

Abstract
Glucose improves memory for a variety of tasks when administered to rats and mice near the time of training. Prior work indicates glucose may enhance memory by increasing the synthesis and release of the neurotransmitter acetylcholine in the brain. To investigate if specific acetylcholine receptor subtypes may mediate some of the memory-enhancing actions of glucose, we examined the effects of subtype-specific nicotinic acetylcholine receptor antagonists on memory in Fischer-344 rats and also examined the ability of glucose to reverse drug-induced impairments. Pre-training peripheral injections of methyllycaconitine (MLA) or dihydro-beta-erythroidine (DHβE), which are specific α7 and α4β2 nicotinic receptor antagonists, respectively, dose-dependently impaired retention latencies in an inhibitory avoidance task when tested 7-days but not 1 hour after training. Immediate post-training glucose injections attenuated the impairments, but were more effective in attenuating the DHβE-induced impairments. Likewise, peripheral or direct intrahippocampal injections of MLA or DHβE dosedependently impaired spatial working memory scores on a spontaneous alternation task. Concurrent administration of glucose reversed DHβE- but not MLA-induced impairments. CREB phosphorylation downstream of cholinergic signaling was assessed 30 minutes after spontaneous alternation testing and intrahippocampal drug infusions. Both MLA and DHβE impaired hippocampal CREB phosphorylation; glucose reversed DHβE but not MLA-induced deficits. The effectiveness of glucose in reversing DHβE- but not MLA- induced impairments in behavioral performance and CREB phosphorylation suggests that activation of α7 receptors may play an important role in memory enhancement by glucose.
Keywords: glucose, memory, acetylcholine, nicotinic, α4β2 receptor, α7 receptor
1. INTRODUCTION
In humans and rodents, rises in blood glucose in response to stressful or emotional stimuli mediate memory improvement for the associated events. Likewise, glucose administration prior to or shortly after an event can increase its salience, converting a relatively unimportant event to a memorable one (Gold and Korol, 2010; Korol and Gold, 2007, 1998; Messier, 2004). In young adult rodents, increases in blood glucose enhance memory in a wide variety of behavioral tasks, ranging from tests of spatial working memory to assessments of memory at long intervals after training (cf. Gold, 2008). Glucose can also reverse age-related memory impairments in these same tasks, improving performance in old rats to the levels seen in young rats (Gold, 2005; McNay and Gold, 2001; Morris et al., 2010; Salinas and Gold, 2005).
Glucose is thought to work directly in the brain to enhance cellular and molecular memory processes, perhaps mediated by enhanced aerobic glycolysis and lactate production in astrocytes (Newman et al., 2011; Suzuki et al., 2011). Previous work suggests that systemically- or centrally-administered glucose may enhance memory by augmenting central release of the neurotransmitter acetylcholine. A number of microdialysis studies show that both peripheral and direct intrahippocampal glucose administration augment training-related acetylcholine release in the hippocampus, concurrent with memory enhancement (Kopf et al., 2001; Morris et al., 2010; Ragozzino and Gold, 1995; Ragozzino et al., 1998, 1996). These effects on acetylcholine release appear to be regionally specific and do not occur in animals at rest (Ragozzino et al., 1998, 1996), suggesting that glucose supports enhanced acetylcholine release in particular brain areas where it is needed to facilitate memory processes.
An open question is whether the memory-improving actions of glucose rely on signaling through some acetylcholine receptor subtypes more than others. Acetylcholine activates both muscarinic and nicotinic acetylcholine receptors. The subtypes of these receptors have diverse functions and variable expression patterns throughout the brain. Previous work indicates that peripheral injections of general muscarinic and nicotinic antagonists impair performance in a number of behavioral memory paradigms, including inhibitory avoidance and spontaneous alternation tasks, and that co-administration of glucose attenuates these impairments (Blanchard and Duncan, 1997; Kopf and Baratti, 1994; Ragozzino and Gold, 1991; Ragozzino et al., 1994; Stone et al., 1995, 1991, 1988). These results imply a lack of regional and receptor specificity in glucose’s actions, suggesting that glucose could compensate for deficits in muscarinic receptors by promoting signaling through nicotinic receptors, and vice versa. However, the results are difficult to interpret because of the wide-ranging effects of these drugs throughout the body and brain, including effects not specific to memory processes.
The emergence of drugs targeting specific acetylcholine receptor subtypes in the brain, as opposed to general muscarinic or nicotinic receptors, makes it possible to begin evaluating which receptor subtypes may be important to the memory-enhancing effects of glucose. Two nicotinic receptor subtypes, α4β2 and α7, possess the functional properties and regional distribution that make them potential mediators of glucose’s effects (Cincotta et al., 2008; Graef et al., 2011; Kenney and Gould, 2008; Levin et al., 2006). Neuronal nicotinic receptors are ionotropic receptors composed of various combinations of five membrane-spanning subunits. Nine α (α2–α10) and three β (β2–β4) subunits have been identified, making a large number of receptor subtype combinations possible. However, the heteromeric α4β2 and homomeric α7 receptor subtypes have garnered the most attention due to their high levels of expression in the brain and roles in a variety of cognitive processes. In many situations, α4β2 and α7 receptors appear to function similarly. For example, a number of studies have utilized direct brain infusions of methyllycaconitine (MLA) or dihydro-β-erythroidine (DHβE), which are competitive antagonists for α7 and α4β2 receptors, respectively, to assess working memory performance in the radial arm maze. Infusions of either MLA or DHβE into the ventral or dorsal hippocampus, amygdala, or frontal cortex induced working memory impairments, with only minor dose-related differences between the drugs (Addy et al., 2003; Bettany and Levin, 2001; Chan et al., 2007; Levin et al., 2002; Nott and Levin, 2006). In other cases, there are distinct functional differences between the α4β2 and α7 receptor subtypes. For instance, there are several studies indicating a specific role for α4β2 but not α7 receptors in mediating the effects of nicotine on enhancing memory for contextual fear conditioning (Davis and Gould, 2006; Davis and Gould, 2007; Davis et al., 2007; Kenney et al., 2012).
Recent evidence indicates that glucose and nicotinic acetylcholine receptors activate a similar downstream molecular signaling pathway related to memory formation. Specifically, cAMP response element binding protein (CREB) is a transcription factor widely implicated in the formation of long-lasting memory and long-term changes in synaptic plasticity (Alberini, 2009; Benito and Barco, 2010; Carlezon et al., 2005; Morris and Gold, 2012a; Silva, 1998; Yin and Tully, 1996). A number of studies indicate that nicotine administration activates CREB in a variety of neuronal cell types in both in vitro and in vivo models (Chang and Berg, 2001; Hu et al., 2002; Nakayama et al., 2001; Pascual et al., 2009; Tang et al., 1998). Likewise, glucose administration following inhibitory avoidance training reverses age-related memory impairments, with parallel activation of CREB phosphorylation. Consistent with the view of regional selectivity in the memory-enhancing actions of glucose, there is a high degree of regional variability in glucose-mediated increases in CREB phosphorylation, including within subregions of the hippocampus (Morris and Gold, 2012b).
The following work examines the effects of the α7 antagonist MLA and the α4β2 antagonist DHβE on memory processes in the inhibitory avoidance and spontaneous alternation tasks. Given that the general nicotinic antagonist mecamylamine impairs behavioral performance in both of these tasks, a major prediction was that one or both of these subtype-specific antagonists would impair performance similar to mecamylamine, perhaps distinguishing which nicotinic receptor subtype mediates the effects of mecamylamine. We hypothesized that any differences in the effects of MLA and DHβE may help determine which nicotinic receptor signaling processes are important in facilitating memory formation in these tasks.
We also tested the ability of glucose to attenuate any drug-induced impairments. Based on the convergent behavioral, neurochemical, and molecular evidence of regional selectivity in the memory-enhancing actions of glucose, a major hypothesis was that glucose may enhance memory by facilitating signaling through specific cholinergic receptor subtypes. Thus, examining interactions between the effects of glucose and subtype-specific antagonists may help identify certain nicotinic receptor subtypes that are important to the memory-enhancing effects of glucose.
