

Abstract
In bacteria, P1-type ATPases are responsible for resistance to di- and monovalent toxic heavy metals by taking them out of the cell. These ATPases have a cytoplasmic N terminus comprising metal binding domains defined by a βαββαβ fold and a CXXC metal binding motif. To check how the structural properties of the metal binding site in the N terminus can influence the metal specificity of the ATPase, the first structure of a Cd(II)-ATPase N terminus was determined by NMR and its coordination sphere was investigated by X-ray absorption spectroscopy. A novel metal binding environment was found, comprising the two conserved Cys residues of the metal binding motif and a Glu in loop 5. A bioinformatic search identifies an ensemble of highly homologous sequences presumably with the same function. Another group of highly homologous sequences is found which can be referred to as zinc-detoxifying P1-type ATPases with the metal binding pattern DCXXC in the N terminus. Because no carboxylate groups participate in Cu(I) or Ag(I) binding sites, we suggest that the acidic residue plays a key role in the coordination properties of divalent cations, hence conferring a function to the N terminus in the metal specificity of the ATPase.
Keywords: CadA, NMR, P1-type ATPase, cadmium detoxification, XAS
Introduction
Bacterial resistance to metal ions has been studied for several years and no common mechanism for resistance toward toxic heavy metals such as Cd(II), Zn(II), Hg(II), Co(II), Cu(I) or Ni(II) has been found. The various mechanisms described are chelation of metals by cytoplasmic proteins, conversion to less toxic species by redox reactions, or extrusion out of the cell by membrane transporters.1,2
Among the outflux systems, P1-type ATPases were found to be responsible for Cd(II) detoxification in Staphylococcus aureus3 and Listeria monocytogenes,4 Zn(II) detoxification in Escherichia coli,5 and Cu(I) detoxification in Enterococcus hirae6 and E. coli.7 Phylogenetic studies of the P1-type ATPase family has led to a sub-division into two groups, one including the Cu(I) and Ag(I)-ATPases and the other, the Cd(II) and Zn(II)-ATPases.8 Biochemical studies of E. coli ZntA and L. monocytogenes CadA have confirmed that these two proteins were able to transport both Cd(II) and Zn(II)9 (unpublished data from our lab on CadA).
P1-type ATPases10 comprise eight transmembrane helices presumably forming the ionic pathway across the membrane. Among these, the sixth helix bears a CPC motif which is thought to ligate the metal to be transported.11 In addition to the membrane domain, a cytoplasmic domain of about 400 amino acids possesses ATPase activity and the cytoplasmic N terminus comprises one or two metal binding domains in prokaryotes and up to six in eukaryotes. Each metal binding domain is made of about 70 amino acid residues which are predicted to have a β1α1β2β3α2β4 ferredoxin-like fold.12 Each metal binding domain bears a CXXC consensus sequence in loop 1 (between β1 and α1), whose Cys residues are involved in coordination of Cu(I)13,14 and of Zn(II).15 Although not necessary for the ion transport activity, the N-terminal metal binding domain(s) seem to interact with the rest of the protein and modulate its activity.16–19
The P1-ATPases ionic specificity most probably relies on the two types of metal binding sites, the membrane transport sites and the N-terminal metal binding sites.20,21 Here, we investigate the role of the N-terminal domain in P1-ATPase metal specificity by determining the first structure of a Cd(II)-ATPase N terminus, and analysing the Cd(II) binding site by comparison with the metal binding sites of other monovalent and divalent P1-type ATPase N termini. Structural data on the N-terminal domain of ZntA from E. coli indicate that an Asp residue, right before the CXXC motif and conserved in some Cd(II) and Zn(II)-ATPases, facilitates the binding of a divalent cation such as Zn(II).15 Because there are no such acidic residues close to the CXXC motif of Cu(I)-ATPases, it was suggested that the presence of this Asp makes the metal binding site specific for Zn(II), or at least for divalent cations, as opposed to Cu(I) and Ag(I).12 The solution structure of the N terminus of the Cd(II)-ATPase from L. monocytogenes is here characterized in the absence and presence of Cd(II) using both NMR and X-ray absorption spectroscopy. All the data are consistent with the participation of a fully conserved Glu residue in Cd(II) binding specificity. In contrast to E. coli ZntA N terminus, in NTKII this Glu is in loop 5 (between α2 and β4), i.e. far from the CXXC motif in the sequence, thus determining a different arrangement of the metal binding residues for these two proteins.
Results
Solution structure and dynamics of apoNTKII
The N-terminal domain of the CadA ATPase from L. monocytogenes (71 amino acid residues, called NTKII hereinafter) is a soluble monomeric protein. It is well folded, as indicated by the good dispersion of the NH signals in the 1H–15N HSQC spectrum (Figure 1(a)). Based on almost complete spectral assignment of the backbone and side-chain resonances (98% for nitrogen and 97% for proton) and on the determination of structural constraints as inter-proton distances and dihedral angles, the structure of apoNTKII was calculated. It has a high resolution over all its sequence, with the exception of a few residues at the C and N termini (average RMSD to the mean structure (residues 4–68) of 0.42(±0.09) Å for the backbone and 1.17(±0.07) Å for all heavy atoms, with a total penalty function of 0.44(± 0.04) Å2). The conformational and energetic analysis of apoNTKII structure is reported in Table S1 of Supplementary Data.
Figure 1.

Effect of Cd(II) binding to NTKII on 1H–15N HSQC spectra. (a) The 2D 15N–1H HSQC spectra (600 MHz, 298 K) of CdNTKII (blue) and of apoNTKII (red). Residues whose 1H–15N cross-peaks disappear upon Cd(II) binding are indicated in light grey. For both samples the protein concentration was about 1.5 mM, in 350 mM phosphate buffer (pH 7). (b) The weighted average chemical shift differences Δavg(HN) (i.e. [(ΔH)2 + (ΔN/10)2]1/2, where ΔH and ΔN are chemical shift differences for 1H and 15N, respectively) are shown. Chemical shift differences are not reported for residues 12–18, 21 or 60, as their 1H–15N cross-peaks are not observed for Cd(II) form. The secondary structure elements of apoNTKII are reported at the top.
ApoNTKII has the classical βαββαβ ferredoxin-like fold (Figure 2), where the four β-strands form an antiparallel twisted β-sheet, and the two helices are located on the same side of the β-sheet. Short loops connect the secondary structure elements. This fold is similar to that observed for the soluble N-terminal domains of copper(I) P1-type ATPases,13,14 and for soluble Cu(I)-chaperones, like Atx1 from yeast22 and the corresponding Hah1 human protein.23 The two metal binding cysteine residues of the CXXC motif are located in loop 1 (Cys14) and at the beginning of helix α1 (Cys17), as already observed in several copper binding domains of P1-type ATPases.13,14 Furthermore, in the apoNTKII structure a potential metal ligand, Glu61, located in loop 5, is spatially close to the metal binding CXXC motif (Figure 2), thus suggesting its possible involvement in cadmium binding.
Figure 2.
