

Abstract
Farnesylation is an important post-translational modification essential for proper localization and function of many proteins. Transfer of the farnesyl group from farnesyl diphosphate (FPP) to proteins is catalyzed by protein farnesyltransferase (FTase). We employed a library of FPP analogues with a range of aryl groups substituting for individual isoprene moieties to examine some of the structural and electronic properties of analogue transfer to peptide catalyzed by FTase. Analysis of steady-state kinetics for modification of peptide substrates revealed that the multiple turnover activity depends on the analogue structure. Analogues where the first isoprene is replaced by a benzyl group and an analogue where each isoprene is replaced by an aryl group are good substrates. In sharp contrast with the steady-state reaction, the single turnover rate constant for dansyl-GCVLS alkylation was found to be the same for all analogues, despite the increased chemical reactivity of the benzyl analogues and the increased steric bulk of other analogues. However, the single turnover rate constant for alkylation does depend on the Ca1a2X peptide sequence. These results suggest that the isoprenoid transition state conformation is preferred over the inactive E•FPP• Ca1a2X ternary complex conformation. Furthermore, these data suggest that the farnesyl binding site in the exit groove may be significantly more selective for the farnesyl diphosphate substrate than the active site binding pocket and therefore might be a useful site for design of novel inhibitors.
Keywords: FPP, substrate analogue, FTase, protein prenylation, enzymology
Numerous membrane associated proteins require posttranslational farnesylation catalyzed by protein farnesyltransferase (FTase) for proper function. FTase catalyzes transfer of a 15-carbon farnesyl group from farnesyl diphosphate 1 (FPP) to a conserved cysteine in the C-terminal Ca1a2X motif of a range of proteins, including the oncoprotein H-Ras (“C” refers to the cysteine, “a” to any aliphatic amino acid, and “X” to any amino acid).(1–8) The covalently attached isoprenoid increases the protein’s hydrophobicity and promotes membrane localization and enhances protein-protein interactions.(9–11) Clinical development of farnesyltransferase inhibitors (FTIs) as anticancer therapeutics has been hampered by alternative prenylation of some FTase substrates by the related prenyltransferase geranylgeranyl transferase type I (GGTase I) when FTase activity is limited.(12) This has spurred development of alternative FTase-transferable lipids incapable of supporting normal prenyl group functions (Prenyl Function Inhibitors, PFIs).(13–16) Defining the isoprenoid chemical features that affect each step of the transferase reaction mechanism may provide useful insights for developing PFIs.(17)
The FTase kinetic mechanism is complex (Figure 1) and the enzyme rarely exists in the free, unbound form during the catalytic cycle. The FTase kinetic mechanism is thought to be functionally ordered and efficient catalysis occurs when FPP first binds to the enzyme forming the enzyme•FPP complex (E•FPP) followed by Ca1a2X substrate association where the active site zinc ion directly coordinates the cysteine thiolate to form a ternary complex (E•FPP•Ca1a2X) that is inactive based on the crystal structure. (18–21)
Figure 1.
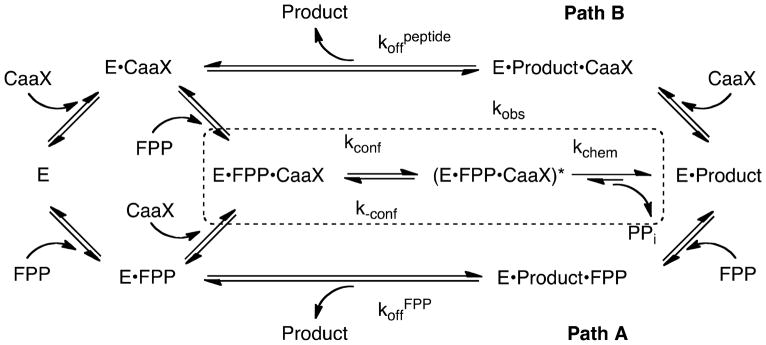
Basic kinetic pathway for FTase. Path A refers to FPP stimulated product release and Path B refers to Ca1a2X peptide stimulated product release. The dashed box encloses rate constants included in the observed single-turnover rate constant kobs.
Models based on structural, mutagenesis and computational studies have been proposed where a conformational rearrangement of the first two FPP isoprene units translocates the reactive isoprenoid C1 5.4 Å across the active site into reacting distance (2.4 Å) of the thiolate to form an active substrate conformation (E•FPP•Ca1a2X)*.(22–24) A variety of experiments suggest that farnesyl transfer to Ca1a2X thiol proceeds by a nucleophilic (SN2, associative) mechanism with electrophilic (SN1, dissociative) character, proceeding through a highly polar “exploded” transition state with considerable (C-1)-O bond cleavage and modest (C1)-S bond formation, partial positive charge on C-1, and partial negative charges on the zinc-coordinated sulfur and on the Mg2+ coordinated diphosphate leaving group (Figure 2).(25–27) Recent computational studies suggest that the transition state structure may depend on the structure of the peptide substrate.(28) The final step in the reaction cycle is product release which is stimulated by binding of either a Ca1a2X peptide (path B) or FPP (path A) to form either the E•peptide or E•FPP complex.(29) The product release pathway depends on the sequence of the peptide.(22, 23, 30)
Figure 2.

FTase catalyzed peptide alkylation by FPP. The transition state is stabilized by positive charge delocalization on the allylic and benzylic systems of FPP and the analogues.
Since FPP is capable of binding to the free enzyme, the E• Ca1a2X complex and the E•product complex, a better understanding of the binding interactions between the active site and FPP is needed and is of particular interest for designing PFIs. FPP analogues have been employed to study the FTase mechanism and the interactions between the isoprenoid, enzymes and the Ca1a2X peptide as well as the biological function of the post-translational modification. The reactivity of FPP analogues depends both on the isoprenoid structure and the peptide substrate sequence. (15, 31–33)
We report the synthesis and reactivity of FPP analogues with a range of steric demands and increased intrinsic chemical reactivity to investigate isoprenoid molecular features that contribute to substrate binding, recognition and catalysis by FTase (Figure 3, Table 1). The analogues vary in the size and electronic properties of one, two, or all three isoprene groups. We used analogues that stabilize the car-bocationic character at C1 to further examine the dissociative character of the transition state.
Figure 3.

FPP and analogue structures
Table 1.
Kinetic constants for FTase catalyzed reaction of FPP and analogues with Ca1a2X peptides
| Isoprenoid | Steady-state kinetic parameters for alkylation of dns-GCVLSa | Single turnover rate constant kobs (s−1)b | ||||
|---|---|---|---|---|---|---|
| kcat (s−1) | KM(μM) | kcat/KM (μM−1s−1) | GCVLS | TKCVIM | TKCVIF | |
| 1 (FPP) | 0.29 ± 0.01 | 1.5 ± 0.2 | 192 ± 4 | 3.8 | 6.8 | 0.26 |
| 2 | 0.0021 ± 0.0001 | 0.49 ± 0.08 | 4.3 ± 0.5 | 4.1 | 5.8 | NDc |
| 3 | 0.43 ± 0.01 | 6.0 ± 0.7 | 72 ± 16 | 4.0 | 5.4 | NDc |
| 4 | 0.03 ± 0.002d | NDc | NDc | 3.1 | 4.3 | 0.21 |
| 5 | 0.20 ± 0.01d | NDc | NDc | 3.1 | 4.7 | 0.22 |
| 6 | 0.12 ± 0.01 | 5 ± 2 | 30 ± 10 | 3.7 | 5.0 | NDc |
| 7 | 0.017 ± 0.003 | 0.55 ± 0.09 | 30 ± 5 | 4.0 | 6.0 | 0.28 |
| 8 | 0.05 ± 0.01d | NDc | NDc | 2.8 | 4.1 | 0.24 |
| 9 | 0.22 ± 0.04 | 5 ± 2 | 40 ± 20 | 3.5 | 5.0 | NDc |
| 10 | > 0.05 c | > 15 c | 1.4 ± 0.2 | 4.2 | 6.4 | NDc |
| 11 | 0.0013 ± 0.0001 | 3.1 ± 0.8 | 0.4 ± 0.01 | 4.0 | 6.3 | NDc |
| 12 | NDc,e | NDc | NDc | 3.8 | 6.2 | 0.29 |
| 13 | 0.0031 ± 0.0001 | 0.2 ± 0.1 | 15 ± 0 | 4.0 | 6.4 | NDc |
| 14 | 0.053 ± 0.002 | 0.5 ± 0.1 | 110 ± 20 | 3.9 | 6.0 | NDc |
| 15 | 0.29 ± 0.01 | 1.5 ± 0.2 | 300 ± 100 | 4.0 | 6.2 | NDc |
Steady-state experiments were performed using dansylated peptides and measuring the time dependent change in fluorescence.(44) Final solutions contained 25 nM FTase, 5 μM dns-GCVLS, varying (1–20 μM) FPP/analogue, 50 mM Heppso, pH 7.8, 5 mM MgCl2, and 2 mM TCEP. Assays were conducted at 25 °C.
Single turnover rate constant, measured using MDCC-PBP/PPiase(58) assay on stopped-flow fluorimeter. Measurements were repeated 2 to 3 times with <10% error. Final solutions contained 400 nM FTase, 100 nM FPP/analog, 25 μM peptide, 5 μM MDCC-PBP, 34 units ml−1 PPiase, 50 mM Heppso, pH 7.8, 5 mM MgCl2, and 2 mM TCEP. Assays were conducted at 25 °C.
ND = Not determined.
Steady-state experiments were performed as described in footnote a except that kcat was determined by varying dns-GCVLS (1 – 10 μM) at saturating analogue (20 μM).
Reaction was too slow to measure by the steady-state assay.
We have characterized FTase kinetic properties with these analogues and describe unexpected results regarding their effect on catalysis. We measured single turnover (STO) rate constants to examine the role of intrinsic chemical reactivity and steric bulk on the FPP conformational rearrangement and the chemical step (kobs, Figure 1). Surprisingly, the STO rate constant is the same as FPP for all analogues tested indicating that the FTase-catalyzed peptide alkylation is very tolerant of increased steric bulk in all three isoprene positions and that the rate of peptide alkylation is not limited by the chemical reactivity of the first isoprene in FPP. Within the series tested here, the hydrophobicity of the analogues does not limit the observed rate of thioether formation. However, the rate of peptide alkylation is dependent on the sequence of the C-terminal residue in the Ca1a2X motif. The steady-state data show that while some analogues are fairly good FTase substrates, others have kcat/KMisoprenoid values that are decreased up to 475–fold. These data indicate that a step subsequent to farnesylation, such as product dissociation, is sensitive to the structure of the farnesyl moiety.
EXPERIMENTAL
Materials and Methods
The peptides TKCVIM, GCVLS and dansylated GCVLS were synthesized and purified by high-pressure liquid chromatography to more than 90% purity by Sigma-Genosys (The Woodlands, TX), and the molecular masses of peptides were confirmed by electrospray mass spectrometry. 7-Diethylamino-3-(((2-maleimidyl)ethyl)amino) carbonyl)coumarin (MDCC) was purchased from Molecular Probes (Eugene, OR). Farnesyl diphosphate (FPP), purine nucleoside phosphorylase (PNPase), 7-methylguanosine (MEG), and inorganic pyrophosphatase from bakers’ yeast (PPiase) were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals used were reagent grade.
