estimated by a classical Maximum Likelihood approach. Indeed, maximizing the (log)likelihood of 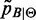 :
:
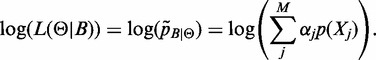 |
is equivalent to a mixture-density parameter estimation problem and can be efficiently treated with the Expectation Maximization algorithm (EM) (19).
This algorithm works in a way that, at the t-esim iteration, the expectation function 
 is maximized (Y is an auxiliary random variable), that is,
is maximized (Y is an auxiliary random variable), that is, 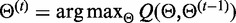 . As previously derived in (19), by introducing the probability that value k arises from the j-esim distribution,
. As previously derived in (19), by introducing the probability that value k arises from the j-esim distribution,
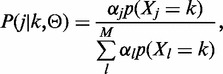 |
(4) |
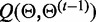 can be written as
can be written as
| (5) |
The mixture coefficient vector  is evaluated by maximizing (5) by the Lagrange multiplier
is evaluated by maximizing (5) by the Lagrange multiplier 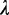 with constraints
with constraints 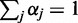 and (4):
and (4):
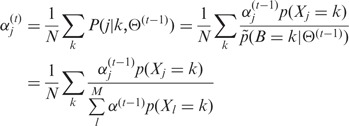 |
(6) |
where N is the number of discrete genomic loci all densities have support.
From (3) the following identity holds:
Introducing the concept of Sensitivity (Recall), defined as 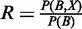 , equation (6) can be simply written as:
, equation (6) can be simply written as:
| (7) |
In other words, each factor is weighted proportionally to the respective Sensitivity.
The spreading error 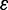 can be interpreted as the affinity of factor
can be interpreted as the affinity of factor 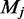 for a control dataset C, defined as the set of random loci where
for a control dataset C, defined as the set of random loci where 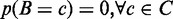 .
.
Indeed, considering a control dataset with 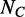 control loci, the spreading error could be estimated as:
control loci, the spreading error could be estimated as:
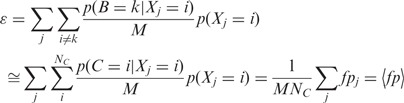 |
where 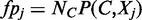 is the number of false positives, that is, the number of control loci localizing in the region where factor
is the number of false positives, that is, the number of control loci localizing in the region where factor 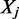 is bound. Therefore, minimizing
is bound. Therefore, minimizing 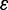 implies the minimization of false positives.
implies the minimization of false positives.
The strategy adopted here to approximate 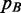 is a two-step procedure where
is a two-step procedure where 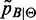 is calculated by (6) and low-probability peaks of
is calculated by (6) and low-probability peaks of 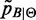 are trimmed out to reduce the number of false positives (the whole procedure is sketched in Figure 1). Obviously this could also reduce the Sensitivity, that is, the number of peaks associated to B. To measure the quality of the approximation, the
are trimmed out to reduce the number of false positives (the whole procedure is sketched in Figure 1). Obviously this could also reduce the Sensitivity, that is, the number of peaks associated to B. To measure the quality of the approximation, the 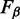 score is evaluated after trimming to find the optimal tradeoff between Sensitivity and false positives reduction (Precision).
score is evaluated after trimming to find the optimal tradeoff between Sensitivity and false positives reduction (Precision).
Figure 1.

(1) EMdeCODE reconstructs mark density distributions by placing the appropriate number of nucleosomes into each peak region and modeling nucleosome occupancy by a Gaussian distribution with variance 200 bp. (2) Expectation Maximization evaluates the sensitivity of each mark to true positives (filled round dots). Marks are weighted accordingly and linearly combined to generate the raw supermark. (3) Peaks of the supermark accounting for random loci (crosses: false positives) are trimmed off the supermark to optimize the F score.
Formally, the 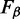 score is defined as the β-weighted harmonic mean of Precision(P) and Sensitivity(R):
score is defined as the β-weighted harmonic mean of Precision(P) and Sensitivity(R): 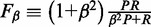 .
.