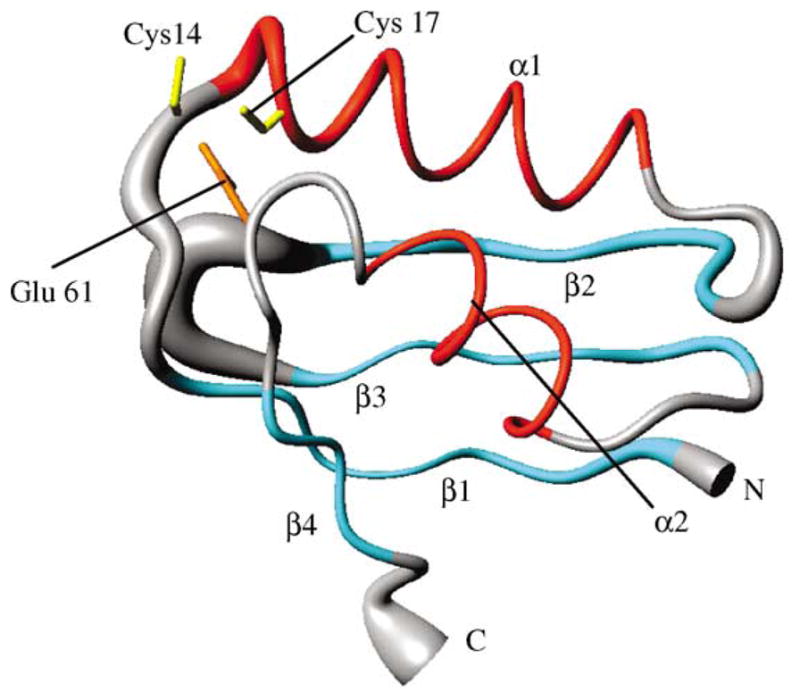
Solution structure of apoNTKII. Backbone atoms are represented as a tube with variable radius, proportional to the backbone RMSD value of each residue. The side-chains of Cys14, Cys17 and Glu61 are also shown. The secondary structure elements are also indicated: β-strands are in cyan and α-helices in red.
Heteronuclear relaxation rates can monitor protein internal motions as well as its overall tumbling rate. The experimental rates are essentially homogeneous along the entire polypeptide sequence (Figure S1 of Supplementary Data; average values 2.15(±0.06) s−1, 7.14(±0.07)s−1 and 0.70(±0.04) for 15N R1, R2 and 1H–15N nuclear Overhauser effects (NOEs), respectively), with the exception of some residues located in loop regions and at the N and C termini. The spectral density function analysis (Figure S2 of Supplementary Data) indicates that some residues around the metal binding CXXC motif (Ser13, Cys14, Ala18, Phe38, Gly39, Ser41, Lys42 and Phe60) experience J(0) values higher than the average indicating the presence of backbone fluctuations on a micro- to millisecond timescale24,25 in the metal binding region. The high J(ωH) values found for the residues at the N and C termini indicate that they undergo backbone motions on the nano- to picosecond timescale,24,25 i.e. faster than the overall protein tumbling rate (Figure S2 of Supplementary Data).
The overall rotational correlation time (τc), as estimated from the R2/R1 ratio, is 4.8(±0.2) ns, consistent with what is expected from an isotropic molecule of about 8 kDa, thus indicating the absence of aggregation. It is also very similar to that estimated, using the same approach, for the other proteins with the same fold.15,26
Sequence analysis of the N-terminal domain of cadmium, zinc and copper ATPases
Starting from the 71 amino acid residue sequence of NTKII, the GenBank was browsed to search for sequences sharing the CXXC motif and at least 50% sequence identity. In this way, 15 sequences (including the query sequence NTKII) belonging to 13 different proteins (hereinafter named CadA-like) were found. Their sequences, which share, on average, 63% sequence identity, are aligned (Figure 3). Glu61, located in loop 5 and spatially close to the CXXC motif in the apoNTKII structure, is fully conserved in all CadA-like sequences (Figure 3). A similar search was performed, with the same criteria, using as query sequence the amino acid sequence of the N-terminal domain of ZntA from E. coli. In this way, 18 sequences were retrieved which, on average, share 56% sequence identity (hereinafter named ZntA-like). The Glu residue in loop 5 is absent from all ATPase sequences found in the latter search. However, ZntA-like sequences have, in loop 1 close to the CXXC motif, a conserved Asp residue (Asp58 in E. coli ZntA sequence) which is known to be involved in zinc binding15 (Figure 3). Sequence and structural analysis of the N-terminal domains of Cu(I)-ATPases clearly shows that no acidic residues are conserved in the vicinity of the CXXC motif nor in loop 5, i.e. in the sequence segments which are spatially close to the metal binding site.12 Specifically, the acidic residues, Asp in loop 1 or Glu in loop 5, present in ZntA-like and CadA-like, are absent from all Cu(I)-ATPase N-terminal domains.12 Therefore, the N-terminal domains of divalent cation transporting P1-type ATPases are always characterized by the presence of an acidic group in the vicinity of the metal binding site. This feature suggests a role of the carboxylate moiety in providing the appropriate coordination sphere for the binding of divalent cations. Finally, this simple bioinformatic search defines two classes of proteins, one, which refers to ZntA N terminus, and another one, which refers to CadA N terminus. The two classes have an identity, which varies from 30% to 40%.
Figure 3.
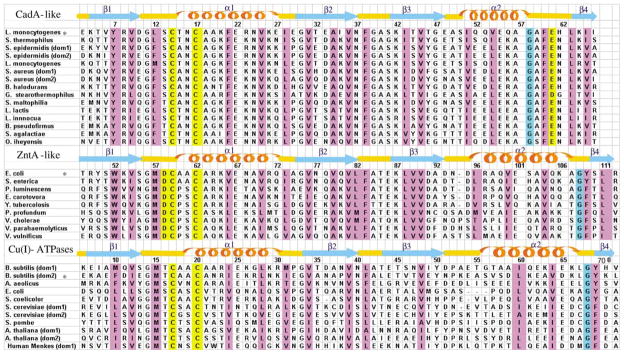
Structure-based sequence alignments of the N termini of various P1-type ATPases. The N-terminal binding domain of CadA from L. monocytogenes, that of ZntA from E. coli and the second domain of CopA from B. subtilis were aligned with other N-terminal binding domains. Secondary structure of NTKII for the CadA-like (this work), ZntA N terminus for the ZntA-like (1MWY) and the second metal-binding site of CopA N terminus for the Cu(I)-ATPases (1JWW) are also shown, together with their amino acid numbering. The asterisk refers to proteins whose secondary structure is shown. Residues that are known or predicted to bind a metal ion are highlighted in yellow. Positions where hydrophobic residues are conserved are highlighted in magenta.
Another conserved pattern of the N-terminal soluble domain of ZntA-like and Cu(I)-ATPases is a highly conserved Gly residue (shaded in cyan in Figure 3) followed by a completely conserved aromatic (Tyr/Phe) residue, both located in loop 5.12,15 Conserved hydrophobic contacts are found in ZntA-like and Cu(I)-ATPase domains between the aromatic ring of the latter residue, the side-chain of the highly conserved Met in loop 1 and the second metal binding Cys.12,15 This sequence arrangement is not present in CadA-like proteins. Still, a Gly residue (shaded in cyan in Figure 3) is fully conserved in CadA-like but in a sequence position which structurally corresponds to a hydrophobic residue conserved in all Cu(I)-ATPases and ZntA-like (Ile64 in the Bacillus subtilis CopA sequence and Val104 in the E. coli ZntA sequence). This Gly residue is then followed, in CadA-like sequences, by a highly conserved Ala-Phe-Glu pattern (Figure 3), where the Glu is Glu61 in NTKII, the potential metal binding residue discussed above. Therefore, a different pattern of amino acid residues is present in this region of CadA-like sequences with respect to that in ZntA-like and Cu(I)-ATPases. This different pattern gives rise to a different organization of the hydrophobic contacts and to local structural changes in CadA-like with respect to ZntA-like and Cu(I)-ATPases (see Discussion).