All synthetic organic reactions except for resin preparation were performed in PTFE tubes using a Quest 210 apparatus manufactured by Argonaut Technologies. All RP-HPLC was performed on an Agilent 1100 HPLC system equipped with a microplate autosampler, diode array and fluorescence detector. N-dansyl-GCVLS was purchased from Peptidogenics (San Jose, CA, USA). Spectrofluorometric analyses were performed in 96-well flat bottom, non-binding surface, black polystyrene plates (Corning, Excitation wavelength, 340 nm; emission wavelength 505 nm with a 10 nm cutoff) with a SpectraMax GEMINI XPS fluorescence well-plate reader. Absorbance readings were determined using a Cary UV/Vis spectrophotometer. All assays were performed at minimum in triplicate where the average values are reported with a one standard of deviation error. Reaction temperature refers to the external bath. All solvents and reagents were purchased from VWR (EM Science-Omnisolv high purity) and Aldrich respectively and used as received. Merrifield-Cl resin was purchased from Argonaut technologies. Synthetic products were purified by silica gel flash chromatography (EtOAc/hexane) unless otherwise noted. RP-HPLC purification of lipid diphosphates was carried out using a Varian Dynamax, 10 μm, 300 Å, C-18 (10mm × 250 mm) column and eluted with a gradient mobile phase and flow rate of 4 mL/min: 0–3 min, 90% A; 3–18 min, 0% A; 18–20 min, 0% A; 20–23 min, 90% A; and monitored at 254 nm & 210 nm. 1H NMR and 13C NMR spectra of alcohols were obtained in CDCl3 and 1H and 31P of diphosphates in D2O with a Varian Inova spectrometer operating at 400 MHz (1H) and 161.8 MHz (31P). Chemical shifts are reported in ppm from CDCl3 internal peak at 7.27 ppm for 1H; D2O (TSP, 0 ppm for 1H; H3PO4 as an external reference, 0 ppm for 31P). ESI-MS were performed at the University of Kentucky Mass Spectra Facility. Positive and negative ion electrospray ionization (ESI) mass spectra were obtained on a Thermofinnigan LCQ with sample introduction by direct infusion.
General procedure for Mitsunobu coupling (18a–b, 22a–c, 25a–g, 29a–b)
DEAD (40% in toluene, 1.2 equiv.) was added drop wise to a stirred solution of alcohol (1equiv.), phenol (1.2 equiv.), Ph3P (1.2 equiv.) in THF at 0°C and stirred for 1 hr. After allowing the reaction to warm to room temperature and stir overnight, it was diluted with sat. NaHCO3, concentrated, and extracted with CH2Cl2 (2X). The organic extracts were dried (MgSO4), filtered and concentrated. Silica gel column chromatography of the oily residue gave the desired compounds.
(E)-2-((3-methyl-4-(3-phenoxyphenoxy)but-2-en-1-yl)oxy)tetrahydro-2H-pyran 18a
1.0 gm of 16 gave 1.6 gm of 18a (84% yield). 1H NMR (CDCl3) δ 1.43–1.59 (m, 5H), 1.66–1.72 (m, 1H), 1.76 (s, 3H), 1.77–1.85 (m, 1H), 3.47–3.50 (m, 1H), 3.83–3.88 (m, 1H), 4.06–4.12 (m, 1H), 4.26–4.31 (m, 1H), 4.40 (s, 2H), 4.61 (t, J = 3.6 Hz, 1H), 4.75 (s, 2H), 5.74 (t, J = 6.4 Hz, 1H), 6.79–6.81 (m, 2H), 6.88–6.98 (m, 3H), 7.20–7.28 (m, 4H).
(E)-2-((3-methyl-4-(4-phenoxyphenoxy)but-2-en-1-yl)oxy)tetrahydro-2H-pyran 18b
1.0 gm of 16 gave 1.72 gm of 18b (91% yield). 1H NMR (CDCl3) δ 1.48–1.60 (m, 4H), 1.67–1.74 (m, 1H), 1.76 (s, 3H), 1.79–1.84 (m, 1H), 3.46–3.51 (m, 1H), 3.83–3.88 (m, 1H), 4.07–4.12 (m, 1H), 4.20–4.32 (m, 1H), 4.38 (s, 2H), 4.60 (t, J = 3.2 Hz, 1H), 5.72–5.75 (m, 1H), 6.84–6.95(m, 6H), 6.99–7.03 (m, 1H).
(E)-3-((2-methyl-4-((tetrahydro-2H-pyran-2-yl)oxy)but-2-en-1-yl)oxy)benzaldehyde, 22a
500 mg of 16 gave 630 mg of 22a (81% yield). 1H NMR (CDCl3) δ 1.46–1.60 (m, 4H), 1.60 –1.72 (m, 1H), 1.76 (s, 3H), 1.77–1.81 (m, 1H), 3.45–3.51 (m, 1H), 3.82–3.87 (m, 1H), 4.06–4.11 (m, 1H), 4.26–4.31 (m, 1H), 4.45 (s, 2H), 4.60 (t, J = 6.8 Hz, 1H), 5.73–5.77 (m, 1H), 7.15–7.18 (m, 1H), 7.36–7.37 (m, 1H), 7.40–7.44 (m, 2H), 9.79 (s, 1H).
(E)-4-((2-methyl-4-((tetrahydro-2H-pyran-2-yl)oxy)but-2-en-1-yl)oxy)benzaldehyde 22b
500 mg of 16 gave 619 mg of 22b (69% yield). 1H NMR (CDCl3) δ 1.42–1.55 (m, 7H), 1.57–1.70 (m, 1H), 1.75 (s, 3H), 1.77–1.84 (m, 1H), 3.44–3.49 (m, 1H), 3.80–3.86 (m, 1H), 4.08–4.18 (m, 3H), 4.24–4.29 (m, 1H), 4.54 (s, 2H), 4.57 (t, J = 7.6 Hz, 1H), 5.71–5.75 (m, 1H), 6.91–7.00 (m, 1H), 7.34–7.38 (m, 2H), 9.79 (s, 1H).
(E)-4-ethoxy-3-((2-methyl-4-((tetrahydro-2H-pyran-2-yl)oxy)but-2-en-1-yl)oxy)benzaldehyde 22c
500 mg of 16 gave 690 mg of 22c (85% yield). 1H NMR (CDCl3) δ 1.46–1.60 (m, 4H), 1.61–1.72 (m, 1H), 1.76 (s, 3H), 1.77–1.84 (m, 1H), 3.45–3.51 (m, 1H), 3.81–3.87 (m, 1H), 4.05–4.10 (m, 1H), 4.27–4.31(m, 1H), 4.47 (s, 2H), 4.59 (t, J = 3.6 Hz, 1H), 5.72–5.76 (m, 1H), 6.98 (d, J = 8.8 Hz, 2H), 7.79 (d, J = 8.8 Hz, 2H), 9.84 (s, 1H).
(E)-2-((4-(4-((3-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-yl)oxy)tetrahydro-2H-pyran 25a
100 mg of 23b gave 104 mg of 25a (82.5% yield). 1H NMR (CDCl3) δ 1.40–1.70 (m, 5H), 1.77–1.81 (m, 1H), 1.78 (s, 3H), 3.47–3.50 (m, 1H), 3.83–8.88 (m, 1H), 4.08–4.12 (m, 1H), 4.30 (m, 1H), 4.41 (s, 2H), 4.61 (t, J = 3.6 Hz, 2H), 4.92 (s, 2H), 5.73 (t, J = 6.0 Hz, 1H), 6.90 (d, J = 8.8 Hz, 2H), 6.95–7.10 (m, 5H), 7.15–7.20(2H).
(E)-2-((3-methyl-4-(3-(phenoxymethyl)phenoxy)but-2-en-1-yl)oxy)tetrahydro-2H-pyran 25b
100 mg of 23a gave 117 mg of 25b (87% yield). 1H NMR (CDCl3) δ 1.49–1.59 (m, 5H), 1.72 (s, 3H), 1.77–1.85 (m, 1H), 3.46–3.52 (m, 1H), 3.83–3.88 (m, 1H), 4.06–4.12 (m, 1H), 4.26–4.31 (m, 1H), 4.40 (s, 2H), 4.60 (t, J = 4.0 Hz, 1H), 5.00 (s, 2H), 5.72–5.75 (m, 1H), 6.79–6.85 (m, 1H), 6.90–6.98 (m, 4H), 7.20–7.28 (m, 4H).
(E)-2-((4-(3-((4-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-yl)oxy)tetrahydro-2H-pyran 25c
100 mg of 23a gave 117 mg of 25c. (82% yield). 1H NMR (CDCl3) δ 1.48–1.69 (m, 5H), 1.77–1.81(m, 1H), 1.78 (s, 3H), 3.47–3.50 (m, 1H), 3.83–3.88 (m, 1H), 4.08–4.11 (m, 1H), 4.28 (dd, J = 6.4, 12.4 Hz, 1H), 4.41 (s, 2H), 4.60 (t, J = 3.6 Hz, 2H), 4.93 (s, 2H), 5.73 (t, J = 6.0 Hz, 1H), 6.90 (d, J = 8.8 Hz, 2H), 7.14–7.18 (m, 3H), 7.30 (d, J = 8.8 Hz, 2H).
(E)-2-((4-(3-((3-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-yl)oxy)tetrahydro-2H-pyran 25d
100 mg of 23a gave 117 mg of 25d. (81% yield). 1H NMR (CDCl3) δ 1.48–1.69 (m, 5H), 1.77–1.81(m, 1H), 1.76 (s, 3H), 3.46–3.51 (m, 1H), 3.83–3.88 (m, 1H), 4.06–4.11 (m, 1H), 4.26–4.31 (m, 1H), 4.40 (s, 2H), 4.60 (t, J = 4.0 Hz, 2H), 4.97 (s, 2H), 5.72–5.75 (m, 1H), 6.72–6.75 (m, 1H), 6.83–6.97 (m, 6H), 7.24–7.27 (m, 1H).
(E)-2-((4-(2-ethoxy-5-(phenoxymethyl)phenoxy)-3-methylbut-2-en-1-yl)oxy)tetrahydro-2H-pyran 25e
100 mg of 23c gave 77 mg of 25e. (77% yield). 1H NMR (CDCl3) δ 1.41 (t, J = 7.2 Hz, 3H), 1.48–1.69 (m, 5H), 1.76 (s, 3H), 1.77–1.81(m, 1H), 3.46–3.51 (m, 1H), 3.82–3.88 (m, 1H), 4.06–4.11 (m, 1H), 4.26 (dd, J = 6.4, 12.4 Hz, 1H), 4.46 (s, 2H), 4.58 (t, J=2.8 Hz, 1H), 4.93 (s, 2H), 5.70–5.73 (m, 1H), 6.84–6.96 (m, 6H), 7.23–7.28 (m, 2H).
(E)-2-((4-(2-ethoxy-5-((2-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-yl)oxy)tetrahydro-2H-pyran 25f
100 mg of 23c gave 120 mg of 25f (80% yield). 1H NMR (CDCl3) δ 1.42 (t, J=7.2 Hz, 3H), 1.48–1.69 (m, 5H), 1.76 (s, 3H), 1.77–1.81(m, 1H), 3.46 (dd, J = 4.4, 10.4 Hz, 1H), 3.82–3.87 (m, 1H), 4.05–4.12 (m, 3H), 4.25 (dd, J = 6, 12.4 Hz, 1H), 4.46 (s, 2H), 4.58 (t, J = 2.4 Hz, 1H), 5.02 (s, 2H), 5.71 (t, J = 7.6 Hz, 1H), 6.95–7.10 (m, 4H), 6.82–6.90 (m, 3H).