These experiments utilize both peripheral and direct intrahippocampal injections and include correlations with the expression of phosphorylated CREB (pCREB) in the hippocampus. Although the focus here was on nicotinic receptors, some muscarinic receptor subtypes (especially M1 receptors) share similar distributions, behavioral effects, molecular signaling pathways, etc. to α7 and α4β2 receptors, and thus may also be important in these contexts. However, muscarinic antagonists tend to have more peripheral side effects compared to MLA and DHβE, which are centrally-acting drugs. In addition, the effects of MLA and DHβE have been examined together in a variety of behavioral studies, including aversive and spatial working memory tasks, allowing for direct comparisons with the present experiments and supporting this investigation of the effects of nicotinic receptor antagonists on memory and on glucose enhancement of memory.
2. METHODS
2.1. Subjects
Young adult (3 to 4 mo.) male Fischer-344 rats ordered from Taconic Farms (Germantown, NY) were individually housed in translucent cages with a 12-h light/dark cycle (lights on at 07:00 h) and ad libitum access to food and water. Animal pain and discomfort were minimized, and all experiments were conducted in accordance with animal care guidelines established by the National Institute of Health and the University of Illinois, which is fully accredited (AAALAC). Rats were handled for 2–3 min each day on 5 consecutive days prior to behavioral training.
2.2. Preparation of drugs
Methyllycaconitine citrate salt hydrate (MLA) was obtained from Sigma-Aldrich (St. Louis, MO). Dihydro-β-erythroidine hydrobromide (DHβE) was obtained from Tocris Bioscience (Minneapolis, MN). MLA, DHβE, and glucose were dissolved in saline for intraperitoneal (i.p.) administration or in artificial cerebral spinal fluid (aCSF; 128 mM NaCl, 2.5 mM KCl, 1.3 mM CaCl2, 2.1 mM MgCl2, 0.9 mM NaH2PO4, 2.0 mM Na2HPO4, 1 mM glucose, pH 7.4) for intrahippocampal administration. The 1.0 mM glucose concentration in the aCSF is based on evidence that this level is seen at baseline in hippocampal extracellular fluid (McNay and Gold, 1999).
2.3. Surgery and intrahippocampal drug infusions
Rats were deeply anesthetized with isoflurane and placed in a stereotaxic apparatus. Stainless steel guide cannulae (Plastics One, Roanoke, VA) were implanted bilaterally into the dorsal hippocampus [coordinates: − 3.2 mm from bregma; ± 2.5 mm lateral; − 1.4 mm deep from dura], according to the atlas of Paxinos and Watson (2003). Rats were monitored and allowed to recover for 7 days after surgery. Prior to training, microinfusion probes (Plastics One) were inserted through the guide cannulae to a point 1 mm below the guide cannulae tips. Compounds were infused bilaterally into the dorsal hippocampus at a rate of 0.25 μl/min for 2 min. Infusion probes were left in place for an additional 1 min to allow solutions to diffuse away from the probe tips.
2.4. Inhibitory Avoidance Training
All training and testing took place between 12:00 and 16:00 h. The inhibitory avoidance apparatus was a trough-shaped alleyway (91 cm long, 22.9 cm wide at the top, 7.6 cm wide at the bottom, and 15.2 cm deep) divided into lit (31 cm) and dark (60 cm) compartments by a sliding door that could be lowered through the floor. Each rat was placed in the lit chamber facing the door. When the rat turned completely around, the door was lowered to a height 2 cm above the floor. When the rat again turned toward the door, a timer was started to record the latency to enter the dark chamber (i.e. the training latency). Upon entering the dark chamber, the rat received a brief foot shock (0.2 mA, 0.4 sec) and the door was closed to prevent reentry into the lit chamber. After training or post-training injections, rats were returned to the holding cage. Retention latencies (max of 600 sec) were tested 7 days later using a similar procedure, but without the foot shock. MLA (0.5, 2, or 5 mg/kg) or DHβE (0.5, 2, or 5 mg/kg) were given i.p. 10 min prior to training. Glucose (250 mg/kg) was given i.p. immediately after training.
2.5. Foot Shock Perceptual Thresholds
10 min after i.p. injections of saline or 2 mg/kg MLA or DHβE, rats received a series of foot shocks (each 0.5-sec duration) beginning at 0.1 mA and increasing at 0.05 mA increments with a 30 sec inter-shock interval until a jump response was induced. The lowest shock intensities that caused the rats to flinch and jump were recorded by two independent observers.
2.6. Spontaneous Alternation Testing
All training took place between 12:00 and 16:00 h. Spontaneous alternation performance was assessed in a 4-arm plus-shaped black Plexiglas maze with an open ceiling. The dimensions of each arm were 45 cm L × 13 cm W × 7 cm H. The dimensions of the central square-shaped area were 13 cm L × 13 cm W × 7 cm H. The training room contained numerous extramaze visual cues in the form of objects and wall decorations. Rats were placed in a random start arm and were allowed to freely traverse the maze for 20 min. The number and sequence of entries were recorded and alternation performance was calculated. An alternation consisted of visiting all four arms within overlapping sets of five arm visits. Using this procedure, the total number of possible alternations was equal to the number of arm entries minus 4. The percent alternation score is equal to (actual alternations/possible alternations) × 100. Chance performance on this task is 44%. Only data from rats that made at least 10 arm choices (6 possible alternations) were included in the analyses. For experiments with i.p. injections, MLA (0.5, 2, or 5 mg/kg) or DHβE (0.5, 2, or 5 mg/kg) were administered alone or in combination with glucose (250 mg/kg) 30 min prior to testing. For experiments with intrahippocampal injections, MLA (6.75, 13.5, or 27 μg/side) or DHβE (4, 8, or 20 μg/side) were infused alone or in combination with glucose (16.7 nmol/side) beginning 10 min prior to training. Some of the experiments with intrahippocampal injections (i.e. those illustrated in Figure 5) utilized a Latin square design in which each rat was tested on four separate occasions with a 3-day rest in between each test. Prior to each test, each rat received a single infusion of a drug, with doses administered in a counterbalanced order.
Figure 5.

Effects of intrahippocampally-administered nicotinic antagonists on working memory assessed in a spontaneous alternation task. (A) MLA doses of 6.75 and 13.5, but not 27 μg/side, significantly impaired alternation scores. (*) ps < .05 vs. aCSF. N = 9. (B) DHβE doses of 8 and 20, but not 4 μg/side, significantly impaired alternation scores. (*) ps < .05 vs. aCSF. N = 9. To the right of each graph is an illustration of the infusion sites targeting the dorsal hippocampus for each experiment. Filled circles represent tips of infusion tracts. Numbers refer to distance in mm posterior to bregma. Adapted with permission from Paxinos and Watson (2003).
2.7. Perfusion and Brain Slicing
For analysis of infusion probe placement, brains were collected within two days of the end of behavioral testing. For pCREB immunohistochemistry, brains were collected 30 min after spontaneous alternation testing. Rats were deeply anesthetized with an overdose injection of sodium pentobarbital (i.p.; Sigma-Aldrich) and then perfused intracardially with 80 ml of 0.1 M phosphate-buffered saline followed by 80 ml of 4% paraformaldehyde in 0.1 M phosphate buffer. Rats were decapitated and the brains were removed and placed into 4% paraformaldehyde in 0.1 M PB for ~72 hrs. The brains were transferred to 20% glycerol in 0.1 M PBS for ~48 hrs. For probe placement analysis, frozen sections (40 μM) containing the probe infusion tracts were collected at −30°C with a Leica 1800 cryostat (Leica Microsystems, Wetzlar, Germany). Sections were mounted onto gelatin-coated slides and stained with cresyl violet to visualize probe placements, which were determined using a dissection microscope. For immunohistochemistry, frozen sections (40 μM) just posterior to the injection site were collected at −30°C with a Leica 1800 cryostat. Slices were stored in a cryopreservative solution (250 mM 40 KD polyvinylpyrrolidone, 880 mM sucrose, 30% v/v ethylene glycol, 50 mM sodium phosphate) at −20°C.