CdNTKII is a homodimeric complex in vitro
In the presence of equivalent amounts of Cd(II) and apoNTKII, the latter binds one cadmium atom per monomer, as revealed by dialysis at equilibrium (see Materials and Methods). Cd(II) coordination to thiols involves S to Cd charge transfer which is known to absorb light in the UV region. Therefore binding of Cd(II) to apoNTKII was followed by absorbance spectroscopy analysing the main absorption band at 231 nm. For this purpose, CdCl2 was added by 0.1 equivalent step up to three equivalents and absorbance spectra were recorded at each step. The spectra shown in Figure 4(a) were obtained after subtracting the apoNTKII spectrum. Simple analysis shows that the absorption levels off when one equivalent of Cd(II) is added (Figure 4(a), inset). Further analysis by the Specfit program (Spectrum Software Associates) suggests that at substoichiometric amounts of Cd(II) two different species contribute to this absorption band, which were identified as NTKII dimers that bind one or two Cd(II) ions (Figure 4(b)). At Cd(II) concentrations lower than 0.5 equivalent, absorption is mainly due to a monometallic dimer (Figure 4(c)). Above 0.5 equivalent, a new species of higher absorption coefficient is formed at the detriment of the monometallic dimer (Figure 4(c)). This new species is a dimetallic dimer, which is the final and only form at one equivalent Cd(II). This description allows us to fit the data as shown in the inset to Figure 4(a). The stability constants estimated from these data are for the monometallic dimer log βm=13.7±0.5 and for the dimetallic dimer log βd=20.7±0.5, where βm=[Cd(NTKII)2]/[Cd][NTKII]2 and βd= [Cd2(NTKII)2]/[Cd]2[NTKII]2.
Figure 4.

Difference absorbance spectra of Cd(II) binding to NTKII. (a) 10 μM NTKII was incubated in 20 mM Mes–NaOH (pH 6), 400 mM NaCl and the reference spectrum was stored. CdCl2 was then added by 1 μM steps and the free NTKII spectrum was subtracted. The inset shows saturation of the signal at 231 nm. (b) Absorption coefficients of the monometallic dimer Cd(NTKII)2 and the dimetallic dimer Cd2(NTKII)2; the numbers (0, 1 2) refer to the number of Cd(II) ions bound to each species. (c) Cd(II)-dependent formation of the mono- and dimetallic dimers of NTKII, at the detriment of the apoNTKII.
The 1H–15N HSQC maps show that a few signals exhibit large shift variations upon Cd(II) addition (Figure 1(a)), while only small changes are observed for the β-sheet and helix α2 (Figure 1(b)), indicating that the overall protein fold is not affected by cadmium binding. The most dramatic changes are observed for residues 12–25, which experience either large chemical shift variations (19, 20, 22–25) (Figure 1(b)) or the disappearance of their NH signals (12–18 and 21), possibly because of line broadening due to conformational exchange processes. Also the region 58–63 exhibits significant chemical shift changes upon Cd(II) binding. In particular, residues 58, 59, 61 and 63 experience large chemical shift changes and the NH cross-peak of Phe60 disappears (Figure 1(b)). All the observed spectral changes are essentially restricted to residues surrounding the CXXC metal binding motif and the conserved Glu61 in loop 5.
Cd(II) binding has also a profound effect on the dynamic properties of NTKII. 15N R1 values are significantly lower for CdNTKII than for apoNTKII (average values: 1.41(±0.05) s−1 (CdNTKII); 2.15(±0.06) s−1 (apoNTKII)), whereas the 15N R2 values are significantly higher for CdNTKII (average values: 11.9(±0.5) s−1 (CdNTKII); 7.14(±0.07) s−1 (apoNTKII)) (Figure S1 of Supplementary Data). The increase in the R2/R1 ratio is a direct measure of the increased overall rotational correlation time of the protein, which is 8.8(±0.2) ns for CdNTKII, whereas it is 4.8(±0.2) ns for apoNTKII. These values indicate the formation of dimeric species upon Cd(II) binding. A similar behaviour, i.e. dimerization upon metal binding, is commonly observed for a variety of metal-transporting proteins. For instance, it was observed with CopZ from E. hirae, for which Cu(I) addition produced a dimeric state where the NH resonances of all residues close to the metal binding region disappear or broaden as a consequence of conformational exchange processes,27 and with Cox11 from S. meliloti, for which Cu(I) addition produced the same NH broadening phenomenon for the CFCF metal-binding region.28
From spectral density function analysis of CdNTKII (Figure S2 of Supplementary Data), it also emerges that several residues (19–25 and 58–63) in the vicinity of the metal binding motif CXXC and of Glu61 undergo conformational exchange processes, which are not present in the apo form (Figure S2 of Supplementary Data). These residues are those experiencing also sizeable chemical shift differences between the two species. The NH signals of Phe60 and Phe21 broaden beyond detection. All these data indicate that two subunits of the dimer interact through the metal binding region (residues 12–25 and 58–63), and that this interaction induces conformational dynamic processes.
The coordination sphere of cadmium
We investigated Cd XAS to provide information about the coordination sphere of cadmium in NTKII. Cd K edge, EXAFS, and Fourier transforms for NTKII either with or without TCEP are displayed in Figure 5 and the results of curve-fitting of Cd EXAFS are provided in Table 1. Edge, EXAFS, and FTs are very similar for both NTKII samples. Cd K edges are quite broad owing to core-hole lifetime broadening, but the edges in Figure 5 are indicative of Cd in a predominantly S environment.29 EXAFS and FTs also indicate the presence of a first shell of ligands at a distance (2.4–2.5 Å) that is expected for Cd–S bonds and curve-fitting results show that the first coordination sphere is dominated by S-containing ligands at ca 2.5 Å (Table 1). Compared in Table 1 are potential first-coordination spheres of CdSxO4−x (x=4–2). The Cd–S Debye–Waller factor ( ) for CdS4 fits is significantly larger than expected (0.0071 versus 0.0026 Å2 for Cd-substituted rubredoxin (F. Bonomi, M. K. Eidsness & R.A.S, unpublished observations)) and the bond-valence sum (BVS) parameter is significantly greater than the expected valence of 2.30 Comparing CdS3O1 and CdS2O2 fits, Cd–S Debye–Waller factors are lower and BVS is closer to 2 for fits with three S-containing ligands.
Figure 5.

(a) Cd K edge spectra and (b) Fourier transforms (k3 weighting, k=2–13 Å−1) for CadA NTKII (continuous) and CadA NTKII+TCEP (broken). k3-weighted EXAFS data are presented in the inset to (b).
Table 1.
Curve fitting results for [Cd] CadA NTKII EXAFS
| Sample filename (k range) Δk3χ | Fit | Shell | Ras (Å) |
|
ΔE0 (eV) | f′a | BVSb | |
|---|---|---|---|---|---|---|---|---|
| CdNTK DNK0 (2–13 Å−1) Δk3χ=8.70 |
1 | Cd–S3 | 2.49 | 0.0054 | −11.97 | 0.129 | 1.81 | |
| 2 | Cd–S4 | 2.49 | 0.0071 | −10.51 | 0.112 | 2.42 | ||
| 3 | Cd–S3 | 2.51 | 0.0054 | −7.82 | 0.111 | 2.10 | ||
| Cd–O1 | 2.26 | 0.0023 | ||||||
| 4 | Cd–S2 | 2.54 | 0.0035 | −4.74 | 0.111 | 1.76 | ||
| Cd–O2 | 2.29 | 0.0028 | ||||||
| CdNTK + TCEP DNKP (2–13 Å−1) Δk3χ=8.02 |
5 | Cd–S3 | 2.50 | 0.0048 | −8.00 | 0.093 | 1.77 | |
| 6 | Cd–S4 | 2.50 | 0.0065 | −8.34 | 0.085 | 2.36 | ||
| 7 | Cd–S3 | 2.51 | 0.0048 | −7.36 | 0.085 | 2.12 | ||
| Cd–O1 | 2.24 | 0.0062 | ||||||
| 8 | Cd–S2 | 2.53 | 0.0032 | −3.50 | 0.087 | 1.79 | ||
| Cd–O2 | 2.29 | 0.0045 | ||||||
| 9 | Cd–S3 | 2.51 | 0.0046 | −7.07 | 0.077 | 2.12 | ||
| Cd–O1 | 2.24 | 0.0064 | ||||||
| Cd–Cd | 3.66 | 0.0059 |
Shell is the chemical unit defined for the multiple scattering calculation. Subscripts denote the number of scatterers per metal-ion. Ras is the metal-scatterer distance. is a mean-square deviation in Ras. ΔE0 is the shift in E0 for the theoretical scattering functions.