Here, β = 0.5 to give more weight to Precision than to Sensitivity. This balances type I and type II errors by adjusting for the high rate of False Positives (fp) inherent to the examination of large datasets for genome-wide binding sites according to statistical significance. Moreover, to make the F score insensitive to data size, the number of false positives fp has been normalized with respect to N [F score–based statistics and comparison with other measures have been extensively discussed in (5)].
Eventually, the algorithm is lined out as follows:
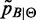 is evaluated by (6). This corresponds to weighting each factor distribution with the respective Sensitivity.
is evaluated by (6). This corresponds to weighting each factor distribution with the respective Sensitivity.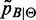 is trimmed to reduce the number of false positives by removing small peaks until F0.5 score is maximized. Accordingly, the support of mass probability functions
is trimmed to reduce the number of false positives by removing small peaks until F0.5 score is maximized. Accordingly, the support of mass probability functions 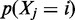 is reduced.
is reduced.If F0.5 score has increased, step 1) is repeated or else it ends.
EMdeCODE has been implemented in Python and can be downloaded from http://seaseq.unige.ch/∼fsantoni/EMdeCODE.
RESULTS
Comparison with previous methods
Cross validation
To compare the performance of EMdeCODE with previously published algorithms, a 5-fold cross validation has been implemented over a set of 74 well-defined enhancers according to the experimental settings proposed in (20), but replacing ChIP-ChIP signals for H3K4me3 and H3K4me1 with the corresponding ChIPSeq profiles (21). As background, 740 random genomic sites have been selected. EMdeCODE scored a PPV of 96.9% ± 1.1 with an optimal window of 300 bp. Notably, this window is considerably smaller than 2000 bp reported elsewhere (12,13,20), probably owing to the higher resolution provided by ChIPSeq data. Comparison with previous methods is reported in Table 1.
Table 1.
Positive Predicted Value comparison among enhancer predicting algorithms in a 5-fold cross-validation test
| Method (H3K4me1, H3K4me3) | Enhancer PPV(±SD)a |
|---|---|
| EMdeCODE | 96.9% ± 1.10 |
| CSI-ANN (12) | 96.22% ± 2.14 |
| HMM (20) | 94.1% ± 0.89 |
| Heintzmann et al. (13) | 85% |
aWhere available.
Enhancer identification
EMDdeCODE has also been used to reconstruct the p300 binding sites distribution in CD4+ T cells, as a strong marker for enhancer identification (22). Thirty-nine ChIPSeq histone modifications (acetylation and methylation) profiles have been used as input (see ‘Materials and Methods’ section). Similarly to the analysis proposed by Firpi et al. (12), EMdeCODE was trained with 213 p300 distal binding sites [at least 2.5 Kbp from a known Transcription Starting Site (TSS)] overlapping PreMod (23) predicted enhancers and 2130 random genomic sites. Here, two loci are considered as overlapping if their genomic distance is <2 Kbp.
EMdeCODE generated 22 878 whole genome putative enhancer sites, 53% overlapping (12 165) with the 36 796 sites generated by CSI-ANN [(12), data in Supplementary Material]. To validate this prediction, the EMdeCODE generated enhancer dataset has been compared with the whole genome distal p300-enriched regions [>2.5 Kbp from TSS, calculated by SICER (24)]. In all, 74% (6069/8245) of p300 regions overlap with EMdeCODE predictions, 29% more than CSI-ANN (45%, 3740/8245). When compared with a background dataset to evaluate the number of false positives (70 000 random selected genomic sites), EMdeCODE predictions obtained an F score of 0.91 (Precision = PPV = 96%, Recall = Sensitivity = 74%), whereas the CSI-ANN F score was 0.74 (Precision = PPV = 89%, Recall = Sensitivity = 45%). This is somehow expected because EMdeCODE aims to maximize the F score. A visual comparison between EMdeCODE and CSI-ANN is reported in Figure 2 by Chromosome Projection Mandalas (5). It is interesting to observe that, consequently, EMdeCODE improves both PPV and Sensitivity. Previously it has been shown that enhancers are enriched with DNase I sensitive sites (13,22,25). Indeed, 80% of EMdeCODE predictions is supported by at least one among p300, DNase sites (26) and PreMod predicted functional sites, 20% more than CSI-ANN (60%). As additional comparison, 49 746 p300 distal binding sites were obtained by peak calling from the original p300 ChIPSeq data. In all, 18 759 (82%) EMdeCODE-generated sites were supported by p300 peaks, 45% more than CSI-ANN (37%). Together, these results indicate that EMdeCODE is a more effective algorithm to approximate the p300 distribution and a superior enhancer predictor.