2.8. Immunohistochemistry
All steps took place at room temperature and all reactions were performed in duplicate. Slices were washed three times for 10 min each time in 0.05 M PBS initially and between all subsequent steps. Slices were first incubated in blocking solution (1% H2O2, 1% normal goat serum, 0.02% triton x-100, 0.05 M PBS) for 10 min. They were transferred to a pre-incubation solution (2% NGS, 0.4% triton x-100, 0.05 M PBS) for 20 min and then incubated overnight in a solution (1% NGS, 0.4% triton x-100, 0.05 M PBS) containing a rabbit primary antibody for Ser-133 phosphorylated CREB (Millipore, Billerica, MA) diluted 1:4000. The next day, the slices were placed for 1 hr in a solution (1% NGS, 0.2% triton x-100, 0.05 M PBS) containing a goat anti-rabbit biotinylated secondary antibody (Santa Cruz, Santa Cruz, CA). They were next incubated for 30 min with ABC reagent (Vector, Burlingame, CA) in 0.05 M PBS, followed by incubation with DAB substrate (Vector) for 4 min. Slices were mounted onto slides and allowed to dry overnight. The next morning, slices were dehydrated with a graded ethanol series of washes, then coverslipped using DPX mountant (Sigma-Aldrich).
2.9. Image Acquisition and Analysis
Sections were imaged using a Leica DM 6000B/CTR6000 light microscope and a Leica DFC350 FX video camera, which was interfaced to a PC computer. This system was used in conjunction with Image-Pro software (Media Cybernetics, Inc., Bethesda, MD) for image acquisition and for correction of unevenness in illumination across images. Image J software (NIH, Bethesda, MA) was used to quantify the integrated optical density of pCREB staining in area CA1 and the dentate gyrus of the hippocampus. A statistical thresholding method in Image J was used to ensure that only specifically labeled cells were being measured. For each image, the optical density of a nearby region with no or little specific staining was calculated and used for background subtraction.
2.10. Statistical Analyses
All analyses were performed using Statview software. Inhibitory avoidance behavioral results were analyzed using a non-parametric Kruskal-Wallis one-way analysis of variance, followed by Mann-Whitney tests for individual comparisons.
The spontaneous alternation behavioral results (except those noted below) and the optical densities of pCREB immunostaining were analyzed using one-way ANOVAs with post hoc Fisher PLSD tests where appropriate. The spontaneous alternation behavioral results that utilized a Latin square design were analyzed using repeated measures ANOVA with post hoc Fisher PLSD tests where appropriate. Correlations between behavioral results and protein expression were determined by simple linear regression.
3. RESULTS
3.1. Inhibitory Avoidance Training with Peripheral Injections of Drugs
Figure 1 shows the effects of peripherally-administered nicotinic antagonists on retention of inhibitory avoidance training. Drugs were administered 10 min prior to training. There was a significant main effect of MLA on 7-day retention latencies (H(3,28) = 12.02, p < .01). Both the 2 and 5 mg/kg doses of MLA significantly depressed 7-day retention latencies compared to those of saline-injected controls (ps < .05). The 2 mg/kg dose was more effective, significantly depressing 7-day retention latencies compared to the 5 mg/kg dose (p < .05). There was also a significant main effect of DHβE on 7-day retention latencies (H(3,28) = 9.60, p < .05). The 2 and 5 mg/kg doses of DHβE were similarly effective at depressing 7-day retention latencies compared to saline controls (ps < .05). Neither MLA nor DHβE significantly altered training latencies.
Figure 1.

Effects of peripherally-administered nicotinic antagonists on inhibitory avoidance memory tested 7 days after training. (A) MLA doses of 2 and 5, but not 0.5 mg/kg, significantly impaired memory. (*) ps < .05 vs. saline. Ns = 8. (B) DHβE doses of 2 and 5, but not 0.5 mg/kg, significantly impaired memory. (*) ps < .05 vs. saline. Ns = 8.
To examine if glucose could attenuate drug-induced impairments in 7-day retention latencies, saline or 2 mg/kg MLA or DHβE were administered 10 min before inhibitory avoidance training in combination with immediate post-training injections of saline or glucose (250 mg/kg). The results are shown in Figure 2. Both MLA-saline and DHβE-saline significantly reduced 7-day retention latencies compared to those of saline-saline controls (ps < .01). Peripheral glucose administration attenuated these drug-induced impairments, but was more effective in reversing DHβE-induced deficits. In the MLA-glucose group, retention latencies were significantly higher than those in the MLA-saline group, but still significantly reduced compared to those in saline-saline controls (ps < .05). By comparison, retention latencies in the DHβE-glucose group were similar to those in the saline-saline group (p > 0.5), with the group median reaching the 600 sec maximum. Training latencies were not significantly different across groups.
Figure 2.
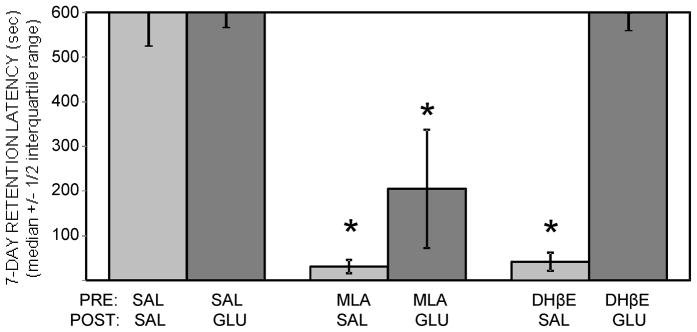
Glucose attenuation of drug-induced memory impairments after inhibitory avoidance training. Pre-training administration of MLA or DHβE (2 mg/kg), together with post-training saline injections, produced deficits in memory tested at 7 days after training. Post-training glucose attenuated the drug-induced memory impairments, but was more effective at reversing the impairments induced by DHβE. (*) ps < .05 vs. saline-saline. Ns = 8.
MLA or DHβE-induced impairments in retention latencies may reflect an inability of rats to acquire the learned response. To examine this possibility, two experiments were performed. The first experiment assessed retention latencies using a shorter training-testing interval. Saline or 2 mg/kg MLA or DHβE was injected 10 min prior to training, and retention latencies were tested 1 hr later. All three groups had median 1-hr retention latencies at the maximum of 600 sec, with no significant differences among groups (data not shown). The second experiment examined the effects of the drugs on foot shock perceptual thresholds. Injections of 2 mg/kg MLA or DHβE 10 min prior to foot shock did not significantly alter flinch or jump thresholds compared to those of saline-injected rats. The flinch thresholds were 0.26 ± 0.01 mA (mean ± s.e.m.) for all groups. Jump thresholds ranged from 0.63 ± 0.03 mA in the saline and DHβE groups to 0.65 ± 0.03 in the MLA group.
3.2. Spontaneous Alternation Testing
3.2.1. Peripheral Injections of Drugs
Figure 3 shows the effects of peripherally-administered nicotinic antagonists on spontaneous alternation scores. MLA or DHβE given 30 min before testing impaired alternation scores, with effective doses similar to those observed in the inhibitory avoidance task (F(3,28) = 3.09, p < .05 for MLA; F(3,28) = 3.34, p < .05 for DHβE). For the MLA experiment, the intermediate dose of 2 mg/kg, but not lower or higher doses, significantly reduced alternation scores compared to saline-injected controls (p < .05). In contrast, MLA had no significant effect on the number of arm entries during testing, which ranged from 24.6 ± 2.8 (mean ± s.e.m.) in the saline group to 26.1 ± 2.6 in the 2 mg/kg MLA group. For the DHβE experiment, doses of 2 and 5 mg/kg were about equally effective at impairing alternation scores compared to saline controls (ps < .05). The number of arm entries decreased with increasing doses of DHβE, ranging from mean values of 23.3 ± 1.3 in the saline group to 18.5 ± 1.9 in the 5 mg/kg DHβE group. However, this was not a statistically significant effect.
Figure 3.

Effects of peripherally-administered nicotinic antagonists on spontaneous alternation spatial working memory scores. (A) An MLA dose of 2, but not 0.5 or 5 mg/kg, significantly impaired alternation scores. (*) p < .05 vs. saline. Ns = 8. (B) DHβE doses of 2 and 5, but not 0.5 mg/kg, significantly impaired alternation scores. (*) ps < .05 vs. saline. Ns = 8.