BVS = Σexp[ro −Ras], B=0.37, r0(Cd(II)–S)=2.304, r0(Cd(II)–O)=1.904.57
The chemical shift of 113Cd nuclei is sensitive to the nature, number and geometric arrangement of the coordinating ligands,31,32 and therefore, is used as fingerprint of the donor atoms. In the 1D 113Cd NMR spectrum, a single broad resonance at 687 ppm was observed, a value typical for a cadmium ion predominantly bound to thiolate donor ligands (Figure 6). A correlation is observed between the number of isolated sulfur atoms present as donors and the increasing downfield chemical shifts experienced by the 113Cd nucleus.31 The chemical shift of 687 ppm of CdNTKII is intermediate between that observed for isolated complexes containing two thiolate sulfur atoms and that for complexes containing four of them.31 Thus, the 113Cd chemical shift observed for CdNTKII is consistent with three sulfur atoms participating in cadmium binding in agreement with XAS data.
Figure 6.

113Cd NMR spectrum of CdNTKII. The protein was in 350 mM phosphate buffer (pH 7) at 298 K. A 100 Hz line broadening was applied. Chemical shift is plotted relative to that of 0.1 M Cd(ClO4)2, δ =0 ppm. The proposed metal binding model is shown in the inset, where the broken lines indicate long Cd–S distances in a distorted dinuclear site.
As only two thiolate ligands are present in the sequence at Cys14 and Cys17 and CdNTKII forms a homodimeric complex containing one cadmium atom per monomer, we propose, on the basis of XAS and 113Cd NMR data, that the two cadmium ions share two thiolate ligands, one from each NTKII monomer, creating a dinuclear cluster (Figure 6 inset). In this model, the other Cys on each NTKII monomer is the third thiolate ligand. In agreement with this model, attempts to fit a Cd–Cd interaction converge for the TCEP sample only (cf. small FT peak in Figure 5(b)) with a Cd–Cd distance of ca 3.7 Å and a Debye–Waller factor of 0.0059 Å2. In addition, 113Cd chemical shift values similar to that of CdNTKII, i.e. ranging from 710 ppm to 660 ppm, have been found for similar Cd2Cys6 binuclear cluster sites in Gal4 and Lac9 transcription factors.33,34 Finally, considering that the most common coordination number for Cd(II) is four when at least three Cys are the metal ligands†, that the fully conserved potential Cd(II) ligand, Glu61, is structurally close to the CXXC motif (Figure 2), that its NH NMR cross-peak largely shifts upon Cd(II) binding (Figure 1(a)), and that XAS data indicate the presence of one oxygen atom in the coordination sphere, we propose that Glu61 is the fourth ligand whose carboxylic oxygen atom saturates the coordination sphere of Cd(II) (Figure 6, inset). Cadmium inorganic complexes with arenephosphinothiol ligands35 show that this Cd(II) coordination environment (Figure 6, inset) is feasible.
Discussion
Original features of NTKII structure
NTKII, similarly to the N-terminal soluble domains of other P1-type ATPases characterized up to now,13–15,36 has a ferredoxin-like fold, comprising the secondary structure elements β1α1β2β3α2β4.Comparison of its structure with those of ZntA (a Zn(II)-ATPase) and CopA (a Cu(I)-ATPase) N termini reveals that, despite sharing the same folding, the coordination spheres of Cd(II) and Zn(II) differ from that of Cu(I) by the presence of an acidic residue, Glu in the case of NTKII and Asp in the case of ZntA. This additional acidic residue could help to stabilize the binding of a divalent cation. The carboxylate moiety is just adjacent to one Cys residue of the consensus motif in the ZntA-like sequences. By contrast, in the CadA-like sequences, it is located far away in sequence, in loop 5 (Figure 3). Note that, although in Cu(I)-ATPases acidic residues are present close in sequence to Glu61 of NTKII (i.e. at positions 62, 65 or 66), they are not located in loop 5 but in helix α2 (Figure 3). This novel organization of the metal ligands in NTKII becomes clear by comparing the structure of NTKII to those of ZntA and CopA N termini (Figure 7). In NTKII, helix α2 is four residues shorter on its C-terminal end than in ZntA and CopA N-terminal domains, because its eighth residue is a Gly (shaded in cyan in Figure 3). In soluble proteins, glycine residues are considered as “helix-breakers”37 because of their inability to form steric interactions and because of the entropic cost of tethering glycine in helical secondary structure. Therefore, they are often found in loop regions and β-turns. The Cd(II) binding region in apoNTKII is stabilized by hydrophobic interactions involving helix α1 (Phe21) and loops 1 (Leu12), 3 (Phe38) and 5 (Phe60 and Leu63) (Figure 7). At variance with NTKII, the hydrophobic interactions, in ZntA and CopA N-terminal domains hydrophobic interactions involve helix α1 (Val66 or Ile24), helix α2 (Val104 or Val64), loop 1 (Met57 or Met15) and loop 5 (Tyr109 or Tyr69) (Figure 7). In NTKII, the hydrophobic contacts are determined by a cluster of three Phe residues, with extensive stacking between the Phe rings, and by two Leu, while it is determined by one Met, one Tyr and two Val/Ile in ZntA and CopA N termini. One of the three Phe residues of NTKII (Phe38) is completely conserved also among the ZntA-like proteins (Figure 3), but it has a different conformation and points towards the solvent in the latter protein thus not contributing extensively to the hydrophobic core. The major difference between the NTKII hydrophobic contacts and those of ZntA and CopA N termini is determined by the presence, in NTKII loop 5, of two hydrophobic residues, Phe60 and Leu63, while only one (Tyr109 or Tyr69) is present in loop 5 of ZntA and CopA N termini (Figure 3). Both residues, fully conserved in CadA-like proteins but not present in ZntA-like proteins nor in Cu(I)-ATPases, form extensive packing interactions with the hydrophobic residues of helix α1, loop 1 and loop 3, with the result of “sticking” loop 5, containing the third cadmium ligand Glu61, close to the CXXC motif (Figure 7). Therefore, we can suggest that NTKII utilizes slightly different hydrophobic contacts and structural arrangements with respect to ZntA and CopA N termini to correctly orient Glu61 towards the CXXC motif and therefore efficiently bind the Cd(II) ion.
Figure 7.

Solution structures of N-terminal metal-binding domains of L. monocytogenes CadA, E. coli ZntA (1MWY) and B. subtilis CopA (1JWW). Apo forms of the proteins, marked with an asterisk in Figure 3, are compared here. van der Waals contacts involving some hydrophobic residues are shown in blue, the metal ligands are shown in yellow and some highly conserved residues are in red. The Gly residues, shaded in cyan in Figure 3, are mapped in cyan on the backbone of the structures.
Cd(II)-induced dimerization in vitro
Cd(II) binding to NTKII induces protein dimerization in vitro with a Cd(II)/monomer stoichiometry of 1:1. XAS and NMR data suggest that the Cd(II) ions participate directly in dimerization as shown in the inset of Figure 6. This is favoured by the coordination properties of Cd(II), which tends to prefer high coordination numbers, usually between four and six.38 In particular, with increasing numbers of Cys thiolates as ligands, the coordination number of four becomes predominant (Cambridge Structural Database†). Indeed, Cysrich metalloproteins such as zinc and cadmium metallothioneins always bind these metal ions in a tetrahedral coordination environment.39 Even if the homodimeric complex were not physiologically relevant, the dimerization would occur in vitro to better satisfy the coordination chemistry requirements of Cd(II) and the extensive solvent accessibility of the metal binding site makes this feasible. The local structural environment of the Cd(II) ions, as characterized through Cd K edge X-ray absorption spectroscopy, supports that oxygen participates in the metal coordination sphere. Indeed, assuming four-coordinate Cd(II), a CdS3O1 first shell should be observed in a dinuclear cluster, whereas isolated monomeric Cd(II) sites would exhibit a CdS2O2 first shell (assuming a solvent (H2O)-derived species as fourth ligand) and dimerization would have to occur by some undefined allosteric effect of Cd(II) binding. Our results (Table 1) suggest that either of these first coordination spheres is possible, although CdS3O1 exhibits more reasonable BVS values.