(E)-2-((3-methyl-4-(3-((4-phenoxyphenoxy)methyl)phenoxy)but-2-en-1-yl)oxy)tetrahydro-2H-pyran 25g
100 mg of 23c gave 123 mg of 25g (78% yield). 1H NMR (CDCl3) δ 1.45–1.60 (m, 6H), 1.65–1.75 (m, 2H), 1.78 (s, 3H), 1.79–1.82 (m, 1H), 3.44–3.50 (m, 1H), 3.82–3.90 (m, 1H), 4.06–4.12(m, 1H), 4.26–4.34 (m, 1H), 4.41 (s, 2H), 4.58–4.62 (m, 1H), 4.98 (s, 2H), 5.72–5.78 (m, 1H), 6.78–6.80 (m, 1H), 6.83–7.06 (m, 8H), 7.20–7.30 (m, 4H).
(E)-3-((3,7-dimethylocta-2,6-dien-1-yl)oxy)benzaldehyde 29a
1 gm of geraniol gave 1.6 gm of 29a (95% yield). 1H NMR (CDCl3) δ 1.57 (s, 3H), 1.64 (s, 3H), 1.72 (s, 3H), 2.03–2.11(m, 4H), 4.56 (s, 2H), 4.58 (s, 2H), 5.04–5.08 (m, 1H), 5.44–5.48 (m, 1H), 7.15–7.18 (m, 1H), 7.37–7.42 (m, 3H), 9.94 (s, 1H).
(E)-4-((3,7-dimethylocta-2,6-dien-1-yl)oxy)benzaldehyde 29b
1 gm of geraniol gave 1.6 gm of 29b (78% yield). 1H NMR (CDCl3) δ 1.57 (s, 3H), 1.64 (s, 3H), 1.72 (s, 3H), 2.02–2.14 (m, 4H), 4.60 (d, J = 6.8 Hz, 2H), 5.03–5.06 (m, 1H), 5.43–5.46 (m, 1H), 6.97 (d, J = 8.8 Hz, 2H), 7.80 (d, J = 8.8 Hz, 2H), 9.85 (s, 1H).
General Procedure for THP ether removal
THP ether and 5% w/v PPTS were stirred in dry MeOH overnight. The reaction mixture was evaporated, taken up in ethyl acetate, washed with sat. NaHCO3, brine, dried (MgSO4), filtered and evaporated. Chromatographic purification of the residue gave alcohol in quantitative yield.
(E)-3-methyl-4-(3-phenoxyphenoxy)but-2-en-1-ol 19a
1H NMR (CDCl3) δ 1.75 (s, 3H), 4.25 (d, J = 6.8 Hz, 2H), 4.40 (s, 2H), 5.72–5.77 (m, 1H), 6.55–6.60 (m, 2H), 6.64 (ddd, J = 0.8, 2.4, 8.4 Hz, 1H), 7.00–7.03 (m, 2H), 7.08–7.12 (m, 1H), 7.18–7.22 (m, 1H), 7.30–7.35(m, 2H).
(E)-3-methyl-4-(4-phenoxyphenoxy)but-2-en-1-ol, 19b
1H NMR (CDCl3) δ 1.75 (s, 3H), 4.22(d, J = 6.8 Hz, 2H), 4.36 (s, 2H), 5.72–5.77(m, 1H), 6.55–6.60 (m, 2H), 6.64 (ddd, J = 0.8, 2.4, 8.4 Hz, 1H), 6.85–6.90 (m, 4H), (7.00–7.03 (m, 1H), 7.08–7.12 (m, 1H), 7.30–7.35(m, 2H).
(E)-4-(4-((3-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-ol 26a
1H NMR (CDCl3) δ 1.78 (s, 3H), 4.20 (d, J=5.6 Hz, 2H), 4.42 (s, 2H), 5.05 (s, 2H), 5.78–5.80 (m, 1H), 6.81–6.90 (m, 1H), 6.90–7.10 (m, 5H), 7.28–7.35 (m, 3H).
(E)-3-methyl-4-(3-(phenoxymethyl)phenoxy)but-2-en-1-ol 26b
1H NMR (CDCl3) δ 1.78 (s, 3H), 4.20 (d, J = 5.6 Hz, 2H), 4.42 (s, 2H), 5.05 (s, 2H), 5.78–5.80 (m, 1H), 6.81–6.90 (m, 1H), 6.90–7.10 (m, 5H), 7.28–7.35 (m, 3H).
(E)-4-(3-((4-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-ol 26c
1H NMR (CDCl3) δ 1.54 (s, 3H), 1.76 (brs, 1H), 4.22 (d, J = 5.6 Hz, 2H), 4.40 (s, 2H), 4.97 (s, 2H), 5.74–5.79 (m, 1H), 6.83–7.00 (m, 7H), 7.23–7.27 (m, 1H).
(E)-4-(3-((3-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-ol 26d
1H NMR (CDCl3) δ 1.42 (t, J = 7.2 Hz, 3H), 1.77 (s, 3H), 4.07 (q, J = 6.8, 14 Hz, 2H), 4.21 (t, J = 5.6Hz, 2H), 4.45 (s, 2H), 4.94 (s, 2H), 5.74–5.78 (m, 1H), 6.83–6.96 (m, 6H), 7.23–7.28 (m, 2H).
(E)-4-(2-ethoxy-5-(phenoxymethyl)phenoxy)-3-methylbut-2-en-1-ol 26e
1H NMR (CDCl3) δ 1.41 (t, J = 7.2 Hz, 3H), 1.76 (s, 3H), 4.07(q, J = 6.8, 14 Hz, 2H), 4.20 (t, J = 6.0 Hz, 2H), 4.45 (s, 2H), 5.02 (s, 2H), 5.75–5.78 (m, 1H), 6.82–6.91 (m, 3H), 6.95–7.08 (m, 4H).
(E)-4-(2-ethoxy-5-((2-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-ol 26f
1H NMR (CDCl3) δ 1.40 (t, J =7.2 Hz, 3H), 1.80 (s, 3H), 4.0 (q, J = 6.8, 14 Hz, 2H), 4.10 (t, J = 6.0 Hz, 2H), 4.40 (s, 2H), 5.00 (s, 2H), 5.78 (t, J = 14 Hz, 1H), 6.80–7.05 (m, 7H).
(E)-3-methyl-4-(3-((4-phenoxyphenoxy)methyl)phenoxy)but-2-en-1-ol 26g
1H NMR (CDCl3) δ 1.76 (s, 3H), 4.22 (t, J=5.6 Hz, 2H), 4.42 (s, 2H), 5.00 (s, 2H), 5.75–5.80 (m, 1H), 6.84–6.87 (m, 1H), 6.91–7.04 (m, 9H), 7.23–7.29 (m, 3H).
(3-((3-phenoxyphenoxy)methyl)phenyl)methanol 36
1H NMR (CDCl3) δ 4.70 (s, 2H), 5.02 (s, 2H), 6.90–6.96 (m, 5H), 7.00–7.04 (m, 1H), 7.23–7.25 (m, 2H), 7.36–7.42 (m, 4H).
General procedure for aldehyde reduction
NaBH4 (2equiv.) was added to aldehyde (1equiv.) in EtOH at 0°C and stirred for 3 – 6 h. The mixture was diluted with water and extracted with CH2Cl2 (2X). The organic extracts were dried (MgSO4), filtered and evaporated. The silica gel column chromatographic purification of the oily residue gave the alcohol in quantitative yield.
(E)-(3-((4-((tetrahydro-2H-pyran-2-yl)oxy)but-2-en-1-yl)oxy)phenyl)methanol 23a
1H NMR (CDCl3) δ 1.48–1.57 (m, 5H), 1.62–1.72 (m, 2H), 1.76 (s, 3H), 1.79–1.84 (m, 1H), 3.46–3.51 (m, 1H), 3.82–3.87 (m, 1H), 4.06–4.11 (m, 1H), 4.26–4.31 (m, 1H), 4.41 (s, 2H), 4.59 (t, J = 3.6 Hz, 1H), 4.64 (d, J = 4.4 Hz, 2H), 5.72–5.75 (m, 1H), 6.80–6.92 (m, 1H), 6.90–6.92 (m, 2H), 7.21–7.25 (m, 1H).
(E)-(4-((2-methyl-4-((tetrahydro-2H-pyran-2-yl)oxy)but-2-en-1-yl)oxy)phenyl)methanol 23b
1H NMR (CDCl3) δ 1.40–1.44 (m, 3H), 1.48–1.56 (m, 5H), 1.65–1.71 (m, 1H), 1.76 (s, 3H), 3.45–3.48 (m, 1H), 3.82–3.87 (m, 1H), 4.02–4.13 (m, 2H), 4.24–4.28 (m, 1H), 4.45 (s, 2H), 4.56–4.58 (m, 3H), 5.71 (t, J = 7.2 Hz, 1H), 6.79–6.90 (m, 4H).
(E)-(4-ethoxy-3-((2-methyl-4-((tetrahydro-2H-pyran-2-yl)oxy)but-2-en-1-yl)oxy)phenyl)methanol 23c
1H NMR (CDCl3) δ 1.48–1.56 (m, 5H), 1.68–1.73 (m, 2H), 1.76 (s, 3H), 1.77–1.85 (m, 1H), 3.46–3.51 (m, 1H), 3.83–3.88 (m, 1H), 4.06–4.12 (m, 1H), 4.27–4.32 (m, 1H), 4.40 (s, 2H), 4.59–4.61 (m, 3H), 5.72–5.75 (m, 1H), 6.87 (d, J = 8.8 Hz, 2H), 7.25 (d, J = 8.8 Hz, 2H).
(E)-(3-((3,7-dimethylocta-2,6-dien-1-yl)oxy)phenyl)methanol 30a
1H NMR (CDCl3) δ 1.58 (s, 3H), 1.65 (s, 3H), 1.71 (s, 3H), 2.05–2.09 (m, 4H), 4.52 (d, J = 6.4 Hz, 2H), 4.64 (s, 2H), 5.07 (t, J = 6.8 Hz, 1H), 5.46 (t, J = 8.0 Hz, 1H), 6.81–6.83 (m, 1H), 6.89–6.92 (m, 2H), 7.22–7.26 (m, 1H).
(E)-(4-((3,7-dimethylocta-2,6-dien-1-yl)oxy)phenyl)methanol 30b
1H NMR (CDCl3) δ 1.58 (s, 3H), 1.65 (s, 3H), 1.71 (s, 3H), 2.04–2.11 (m, 4H), 4.51(d, J = 7.2 Hz, 2H), 4.59 (d, J = 6.0 Hz, 2H), 5.05–5.08 (m, 1H), 5.44–5.48 (m, 1H), 6.88 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 8.8 Hz, 2H).