To determine if glucose could attenuate drug-induced impairments in spontaneous alternation scores, MLA or DHβE (2 mg/kg) was administered alone or in combination with glucose (250 mg/kg) 30 min before testing (Figure 4). MLA and DHβE significantly impaired alternation scores compared to saline-injected controls (ps < .01). Glucose had no significant effect on attenuating MLA-induced deficits; rats treated with MLA-glucose had alternation scores similar to those in the MLA group (p > 0.5) and significantly lower than those of saline controls (p < .05). However, glucose reversed DHβE-induced impairments in alternation performance, raising scores well above those of rats administered DHβE alone (p < .05). The numbers of arm entries were significantly reduced in the DHβE (17.5 ± 1.5) and DHβE-glucose (17.1 ± 0.8) groups compared to saline controls (23.4 ± 1.4; ps < .01). Therefore, the effect of glucose was restricted to DHβE effects on alternation scores and did not extend to drug effects on locomotor activity. The numbers of arm entries in the other groups were not significantly different from those in the saline group.
Figure 4.
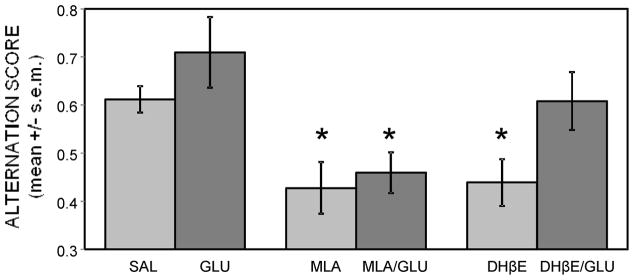
Glucose attenuation of drug-induced working memory impairments in the spontaneous alternation task. MLA or DHβE (2 mg/kg) produced deficits in alternation scores. Co-administration of glucose with the antagonists attenuated DHβE but not MLA-induced deficits. (*) ps < .05 vs. saline. Ns = 8.
3.2.2. Intrahippocampal Injections of Drugs
Figure 5 shows the effects of intrahippocampally-administered nicotinic antagonists on spontaneous alternation scores. To reduce the number of rats subjected to surgical procedures, a Latin square design was used here but not in subsequent experiments with glucose. Drugs were infused beginning 10 min before testing. Both MLA and DHβE infusions significantly impaired alternation scores (F(3,24) = 3.02, p < .05 for MLA; F(3,24) = 3.17, p < .05 for DHβE). MLA doses of 6.75 and 13.5 μg/side were equally effective at impairing alternation scores compared to those of aCSF-injected controls (ps < .05). However, MLA lost its effectiveness in impairing alternation scores at the highest dose of 27 μg/side. DHβE was ineffective at the lowest dose of 4 μg/side, but significantly impaired alternation performance at higher doses of 8 and 20 μg/side (ps < .05 vs. aCSF). Neither MLA nor DHβE infusions significantly altered the number of arm entries. The mean numbers of arm entries were slightly lower than those seen with peripheral injections, ranging from 19.4 ± 2.8 to 23.1 ± 2.7 in the MLA experiment and 19.1 ± 2.4 to 21.2 ± 1.4 in the DHβE experiment.
Using the Latin square design, each rat received an intrahippocampal injection on four separate occasions, which could have progressively increased damage at the infusion site. To examine this possibility, repeated measures ANOVAs were conducted to determine if the number of injections affected alternation scores, revealing no main effects in either the MLA (F(3,24) = 1.02, p > 0.2) or DHβE (F(3,24) = 2.23, p > 0.1) experiments.
All infusion cannulae were placed in the dorsal hippocampus, as shown schematically in Figure 5. After the four infusions, the area near the tip did not display unusual damage and appeared similar to that seen in other groups in this and in past experiments.
The effects of intrahippocampal glucose on attenuating drug-induced deficits largely paralleled the results seen with peripheral injections. Glucose (16.7 nmol/side) was combined with the lowest effective doses of the nicotinic antagonists and infused 10 min before alternation testing. As shown in Figure 6, glucose had no effect on attenuating MLA-induced impairments in alternation performance. Both the MLA and MLA-glucose groups had significantly depressed alternation scores (ps < .05 vs. aCSF), which were not significantly different from each other (p > 0.5). In contrast, glucose reversed DHβE-induced impairments in alternation scores. Although DHβE alone produced significant deficits in alternation scores (p < .05 vs. aCSF), the DHβE-glucose group had similar scores compared to aCSF (p > 0.5). Unlike the pattern of results seen with peripheral injections of drugs, there was a trend for enhancement in alternation scores following infusion of intrahippocampal glucose alone (p < .06 vs. aCSF). The number of arm entries were similar across groups, ranging from 19.1 ± 3.0 in the DHβE-glucose grouop to 24.6 ± 3.8 in the MLA-glucose group.
Figure 6.
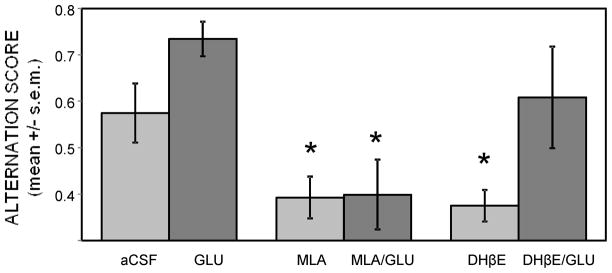
Glucose attenuation of impairments in spontaneous alternation memory scores produced by intrahippocampally-administered antagonists. MLA (6.75 μg/side) or DHβE (8 μg/side) produced deficits in alternation scores. Co-administration of glucose with the antagonists attenuated DHβE but not MLA-induced deficits. There was a trend for enhancement in alternation scores following administration of glucose alone. (*) ps < .05 vs. aCSF. Ns = 8 for aCSF and GLU. Ns = 7 for all other groups.
3.3. pCREB Staining
Immunostaining levels of pCREB were examined 30 min after spontaneous alternation testing in rats receiving intrahippocampal infusions of drugs (corresponding to Figure 6). As shown in Figure 7, pCREB staining levels in area CA1 paralleled the behavioral results. Both MLA and DHβE impaired pCREB staining compared to aCSF-injected controls (ps < .05). Glucose reversed the DHβE- but not MLA-induced pCREB deficits (p > 0.5 DHβE vs. aCSF; p < .05 MLA vs. aCSF). Unlike the behavioral results, there was no trend for enhancement of pCREB staining by intrahippocampal glucose alone (p > 0.5). In contrast to the results for area CA1, there were no significant effects of drug treatments on pCREB staining levels in the dentate gyrus (data not shown).
Figure 7.

Differences in pCREB immunoreactivity in area CA1 following spontaneous alternation testing with intrahippocampal administration of drugs. (A) MLA (6.75 μg/side) or DHβE (8 μg/side) reduced pCREB levels. Co-administration of glucose with the antagonists attenuated DHβE but not MLA-induced pCREB deficits. (*) ps < .05 vs. aCSF. Ns = 8 for aCSF and GLU. Ns = 7 for all other groups. (B) Representative photomicrographs of pCREB immunostaining in area CA1. Scale bar = 100 microns.
As shown in Figure 8, there was a significant positive correlation between area CA1 pCREB staining levels and spontaneous alternation scores in rats receiving MLA-glucose injections (r = 0.90, p < .01), and a trend for significance in rats receiving MLA alone (r = 0.69, p = .09). There were no correlations or trends in any of the other groups (ps > 0.2).
Figure 8.
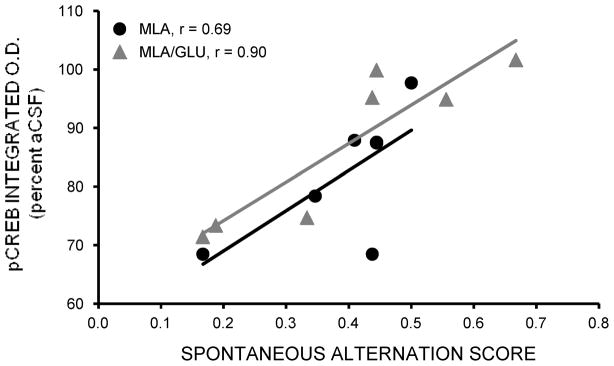
Correlations between area CA1 pCREB immunostaining and spontaneous alternation scores. There was a significant correlation in rats receiving intrahippocampal injections of MLA-glucose (p < .01), and a trend for a correlation in rats receiving MLA alone (p = .09).