Another expected consequence of the proposed dinuclear site is the presence of a Cd–Cd FT peak. Cd–Cd distances for bis-thiolate bridged Cd(II) dinuclear clusters with predominant thiolate ligation range from 3.48 Å −3.69 Å.40 The only possible reflection of such an interaction is the small FT peak (Figure 5(b)) at ~3.8 Å and this can be fitted with a 3.7 Å Cd–Cd distance in the TCEP-containing sample only. The Debye–Waller value for the Cd–Cd shell in the fit of the TCEP-containing sample EXAFS data is 0.0059 Å2, close to 0.0070 Å2 reported for Cd–Cd in [Cd4(SPh)10]2−,29 but the latter Cd–Cd distance is 4.3 Å and the bridging is different from that proposed here. Other Cd(II) dinuclear models display 3.8 Å–3.9 Å Cd–Cd distances,35 allowed by a less symmetrical bridging environment with one Cd-bridging thiolate bond longer than the other (for each Cd(II)). This would imply a Cd(II) first-shell environment with significant static disorder in the Cd–S shell. Assuming that the Cd–S Debye–Waller factor for Cd-substituted rubredoxin ( ; other reported Cd-S values are slightly higher, for example, for [Cd(SPh)4]2−)29 represents the vibrational component (four identical Cd–S bond lengths), we can use the observed Cd–S (0.0054 Å2) for NTKII samples to extract the contribution of static bond length disorder to the Cd–S shell,41 yielding ΔR≈0.1 Å. This would represent the largest difference consistent with our data between the one long and two short Cd–S distances in a distorted dinuclear site and could result in a longer Cd–Cd distance depending on Cd–S–Cd angles. At longer Cd–Cd distances, the Debye–Waller factor would increase further and the Cd–Cd FT peak could easily become undetectable. It is also possible that the dynamics observed by NMR (vide supra) contain a component of dinuclear site dynamics that could contribute an additional vibrational Debye–Waller factor component to the Cd–Cd interaction. Thus, although the dinuclear structure proposed in Figure 6 is not required by the EXAFS data, it is definitely consistent with these data.
NMR and sequence analysis data suggest that Glu61 is the best candidate as oxygen donor in the proposed binuclear cluster. Therefore, Glu61 in loop 5 can play a key role in the metal coordination properties of the N-terminal domains of CadA-like P1-type ATPases, providing the appropriate coordination sphere for stabilizing a divalent cation better than a monovalent cation.
Concluding remarks
The present work identified two classes of N-terminal domains, one related to the ZntA-like ATPases and the other to CadA-like ATPases. In vivo studies on ZntA bearing the N terminus of the human Cu(I)-ATPase ATP7B instead of its genuine N terminus showed that the chimera still confers resistance to Zn(II) although with lower activity, but not to Cu(I),20 suggesting that the membrane site predominantly determines the type of metal ion that is transported. However, the same metal binding specificity in the N terminus and in the transmembrane segments of the ATPase could be important to obtain maximal activity. Indeed, although the role of the N terminus has not yet been clearly established, this domain is thought to modulate the activity of the ATPase through an interaction with the rest of the protein which depends on the presence of the metal.18,19 Accordingly, a certain degree of specificity of the N-terminal domain was first shown by the result that chimeras of ATP7B and ZntA could not restore the full activity of ZntA20 and, more recently, by in vivo data obtained with the cyanobacterium Synechocystis PCC 6803.21 In the latter organism a chimera of CoaT, a Co(II)-ATPase, bearing the N terminus of ZiaA, a Zn(II)-ATPase, confers resistance to Zn(II), showing that the membrane site of CoaT can transport Zn(II) and that the ZiaA N terminus participates in the selectivity of CoaT, inasmuch as it determines that among various cations, Zn(II) will be transported. From our structural and bioinformatic analysis of the cytoplasmic N-terminal domain it emerges that the same ferredoxin-like fold, comprising the secondary structure elements βαββαβ, is shared by Cu(I), Zn(II), and Cd(II)-ATPases (Figure 7) and that a single amino acid change may represent a relevant contribution to the metal specificity of heavy metal P1-type ATPases. Indeed, although the different metal binding patterns can bind several metal ions,42 a different pump efficiency for divalent cations as opposed to monovalent cations has been observed.9,43,44 Moreover, ZntA and CadA ATPases showed the maximal transport activity for zinc and cadmium, respectively5 (unpublished data from R.M., N.B., E.M. & P.C. on CadA and NTKII). Therefore, the presence of acidic residues at specific positions in the sequence but spatially close to the CXXC metal binding motif can be relevant in the ATPase pumping activity. It is possible that in the N terminus the different location of the carboxylate moiety with respect to the CXXC metal binding motif confers a kinetic advantage to the ATPase. Indeed, as the carboxylate ligand is provided by a distant loop, the metal binding site in CadA N terminus could better fit the larger Cd(II) ion than Zn(II). The same metal binding specificity in both the soluble N-terminal site and the membrane site of the ATPase could have a key role in optimizing the overall efficiency of the metal detoxification mechanism for a specific metal ion with respect to others, even if the ATPase can pump different heavy metal ions. The alignment of the eighth helix of P1-type ATPases shows that a Met conserved among the Cu(I)-ATPases is changed to an Asp in the Cd(II) or Zn(II)-ATPases,45 suggesting that the membrane site selectivity may also be based on the presence of an acidic residue close to the metal binding site.
Materials and Methods
Sequence searching
ATPase sequences were obtained by searching the GenBank Databases (CDS translations+PDB+SwissProt+PIR+PRF) using sequence similarity criteria. This was accomplished by starting from sequences of proteins of known function (the soluble N-terminal domain of CadA from L. monocytogenes and of ZntA from E. coli) and performing a BLAST† search using the sequence length as search criterion and the CXXC metal-binding motif. Then, those sequences having a sequence identity above 50% with the reference sequences were selected and alignment was performed with CLUSTALW program.46 For P1-type ATPases with more than one N-terminal metal-binding domain, the sequences were subdivided into individual metal-binding domains. The sequence of each domain was used for alignment, neglecting the rest of the sequence.
NTKII cloning and purification
NTKII corresponds to the first 71 amino acid residues of CadA, where the original serine 71 has been replaced by an alanine. The sequence encoding NTKII was first amplified by PCR from the original sequence of CadA, using the primers NTKPQ1 (5′-GAAGGA GTGGGAGCTCCCA TGGCAGAAAAGACTG-3′) and NTKPQ2 (5′-GCTCTGGATC AGTAAAAGCTTCT TTC TCTGGAATG-3′). The PCR product was further sub-cloned into the NcoI/BglII sites of the pQE-60 vector (Qiagen). The construction was checked by sequencing (Genome Express, France).
NTKII production was performed in the E. coli strain M15 (pREP4). Bacteria transformed with the NTKII expression vector were grown at 37 °C in 1l of LB medium for unlabelled protein or minimal M9 medium supplemented with [15N]ammonium chloride, containing 100 μg/ml of ampicillin and 25 μg/ml of kanamycin. After 24 h growth, the culture was cooled by addition of 1l of cold fresh medium. Production of NTKII was induced by addition of 2 mM IPTG and kept at 15 °C for 24 h.