General procedure for diphosphate synthesis
Ph3PCl2 (2 equiv.) in dry CH3CN was added dropwise to a cooled (0°C) solution of the alcohol (1 equiv.) in CH3CN (5 mL) and stirred for 2 h. The reaction mixture was concentrated and filtered through a pad of silica gel. The chlorides were sufficiently pure to proceed to the next step. ((n-Bu)4N)3HP2O7 (5 equiv.) in CH3CN was then added to the solution of chloride in CH3CN at 0°C and the solution was allowed to warm up to room temperature and stirred over-night. The reaction mixture was concentrated and washed with Et2O. The organic extracts were discarded and the residue suspended in 4 mL ion exchange buffer (25 mM NH4HCO3 in 2% (v/v) i-PrOH/water). The resultant white solution was loaded onto a preequlibrated 6×50 cm column of Dowex AG 50W-X8 (100–200 mesh) cation-exchange resin (NH4+ form). The flask was washed with buffer (2 × 2 mL) and loaded onto the column before eluting with 150 mL of ion exchange buffer. The eluent was lyophilized to yield a white solid. This solid was dissolved in 25 mM solution of NH4HCO3 buffer (4 mL), purified by RP-HPLC (retention time about 7 min) and lyophilized to give the diphosphate as a white powder.
(2E,6E)-3,7-dimethyl-8-(2,3,5,6-tetrafluorophenoxy)octa-2,6-dien-1-diphosphate 3
(26% in two steps from the Mitsunobu reaction product).(14) 1H NMR (D2O) δ 1.50 (s, 3H), 1.60 (s, 3H), 1.83–1.87 (m, 2H), 1.98–2.04 (m, 2H), 4.27 (t, J = 6.4Hz, 2H), 4.49 (s, 2H), 5.23 (t, J = 7.6Hz, 1H), 5.36 (t, J = 6.8Hz, 1H), 6.87–6.95 (m, 1H). 31P (D2O) δ −5.90 (d, J = 22Hz, 1P), −9.49 (d, J = 22Hz, 1P). LRMS(EI) (M+-H+) 477 (M+) 478
(E)-3-methyl-4-(3-phenoxyphenoxy)but-2-en-1-diphosphate 4
100 mg of 19a gave 42.7 mg of 4, (23% yield). δ 1H NMR (D2O) δ 1.63 (s, 3H), 4.40 (s, 2H), 4.64 (s, 2H), 5.63–5.69 (m, 1H), 6.85–6.89 (m, 1H), 6.97–7.01 (m, 1H), 7.21–7.25 (m, 2H). 31P (D2O) δ −9.7 (d, J = 22 Hz, 1P), 8.76 (d, J = 22 Hz, 1P). LRMS(EI) (M+-H+) 429 (M+) 430.
(E)-3-methyl-4-(4-phenoxyphenoxy)but-2-en-1-diphosphate 5
100 mg of 19b gave 53 mg of 5 (28% yield). 1H NMR (D2O) δ 1.58 (s, 3H), 4.35 (bs, 2H), 4.62 (2H, s), 5.58 (t, J = 5.6Hz, 1H), 6.50–6.54 (m, 2H), 6.65 (ddd, J = 0.8, 2.4, 8.4Hz, 1H), 6.91–6.94 (m, 2H), 7.03–7.07 (m, 1H), 7.14–7.18 (m, 1H), 7.25–7.30 (m, 2H). 31P (D2O) δ −9.7 (d, J = 20.8 Hz, 1P), −8.51(d, J = 20.8 Hz, 1P). LRMS(EI) (M+-H+) 429 (M+) 430.
(E)-4-(4-((3-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-diphosphate 6
50 mg of 26a gave 20mg 6 (22% yield). 1H NMR (D2O) δ 1.59 (s, 3H), 4.35 (t, J = 7.2 Hz, 2H), 4.40 (s, 2H), 5.00 (s, 2H), 5.61 (t, J = 6.8 Hz, 1H), 6.83–7.00 (m, 6H), 7.17–7.21 (m, 3H). 31P (D2O) δ −9.35 (d, J = 22.0 Hz, 1P), −9.82 (d, J = 22.0 Hz, 1P). LRMS(EI) (M+-H+) 429 (M+) 430.
(E)-3-methyl-4-(3-(phenoxymethyl)phenoxy)but-2-en-1-diphosphate 7
50 mg of 26b gave 24mg of 7 (28% yield). 1H NMR (D2O) δ 1.58 (s, 3H), 4.33 (t, J = 5.6Hz, 2H), 4.35 (s, 2H), 4.94 (s, 2H), 5.61 (t, J = 6.4 Hz, 1H), 6.80–6.92 (m, 2H), 7.15–7.19 (m, 2H). 31P (D2O) δ −5.8 (d, J = 20.0 Hz, 1P), −9.39 (d, J = 20.0 Hz, 1P). LRMS(EI) (M+-H+) 443 (M+) 444.
(E)-4-(3-((4-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-diphosphate 8
50 mg of 26c gave 21mg of 8 (25% yield). 1H NMR (D2O) δ 1.60 (s, 3H), 4.37 (t, J = 6.8 Hz, 1H), 4.40 (s, 2H), 5.0 (s, 2H), 5.63 (t, J = 7.2 Hz, 1H), 6.84 – 7.14 (m, 7H), 7.18–7.22 (m, 2H). 31P (D2O) δ −7.48 (d, J = 20.0 Hz, 1P), −9.55 (d, J = 20.0 Hz, 1P). LRMS(EI) (M+-H+) 461 (M+) 462.
(E)-4-(3-((3-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-diphosphate 9
50mg of 26d gave 22mg of 9 (28% yield). 1H NMR (D2O) δ 1.58 (s, 3H), 4.33–4.35 (m, 4H), 4.94 (s, 2H), 5.61 (t, J = 6.0Hz, 1H), 6.80–6.92 (m, 5H), 7.15–7.19 (m, 3H). 31P (D2O) δ −5.74 (d, J = 19.6 Hz, 1P), −9.40 (d, J = 19.6 Hz, 1P). LRMS(EI) (M+-H+) 461 (M+) 462.
(E)-4-(2-ethoxy-5-(phenoxymethyl)phenoxy)-3-methylbut-2-en-1-diphosphate 10
50 mg of 26e gave 31mg of 10 (36% yield) 1H NMR (D2O) δ 1.15 (t, J = 7.2 Hz, 3H), 1.56 (s, 3H), 3.92 (q, 6.8, 14 Hz, 2H), 4.33 (t, J = 6.4 Hz, 2H), 4.37 (s, 2H), 4.86 (s, 2H), 5.56 (t, J = 6.0 Hz, 1H), 6.82–6.93 (m, 6H), 7.13–7.17 (m, 2H). 31P (D2O) δ −7.03 (d, J = 19.5 Hz, 1P), −9.39 (d, J = 19.5 Hz, 1P). LRMS(EI) (M+-H+) 487 (M+) 488.
(E)-4-(2-ethoxy-5-((2-fluorophenoxy)methyl)phenoxy)-3-methylbut-2-en-1-diphosphate 11
50 mg of 26f gave 18mg of 11 (22% yield). 1H NMR (D2O) δ 1.16 (t, J = 7.2 Hz, 3H), 1.57 (s, 3H), 3.91 (q, J=6.8, 14.0 Hz, 3H), 4.33 (t, J = 6.4 Hz, 2H), 4.37 (s, 2H), 4.86 (s, 2H), 5.56 (t, J = 6.4 Hz, 1H), 6.82–6.85 (m, 5H), 6.93 (s, 1H), 7.13–7.17 (m, 2H). 31P (D2O) δ −7.03 (brs, 1P), −9.59 (brs, 1P). LRMS(EI) (M+-H+) 505 (M+) 506.
(E)-3-methyl-4-(3-((4-phenoxyphenoxy)methyl)phenoxy)but-2-en-1-diphosphate 12
50mg of 26g gave 21mg of 12 (26% yield). 1H NMR (CDCl3) δ 1.20 (t, J = 7.6 Hz, 3), 1.60 (s, 3H), 3.93 (q, J = 7.2, 14 Hz, 2H), 4.37 (t, J = 6.4 Hz, 2H), 4.40 (s, 2H), 4.94 (s, 2H), 5.59 (t, J = 6.0 Hz, 1H), 6.80–6.84 (m, 3H), 6.92–7.10 (m, 4H). 31P (D2O) δ −9.59 (brs, 1P), −7.81 (brs, 1P). LRMS(EI) (M+-H+) 535 (M+) 536.
(E)-(3-((3,7-dimethylocta-2,6-dien-1-yl)oxy)phenyl)methanyl-diphosphate 13
100 mg of 30a gave 34.5mg of 13 (19% yield). 1H NMR (D2O) δ 1.42 (s, 3H), 1.47(s, 3H), 1.93–1.99 (m, 4H), 4.47 (d, J = 6.8, 2H), 5.31 (t, J = 6.0 Hz, 1H), 4.97 (t, J = 6.4 Hz, 1H), 6.80 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 8.8 Hz, 2H). 31P (D2O) δ −10.02 (d, J = 22.0 Hz, 1P), −8.00 (d, J = 22.0 Hz, 1P). LRMS(EI) (M+-H+) 419 (M+) 420.
(E)-(4-((3,7-dimethylocta-2,6-dien-1-yl)oxy)phenyl)methanyl-diphosphate 14
100 mg of 30b gave 32.0 mg of 14 (18% yield). 1H NMR (D2O) δ 1.42 (s, 3H), 1.47 (s, 3H), 1.56 (s, 3H), 1.94–2.02 (m, 4H), 4.49 (d, J = 8.0 Hz, 2H), 4.80 (d, J = 6.0 Hz, 2H), 4.98 (t, J = 7.2Hz, 1H), 5.33 (t, J = 6.0 Hz, 1H), 6.76–6.78 (m, 1H), 6.94–6.96 (m, 2H), 7.16–7.20 (m, 1H). 31P (D2O) δ −9.78 (d, 22.0 Hz, 1P), −5.93 (d, J = 22.0 Hz, 1P). LRMS(EI) (M+-H+) 419 (M+) 420.
(3-((3-phenoxyphenoxy)methyl)phenyl)methanyl-diphosphate 15
50mg of 36 gave 23mg of 15 (27% yield). 1H NMR (D2O) δ 1.73 (s, 3H), 4.97 (s, 2H), 5.82 (d, J = 6.4 Hz, 2H), 4.97 (s, 2H), 6.51–6.53 (m, 2H), 6.68 (dd, J = 2.0, 9.6 Hz, 2H), 6.87–6.89 (m, 2H), 7.05 (t, J = 7.6 Hz, 1H), 7.15–7.34 (m, 7H). 31P (D2O) δ −7.10 (d, J = 22 Hz, 1P), 9.87 (d, J = 22 Hz, 1P). LRMS(EI) (M+-H+) 465 (M+) 466.