Figure 2.

Chromosome projection mandalas calculated within 2 Kbp for EMdeCODE and CSI-ANN enhancer predictions versus p300 enriched regions on chr1. Each dot on the mandala represents the center of a p300 region in polar coordinates. Angular distance corresponds to genomic location, and radial distance from the outer circle indicates the log-scaled distance in nucleotides from the closest predicted enhancer. Inner circles mark 1, 2 (in blue), 10, 20 Kbp and so on. Blue filled circles represent p300 sites within 2 Kbp from the nearest predicted enhancer.
Enhancer ‘Code’
The composition of the enhancer ‘code’ generated by EMdeCODE (reported in Supplementary Figure S1A) is dominated by H3K36ac, an highly conserved histone modification already reported to be strongly enriched at enhancer sites (27,28) followed by H2BK120 and H3K4ac, two marks associated with active enhancers and promoters (29). Notably, EMdeCODE enhancers are also enriched in H3K4me3 (76%) and H3K27ac (70%) and depleted in H3K27me3 (<1%), indicating that most of them are likely active enhancers (30,31).
Interestingly, this particular composition depends on modeling ChIP-Seq profiles as probability mass distributions and specifically on the normalization 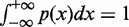 . When min-max normalization is applied (i.e.
. When min-max normalization is applied (i.e. 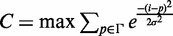 in eq. 0), EMdeCODE generates 45 616 putative enhancers, 16 961 (out of 22 878, 75%) overlapping with those ones produced by standard normalization. In this case the composition of this supermark is dominated by the well known marks H2AZ, H3K9me1, H3K4me1 and H3K4me3 (Supplementary Figure S1B). Notably, the performance of this prediction is slightly better in Sensitivity (77%, 6374/8244 of p300 regions overlap with this prediction) but not in Precision (89%), leading to a lower F score = 0.86. For this reason, the standard normalization has been preferred in EMdeCODE implementation.
in eq. 0), EMdeCODE generates 45 616 putative enhancers, 16 961 (out of 22 878, 75%) overlapping with those ones produced by standard normalization. In this case the composition of this supermark is dominated by the well known marks H2AZ, H3K9me1, H3K4me1 and H3K4me3 (Supplementary Figure S1B). Notably, the performance of this prediction is slightly better in Sensitivity (77%, 6374/8244 of p300 regions overlap with this prediction) but not in Precision (89%), leading to a lower F score = 0.86. For this reason, the standard normalization has been preferred in EMdeCODE implementation.
Retroviral integration site prediction
EMdeCODE has been used to generate new virtual marks by combining 39 different histone marks previously extracted from CD4+ T cells [methylation (32) and acetylations (29)] with 6 histone marks from HeLa cells (21). These new marks have been tested for association with several gammaretroviral integration datasets with respect to randomly generated matched control datasets [similarly to (5)].
Indeed, EMdeCODE improved the prediction of retroviral integration sites with respect to (5). The Sensitivity (Recall) increases 5% (to 10%) explaining up to 85% of integration sites in CD4+ T cells. Accordingly, the related F score increases 3% (to 7%, Figure 3). The mixture coefficient vector  , calculated for each chromosome, stores the contribution of each mark to the final association. As expected, the optimal receipt includes all marks related to active transcription and open chromatin, whereas H3K27me3 and other heterochromatin-related marks give no contribution (Supplementary Figure S2). All results have been checked using a 5-fold cross-validation strategy similar to what presented in (5).