For NTKII purification, cells collected by centrifugation (10 min, 13,000g, 4 °C) were suspended in 50 ml of 100 mM NaH2PO4–NaOH (pH 6.5), 1 mM PMSF, 1 mM EDTA, one tablet of antiprotease (Roche Molecular Biochemicals). Cells were lysed by sonication and then centrifuged (50,000g, 30 min, 4 °C). The resulting supernatant was brought to pH 3.2 by addition of HCl, incubated for 30 min on ice under continuous stirring and then centrifuged (50,000g, 30 min, 4 °C). The supernatant, enriched in NTKII, was brought to pH 6.5 by addition of NaOH and to 200 mM NaCl. At that step, 5 mM TCEP1 was added to protect thiol groups from oxidation. After volume reduction to 800 μl (Vivaspin concentrator, 5000 MWCO PES; Vivascience), the sample was fractionated on a Sephadex G50 column previously equilibrated with 100 mM Mops–NaOH (pH 7), 200 mM NaCl, 1 mM TCEP. Fractions containing highly purified NTKII, as checked using silver-stained polyacrylamide gels, were concentrated and either lyophilized or stored at −85 °C. Protein concentration was measured using the MicroBC Assay (Pierce). A yield of 10 mg of purified NTKII per liter of culture was usually obtained.
Cadmium binding to NTKII
Purified NTKII was thawed and two gel-filtration steps were performed under anaerobic conditions to remove TCEP using NAP-10 columns (Amersham Biosciences). The protein concentration was checked by the MicroBC assay. Cd(II) binding was measured in 50 mM Mes–NaOH (pH 6), 400 mM NaCl and at 2 °C to slow oxidation of the cysteine residues. In one series of experiments, 100 μM NTKII mixed with one, two or four equivalents of Cd(II) was dialyzed against the buffer for 2 h. The amount of Cd(II) in the bath was measured with the colorimetric dye PAR at 494 nm. In another series of experiments, 85 μM NTKII mixed with 0.6, 1.2 and 2.4 equivalents of 109Cd(II) in 50 mM Hepes–NaOH (pH7), 1 mM TCEP were concentrated by filtration (Vivaspin concentrator; Vivascience). The amount of 109Cd(II) in the filtrate was counted by scintillation. Because we had evidence that the Cd-bound NTKII was a dimer, another series of experiments were performed to get insights into the steps leading to NTKII dimerization. For this purpose, CdCl2 was added by 0.1 equivalent step up to three equivalents and UV-absorbance spectra were recorded in a U-3010 spectrophotometer (Hitachi) at each step.
NMR sample preparation
As a precaution against disulfide formation in proteins that contain a CXXC motif, these samples were prepared in a Vac atmospheres nitrogen chamber. For NMR experiments, the storage buffer was changed to 50 mM sodium phosphate (pH 7), 700 μM TCEP and the preparation was freeze-dried. Hydration just before the NMR experiment led to about 2 mM NTKII in the buffer described below. To prepare the cadmium-loaded form, apoNTKII in Hepes buffer (pH 7.5) was first treated with an excess of TCEP to fully reduce Cys residues and then 2.5 equivalents of CdCl2 or 113CdCl2 were added. The unbound metal was finally removed by a PD10 desalting column. The NMR samples of apo and metal loaded NTKII were in 350 mM sodium phosphate buffer (pH 7), 90% H2O/10% 2H2O in the presence of 5 mM TCEP.
XAS sample preparation
ApoNTKII was anaerobically reduced with 5 mM TCEP in 50 mM Hepes buffer (pH 7). The protein sample was then split into two: (A) 1.2 equivalents of CdCl2 was directly added to the solution containing 5 mM TCEP; (B) TCEP was removed by a PD10 desalting column and 1.2 equivalents of CdCl2 were then added. The unbound Cd(II) ions were removed by extensive washing in a glove-box. The final samples were ca 3.0 mM in Cd.
NMR experiments and structure calculations
NMR spectra were recorded on Avance 700, 600 and 500 Bruker spectrometers, the latter being equipped with a triple resonance cryo-probe. All the triple resonance (TXI 5-mm) probes used were equipped with pulsed field gradients along the z-axis.
All the NMR experiments, summarized in Table S2 of Supplementary Data, were recorded on unlabelled and 15N-labelled samples at 298 K, processed with the standard Bruker software (XWINNMR) and analysed with the XEASY program. The 2D NOESY and 3D NOESY-15N HSQC maps were acquired with a mixing time of 100 ms and a recycle time of 1.5 s. Resonance assignment and structural restraints (i.e. proton–proton distances and dihedral angles (φ, ψ and χ1) were determined through the experiments listed in Table S2 of Supplementary Data. 1H–15N HSQC spectra do not show chemical shift changes in phosphate buffer (pH 7) with concentrations ranging from 100 mM to 350 mM.
Structure calculations were performed through iterative cycles of DYANA47 followed by restrained energy minimization with AMBER 5.048 applied to each member of the family. The structure displays the following secondary structure elements: 4–10 (β1), 15–27 (α1), 31–37 (β2), 42–47 (β3), 51–58 (α2), 64–66 (β4). The quality of the structures was evaluated using the programs PROCHECK and PROCHECK-NMR.49 Figures were generated with the program MOLMOL.50
Relaxation measurements and analysis
Relaxation experiments were performed on Bruker Avance 600 MHz spectrometers at 298 K. 15N R1, R2, and steady-state heteronuclear 1H–15N NOEs were measured with pulse sequences as described by Farrow et al.51 and reported in Table S2 of Supplementary Data. Integration of cross-peaks for all spectra was performed by using the standard routine of the XWINNMR program. Relaxation rates R1 and R2 were determined by fitting the cross-peak intensities measured as a function of the delay within the pulse sequence, to a single-exponential decay.52 Errors in the rates were estimated through a Monte Carlo approach.25,53 The heteronuclear NOE values were obtained from the ratio of the peak intensity for 1H-saturated and unsaturated spectra. The heteronuclear NOE values and their errors were estimated by calculating the mean ratio and the standard error from the available data sets. The experimental relaxation rates were used to map the spectral density function values, J(ωH), J(ωN), J(0), following a procedure available in the literature.24,25 An estimate of the overall rotational correlation time was derived from the measured R2/R1 ratio. In this analysis, care was taken to remove from the input relaxation data those NH groups having an exchange contribution to the R2 value or exhibiting large-amplitude internal motions on a timescale longer than a few hundred picoseconds, identified from low NOE values, as inclusion of these data would bias the τc calculation.54,55
One-dimensional 113Cd NMR spectra were acquired on a Bruker Avance 600 MHz equipped with a BBO (direct observe) probe head with 480,000 scans (acquisition time 0.09 s, recycle delay 0.41 s, 8000 complex points). Chemical shifts are referred to the resonance position of 0.1 M Cd(ClO4)2.
X-ray absorption spectroscopy measurements
Cd K edge XAS data were collected at the Stanford Synchrotron Radiation Laboratory Beamline 9-3 with SPEAR3 operating at 3.0 GeV and 85–100 mA. Focusing optics were configured for 22 keV mirror cutoff operations, allowing sufficient flux at the Cd K edge (~26.7 keV) for these experiments. Si[220] monochromator crystals provided a 1 mm vertically apertured beam to the sample contained in a LHe-flow cryostat, holding the sample at 10 K throughout data collection. Two independent XAS samples were prepared from each NTKII sample provided and data were averaged for analysis using a total of six and 11 20 min scans for the NTKII and NTKII+TCEP data sets, respectively. Data were collected by fluorescence excitation using a 30 element intrinsic Ge detector with each detector windowed on the Cd Kα emission peak (at 23170 eV). A Cd foil was used for internal calibration (first inflection point at 26714.0 eV), a polynomial (−1 order) background was subtracted, and third-order spline subtraction (spline points at 26,720, 26,988, 27,256, 27,524 eV) generated EXAFS data with E0=26720 eV. k3χ EXAFS data from k=2–13 Å−1 were Fourier-transformed using S-based phase correction. Curve-fitting was performed with EXAFSPAK software† using feff 8.2056 for calculation of scattering functions. The bond-valence sum (BVS) method30 was used to check for chemical sensibility of bond lengths and coordination spheres.