2-((3-((3-phenoxyphenoxy)methyl)benzyl)oxy) tetrahydro-2H-pyran 35
To the cooled 0°C solution of m-hydroxyphenol 33 (500 mg, 2.68 mmol) in dry DMF was added 60% sodium hydride in mineral oil (129 mg, 3.2 mmol). The resultant solution was stirred at 0°C for 1hr and 2hr at room temperature. 2-((3-(chloromethyl)benzyl)oxy)tetrahydro-2H-pyran (645 mg, 2.68 mmol) was added dropwise and stirred over night. The reaction mixture was quenched with water, extracted with methylene chloride, and the organic phase washed with water (3X). The combined organic extracts were washed with brine, dried over MgSO4, concentrated and purified by silica gel column chromatography (hexanes, ethyl acetate) to obtain oily residue gave 35 (980 mg 93%). 1H NMR (CDCl3) δ 1.47–1.81 (m, 2H), 1.80–1.88 (m, 1H), 3.49–3.55 (m, 1H), 3.86–3.92 (m, 1H), 4.49 (d, J = 12.0 Hz, 2H), 4.68 (t, J = 3.6 Hz, 1H), 4.78 (d, J = 12.4 Hz, 2H), 5.00 (s, 2H), 5.57–6.62 (m, 2H), 6.70 (dd, J = 2.4, 8.4 Hz, 1H), 6.98–7.01(m, 2H), 7.06–7.10 (m, 1H), 7.17–7.28 (m, 1H), 7.29–7.39 (m, 6H).
Preparation of MDCC-PBP
The purification and labeling of the His6-tagged A197C phosphate binding protein (PBP) with the coumarin fluorophore MDCC was performed as described.(27, 34) The final MDCC-labeled PBP was dialyzed against 50 mM Hepes, pH 7.8, 2 mM TCEP, 0.5 units mL−1 purine nucleoside phosphorylase, and 15 mM 7-methylguanosine (“phosphate mop”) to remove any residual phosphate by forming ribose-1-phosphate. The low molecular weight species of the “Pi mop” are removed by exchanging the buffer to 50 mM Hepes, pH 7.8, and 2 mM TCEP using Amicon Ultra centrifugal filter devices (10,000 MWCO). The purity of the labeled protein was confirmed by SDS-PAGE. Protein concentration and yield are determined by absorbance at 280 nm using a molecular weight of 35276 g mol−1 and a calculated extinction coefficient of 64204 M−1cm−1, and protein stocks were stored at −80 °C.
Preparation of WT FTase
Recombinant rat protein FTase expression and purification were carried out in E. coli BL21(DE3) FPT/pET23a cells as described previously.(23, 35) The purified FTase was determined by SDS-PAGE to be >90% pure. The protein was dialyzed at 4 °C against 50 mM Hepes, pH 7.8, and 2 mM TCEP, and stored at −80 °C. The concentration of FTase was determined by active site titration as previously described. (23)
Single Turnover Kinetics
The single turnover rate constant was determined by measuring the release of diphosphate (PPi) using a fluorescently labeled phosphate binding protein (MDCC-PBP) coupled with PPi cleavage by inorganic pyrophosphatase (PPiase), as described in Pais et al. FTase was preincubated with FPP or analogue for >15 minutes at room temperature, and then rapidly mixed with a peptide solution containing MDCC-PBP and PPiase. The final concentrations used was 800 nM FTase, 200 nM FPP/analogue, 25 μM peptide, 5 μM MDCC-PBP, 34 units mL−1 PPiase, 50 mM Heppso, pH 7.8, 5 mM MgCl2 and 2 mM TCEP. Experiments were conducted at 25 °C using a KinTek model SF-2001 stopped-flow apparatus (KinTek Corporation, Austin, TX) to detect an increase in fluorescence upon binding of inorganic phosphate to MDCC-PBP (λex = 430 nm, λem = 450 nm Corion LL-450-F cutoff filter). The stopped-flow syringes and mixing chamber were preincubated prior to experiments in buffer containing a “Pi mop” (50 mM Heppso, pH 7.8, 5 mM MgCl2, 2 mM TCEP, 0.5 units mL−1 PNPase and 15 μM MEG). At least five kinetic traces were averaged and the single turnover rate constant (kobs) was determined by fitting Eq. 1 to the data, where Fl is the observed fluorescence at <450 nm at time t, ΔFl is the amplitude, and Flmax is the fluorescence endpoint. The single turnover rate constant (kobs) was derived from a reversible two-step kinetic mechanism proceeding from the E•FPP•CaaX complex shown in the dashed box in Figure 1 using equation 1.
| Eq. (1) |
| Eq. (2) |
Steady-state kinetics
The steady-state kinetic parameters kcat, KMisoprenoid, and kcat/KMisoprenoid, were determined from the dependence of the initial velocity on the concentration of FPP or analogue at saturating dansylated peptide (dns-GCVLS) concentration or kcat was determined from the dependence of the initial velocity on the concentration of dns-GCVLS at saturating isoprenoid concentration. The initial velocity was measured from the time-dependent increase in fluorescence intensity (λex = 340 nm, λem = 520 nm) upon farnesylation of dansylated GCVLS, as described previously. Reactions are initiated by the addition of FTase (25 nM final concentration) to solutions containing 5 μM dns-GCVLS, varying (1–20 μM) FPP/analogue, 50 mM Heppso, pH 7.8, 5 mM MgCl2, and 2 mM TCEP at 25 °C. For measurements at fixed isoprenoid concentration, reactions are initiated by the addition of FTase (25 nM final concentration) to solutions containing varying (1–10 μM) dns-GCVLS, varying 20 μM isoprenoid, 50 mM Heppso, pH 7.8, 5 mM MgCl2, and 2 mM TCEP at 25 °C The fluorescence intensity over time is measured for the first 10% of the reaction, using a Polarstar Galaxy fluorescence plate reader (BMG Laboratory Technologies, Durham, NC). The initial velocity of the reaction in fluorescence units s−1 (R) is converted to the velocity of the product formed in μM s−1 (V) using Eq. 3, where P is the concentration of the limiting substrate and Fmax is the amplitude in fluorescence measured from the endpoint of each experiment.
| Eq. (3) |
The values of the steady-state kinetic parameters kcat, KMisoprenoid, and kcat/KMisoprenoid are calculated from a fit of the Michaelis-Menten equation to the initial V versus [S] data.
RESULTS
Design And Synthesis Of Aryl Substituted Isoprenoid Diphosphate
FPP analogues 2–15 (Figure 3) were designed to probe aspects of isoprenoid C1 reactivity, the isoprene conformational rearrangement and the steric constraints of the FTase mechanism. We had previously established that an aniline or phenoxy group is an isostere for the terminal isoprene of FPP and that a range of substituent groups are tolerated by FTase to give transferable analogues under steady state conditions. Analogues 2–15 were designed to test the extent that FPP could be altered and still allow transfer catalyzed by FTase. The chemical reactivity of C1 was increased by replacing the first isoprene with a benzyl moiety in analogues 13, 14 and 15 (Table 1, Schemes 1 & 2). Solvolysis studies suggest benzyl isoprenoids 13, 14 and 15 are expected to be respectively, 5 and 20 times more reactive than the corresponding allylic isoprenoids in SN1 and SN2 type reactions.(36, 37) The terminal isoprene was replaced by an aromatic group in analogues 2–12 and 15 but retained in analogues 13 and 14. The second isoprene in analogues 4–12 and 15 was replaced by a series of substituted phenyl groups. In analogues 4–12, the first isoprene unit was retained and the steric demands and number of rotatable bonds relative to FPP were varied. Analogue 12 is longer than the other FPP analogues, and was designed to mimic the 20-carbon geranylgeranyl diphosphate (GGPP). GGPP is an alternative FTase substrate for some peptide sequences, but is a nano-molar inhibitor of the enzyme with others. (38–40)
Scheme 1.
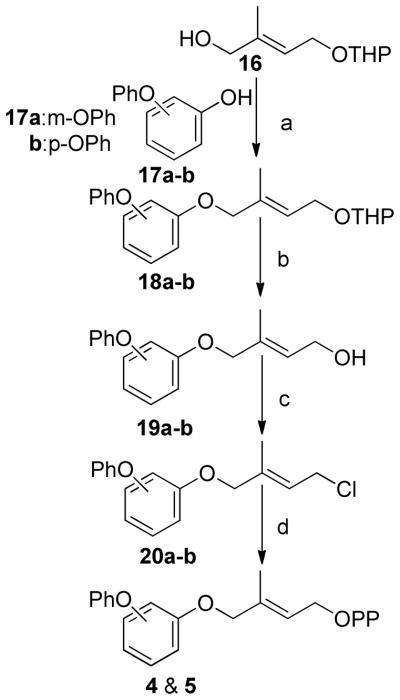
(a) DEAD, Ph3P, THF, (b)PPTS, MeOH, (c) Ph3PCl2, CH3CN, (d) (n-Bu4)3HP2O7, CH3CN
Scheme 2.
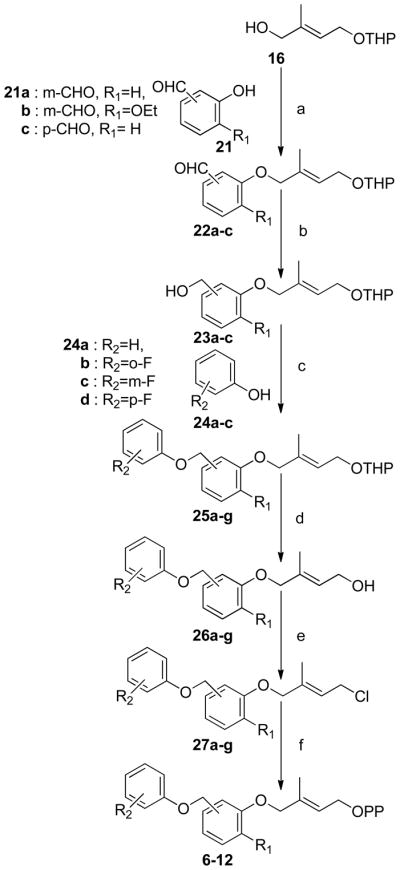
(a) DEAD, Ph3P, THF, (b) NaBH4, EtOH, (c) DEAD, Ph3P, THF, (d) PPTS, MeOH, (e) Ph3PCl2, CH3CN, (f) (n-Bu4)3HP2O7, CH3CN
Analogues 2 and 3 were prepared as previously reported. (14, 41)Analogues 4 and 5 were prepared by Mitsunobu coupling of either m- or p-phenoxyphenol with alcohol 16 to give THP protected isoprenols 18a–b (Scheme 1). Removal of the THP ether with PPTS in methanol afforded alcohols 19a–b which were converted to the corresponding chlorides 20a–b using Ph3PCl2 in acetonitrile.(14) The allylic chlorides were diphosphorylated with (n-Bu4)3HP2O7 to give diphosphates 4 and 5 in moderate yield.
Preparation of FPP analogues 6–12 is shown in Scheme 2 and involved Mitsunobu coupling of alcohol 16 with the appropriate hydroxybenzaldehyde to give aldehydes 22a–c which were reduced with NaBH4 to the corresponding benzyl alcohols 23a–c. A second Mitsunobu reaction with the requisite substituted phenols then afforded THP ethers 25a–g. These protected isoprenoid analogues were converted to the desired diphosphates 6–12 as described above.
Synthesis of FPP analogues 13 and 14 where the first isoprene moiety was replaced by a benzyl group is shown in Scheme 3. Mitsunobu coupling of geraniol with either m- or p-hydroxybenzaldehyde followed by reduction of the aldehydes 29a–b with NaBH4 gave the corresponding alcohols 30a–b. The desired diphosphates 13 and 14 were obtained by conversion of these alcohols as described above.
Scheme 3.