4. DISCUSSION
4.1. Effects of Nicotinic Antagonists on Memory
Peripheral injections of MLA or DHβE impaired memory when tested 7 days after inhibitory avoidance training, supporting the view that inhibition of cholinergic signaling through either α7 or α4β2 nicotinic receptors inhibits the formation of long-lasting memories. Previous studies in rats and mice have shown that pre-training peripheral administration of the centrally-acting nicotinic antagonist mecamylamine impairs later retention for inhibitory avoidance training, whereas the peripherally-acting nicotinic antagonist hexamethonium has no effect (Chiappetta and Jarvik, 1969; Glick and Greenstein, 1972; Goldberg et al., 1971; Ragozzino and Gold, 1991; Rush and Streit, 1992). The present results extend these findings, suggesting that mecamylamine could exert its behavioral effects through inhibition of α4β2 and/or α7 receptors in the brain.
In the present study, the administration of the drugs prior to instead of after inhibitory avoidance training leaves open the possibility that the drugs produced apparent amnesia by altering factors other than memory, such as attention, motivation, or sensorimotor processes. However, a few pieces of evidence indicate that behavioral differences due to effects on non-mnemonic variables are unlikely. First, neither MLA nor DHβE altered training latencies or thresholds for foot shock sensitivities, suggesting that these measures of sensorimotor processes were intact during training. Second, the high memory scores in MLA- or DHβE-injected rats tested 1 hr after training indicate the drugs did not interfere with the ability of the rats to learn and initially remember the training experience. However, differences between saline- and drug-injected groups could have been masked by a ceiling effect, given that the median 1-hr retention latencies in all groups reached the maximum of 600 sec.
Peripheral administration of MLA or DHβE also impaired performance in the spontaneous alternation task. These results were remarkably similar to the effects of the antagonists on retention performance in the inhibitory avoidance task, and were consistent with past findings showing that peripheral injections of mecamylamine, but not the peripherally acting hexamethonium, impair alternation performance (Ragozzino and Gold, 1991). In both the spontaneous alternation and inhibitory avoidance tasks, MLA-induced impairments followed a U-shaped pattern, in which the middle dose of 2 mg/kg was most effective at impairing behavioral performance. In contrast, both 2 and 5 mg/kg doses of DHβE were similarly effective at impairing performance in these behavioral tasks. The U-shaped pattern observed for MLA-induced deficits may relate to non-specific effects of the drug at higher doses.
Direct infusions of MLA or DHβE into the dorsal hippocampus also impaired performance in the spontaneous alternation task, suggesting that the drugs work directly in the brain to impair spatial working memory processes. These results are consistent with prior behavioral studies of spatial working memory in the appetitive radial arm maze task, where infusions of MLA or DHβE directly into the hippocampus, amygdala, or frontal cortex, significantly increased working memory errors (Addy et al., 2003; Bettany and Levin, 2001; Chan et al., 2007; Levin et al., 2002; Nott and Levin, 2006). Interestingly, infusions of DHβE into the mediodorsal thalamic nucleus actually reduced working memory errors during radial arm maze testing (Cannady et al., 2009). Opposing effects in different brain regions of MLA or DHβE have also been observed in trace fear conditioning (Raybuck and Gould, 2010), suggesting some regional variability in the actions of these drugs on learning and memory. In the present study, the effects of peripheral administration of MLA or DHβE on alternation scores were similar to those seen with intrahippocampal infusions, consistent with the possibility that the effects on memory of the peripherally-injected drugs may be mediated by hippocampal actions.
An interesting and unexpected result was the lack of any dissociation between the effects of MLA and DHβE on behavioral performance. Similar doses of each drug impaired performance in both the inhibitory avoidance and spontaneous alternation tasks, suggesting that both α7 and α4β2 receptors may be important in modulating memory in these tasks. These results underscore the diverse role of nicotinic acetylcholine receptors in learning and memory processes, supporting the view that multiple interconnected or parallel nicotinic signaling mechanisms can support memory formation.
4.2. Ability of Glucose to Reverse Drug-Induced Memory Impairments
In the inhibitory avoidance task, post-training injections of glucose were more effective at reversing DHβE- than at reversing MLA-induced memory deficits. Likewise, in the spontaneous alternation task, co-administration of glucose with the antagonists reversed DHβE- but not MLA-induced memory impairments, regardless of whether the drug/glucose combinations were administered peripherally or directly into the hippocampus. Previous work has shown that glucose reverses the memory-impairing effects of mecamylamine in both the inhibitory avoidance and spontaneous alternation tasks. However, glucose does not attenuate mecamylamine-induced decreases in locomotor activity, such as reductions in the number of arm entries during spontaneous alternation testing (Ragozzino and Gold, 1991; Ragozzino et al., 1994). In the present study, the results obtained with peripheral DHβE administration parallel those seen previously with mecamylamine. DHβE produced memory impairments and decreased the number of arm visits during spontaneous alternation testing; glucose attenuated the memory but not motor deficits. In contrast, MLA-induced memory impairments were not attenuated by glucose and followed a U-shaped dose-response function not seen previously with mecamylamine. MLA also had no significant effect on spontaneous alternation arm entries. The similarities in the behavioral effects of mecamylamine and DHβE but not MLA may be a consequence of the functional properties of mecamylamine. Although mecamylamine is a nonselective nicotinic antagonist, it is a more potent and long-lasting inhibitor of β subunit-containing receptors (Papke et al., 2001). Therefore, the behavioral effects of mecamylamine are more likely mediated by inhibition of α4β2 and/or other β subunit-containing receptors, as opposed to α7 receptors.
The present findings indicate that glucose may enhance memory by differentially modulating nicotinic receptor signaling through distinct receptor subtypes. Previous studies indicate that glucose is remarkably effective at attenuating or reversing memory impairments produced by a variety of centrally-acting pharmacological agents, including mecamylamine, scopolamine, morphine, and muscimol (Blanchard and Duncan, 1997; Jafari et al., 2004; Kopf and Baratti, 1994; Krebs and Parent, 2005; Krebs-Kraft and Parent, 2008; Ragozzino and Gold, 1991; Ragozzino et al., 1994, 1992; Stone et al., 1995, 1991, 1988). Glucose also reverses age-related memory impairments in inhibitory avoidance, spontaneous alternation, and other tasks (McNay and Gold, 2001; Morris et al., 2010; Salinas and Gold, 2005). The present study illustrates a rare case in which glucose was ineffective at reversing a pharmacologically-induced memory deficit. The ineffectiveness of glucose at reversing MLA-induced behavioral deficits suggests that the memory-improving actions of glucose may rely more on activation of α7- as opposed to α4β2-mediated signaling processes. For example, glucose may selectively up-regulate acetylcholine release in regions with high densities of α7 receptors. Previous studies suggest that glucose is targeted to particular neural systems in response to task demands, where it may enhance local acetylcholine release (cf. Gold, 2004). There are high concentrations of α7 receptors in brain areas in which direct glucose injections lead to memory improvement. The hippocampus has particularly dense levels of α7 receptors, which are found both pre- and post-synaptically throughout the strata of the CA subfields and dentate gyrus. In the hippocampus, α7 receptors often form clusters at GABAergic and other synaptic sites, and have a distinct regional localization and cell type distribution compared to α4β2receptors, which tend to be more diffusely distributed (Fabian-Fine et al., 2001; Gotti and Clementi, 2004; Kawai et al., 2002; Tribollet et al., 2004; Zarei et al., 1999). By contrast, Pych et al. (2006) showed that injections of glucose into the dorsal striatum, which has relatively low levels of α7 receptors but an abundance of α4β2 receptors (Gotti and Clementi, 2004; Tribollet et al., 2004), failed to enhance learning of a striatum-sensitive maze in which learning is based on egocentric responses.
Glucose may also selectively modulate α7-mediated signaling processes due to the unique functional properties of these receptors. α7 receptors have the highest calcium permeability of any known nicotinic receptor subtype (Castro and Albuquerque, 1995; Séguéla et al., 1993; Shen and Yakel, 2009). Calcium influx through these receptors plays an important role in the presynaptic release of GABA, glutamate, and other neurotransmitters in the brain (Shen and Yakel, 2009; Sher et al., 2004; Wonnacott et al., 2006). Glucose may potentiate these responses through actions mediated by presynaptic ATP-dependent potassium (K-ATP) channels. A number of studies indicate that regulation of K-ATP channel activity is important to the memory-enhancing effects of glucose (Stefani and Gold, 2001, 1998; Stefani et al., 1999; Rashidy-Pour, 2001). Another unique property of α7 receptors is that they have a high affinity for choline, which functions as a full and selective agonist of these receptors (Alkondon et al., 1997; Mike et al., 2000; Uteshev et al., 2003). Glucose may modulate the activity of α7 receptors by regulating local extracellular choline levels. Previous studies indicate that glucose regulates high-affinity choline uptake and can act synergistically to improve memory when administered together with choline (Kopf et al., 2001; Micheau et al., 1995; Messier et al., 1990). However, the effects of glucose on extracellular choline levels, in the context of memory enhancement, have not been directly investigated.