Supplementary Material
Acknowledgments
This work was supported by the European Community (SPINE no.QLG2-CT-2002-00988, Structural Proteomics in Europe), by MIUR COFIN 2003 Il ruolo degli ioni metallici nei processi metabolici and by Ente Cassa Risparmio di Firenze. Part of this work was supported by the Programme de Toxicologie Nucléaire du Commissariat à l’Energie Atomique. XAS work in the laboratory of R.A.S. is supported by the National Institutes of Health (GM 42025). Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program.
Abbreviations used
- TCEP
Tris(2-carboxyethyl)phosphine hydrochloride
- NOE
nuclear Overhauser effect
- HSQC
heteronuclear single quantum correlation
- BBO
Broad band observe
- RMSD
root-mean-square deviation
- BSV
bond-valence sum
Footnotes
www-ssrl.slac.stanford.edu/exafspak.html
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j. jmb.2005.11.055
Data Bank accession codes
Atomic coordinates and NMR constraints of apoNTKII have been deposited in the RCSB Protein Data Bank with accession codes 2AJ0 and 2AJ1. Resonance assignments are available at the BioMagResBank (accession number 6811).
References
- 1.Silver S, Phung LT. Bacterial heavy metal resistance: new surprises. Annu Rev Microbiol. 1996;50:753–789. doi: 10.1146/annurev.micro.50.1.753. [DOI] [PubMed] [Google Scholar]
- 2.Nies DH. Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol Rev. 2003;27:313–319. doi: 10.1016/S0168-6445(03)00048-2. [DOI] [PubMed] [Google Scholar]
- 3.Nucifora G, Chu L, Misra TK, Silver S. Cadmium resistance from Staphylococcus aureus plasmid pI258 cadA gene results from a cadmium-efflux ATPase. Proc Natl Acad Sci USA. 1989;86:3544–3548. doi: 10.1073/pnas.86.10.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lebrun M, Audurier A, Cossart P. Plasmid-borne cadmium resistance genes in Listeria monocytogenes are similar to cadA and cadC of Staphylococcus aureus and are induced by cadmium. J Bacteriol. 1994;176:3040–3048. doi: 10.1128/jb.176.10.3040-3048.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rensing C, Mitra B, Rosen BP. The zntA gene of Escherichia coli encodes a Zn(II)-translocating P-type ATPase. Proc Natl Acad Sci USA. 1997;94:14326–14331. doi: 10.1073/pnas.94.26.14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Odermatt A, Suter H, Krapf R, Solioz M. Primary structure of two P-type ATPase involved in copper homeostasis in Enterococcus hirae. J Biol Chem. 1993;268:12775–12779. [PubMed] [Google Scholar]
- 7.Rensing C, Fan B, Sharma R, Mitra B, Rosen BP. CopA: an Escherichia coli Cu(I)-translocating P-type ATPase. Proc Natl Acad Sci USA. 2000;97:652–656. doi: 10.1073/pnas.97.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Axelsen KB, Palmgren MG. Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol. 1998;46:84–101. doi: 10.1007/pl00006286. [DOI] [PubMed] [Google Scholar]
- 9.Sharma R, Rensing C, Rosen BP, Mitra B. The ATP hydrolytic activity of purified ZntA, a Pb(II)/Cd(II)/Zn(II)-translocating ATPase from Escherichia coli. J Biol Chem. 2000;275:3873–3878. doi: 10.1074/jbc.275.6.3873. [DOI] [PubMed] [Google Scholar]
- 10.Lutsenko S, Kaplan JH. Organization of P-type ATPases: significance of structural diversity. Biochemistry. 1995;34:15607–15613. doi: 10.1021/bi00048a001. [DOI] [PubMed] [Google Scholar]
- 11.Solioz M, Vulpe CD. CPx-type ATPases: a class of P-type ATPases that pump heavy metals. Trends Biochem Sci. 1996;21:237–241. [PubMed] [Google Scholar]
- 12.Arnesano F, Banci L, Bertini I, Ciofi-Baffoni S, Molteni E, Huffman DL, O’Halloran TV. Metallochaperones and metal transporting ATPases: a comparative analysis of sequences and structures. Genome Res. 2002;12:255–271. doi: 10.1101/gr.196802. [DOI] [PubMed] [Google Scholar]
- 13.Banci L, Bertini I, Ciofi-Baffoni S, Huffman DL, O’Halloran TV. Solution structure of the yeast copper transporter domain Ccc2a in the apo and Cu(I)-loaded states. J Biol Chem. 2001;276:8415–8426. doi: 10.1074/jbc.M008389200. [DOI] [PubMed] [Google Scholar]
- 14.Banci L, Bertini I, Ciofi-Baffoni S, D’Onofrio M, Gonnelli L, Marhuenda-Egea FC, Ruiz-Dueñas FJ. NMR characterization of the N-terminal domain of a potential copper translocating P-type ATPase from Bacillus subtilis. J Mol Biol. 2002;317:415–429. doi: 10.1006/jmbi.2002.5430. [DOI] [PubMed] [Google Scholar]
- 15.Banci L, Bertini I, Ciofi-Baffoni S, Finney LA, Outten CE, O’Halloran TV. A new zinc-protein coordination site in an intracellular metal trafficking: solution structure of the apo and Zn(II) forms of ZntA(46-118) J Mol Biol. 2002;323:883–897. doi: 10.1016/s0022-2836(02)01007-0. [DOI] [PubMed] [Google Scholar]
- 16.Tsivkovskii R, MacArthur BC, Lutsenko S. The Lys1010-Lys1325 fragment of the Wilson’s disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. J Biol Chem. 2001;276:2234–2242. doi: 10.1074/jbc.M003238200. [DOI] [PubMed] [Google Scholar]
- 17.Bal N, Wu CC, Catty P, Guillain F, Mintz E. Cd2+ and the N-terminal metal-binding domain protect the putative membranous CPC motif of the Cd2+-ATPase of Listeria monocytogenes. Biochem J. 2003;369:681–685. doi: 10.1042/BJ20021416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bal N, Mintz E, Guillain F, Catty P. A possible regulatory role for the metal-binding domain of CadA, the Listeria monocytogenes Cd2+-ATPase. FEBS Letters. 2001;506:249–252. doi: 10.1016/s0014-5793(01)02927-1. [DOI] [PubMed] [Google Scholar]
- 19.Mitra B, Sharma R. The cysteine-rich amino-terminal domain of ZntA, a Pb(II)/Zn(II)/Cd(II)-translocating ATPase from Escherichia coli, is not essential for its function. Biochemistry. 2001;40:7694–7699. doi: 10.1021/bi010576g. [DOI] [PubMed] [Google Scholar]
- 20.Hou ZJ, Narindrasorasak S, Bhushan B, Sarkar B, Mitra B. Functional analysis of chimeric proteins of the Wilson Cu(I)-ATPase (ATP7B) and ZntA, a Pb(II)/Zn(II)/Cd(II)-ATPase from Escherichia coli. J Biol Chem. 2001;276:40858–40863. doi: 10.1074/jbc.M107455200. [DOI] [PubMed] [Google Scholar]
- 21.Borrelly GP, Rondet SA, Tottey S, Robinson NJ. Chimeras of P-type ATPases and their transcriptional regulators: contributions of a cytosolic amino-terminal domain to metal specificity. Mol Microbiol. 2004;53:217–227. doi: 10.1111/j.1365-2958.2004.04106.x. [DOI] [PubMed] [Google Scholar]
- 22.Arnesano F, Banci L, Bertini I, Huffman DL, O’Halloran TV. Solution structure of the Cu(I) and apo forms of the yeast metallochaperone, Atx1. Biochemistry. 2001;40:1528–1539. doi: 10.1021/bi0014711. [DOI] [PubMed] [Google Scholar]