(a) DEAD, Ph3P, THF, (b) NaBH4, EtOH, (c) Ph3PCl2, CH3CN, (d) (n-Bu4)3HP2O7, CH3CN
Synthesis of analogue 15 where all of the isoprene units are replaced by aromatic moieties is shown in Scheme 4. Previously described chloride 32(42) was coupled with the sodium salt of m-phenoxyphenol followed by removal of the THP protecting group, chlorination and diphosphorylation as described above.
Scheme 4.
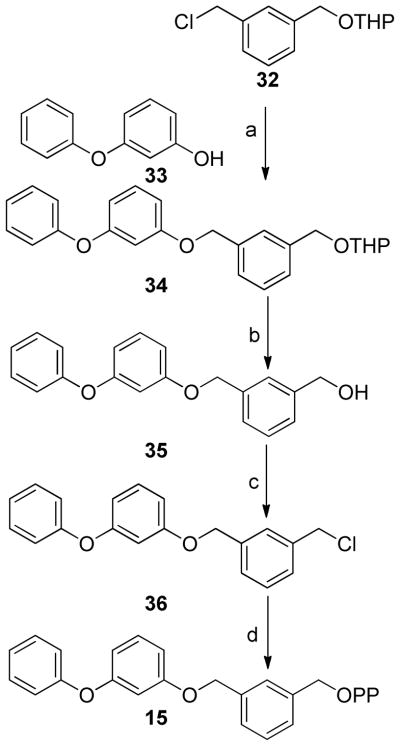
(a) NaH, THF, (b) PPTS, MeOH, (c) Ph3PCl2, CH3CN, (d) (n-Bu4)3HP2O7, CH3CN
FTase-Catalyzed Alkylation of GCVLS Depends On The Isoprenoid Donor Structure
A fluorescent assay was used to determine the effect of the FPP analogue structure on multiple turnover kinetics catalyzed by FTase. The steady-state kinetic parameters kcat for 1–15 and KMisoprenoid of FPP and analogues 2, 3, 6, 7, 9–11, 13–15 with dansylated GCVLS peptide (dns-GCVLS) were measured (Table 1). KMisoprenoid was measured by varying the FPP or analogue concentration in the presence of saturating peptide, and kcat was measured by varying the isoprenoid concentration in the presence of saturating peptide, or by varying the peptide concentration in the presence of saturating analogue.(43) The dns-GCVLS peptide corresponds to the well-characterized Ca1a2X sequence of H-Ras.(21, 39, 44)
FTase catalyzed transfer of the analogues to dns-GCVLS with equal or lower efficiency than FPP, with the exception of the GGPP mimetic 12 for which no turnover was detected. For the FPP analogues where the third isoprene is replaced by substituted aniline groups, the steady state kinetic parameters depend in a complex manner on the size and ring position of the substituents, and the sequence of the Ca1a2X peptide. For example, analogue 2 is a poor donor for modification of the dansyl-GCVLS peptide but is a good substrate with the dansyl-GCVIM peptide. The steady-state rate constant kcat/KMisoprenoid is termed the “specificity constant” for reaction of a peptide with different isoprenoids and can be used to determine how efficiently the FPP analogue is transferred to the peptide.(15, 30, 31) Relative to FPP, the value of kcat/KMisoprenoid for the analogues decreases up to 475-fold. Analysis of the steady-state parameters reveals that the rate constant kcat decreases for the analogues 1.3- to 220-fold relative to FPP, while the value of KMisoprenoid varies from 0.2–9.2 μM, which is within a 7.5-fold difference from FPP (KMisoprenoid = 1.5 μM). Therefore, the decreases in kcat roughly follow the same trends observed for kcat/KMisoprenoid.
The efficiency of the multiple turnover reaction depends on analogue structure. Notably, a meta- or para- linkage geometry of the second isoprene substitution does not appear to alter the the value of kcat/KMisoprenoid for modification of peptides catalyzed by FTase. For example, compounds 9 (meta-) and 6 (para-) have very similar efficiencies. Furthermore, the reactivity of analogues with dns-GCVLS does not depend on having the same number of rotatable bonds as FPP. Analogue 15 has seven rotatable bonds compared with eight in FPP. Clearly, the rotatable bonds in these analogues can adopt conformations sufficient to achieve the reactive ternary complex and the transition state. In contrast, the FTase-catalyzed efficiency of alkylation of peptides with analogues 10 and 11 is severely compromised suggesting that increased steric bulk on the lipid reduces either the peptide affinity or the rate constant for product formation.
Analogues Where The Alpha Isoprene Is Replaced By A Benzyl Group Are Transferable By FTase
Benzyl analogues 13, 14 and 15 are surprisingly good substrates, with values of kcat/KMisoprenoid that are within a factor of three of FPP (Table 1). Remarkably, analogue 15 is almost as good a substrate as FPP for FTase despite having no isoprene units in the structure. Aryl analogue 13 shows that most of the reactivity of FPP is retained by substituting a benzyl group for the first isoprene. This is the first instance where FTase has been shown to catalyze transfer of an isoprenoid with a non-allylic diphosphate to Ca1a2X peptide.
The Single Turnover Rate Constant For GCVLS Alkylation Is The Same For Structurally Diver-gent Isoprenoid Diphosphates
We measured the STO rate constant (kobs) for FTase-catalyzed alkylation of GCVLS by FPP and analogues 2–15 using a fluorescence-based assay that detects the release of pyrophosphate following alkyl transfer under conditions of excess FTase and peptide and limiting isoprenoid diphosphate analogue.(45) The observed rate constant for alkylated peptide product formation (kobs), indicated in Figure 1, measures formation of the E•Product complex from the ternary E•FPP•Peptide complex and includes both the chemical farnesylation step (kchem) and the proposed FPP rotational rearrangement (kconf) to form the active substrate conformation (eq 3).(26, 27, 31) Surprisingly, the observed rate constant under STO conditions (kobs) for all of the analogues is very similar to that measured for FPP (Table 1). The discrepancy between the effects of these analogues on the single turnover and steady-state kinetics suggests that different rate-limiting steps are being measured by the two methods; in particular, product dissociation may contribute significantly to the measured values for the steady-state kinetics.
The Rate Constant For Alkylation Depends On The Ca1a2X Peptide Sequence
To delineate the dependence of the STO rate constants on the peptide structure, the reactivity of analogues 2–15 was also measured for TKCVIM and TKCVIF (Table 1). TKCVIM is the K-Ras Ca1a2X peptide and the STO rate constant for farnesylation is nearly twice as fast (FPP, kobs = 6.5 s−1) as the value for GCVLS (FPP, kobs = 3.4 s−1). These rate constants are in good agreement with those previously reported.(31, 46) TKCVIF is the Ca1a2X peptide from TC21 and is normally a substrate for both FTase and GGTase I. The STO rate constant for farnesylation of TKCVIF catalyzed by FTase is 25-fold slower (FPP, kobs = 0.27 s−1) than the value for TKCVIM. Once again, the STO rate constants for the structurally dissimilar analogues 2–15 were identical to FPP for each of the three peptides examined (Table 1). Notably, the GGPP mimetic 12 reacts as efficiently as FPP with all three peptides even though modification by this analogue under steady state turnover conditions is undetectable. These results indicate that the conformational rearrangement and the farnesylation step are insensitive to the structural differences between FPP and analogues 2–15 (Table 1). Rather, the STO rate constant for peptide alkylation depends on the structure of the peptide, particularly the C-terminal residue in the Ca1a2X motif. The uniformly high reactivity of FPP and analogues 2–15 suggests a facile conformational change in the analogue hydrocarbon chains to achieve the transition state, consistent with recent computational studies. (28) Furthermore, the conformational rearrangement of the isoprenoid diphosphate in the active site to achieve the reactive conformation is independent of the product release step.
DISCUSSION
Increased Isoprenoid Chemical Reactivity Does Not Alter The Rate Constant For Peptide Alkylation
The electronic structure of the first isoprene unit is thought to be critical for FTase catalyzed transfer to peptide. 3-vinyl-FPP is an efficient alternative substrate for FTase and is likely to have increased delocalization of any cationic character in the transition state.(47, 48) The structure of the first isoprenoid is also an important factor in efficient multiple turnovers as both the 2-Z isomer of FPP and 3-allyl-FPP are potent inhibitors of FTase although they are electronically almost identical to FPP. It is unknown whether these two molecules alkylate peptide substrates under STO conditions.(47, 48) The reactivity of C1 in benzyl analogues 13, 14 and 15 is increased relative to FPP. The solvolytic reactivity of p-geranyloxybenzyl diphosphate 13 is higher than the meta substituted 14 and 15 due to enhanced stabilization of the delocalized positive charge that develops on the aromatic ring in the transition state (Figure 2B).(37) The identical STO reactivity for analogues 13–15 indicates that enhanced stabilization of the carbocationic aspects of the transition state does not contribute to an increased observed rate constant for peptide alkylation catalyzed by FTase. These results contrast with the substantial decrease in reactivity that electron withdrawing fluorine substitution at the C3 methyl of FPP have on both steady state turnover and kchem.(26, 27)
Isoprenoid hydrophobicity is uncorrelated with efficiency in multi-turnover reactions,(14) although hydrophobicity of the peptide substrates affects reactivity. (31, 46) The lack of any isoprenoid structural effect for analogues 2–15 on the STO rates suggest that interactions between the isoprenoid and the amino acid residues lining the walls of the active site are not particularly sensitive to the polarity of the chain.
Reaction Steps Involved In FTase Isoprenoid Diphosphate Selectivity
For FTase, the steady-state parameter kcat/KMisoprenoid normally includes the rate constants for reaction steps for formation of the E•FPP•Ca1a2X complex, including peptide binding to E•FPP, and the first irreversible step of diphos-phate dissociation (Figure 1).(30, 31, 45) However, since FPP also enhances product dissociation, the measured value of kcat/KMisoprenoid likely includes these steps as well.(29) In contrast, peptide alkylation catalyzed by FTase under single-turnover conditions (with saturating enzyme) monitors only steps that occur after peptide binding through diphosphate release (kobs, dashed box, Figure 1).(45) Therefore, FTase-catalyzed single-turnover kinetics in the presence of limiting isoprenoid diphosphate isolates the alkyl transfer step (kchem) and presumed isoprenoid conformational change (kconf) from the dissociation of the alkylated peptide (koffCaaX, Figure 1) which is frequently the rate-limiting step for kcat under multi-ple turnover conditions.(18, 26, 49) The lack of any isoprenoid structural effect, yet strong Ca1a2X pep-tide sequence effect on kobs suggests that the chemical step and/or the proposed conformational rear-rangement of the FPP depends on how the Ca1a2X peptide binds to FTase. The clear differences in kobs for the three peptides suggests that subtle differences in the Ca1a2X-FTase binding interactions alter the rate of the proposed isoprenoid conformational rearrangement or alter the chemical reactivity of the thiolate nucleophile.