, calculated for each chromosome, stores the contribution of each mark to the final association. As expected, the optimal receipt includes all marks related to active transcription and open chromatin, whereas H3K27me3 and other heterochromatin-related marks give no contribution (Supplementary Figure S2). All results have been checked using a 5-fold cross-validation strategy similar to what presented in (5).
Figure 3.

Histograms of the F score (upper panel) and the percentage of associated proviruses wi2Kbp of the EMdeCODE-generated supermark (lower panel) with respect to MLV (I and II) proviruses (41,42) and HIVmINmGAG chimera (41) integrated in HeLa in comparison with the ‘supermarker’ previously reported by Santoni et al.
Discriminating between coding and non-coding DNA
Another possible application of EMdeCODE is the identification and the quantification of differential epigenetic profiles between two distinct genomic features. Here, the algorithm has been used to identify epigenetic differences that could characterize coding from non-coding transcripts as, in this case, long non-coding RNA (lncRNA). It is believed that non-coding mRNAs transcribed by the PolII transcriptional machine have the same epigenetic landscape found in promoter regions specific of coding mRNA (33). To investigate this hypothesis, a total of 182 lncRNA, 84 not overlapping with any known expressed transcript within 6 Kbp upstream of the TSS and 98 not overlapping within 6 Kbp downstream of the 3′-end of transcription regions (EoT), all actively transcribed in CD4+ T cells [accordingly to publicly available RNASeq data (28,34)], with a clear PolII peak in the promoter region [data from (32)] and more than 400 nt long have been selected. As a matched control, 84 + 98 coding PolII transcribed RefSeq genes with the same 6 Kbp non-overlapping condition as selected lncRNA and with comparable transcriptional level and length have been randomly chosen (Supplementary Figure S3). EMdeCODE was then fed with the above-mentioned 39 markers to identify possible discriminating factors over promoter regions, gene bodies (Tx) and the 3′-EoT. A comparison with 2000 randomly selected genomic regions has been performed as reference.
Diagrams in Figure 4A report F score curves calculated for coding versus non-coding genes (Gene versus ncRNA), coding versus random (Gene versus Rnd) and non-coding versus random (ncRNA versus Rnd), considering gene bodies and increasing regions from TSS and 3′ EoT respectively. As expected, both coding genes and non-coding RNA (ncRNA) can easily distinguished from random sites in gene bodies (Tx, F score = 0.87 and 0.86), whereas it is more difficult but still possible to discriminate between them (Tx, F score = 0.67). PolII transcribed lncRNA and coding genes do not differ in the epigenetic profile of the promoter region (F score <0.6), accordingly to what previously reported, but they are clearly distinct up to 6 Kbp downstream the EoT (F score = 0.78). Figure 4B reports the contribution of each mark to the final supermark (i.e. the mixture coefficient vector  defined in eq. 2) sorted by their percentage in gene bodies. The leading histone mark here is H3K4me3, unexpectedly followed by H3R2me1 that is also the fourth contributor in downstream region after the well-known marks H2AZ, H3K36me1 and H3K36me3 and in comparison with random sites (Gene versus Rnd, Supplementary Figure S4). No functions in human cells have been associated to this mark so far. Notably, H3R2me1 does not show up in lncRNA versus Rnd analysis (Supplementary Figure S5), indicating that this mark might be specific to coding regions. To support this hypothesis, I calculated H3R2me1 and PolII density plots extending 6 Kbp 5′ and 3′ gene bodies of the two matched dataset. H3R2me1 appears to be indeed enriched in coding gene bodies with respect to expressed lncRNA. Its density increases drastically in the promoter region reaching its maximum immediately after TSS and it decreases smoothly but peaking again around the 3′ EoT region (Figure 5A, black line). Conversely, H3R2me1 is almost flat in lncRNA regions (Figure 5A, red line). This different behavior cannot be explained by differences of length, expression level or by PolII-mediated activation as clearly shown in Figure 5B. Moreover, correlation between level of expression of coding genes and lncRNA with H3R2me1 density in T cell data is rather low: 0.25 and 0.22, respectively. Interestingly, coding PolII density has a ‘bump’ at 3′ EoT [interpreted as a slow release of PolII from DNA (35)], absent in lncRNA and that overlap with H3R2me1 increased density in the same region (Figure 5A and B).