There are a number of other possible explanations for why glucose was ineffective at reversing MLA-induced behavioral impairments. The present study only tested one peripheral and one intrahippocampal dose of glucose. These doses were similar to or the same as those used in previous studies, in which glucose reversed pharmacologically-induced or age-dependent memory impairments. However, testing a range of glucose concentrations may reveal dose-response interactions with MLA or DHβE that would alter the interpretations of results. Another possibility is that MLA but not DHβE impaired behavioral performance by affecting cognitive or other processes that are not modulated by glucose. For example, there is some evidence that systemically administered MLA plays a greater role than DHβE in altering attentional processes (Hahn et al., 2011).
4.3. Effects of Nicotinic Antagonists and Glucose on CREB Activation
Intrahippocampal injections of MLA or DHβE reduced pCREB levels in area CA1 but not the dentate gyrus, perhaps because of insufficient diffusion of the drugs to this hippocampal region. Co-administration of glucose reversed DHβE- but not MLA-induced pCREB deficits in area CA1, paralleling the behavioral results and providing converging evidence for the effects of these antagonists on memory processes. The ability of glucose to reverse DHβE-induced deficits in pCREB activation supports prior work showing that peripheral glucose or intrahippocampal lactate administration modulates hippocampal CREB phosphorylation in the context of memory enhancement (Morris and Gold, 2012b; Suzuki et al., 2011). Glucose infusions into area CA1 have also been shown to activate components of the mTOR pathway while improving memory (Dash et al., 2006).
Glucose did not attenuate MLA-induced deficits in pCREB activation in area CA1, and levels of pCREB in MLA-injected rats were correlated with individual differences in alternation performance. These results support the hypothesis that α7 signaling pathways involving CREB activation may be important to the memory-enhancing effects of glucose. Previous studies have shown that chronic changes in CREB levels were correlated with spatial working memory performance in the radial arm maze (Rendeiro et al., 2012; Williams et al., 2008). However, based on the time of drug administration in the present study, it is unlikely that the acute changes in CREB phosphorylation could initiate transcriptional events quickly enough to alter behavioral performance. More likely, CREB activation here serves as a good marker of upstream molecular processes that may play a more direct role in modulating spatial working memory processes.
4.4. Conclusions
The present results indicate that glucose reverses deficits in memory and CREB phosphorylation produced by the α4β2 receptor antagonist DHβE but not the α7 receptor antagonist MLA. These findings support the view that memory enhancement by glucose involves interactions with certain nicotinic receptor subtypes more than others. The ineffectiveness of glucose at reversing MLA-induced deficits in both behavioral and molecular measures suggests that the memory-enhancing effects of glucose may involve its ability to activate downstream α7 receptor signaling. Although α4β2 receptors appear to be less important in this regard, the current results do not rule out the possibility that other acetylcholine receptor subtypes (e.g. muscarinic receptors) or neurotransmitter systems may also play a role. Given the central function of glucose in brain metabolism, it seems likely that glucose modulates cognition by multiple mechanisms, perhaps dependent on the interaction of brain area, memory task, and other variables.
Highlights.
The α4β2 nicotinic receptor antagonist DHβE impaired memory and CREB activation.
The α7 nicotinic receptor antagonist MLA also impaired memory and CREB activation.
Glucose reversed deficits induced by DHβE but not those induced by MLA.
Thus, the memory-enhancing effects of glucose may rely on α7 receptor activation.
Acknowledgments
Supported by an NIH NRSA F30AG034803 (KAM), by NIH grants AG07648 and NSF grants IOS-08-43175 and IOS 10-52464, and by an award from the Alzheimer’s Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addy NA, Nakijama A, Levin ED. Nicotinic mechanisms of memory: effects of acute local DHbetaE and MLA infusions in the basolateral amygdala. Brain Res Cogn Brain Res. 2003;16:51–57. doi: 10.1016/s0926-6410(02)00209-4. [DOI] [PubMed] [Google Scholar]
- Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX. Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci. 1997;9:2734–2742. doi: 10.1111/j.1460-9568.1997.tb01702.x. [DOI] [PubMed] [Google Scholar]
- Benito E, Barco A. CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230–240. doi: 10.1016/j.tins.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Bettany JH, Levin ED. Ventral hippocampal alpha 7 nicotinic receptor blockade and chronic nicotine effects on memory performance in the radial-arm maze. Pharmacol Biochem Behav. 2001;70:467–474. doi: 10.1016/s0091-3057(01)00643-8. [DOI] [PubMed] [Google Scholar]
- Blanchard JG, Duncan PM. Effect of combinations of insulin, glucose and scopolamine on radial arm maze performance. Pharmacol Biochem Behav. 1997;58:209–214. doi: 10.1016/s0091-3057(97)00064-6. [DOI] [PubMed] [Google Scholar]
- Cannady R, Weir R, Wee B, Gotschlich E, Kolia N, Lau E, Brotherton J, Levin ED. Nicotinic antagonist effects in the mediodorsal thalamic nucleus: regional heterogeneity of nicotinic receptor involvement in cognitive function. Biochem Pharmacol. 2009;78:788–794. doi: 10.1016/j.bcp.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Castro NG, Albuquerque EX. alpha-Bungarotoxin-sensitive hippocampal nicotinic receptor channel has a high calcium permeability. Biophys J. 1995;68:516–524. doi: 10.1016/S0006-3495(95)80213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WK, Wong PT, Sheu FS. Frontal cortical alpha7 and alpha4beta2 nicotinic acetylcholine receptors in working and reference memory. Neuropharmacology. 2007;52:1641–1649. doi: 10.1016/j.neuropharm.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Chang KT, Berg DK. Voltage-gated channels block nicotinic regulation of CREB phosphorylation and gene expression in neurons. Neuron. 2001;32:855–865. doi: 10.1016/s0896-6273(01)00516-5. [DOI] [PubMed] [Google Scholar]
- Chiappetta L, Jarvik ME. Comparison of learning impairment and activity depression produced by two classes of cholinergic blocking agents. Arch Int Pharmacodyn Ther. 1969;179:161–166. [PubMed] [Google Scholar]
- Cincotta SL, Yorek MS, Moschak TM, Lewis SR, Rodefer JS. Selective nicotinic acetylcholine receptor agonists: potential therapies for neuropsychiatric disorders with cognitive dysfunction. Curr Opin Investig Drugs. 2008;9:47–56. [PubMed] [Google Scholar]
- Dash PK, Orsi SA, Moore AN. Spatial memory formation and memory-enhancing effect of glucose involves activation of the tuberous sclerosis complex-Mammalian target of rapamycin pathway. J Neurosci. 2006;26:8048–8056. doi: 10.1523/JNEUROSCI.0671-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JA, Gould TJ. beta2 subunit-containing nicotinic receptors mediate the enhancing effect of nicotine on trace cued fear conditioning in C57BL/6 mice. Psychopharmacology. 2007;190:343–352. doi: 10.1007/s00213-006-0624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JA, Gould TJ. The effects of DHBE and MLA on nicotine-induced enhancement of contextual fear conditioning in C57BL/6 mice. Psychopharmacology. 2006;184:345–352. doi: 10.1007/s00213-005-0047-y. [DOI] [PubMed] [Google Scholar]