Increased Isoprenoid Bulk Does Not Alter The Rate Constant For GCVLS Alkylation
Previous measurements demonstrated that the 2° KIE for reaction of FPP with peptides under STO conditions catalyzed by FTase varies with the structure of the peptide; the α-2° 3H KIE observed for TKCVIF is large (1.13 ± 0.01) but decreases to near unity for GCVLS.(27) The original interpretation of this result was that the conformational change became the rate-limiting step for farnesylation of GCVLS under STO conditions. However, recent computational studies from the Merz group suggest that the ΔG‡ for the conformational step is much smaller than that of the alkylation step, even for GCVLS, and that the change in the 2° KIE reflects alterations in the transition state structure due to changes in the peptide binding mode. The similar STO kobs for all of the analogues indicates that substantial conformational freedom to achieve the transition state is available to the lipid in the confines of the active site and that any barriers to achieving the transition state are small. These observations indicate that the decreased steady-state turnover rate for the analogues most likely reflects alterations in the product dissociation step.
The Isoprenoid Structure Mainly Affects Product Release
The decoupling of isoprenoid structure and intrinsic chemical reactivity from the STO rate constant kobs while retaining a profound isoprenoid structural dependence on the rate of product release, kcat, indicates that the alkylation reaction depends mainly on the structure of the peptide(46) while product release depends on the structure of both the peptide and the isoprenoid.(30, 31) Equation 2 demonstrates the contribution of the conformational rearrangement kconf and the chemical step kchem to the STO rate constant kobs. Inspection of equation 2 suggests that either kconf and k-conf are unchanged by the range of structural modifications explored here, or that kchem is sufficiently large to be the rate-determining step for all of the substrates. This latter conclusion is consistent with computational studies suggesting that the barrier to achieving the transition state going from the E•FPP•Ca1a2X complex to the E•FPP•Ca1a2X* complex is on the order of 1 kcal/mol for CVIM to 2.5 kcal/mol for CVLM compared to ~20 kcal/mol for the chemical step. (24, 28, 50)
Mg2+ binding accelerates FTase-catalyzed alkylation of GCVLS and TKCVIF by FPP by 700-fold and 100-fold, respectively. The Mg2+ cofactor may stabilize both the active substrate conformation and the developing charge on the pyrophosphate leaving group.(51–53) The FPP pyrophosphate leaving group in the inactive (E•FPP•Ca1a2X) complex is bound in the FTase PPi binding pocket comprised of positively charged residues.(19, 22, 54–57) Conformational rearrangement of the FPP isoprenoid chain moves the pyrophosphate leaving group out of the PPi binding pocket and creates a Mg2+ binding site that stabilizes the active complex (E•FPP•Ca1a2X*) which is then followed by attack of the zinc bound thiolate nucleophile on C1. For each CaaX peptide, the observed rate constant, kobs, for analogues 2–15 were identical to the natural substrate FPP. The insensitivity of kobs to the increased chemical reactivity of analogues 13–15 indicates that the rate of the chemical step kchem for each of the three peptides was not limited by the intrinsic reactivity of the allylic C1 electrophile. Furthermore, the insensitivity of kobs to the structure of the analogues for all three peptides suggests that there is abundant room in the active site for the proposed conformational rearrangement of the isoprenoid to take place. Consistent with this model, the uniform observed values of kobs for FPP and analogues 2–15 could be accommodated by correlated changes in chemical reactivity that compensate for an isoprenoid structure-dependent decrease in the ratio of the equilibrium constant for the conformational change (kconf/k-conf). However, there is no increase in kobs relative to FPP for the more reactive benzyl analogues 13, 14 and 15 suggesting that kchem depends on the CaaX peptide sequence and does not vary with isoprenoid structure.
Implications For Development Of Alternative FTase Substrates That Block Prenylated Protein Function (PFIs)
This study tested the sensitivity of the chemical step to changes in electronic structure of the lipid electrophile, and the sensitivity of the conformational rearrangement required to achieve the reactive conformation to changes in the lipid structure. The primary conclusions are that (1) increases in chemical reactivity of the lipid electrophile do not increase kobs and (2) increased steric bulk in the isoprene chain does not decrease kobs, but (3) kobs depends on the Ca1a2X X-residue and (4) in contrast, the steady state rate constants for turnover are highly dependent on lipid structure. The ability of FTase to transfer analogues with no isoprene units opens the door to a much wider range of alternative structures as potential PFIs. These results suggest that as long as the chemical reactivity of the analogue C1 is at least as reactive as the allylic group of FPP, efficient S-alkylation is possible and largely unaffected by the chemical structure and bulk of the isoprenoid chain. There must be limits to the size of transferrable lipid moieties, as the FTase pocket has finite dimensions. However, the limits were not reached by the analogues investigated in this study. In contrast to the structure-independent rate of cysteine alkylation, product release was highly dependent on the structure of the analogue. Previously, our lab and others have noted a Ca1a2X sequence dependent interplay in the turnover of FPP analogues.(30, 32) Despite investigation of almost 200 analogues with a wide range of chemical structures, no clear structure-activity relationship has emerged. Broadly, large analogues like 10–12 are inefficiently turned over. Furthermore, these data suggest that the farnesyl binding site in the exit groove may be significantly more selective for the farnesyl diphosphate substrate than the active site binding pocket and therefore might be a useful site for design of novel inhibitors. Alternatively, the conformation of the isoprenoid in the E•product complex may block efficient binding of the new analogue diphosphate in the exit groove, or is inhibited from rearranging to put the alkylated isoprenoid into the exit groove.
Supplementary Material
Acknowledgments
NIH grants R01 GM66152 to HPS and R01 GM40602 to CAF.
ABBREVIATIONS
- FTase
Farnesyltransferase
- AGPP
8-anilinogeranyldiphosphate
- FPP
Farnesyl diphosphate
- TLC
thin layer chromatography
- GGTase I
Geranylgeranyl transferase
- FTI
Farnesyltransferase
- Ca1a2X
tetrapeptide sequence cysteine aliphatic amino acid-aliphatic amino acid-X(serine, glutamine, or methionine)
- GGPP
Geranylgeranyldiphosphate
- DEAD
Diethylazodicarboxylate
- DMF
Dimethylforma-mide
- THF
Tetrahydrofuran
- PPTS
Pyridinium-p-toluene sulfonate
- H-Ras
Harvey-Ras
- K-Ras
Kirsten-Ras
- DTT
Dithiothreitol
- RP-HPLC
Reverse Phase High Performance Liquid Chromatography
- PBP
phosphate binding protein
- kcat
turnover number
- Kmisoprenoid
Michaelis-Menten constant for FPP or analogue
- MDCC
Diethylamino-3-[N-(2-malemidoethyl)carbamoyl] coumarin
- PNPase
Polynucleotide phosphorylase
- TCEP
Tris(2-carboxyethyl)phosphine hydrochloride
Footnotes
Supporting Information. Detailed experimental procedure for synthesis of all new compounds, 1H NMR of 19a–b, 26a–g, 30a–b 35, 1H, 13P and LR mass spectra of 3–15 are available.
References
- 1.Fu HW, Casey PJ. Enzymology and biology of CaaX protein prenylation. Recent progress in hormone research. 1999;54:315–342. discussion 342–313. [PubMed] [Google Scholar]
- 2.Casey PJ, Seabra MC. Protein prenyltransferases. J Biol Chem. 1996;271:5289–5292. doi: 10.1074/jbc.271.10.5289. [DOI] [PubMed] [Google Scholar]
- 3.Adjei AA. An overview of farnesyltransferase inhibitors and their role in lung cancer therapy. Lung cancer. 2003;41(Suppl 1):S55–62. doi: 10.1016/s0169-5002(03)00143-0. [DOI] [PubMed] [Google Scholar]
- 4.Rowinsky EK, Windle JJ, Von Hoff DD. Ras protein farnesyltransferase: A strategic target for anticancer therapeutic development. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1999;17:3631–3652. doi: 10.1200/JCO.1999.17.11.3631. [DOI] [PubMed] [Google Scholar]
- 5.Zhang FL, Casey PJ. Influence of metal ions on substrate binding and catalytic activity of mammalian protein geranylgeranyltransferase type-I. The Biochemical journal. 1996;320(Pt 3):925–932. doi: 10.1042/bj3200925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sousa SF, Fernandes PA, Ramos MJ. Unraveling the mechanism of the farnesyltransferase enzyme. Journal of biological inorganic chemistry: JBIC: a publication of the Society of Biological Inorganic Chemistry. 2005;10:3–10. doi: 10.1007/s00775-004-0612-6. [DOI] [PubMed] [Google Scholar]
- 7.Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K. Lamin B1 is required for mouse development and nuclear integrity. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10428–10433. doi: 10.1073/pnas.0401424101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. 2007;6:541–555. doi: 10.1038/nrd2221. [DOI] [PubMed] [Google Scholar]
- 9.Reents R, Wagner M, Schlummer S, Kuhlmann J, Waldmann H. Synthesis and application of fluorescent ras proteins for live-cell imaging. Chembiochem. 2005;6:86–94. doi: 10.1002/cbic.200400233. [DOI] [PubMed] [Google Scholar]
- 10.Rando RR. Chemical biology of isoprenylation/methylation. Biochemical Society transactions. 1996;24:682–687. doi: 10.1042/bst0240682. [DOI] [PubMed] [Google Scholar]
- 11.Figueroa C, Taylor J, Vojtek AB. Prenylated Rab acceptor protein is a receptor for prenylated small GTPases. J Biol Chem. 2001;276:28219–28225. doi: 10.1074/jbc.M101763200. [DOI] [PubMed] [Google Scholar]
- 12.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, Bishop WR, Pai JK. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 13.Roberts MJ, Troutman JM, Chehade KA, Cha HC, Kao JP, Huang X, Zhan CG, Peterson YK, Subramanian T, Kamalakkannan S, Andres DA, Spielmann HP. Hydrophilic anilinogeranyl diphosphate prenyl analogues are Ras function inhibitors. Biochemistry. 2006;45:15862–15872. doi: 10.1021/bi061704+. [DOI] [PubMed] [Google Scholar]
- 14.Subramanian T, Liu S, Troutman JM, Andres DA, Spielmann HP. Protein farnesyltransferase-catalyzed isoprenoid transfer to peptide depends on lipid size and shape, not hydrophobicity. Chembiochem. 2008;9:2872–2882. doi: 10.1002/cbic.200800248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Troutman JM, Subramanian T, Andres DA, Spielmann HP. Selective modification of CaaX peptides with ortho-substituted anilinogeranyl lipids by protein farnesyl transferase: competitive substrates and potent inhibitors from a library of farnesyl diphosphate analogues. Biochemistry. 2007;46:11310–11321. doi: 10.1021/bi700516m. [DOI] [PubMed] [Google Scholar]