defined in eq. 2) sorted by their percentage in gene bodies. The leading histone mark here is H3K4me3, unexpectedly followed by H3R2me1 that is also the fourth contributor in downstream region after the well-known marks H2AZ, H3K36me1 and H3K36me3 and in comparison with random sites (Gene versus Rnd, Supplementary Figure S4). No functions in human cells have been associated to this mark so far. Notably, H3R2me1 does not show up in lncRNA versus Rnd analysis (Supplementary Figure S5), indicating that this mark might be specific to coding regions. To support this hypothesis, I calculated H3R2me1 and PolII density plots extending 6 Kbp 5′ and 3′ gene bodies of the two matched dataset. H3R2me1 appears to be indeed enriched in coding gene bodies with respect to expressed lncRNA. Its density increases drastically in the promoter region reaching its maximum immediately after TSS and it decreases smoothly but peaking again around the 3′ EoT region (Figure 5A, black line). Conversely, H3R2me1 is almost flat in lncRNA regions (Figure 5A, red line). This different behavior cannot be explained by differences of length, expression level or by PolII-mediated activation as clearly shown in Figure 5B. Moreover, correlation between level of expression of coding genes and lncRNA with H3R2me1 density in T cell data is rather low: 0.25 and 0.22, respectively. Interestingly, coding PolII density has a ‘bump’ at 3′ EoT [interpreted as a slow release of PolII from DNA (35)], absent in lncRNA and that overlap with H3R2me1 increased density in the same region (Figure 5A and B).
Figure 4.

Coding (Gene), ncRNA and Rnd comparisons. (A) Supermarks and corresponding F scores are calculated for gene bodies (Tx) and for genomic regions considering regions extending upstream from TSS (negative sizes on x axis) and downstream of 3′-end of transcription (EoT, positive sizes). Association is considered significant if F score >0.6. (B) Single-mark contributions (mixture coefficient vector  ) to supermark discriminating Gene versus lncRNA ordered by contribution in gene bodies. Only marks contributing >1% are reported.
) to supermark discriminating Gene versus lncRNA ordered by contribution in gene bodies. Only marks contributing >1% are reported.
Figure 5.

H3R2me1 (A) and PolII (B) read density plots of coding (black) and non-coding (red) gene bodies normalized by length and divided in 50 non-overlapping bins +15 bins for ±6 Kbp extensions. (C) Density plots in untranscribed genes.
DISCUSSION
Understanding the combinatorial complexity of histone post-translational modifications is important to elucidate the mechanisms of gene expression regulation as well as protein–DNA interactions. In this perspective, EMdeCODE recreates the probability mass distribution of observing a specific event on a specific DNA location by aggregating ChIPSeq histone marks profiles into a new associated supermark. The algorithm is based on classical Expectation Maximization approach and mixture modeling with statistical selection. The goal is to maximize the association in term of F score, thus reaching an optimal tradeoff between Precision and Sensitivity.
Compared with previous methods, EMdeCODE significantly improves the identification of putative enhancers, as demonstrated by the good approximation of the experimental p300 distribution in CD4+ T cells. One of the reasons for this increased performance can be ascribed to the choice of reconstructing a minimal profile by considering only significant peaks from ChIPSeq data, thus consistently reducing the associated noise and defining a common background for all the available marks. Moreover, the interpretation of ChIPSeq profiles as probability mass densities and the choice of the F0.5 score as cost function increase the contribution of less abundant but significantly associated marks and, at the same time, reduce the number of false positives.
When applied to the prediction of retroviral integration sites, EMdeCODE outperforms the heuristic method reported in (5) and generates a supermark localized within 2 Kbp of 75–85% of gammaretroviral proviruses, while increasing the corresponding F score. It is worth to observe that the best performance is obtained in CD4+ T cells where the highest number of marks is also available. This again demonstrates that EMdeCODE exploits and properly balances even minor contributions that are underrated by the heuristic approach.