- Davis JA, Kenney JW, Gould TJ. Hippocampal alpha4beta2 nicotinic acetylcholine receptor involvement in the enhancing effect of acute nicotine on contextual fear conditioning. J Neurosci. 2007;27:10870–10877. doi: 10.1523/JNEUROSCI.3242-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A. Ultrastructural distribution of the alpha7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci. 2001;21:7993–8003. doi: 10.1523/JNEUROSCI.21-20-07993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick SD, Greenstein S. Differential effects of scopolamine and mecamylamine on passive avoidance behavior. Life Sci I. 1972;11:169–179. doi: 10.1016/0024-3205(72)90258-5. [DOI] [PubMed] [Google Scholar]
- Gold PE. Memory enhancing drugs. In: Eichenbaum H, Byrne J, editors. Memory Systems (vol. 3 of Learning and Memory: A Comprehensive Reference) Elsevier Science; Oxford: 2008. pp. 555–576. [Google Scholar]
- Gold PE. Glucose and age-related changes in memory. Neurobiol Aging. 2005;26 (Suppl 1):60–64. doi: 10.1016/j.neurobiolaging.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Gold PE. Coordination of multiple memory systems. Neurobiol Learn Mem. 2004;80:194–210. doi: 10.1016/j.nlm.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Gold PE, Korol DL. Hormones and memory. In: Koob G, Le Moal M, Thompson RF, editors. Encyclopedia of Behavioral Neuroscience. Vol. 2. Academic Press; Oxford: 2010. pp. 57–64. [Google Scholar]
- Goldberg ME, Sledge K, Hefner M, Robichaud RC. Learning impairment after three classes of agents which modify cholinergic function. Arch Int Pharmacodyn Ther. 1971;193:226–235. [PubMed] [Google Scholar]
- Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74:363–396. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Graef S, Schönknecht P, Sabri O, Hegerl U. Cholinergic receptor subtypes and their role in cognition, emotion, and vigilance control: an overview of preclinical and clinical findings. Psychopharmacology (Berl) 2011;215:205–229. doi: 10.1007/s00213-010-2153-8. [DOI] [PubMed] [Google Scholar]
- Hahn B, Shoaib M, Stolerman IP. Selective nicotinic receptor antagonists: effects on attention and nicotine-induced attentional enhancement. Psychopharmacology (Berl) 2011;217:75–82. doi: 10.1007/s00213-011-2258-8. [DOI] [PubMed] [Google Scholar]
- Hu M, Liu QS, Chang KT, Berg DK. Nicotinic regulation of CREB activation in hippocampal neurons by glutamatergic and nonglutamatergic pathways. Mol Cell Neurosci. 2002;21:616–625. doi: 10.1006/mcne.2002.1202. [DOI] [PubMed] [Google Scholar]
- Jafari MR, Zarrindast MR, Djahanguiri B. Effects of different doses of glucose and insulin on morphine state-dependent memory of passive avoidance in mice. Psychopharmacology (Berl) 2004;175:457–462. doi: 10.1007/s00213-004-1841-7. [DOI] [PubMed] [Google Scholar]
- Kawai H, Zago W, Berg DK. Nicotinic alpha 7 receptor clusters on hippocampal GABAergic neurons: regulation by synaptic activity and neurotrophins. J Neurosci. 2002;22:7903–7912. doi: 10.1523/JNEUROSCI.22-18-07903.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Gould TJ. Modulation of hippocampus-dependent learning and synaptic plasticity by nicotine. Mol Neurobiol. 2008;38:101–121. doi: 10.1007/s12035-008-8037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Raybuck JD, Gould TJ. Nicotinic receptors in the dorsal and ventral hippocampus differentially modulate contextual fear conditioning. Hippocampus. 2012 doi: 10.1002/hipo.22003. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf SR, Baratti CM. Memory-improving actions of glucose: involvement of a central cholinergic muscarinic mechanism. Behav Neural Biol. 1994;62:237–243. doi: 10.1016/s0163-1047(05)80022-6. [DOI] [PubMed] [Google Scholar]
- Kopf SR, Buchholzer ML, Hilgert M, Loffelholz K, Klein J. Glucose plus choline improve passive avoidance behaviour and increase hippocampal acetylcholine release in mice. Neuroscience. 2001;103:365–371. doi: 10.1016/s0306-4522(01)00007-0. [DOI] [PubMed] [Google Scholar]
- Korol DL, Gold PE. Modulation of learning and memory by adrenal and ovarian hormones. In: Kesner RP, Martinez JL, editors. Neurobiology of Learning and Memory. Elsevier Science; New York: 2007. pp. 243–268. [Google Scholar]
- Korol DL, Gold PE. Glucose, memory, and aging. Am J Clin Nutr. 1998;67:764S–771S. doi: 10.1093/ajcn/67.4.764S. [DOI] [PubMed] [Google Scholar]
- Krebs DL, Parent MB. The enhancing effects of hippocampal infusions of glucose are not restricted to spatial working memory. Neurobiol Learn Mem. 2005;83:168–172. doi: 10.1016/j.nlm.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Krebs-Kraft DL, Parent MB. Hippocampal infusions of glucose reverse memory deficits produced by co-infusions of a GABA receptor agonist. Neurobiol Learn Mem. 2008;89:142–152. doi: 10.1016/j.nlm.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Bradley A, Addy N, Sigurani N. Hippocampal alpha 7 and alpha 4 beta 2 nicotinic receptors and working memory. Neuroscience. 2002;109:757–765. doi: 10.1016/s0306-4522(01)00538-3. [DOI] [PubMed] [Google Scholar]
- Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl) 2006;184:523–539. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- McNay EC, Gold PE. Age-related differences in hippocampal extracellular fluid glucose concentration during behavioral testing and following systemic glucose administration. J Gerontol ABiol Sci Med Sci. 2001;56:B66–B71. doi: 10.1093/gerona/56.2.b66. [DOI] [PubMed] [Google Scholar]
- McNay EC, Gold PE. Extracellular glucose concentrations in the rat hippocampus measured by zero-net-flux: effects of microdialysis flow rate, strain, and age. J Neurochem. 1999;72:785–790. doi: 10.1046/j.1471-4159.1999.720785.x. [DOI] [PubMed] [Google Scholar]
- Messier C. Glucose improvement of memory: a review. Eur J Pharmacol. 2004;19:33–57. doi: 10.1016/j.ejphar.2004.02.043. [DOI] [PubMed] [Google Scholar]
- Messier C, Durkin T, Mrabet O, Destrade C. Memory-improving action of glucose: Indirect evidence for a facilitation of hippocampal acetylcholine synthesis. Behav Brain Res. 1990;39:135–43. doi: 10.1016/0166-4328(90)90100-s. [DOI] [PubMed] [Google Scholar]
- Micheau J, Messier C, Jaffard R. Glucose enhancement of scopolamine-induced increase of hippocampal high-affinity choline uptake in mice: Relation to plasma glucose levels. Brain Res. 1995;685:99–104. doi: 10.1016/0006-8993(95)00415-m. [DOI] [PubMed] [Google Scholar]
- Mike A, Castro NG, Albuquerque EX. Choline and acetylcholine have similar kinetic properties of activation and desensitization on the alpha7 nicotinic receptors in rat hippocampal neurons. Brain Res. 2000;882:155–168. doi: 10.1016/s0006-8993(00)02863-8. [DOI] [PubMed] [Google Scholar]
- Morris KA, Chang Q, Mohler EG, Gold PE. Age-related memory impairments due to reduced blood glucose responses to epinephrine. Neurobiol Aging. 2010;31:2136–2145. doi: 10.1016/j.neurobiolaging.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KA, Gold PE. Age-related impairments in memory and in CREB and pCREB expression in hippocampus and amygdala following inhibitory avoidance training. Mech Ageing Dev. 2012a;133:291–299. doi: 10.1016/j.mad.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KA, Gold PE. Epinephrine and glucose modulate training-related CREB phosphorylation in old rats: relationships to age-related memory impairments. 2012b doi: 10.1016/j.exger.2012.11.010. In preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Numakawa T, Ikeuchi T, Hatanaka H. Nicotine-induced phosphorylation of extracellular signal-regulated protein kinase and CREB in PC12h cells. J Neurochem. 2001;79:489–498. doi: 10.1046/j.1471-4159.2001.00602.x. [DOI] [PubMed] [Google Scholar]
- Newman LA, Korol DL, Gold PE. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One. 2011;6:e28427. doi: 10.1371/journal.pone.0028427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Levin ED. Dorsal hippocampal alpha7 and alpha4beta2 nicotinic receptors and memory. Brain Res. 2006;1081:72–78. doi: 10.1016/j.brainres.2006.01.052. [DOI] [PubMed] [Google Scholar]