- 16.Placzek AT, Hougland JL, Gibbs RA. Synthesis of frame-shifted farnesyl diphosphate analogs. Org Lett. 2012;14:4038–4041. doi: 10.1021/ol300683r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen UT, Goody RS, Alexandrov K. Understanding and exploiting protein prenyltransferases. Chembiochem. 2010;11:1194–1201. doi: 10.1002/cbic.200900727. [DOI] [PubMed] [Google Scholar]
- 18.Furfine ES, Leban JJ, Landavazo A, Moomaw JF, Casey PJ. Protein farnesyltransferase: kinetics of farnesyl pyrophosphate binding and product release. Biochemistry. 1995;34:6857–6862. doi: 10.1021/bi00020a032. [DOI] [PubMed] [Google Scholar]
- 19.Long SB, Casey PJ, Beese LS. Cocrystal structure of protein farnesyltransferase complexed with a farnesyl diphosphate substrate. Biochemistry. 1998;37:9612–9618. doi: 10.1021/bi980708e. [DOI] [PubMed] [Google Scholar]
- 20.Huang CC, Casey PJ, Fierke CA. Evidence for a catalytic role of zinc in protein farnesyltransferase. Spectroscopy of Co2+-farnesyltransferase indicates metal coordination of the substrate thiolate. J Biol Chem. 1997;272:20–23. doi: 10.1074/jbc.272.1.20. [DOI] [PubMed] [Google Scholar]
- 21.Pompliano DL, Rands E, Schaber MD, Mosser SD, Anthony NJ, Gibbs JB. Steady-state kinetic mechanism of Ras farnesyl:protein transferase. Biochemistry. 1992;31:3800–3807. doi: 10.1021/bi00130a010. [DOI] [PubMed] [Google Scholar]
- 22.Long SB, Casey PJ, Beese LS. Reaction path of protein farnesyltransferase at atomic resolution. Nature. 2002;419:645–650. doi: 10.1038/nature00986. [DOI] [PubMed] [Google Scholar]
- 23.Bowers KE, Fierke CA. Positively charged side chains in protein farnesyltransferase enhance catalysis by stabilizing the formation of the diphosphate leaving group. Biochemistry. 2004;43:5256–5265. doi: 10.1021/bi049822p. [DOI] [PubMed] [Google Scholar]
- 24.Cui G, Merz KM., Jr Computational studies of the farnesyltransferase ternary complex part II: the conformational activation of farnesyldiphosphate. Biochemistry. 2007;46:12375–12381. doi: 10.1021/bi701324t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dolence JM, Poulter CD. A mechanism for posttranslational modifications of proteins by yeast protein farnesyltransferase. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:5008–5011. doi: 10.1073/pnas.92.11.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang C, Hightower KE, Fierke CA. Mechanistic studies of rat protein farnesyltransferase indicate an associative transition state. Biochemistry. 2000;39:2593–2602. doi: 10.1021/bi992356x. [DOI] [PubMed] [Google Scholar]
- 27.Pais JE, Bowers KE, Fierke CA. Measurement of the alpha-secondary kinetic isotope effect for the reaction catalyzed by mammalian protein farnesyltransferase. J Am Chem Soc. 2006;128:15086–15087. doi: 10.1021/ja065838m. [DOI] [PubMed] [Google Scholar]
- 28.Yang Y, Wang B, Ucisik MN, Cui G, Fierke CA, Merz KM., Jr Insights into the mechanistic dichotomy of the protein farnesyltransferase peptide substrates CVIM and CVLS. J Am Chem Soc. 2012;134:820–823. doi: 10.1021/ja209650h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tschantz WR, Furfine ES, Casey PJ. Substrate binding is required for release of product from mammalian protein farnesyltransferase. J Biol Chem. 1997;272:9989–9993. doi: 10.1074/jbc.272.15.9989. [DOI] [PubMed] [Google Scholar]
- 30.Troutman JM, Andres DA, Spielmann HP. Protein farnesyl transferase target selectivity is dependent upon peptide stimulated product release. Biochemistry. 2007;46:11299–11309. doi: 10.1021/bi700513n. [DOI] [PubMed] [Google Scholar]
- 31.Hougland JL, Lamphear CL, Scott SA, Gibbs RA, Fierke CA. Context-dependent substrate recognition by protein farnesyltransferase. Biochemistry. 2009;48:1691–1701. doi: 10.1021/bi801710g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reigard SA, Zahn TJ, Haworth KB, Hicks KA, Fierke CA, Gibbs RA. Interplay of isoprenoid and peptide substrate specificity in protein farnesyltransferase. Biochemistry. 2005;44:11214–11223. doi: 10.1021/bi050725l. [DOI] [PubMed] [Google Scholar]
- 33.Krzysiak AJ, Aditya AV, Hougland JL, Fierke CA, Gibbs RA. Synthesis and screening of a CaaL peptide library versus FTase reveals a surprising number of substrates. Bioorganic & medicinal chemistry letters. 2010;20:767–770. doi: 10.1016/j.bmcl.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirshberg M, Henrick K, Haire LL, Vasisht N, Brune M, Corrie JE, Webb MR. Crystal structure of phosphate binding protein labeled with a coumarin fluorophore, a probe for inorganic phosphate. Biochemistry. 1998;37:10381–10385. doi: 10.1021/bi980428z. [DOI] [PubMed] [Google Scholar]
- 35.Zimmerman KK, Scholten JD, Huang CC, Fierke CA, Hupe DJ. High-level expression of rat farnesyl:protein transferase in Escherichia coli as a translationally coupled heterodimer. Protein Expr Purif. 1998;14:395–402. doi: 10.1006/prep.1998.0979. [DOI] [PubMed] [Google Scholar]
- 36.Mu YQ, Omer CA, Gibbs RA. On the stereochemical course of human protein-farnesyl transferase. J Am Chem Soc. 1996;118:1817–1823. [Google Scholar]
- 37.March J. Advanced Organic Chemistry. 2. McGraw-Hill; 1968. [Google Scholar]
- 38.Shao Y, Eummer JT, Gibbs RA. Stereospecific synthesis and biological evaluation of farnesyl diphosphate isomers. Org Lett. 1999;1:627–630. doi: 10.1021/ol990714i. [DOI] [PubMed] [Google Scholar]
- 39.Chehade KA, Kiegiel K, Isaacs RJ, Pickett JS, Bowers KE, Fierke CA, Andres DA, Spielmann HP. Photoaffinity analogues of farnesyl pyrophosphate transferable by protein farnesyl transferase. J Am Chem Soc. 2002;124:8206–8219. doi: 10.1021/ja0124717. [DOI] [PubMed] [Google Scholar]
- 40.Henriksen BS, Zahn TJ, Evanseck JD, Firestine SM, Gibbs RA. Computational and conformational evaluation of FTase alternative substrates: insight into a novel enzyme binding pocket. Journal of chemical information and modeling. 2005;45:1047–1052. doi: 10.1021/ci0496550. [DOI] [PubMed] [Google Scholar]
- 41.Chehade KA, Andres DA, Morimoto H, Spielmann HP. Design and synthesis of a transferable farnesyl pyrophosphate analogue to Ras by protein farnesyltransferase. The Journal of organic chemistry. 2000;65:3027–3033. doi: 10.1021/jo991735t. [DOI] [PubMed] [Google Scholar]
- 42.Naghipour A, Sabounchei SJ, Morales-Morales D, Hernandez-Ortega S, Jensen CM. Synthesis of a new class of unsymmetrical PCP’ pincer ligands and their palladium(II) complexes: X-ray structure determination of PdCl{C6H3-2-CH2PPh2-6-CH2PBu2t} Journal of Organometallic Chemistry. 2004;689:2494–2502. [Google Scholar]
- 43.Micali E, Chehade KA, Isaacs RJ, Andres DA, Spielmann HP. Protein farnesyltransferase isoprenoid substrate discrimination is dependent on isoprene double bonds and branched methyl groups. Biochemistry. 2001;40:12254–12265. doi: 10.1021/bi011133f. [DOI] [PubMed] [Google Scholar]
- 44.Cassidy PB, Dolence JM, Poulter CD. Continuous fluorescence assay for protein prenyltransferases. Methods in enzymology. 1995;250:30–43. doi: 10.1016/0076-6879(95)50060-x. [DOI] [PubMed] [Google Scholar]
- 45.Pais JE, Bowers KE, Stoddard AK, Fierke CA. A continuous fluorescent assay for protein prenyltransferases measuring diphosphate release. Anal Biochem. 2005;345:302–311. doi: 10.1016/j.ab.2005.07.040. [DOI] [PubMed] [Google Scholar]
- 46.Hartman HL, Hicks KA, Fierke CA. Peptide specificity of protein prenyltransferases is determined mainly by reactivity rather than binding affinity. Biochemistry. 2005;44:15314–15324. doi: 10.1021/bi0509503. [DOI] [PubMed] [Google Scholar]
- 47.Zahn TJ, Whitney J, Weinbaum C, Gibbs RA. Synthesis and evaluation of GGPP geometric isomers: divergent substrate specificities of FTase and GGTase I. Bioorganic & medicinal chemistry letters. 2001;11:1605–1608. doi: 10.1016/s0960-894x(01)00292-x. [DOI] [PubMed] [Google Scholar]
- 48.Rawat DS, Krzysiak AJ, Gibbs RA. Synthesis and biochemical evaluation of 3,7-disubstituted farnesyl diphosphate analogues. The Journal of organic chemistry. 2008;73:1881–1887. doi: 10.1021/jo701725b. [DOI] [PubMed] [Google Scholar]
- 49.Mathis JR, Poulter CD. Yeast protein farnesyltransferase: a pre-steady-state kinetic analysis. Biochemistry. 1997;36:6367–6376. doi: 10.1021/bi9629182. [DOI] [PubMed] [Google Scholar]
- 50.Cui G, Wang B, Merz KM., Jr Computational studies of the farnesyltransferase ternary complex part I: substrate binding. Biochemistry. 2005;44:16513–16523. doi: 10.1021/bi051020m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pickett JS, Bowers KE, Fierke CA. Mutagenesis studies of protein farnesyltransferase implicate aspartate beta 352 as a magnesium ligand. J Biol Chem. 2003;278:51243–51250. doi: 10.1074/jbc.M309226200. [DOI] [PubMed] [Google Scholar]
- 52.Pickett JS, Bowers KE, Hartman HL, Fu HW, Embry AC, Casey PJ, Fierke CA. Kinetic studies of protein farnesyltransferase mutants establish active substrate conformation. Biochemistry. 2003;42:9741–9748. doi: 10.1021/bi0346852. [DOI] [PubMed] [Google Scholar]
- 53.Saderholm MJ, Hightower KE, Fierke CA. Role of metals in the reaction catalyzed by protein farnesyltransferase. Biochemistry. 2000;39:12398–12405. doi: 10.1021/bi0011781. [DOI] [PubMed] [Google Scholar]
- 54.Long SB, Hancock PJ, Kral AM, Hellinga HW, Beese LS. The crystal structure of human protein farnesyltransferase reveals the basis for inhibition by CaaX tetrapeptides and their mimetics. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:12948–12953. doi: 10.1073/pnas.241407898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park HW, Boduluri SR, Moomaw JF, Casey PJ, Beese LS. Crystal structure of protein farnesyltransferase at 2.25 angstrom resolution. Science. 1997;275:1800–1804. doi: 10.1126/science.275.5307.1800. [DOI] [PubMed] [Google Scholar]
- 56.Reid TS, Terry KL, Casey PJ, Beese LS. Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. Journal of molecular biology. 2004;343:417–433. doi: 10.1016/j.jmb.2004.08.056. [DOI] [PubMed] [Google Scholar]
- 57.Strickland CL, Windsor WT, Syto R, Wang L, Bond R, Wu Z, Schwartz J, Le HV, Beese LS, Weber PC. Crystal structure of farnesyl protein transferase complexed with a CaaX peptide and farnesyl diphosphate analogue. Biochemistry. 1998;37:16601–16611. doi: 10.1021/bi981197z. [DOI] [PubMed] [Google Scholar]
- 58.Hougland JL, Fierke CA. Getting a handle on protein prenylation. Nat Chem Biol. 2009;5:197–198. doi: 10.1038/nchembio0409-197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.