EMdeCODE can also be used to determine whether some marks can be used to discriminate among genomic events. Indeed, it has been capable of determining supermarks which distinguish coding from non-coding genes, despite substantial epigenetic homogeneity of transcriptional regions having been previously reported (36). This seems to be true only for the promoter region up to 6 Kbp (upstream from the TSS), where no significant differences can be observed. Regions downstream (from 2 to 6 Kbp) of coding genes, instead, are particularly enriched in transcription associated marks, such as H2AZ, H3K4me3 and H3K36me1-3 among others, likely accounting for a complex regulatory structure that is less pronounced for most of non-coding transcripts. The gene body, also, is clearly enriched of H3K36me3 and H3K36me1 in coding genes, especially in comparison with random sites (Supplementary Figure S4), in line with what has been previously reported (37). Perhaps less expected is the strong influence of H3R2me1 in coding gene bodies with respect to lncRNA and random regions as shown in Figure 4B and Supplementary Figure S4. The specificity of this mark for coding regions is confirmed by its absence in lncRNA compared with random regions even if they can be clearly discriminated (F score >0.75, Figure 4A and Supplementary Figure S5). H3R2me1 significantly contributes to coding genes characterization in 3′ EoT regions and 6 Kbp downstream. H3R2me1 read density profile shows a peculiar shape in coding regions of the matched dataset by markedly outlining the gene body. Conversely, H3R2me1 is considerably depleted in non-coding gene bodies, explaining why it has been picked up by EMdeCODE (Figure 5A). H3R2 is methylated by PRMT1 and CARM1 that subsequently coactivate nuclear hormone receptor-mediated transcription, suggesting that the H3R2 methylation may be involved in transcriptional activity (38), as recently observed in Drosophila (39). On the other hand, H3R2me1 is not particularly enriched in active promoter regions of human DNA (32) and it is not strongly correlated with expression levels [see Results and (32)]. Interestingly, a recent study proposed that symmetrical dimethylation of H3R2 may have a role in euchromatin maintenance by mediating histone H3K4 methylation and H3 and H4 acetylation (40). Figure 5C reports PolII and H3R2me1 densities of 150 untranscribed 6 Kbp non-overlapping coding and non-coding genes. Again, monomethylated arginin of histone 3 is denser in coding regions even in complete absence of transcriptional activity (flat PolII density). These observations together with the other results obtained by EMdeCODE support the hypothesis that H3R2 is a key amino acid residue epigenetically involved in the maintenance and the protection of coding regions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–5.
FUNDING
European Research Council [ERC-HuCNC-249968 to F.A.S.]. Funding for open access charge: University of Geneva, Department of Genetic Medicine and Development.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The author thanks Massimo Pizzato and Stylianos Antonarakis for stimulating discussions and suggestions.