- Papke RL, Sanberg PR, Shytle RD. Analysis of mecamylamine stereoisomers on human nicotinic receptor subtypes. J Pharmacol Exp Ther. 2001;297:646–656. [PubMed] [Google Scholar]
- Pascual MM, Pastor V, Bernabeu RO. Nicotine-conditioned place preference induced CREB phosphorylation and Fos expression in the adult rat brain. Psychopharmacology (Berl) 2009;207:57–71. doi: 10.1007/s00213-009-1630-4. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 5. Elsevier Academic Press; Amsterdam: 2003. [Google Scholar]
- Pych JC, Kim M, Gold PE. Effects of injections of glucose into the dorsal striatum on learning of place and response mazes. Behav Brain Res. 2006;167:373–378. doi: 10.1016/j.bbr.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Ragozzino ME, Gold PE. Glucose injections into the medial septum reverse the effects of intraseptal morphine infusions on hippocampal acetylcholine output and memory. Neuroscience. 1995;68:981–988. doi: 10.1016/0306-4522(95)00204-v. [DOI] [PubMed] [Google Scholar]
- Ragozzino ME, Gold PE. Glucose effects on mecamylamine-induced memory deficits and decreases in locomotor activity in mice. Behav Neural Biol. 1991;56:271–282. doi: 10.1016/0163-1047(91)90424-o. [DOI] [PubMed] [Google Scholar]
- Ragozzino ME, Arankowsky-Sandoval G, Gold PE. Glucose attenuates the effect of combined muscarinic-nicotinic receptor blockade on spontaneous alternation. Eur J Pharmacol. 1994;256:31–36. doi: 10.1016/0014-2999(94)90612-2. [DOI] [PubMed] [Google Scholar]
- Ragozzino ME, Parker ME, Gold PE. Spontaneous alternation and inhibitory avoidance impairments with morphine injections into the medial septum. Attenuation by glucose administration Brain Res. 1992;597:241–249. doi: 10.1016/0006-8993(92)91480-3. [DOI] [PubMed] [Google Scholar]
- Ragozzino ME, Unick KE, Gold PE. Hippocampal acetylcholine release during memory testing in rats: augmentation by glucose. Proc Natl Acad Sci USA. 1996;93:4693–4698. doi: 10.1073/pnas.93.10.4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragozzino ME, Pal SN, Unick K, Stefani MR, Gold PE. Modulation of hippocampal acetylcholine release and spontaneous alternation scores by intrahippocampal glucose injections. J Neurosci. 1998;18:1595–1601. doi: 10.1523/JNEUROSCI.18-04-01595.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidi-Pour A. ATP-sensitive potassium channels mediate the effects of a peripheral injection of glucose on memory storage in an inhibitory avoidance task. Behav Brain Res. 2001;126:43–48. doi: 10.1016/s0166-4328(01)00242-x. [DOI] [PubMed] [Google Scholar]
- Raybuck JD, Gould TJ. The role of nicotinic acetylcholine receptors in the medial prefrontal cortex and hippocampus in trace fear conditioning. Neurobiol Learn Mem. 2010;94:353–363. doi: 10.1016/j.nlm.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendeiro C, Vauzour D, Kean RJ, Butler LT, Rattray M, Spencer JP, Williams CM. Blueberry supplementation induces spatial memory improvements and region-specific regulation of hippocampal BDNF mRNA expression in young rats. Psychopharmacology (Berl) 2012 doi: 10.1007/s00213-012-2719-8. In Press. [DOI] [PubMed] [Google Scholar]
- Rush DK, Streit K. Memory modulation with peripherally acting cholinergic drugs. Psychopharmacology (Berl) 1992;106:375–382. doi: 10.1007/BF02245421. [DOI] [PubMed] [Google Scholar]
- Salinas JA, Gold PE. Glucose regulation of memory for reward reduction in young and aged rats. Neurobiol Aging. 2005;26:45–52. doi: 10.1016/j.neurobiolaging.2004.02.015. [DOI] [PubMed] [Google Scholar]
- Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JX, Yakel JL. Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol Sin. 2009;30:673–680. doi: 10.1038/aps.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher E, Chen Y, Sharples TJ, Broad LM, Benedetti G, Zwart R, McPhie GI, Pearson KH, Baldwinson T, De Filippi G. Physiological roles of neuronal nicotinic receptor subtypes: new insights on the nicotinic modulation of neurotransmitter release, synaptic transmission and plasticity. Curr Top Med Chem. 2004;4:283–297. doi: 10.2174/1568026043451393. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Stefani MR, Gold PE. Intrahippocampal infusions of K-ATP channel modulators influence spontaneous alternation performance: Relationships to acetylcholine release in the hippocampus. J Neurosci. 2001;21:609–614. doi: 10.1523/JNEUROSCI.21-02-00609.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani MR, Gold PE. Intra-septal injections of glucose and glibenclamide attenuate galanin-induced spontaneous alternation performance deficits in the rat. Brain Res. 1998;813:50–56. doi: 10.1016/s0006-8993(98)00876-2. [DOI] [PubMed] [Google Scholar]
- Stefani MR, Nicholson GM, Gold PE. ATP-sensitive potassium channel blockade enhances spontaneous alternation performance in the rat: A potential mechanism for glucose-mediated memory enhancement. Neuroscience. 1999;93:557–563. doi: 10.1016/s0306-4522(99)00128-1. [DOI] [PubMed] [Google Scholar]
- Stone WS, Croul CE, Gold PE. Attenuation of scopolamine-induced amnesia in mice. Psychopharmacology (Berl) 1988;96:417–420. doi: 10.1007/BF00216073. [DOI] [PubMed] [Google Scholar]
- Stone WS, Rudd RJ, Gold PE. Glucose attenuation of atropine-induced deficits in paradoxical sleep and memory. Brain Res. 1995;694:133–138. doi: 10.1016/0006-8993(95)00810-d. [DOI] [PubMed] [Google Scholar]
- Stone WS, Walser B, Gold SD, Gold PE. Scopolamine- and morphine-induced impairments of spontaneous alternation performance in mice: reversal with glucose and with cholinergic and adrenergic agonists. Behav Neurosci. 1991;105:264–271. doi: 10.1037//0735-7044.105.2.264. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–823. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Wu H, Mahata SK, O’Connor DT. A crucial role for the mitogen-activated protein kinase pathway in nicotinic cholinergic signaling to secretory protein transcription in pheochromocytoma cells. Mol Pharmacol. 1998;54:59–69. doi: 10.1124/mol.54.1.59. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Bertrand D, Marguerat A, Raggenbass M. Comparative distribution of nicotinic receptor subtypes during development, adulthood and aging: an autoradiographic study in the rat brain. Neuroscience. 2004;124:405–420. doi: 10.1016/j.neuroscience.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Uteshev VV, Meyer EM, Papke RL. Regulation of neuronal function by choline and 4OH-GTS-21 through alpha 7 nicotinic receptors. J Neurophysiol. 2003;89:1797–1806. doi: 10.1152/jn.00943.2002. [DOI] [PubMed] [Google Scholar]
- Williams CM, El Mohsen MA, Vauzour D, Rendeiro C, Butler LT, Ellis JA, Whiteman M, Spencer JP. Blueberry-induced changes in spatial working memory correlate with changes in hippocampal CREB phosphorylation and brain-derived neurotrophic factor (BDNF) levels. Free Radic Biol Med. 2008;45:295–305. doi: 10.1016/j.freeradbiomed.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Wonnacott S, Barik J, Dickinson J, Jones IW. Nicotinic receptors modulate transmitter cross talk in the CNS: nicotinic modulation of transmitters. J Mol Neurosci. 2006;30:137–140. doi: 10.1385/JMN:30:1:137. [DOI] [PubMed] [Google Scholar]
- Yin JC, Tully T. CREB and the formation of long-term memory. Curr Opin Neurobiol. 1996;6:264–268. doi: 10.1016/s0959-4388(96)80082-1. [DOI] [PubMed] [Google Scholar]
- Zarei MM, Radcliffe KA, Chen D, Patrick JW, Dani JA. Distributions of nicotinic acetylcholine receptor alpha7 and beta2 subunits on cultured hippocampal neurons. Neuroscience. 1999;88:755–764. doi: 10.1016/s0306-4522(98)00246-2. [DOI] [PubMed] [Google Scholar]