REFERENCES
- 1.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 2.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spivakov M, Fisher AG. Epigenetic signatures of stem-cell identity. Nat. Rev. Genet. 2007;8:263–271. doi: 10.1038/nrg2046. [DOI] [PubMed] [Google Scholar]
- 4.Nightingale KP, O'Neill LP, Turner BM. Histone modifications: signalling receptors and potential elements of a heritable epigenetic code. Curr. Opin. Genet. Dev. 2006;16:125–136. doi: 10.1016/j.gde.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 5.Santoni FA, Hartley O, Luban J. Deciphering the code for retroviral integration target site selection. PLoS Comput. Biol. 2010;6:e1001008. doi: 10.1371/journal.pcbi.1001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner BM. Defining an epigenetic code. Nat. Cell Biol. 2007;9:2–6. doi: 10.1038/ncb0107-2. [DOI] [PubMed] [Google Scholar]
- 7.Huff JT, Plocik AM, Guthrie C, Yamamoto KR. Reciprocal intronic and exonic histone modification regions in humans. Nat. Struct. Mol. Biol. 2010;17:1495–1499. doi: 10.1038/nsmb.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hon G, Ren B, Wang W. ChromaSig: a probabilistic approach to finding common chromatin signatures in the human genome. PLoS Comput. Biol. 2008;4:e1000201. doi: 10.1371/journal.pcbi.1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods. 2012;9:473–476. doi: 10.1038/nmeth.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Steensel B, Braunschweig U, Filion GJ, Chen M, van Bemmel JG, Ideker T. Bayesian network analysis of targeting interactions in chromatin. Genome Res. 2010;20:190–200. doi: 10.1101/gr.098822.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Firpi HA, Ucar D, Tan K. Discover regulatory DNA elements using chromatin signatures and artificial neural network. Bioinformatics. 2010;26:1579–1586. doi: 10.1093/bioinformatics/btq248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 14.Micsinai M, Parisi F, Strino F, Asp P, Dynlacht BD, Kluger Y. Picking ChIP-seq peak detectors for analyzing chromatin modification experiments. Nucleic Acids Res. 2012;40:e70. doi: 10.1093/nar/gks048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song Q, Smith AD. Identifying dispersed epigenomic domains from ChIP-Seq data. Bioinformatics. 2011;27:870–871. doi: 10.1093/bioinformatics/btr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat. Methods. 2009;6:S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyle AP, Guinney J, Crawford GE, Furey TS. F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics. 2008;24:2537–2538. doi: 10.1093/bioinformatics/btn480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bilmes JA. 1997. A gentle tutorial on the EM algorithm and application to Gaussian Mixtures and Baum-Welch. Technical Report TR-97-021, ICSI. [Google Scholar]
- 20.Won KJ, Chepelev I, Ren B, Wang W. Prediction of regulatory elements in mammalian genomes using chromatin signatures. BMC Bioinformatics. 2008;9:547. doi: 10.1186/1471-2105-9-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 22.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferretti V, Poitras C, Bergeron D, Coulombe B, Robert F, Blanchette M. PReMod: a database of genome-wide mammalian cis-regulatory module predictions. Nucleic Acids Res. 2007;35:D122–D126. doi: 10.1093/nar/gkl879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris SA, Rao B, Garcia BA, Hake SB, Diaz RL, Shabanowitz J, Hunt DF, Allis CD, Lieb JD, Strahl BD. Identification of histone H3 lysine 36 acetylation as a highly conserved histone modification. J. Biol. Chem. 2007;282:7632–7640. doi: 10.1074/jbc.M607909200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells. Nature. 2011;474:516–520. doi: 10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA. 2011;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 33.Ugarkovic D, editor. Long Non-Coding RNAs. New York: Springer-Verlag; 2011. [Google Scholar]
- 34.Barski A, Chepelev I, Liko D, Cuddapah S, Fleming AB, Birch J, Cui K, White RJ, Zhao K. Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes. Nat. Struct. Mol. Biol. 2010;17:629–634. doi: 10.1038/nsmb.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka Y, Katagiri Z, Kawahashi K, Kioussis D, Kitajima S. Trithorax-group protein ASH1 methylates histone H3 lysine 36. Gene. 2007;397:161–168. doi: 10.1016/j.gene.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 38.An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Kirmizis A, Santos-Rosa H, Penkett CJ, Singer MA, Green RD, Kouzarides T. Distinct transcriptional outputs associated with mono- and dimethylated histone H3 arginine 2. Nat. Struct. Mol. Biol. 2009;16:449–451. doi: 10.1038/nsmb.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Migliori V, Muller J, Phalke S, Low D, Bezzi M, Mok WC, Sahu SK, Gunaratne J, Capasso P, Bassi C, et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol. 2012;19:136–144. doi: 10.1038/nsmb.2209. [DOI] [PubMed] [Google Scholar]
- 41.Lewinski MK, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G, Collins F, Shinn P, Leipzig J, Hannenhalli S, et al. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog. 2006;2:e60. doi: 10.1371/journal.ppat.0020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.