- 23.Anastassopoulou J, Banci L, Bertini I, Cantini F, Katsari E, Rosato A. Solution structure of the apo-and copper(I) loaded human metallochaperone HAH1. Biochemistry. 2004;43:13046–13053. doi: 10.1021/bi0487591. [DOI] [PubMed] [Google Scholar]
- 24.Peng JW, Wagner G. Mapping of spectral density function using heteronuclear NMR relaxation measurements. J Magn Reson. 1992;98:308–332. [Google Scholar]
- 25.Peng JW, Wagner G. Mapping of the spectral densities of N–H bond motions in eglin c using heteronuclear relaxation experiments. Biochemistry. 1992;31:8571–8586. doi: 10.1021/bi00151a027. [DOI] [PubMed] [Google Scholar]
- 26.Arnesano F, Banci L, Bertini I, Cantini F, Ciofi-Baffoni S, Huffman DL, O’Halloran TV. Characterization of the binding interface between the copper chaperone Atx1 and the first cytosolic domain of Ccc2 ATPase. J Biol Chem. 2001;276:41365–41376. doi: 10.1074/jbc.M104807200. [DOI] [PubMed] [Google Scholar]
- 27.Wimmer R, Herrmann T, Solioz M, Wüthrich K. NMR structure and metal interactions of the CopZ copper chaperone. J Biol Chem. 1999;274:22597–22603. doi: 10.1074/jbc.274.32.22597. [DOI] [PubMed] [Google Scholar]
- 28.Banci L, Bertini I, Cantini F, Ciofi-Baffoni S, Gonnelli L, Mangani S. Solution structure of Cox11: a novel type of alpha-immunoglobulin-like fold involved in CuB site formation of cytochrome c oxidase. J Biol Chem. 2004;279:34833–34839. doi: 10.1074/jbc.M403655200. [DOI] [PubMed] [Google Scholar]
- 29.Pickering IJ, Prince RC, George GN, Rauser WE, Wickramasinghe WA, Watson AA, et al. X-ray absorption spectroscopy of cadmium phytochelatin and model systems. Biochim Biophys Acta. 1999;1429:351–364. doi: 10.1016/s0167-4838(98)00242-8. [DOI] [PubMed] [Google Scholar]
- 30.Liu WT, Thorp HH. Bond valence sum analysis of metal-ligand bond lengths in metalloenzymes and model complexes. 2 Refined distances and other enzymes. Inorg Chem. 1993;32:4102–4105. [Google Scholar]
- 31.Coleman JE. Cadmium-113 nuclear magnetic resonance applied to metalloproteins. Methods Enzymol. 1993;227:16–43. doi: 10.1016/0076-6879(93)27004-z. [DOI] [PubMed] [Google Scholar]
- 32.Oz G, Pountney DL, Armitage IM. NMR spectroscopic studies of I=1/2 metal ions in biological systems. Biochem Cell Biol. 1998;76:223–234. doi: 10.1139/bcb-76-2-3-223. [DOI] [PubMed] [Google Scholar]
- 33.Pan T, Coleman JE. GAL4 Transcription factor is not a zinc finger but forms a Zn(II)2Cys6 binuclear cluster. Proc Natl Acad Sci USA. 1990;87:2077–2081. doi: 10.1073/pnas.87.6.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan T, Halvorsen Y, Dickson RC, Coleman JE. The transcription factor LAC9 from Kluyveromyces lactis-like GAL4 from Saccharomyces cerevisiae forms a Zn(II)2Cys6 binuclear cluster. J Biol Chem. 1990;265:21427–21429. [PubMed] [Google Scholar]
- 35.Perez-Lourido P, Romero J, Garcia-Vazquez JA, Sousa A, Maresca PK, Zubieta J. Synthesis and characterization of zinc and cadmium compounds with arenephosphinothiol ligands. Crystal and molecular structures of [Cd2{2-(Ph2PO)C6H4S}4], [Zn{2-(Ph2P)-6-(Me3Si)C6H3S}2], [Cd{2-(Ph2PO)-6-(Me3Si)C6H3S}2(CH3OH)], and [Zn{PhPO(C6H4S-2)2}(bipy)] Inorg Chem. 1999;38:3709–3715. doi: 10.1021/ic981405b. [DOI] [PubMed] [Google Scholar]
- 36.Gitschier J, Moffat B, Reilly D, Wood WI, Fairbrother WJ. Solution structure of the fourth metal-binding domain from the Menkes copper-transporting ATPase. Nature Struct Biol. 1998;5:47–54. doi: 10.1038/nsb0198-47. [DOI] [PubMed] [Google Scholar]
- 37.O’Neil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 38.Holm RH, Kennepohl P, Solomon EI. Structural and functional aspects of metal sites in biology. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 39.Henkel G, Krebs B. Metallothioneins: Zinc, cadmium, mercury, and copper thiolates and selenolates mimicking protein active site features–structural aspects and biological implications. Chem Rev. 2004;104:801–824. doi: 10.1021/cr020620d. [DOI] [PubMed] [Google Scholar]
- 40.Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallog sect B. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 41.Scott RA. Measurement of metal-ligand distances by EXAFS. Methods Enzymol. 1985;117:414–459. [Google Scholar]
- 42.Liu J, Stemmler AJ, Fatima J, Mitra B. Metal-binding characteristics of the amino-terminal domain of ZntA: binding of lead is different compared to cadmium and zinc. Biochemistry. 2005;44:5159–5167. doi: 10.1021/bi0476275. [DOI] [PubMed] [Google Scholar]
- 43.Hou Z, Mitra B. The metal specificity and selectivity of ZntA from Escherichia coli using the acylphosphate intermediate. J Biol Chem. 2003;278:28455–28461. doi: 10.1074/jbc.M301415200. [DOI] [PubMed] [Google Scholar]
- 44.Gaballa A, Helmann JD. Bacillus subtilis CPx-type ATPases: characterization of Cd Zn, Co and Cu efflux systems. Biometals. 2003;16:505. doi: 10.1023/a:1023425321617. [DOI] [PubMed] [Google Scholar]
- 45.Argüello JM. Identification of ion-selectivity determinants in heavy-metal transport P1B-type ATPases. J Membr Biol. 2003;195:93–108. doi: 10.1007/s00232-003-2048-2. [DOI] [PubMed] [Google Scholar]
- 46.Thompson JD, Higgins DG, Gibson TJ. CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 48.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Ferguson DM, et al. AMBER 5.0. University of California; San Francisco: 1997. [Google Scholar]
- 49.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 50.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structure. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 51.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, et al. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 52.Press WH, Flannery BP, Teukolsky SA, Vetterling WT. Numerical Recipes in C–The Art of Scientific Computing. Cambridge University Press; New York: 1988. [Google Scholar]
- 53.Zinn-Justin S, Berthault P, Guenneugues M, Desvaux H. Off-resonance RF fields in heteronuclear NMR. Application to the study of slow motions. J Biomol NMR. 1997;10:363–372. doi: 10.1023/A:1018365815186. [DOI] [PubMed] [Google Scholar]
- 54.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 55.Pervushin K, Riek R, Wider G, Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ankudinov A, Ravel B, Rehr JJ, Conradson SD. Real-space multiple-scattering calculation and interpretation of X-ray-absorption near-edge structure. Phys Rev ser B. 1998;58:7565–7575. [Google Scholar]
- 57.Brown ID, Altermatt D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crysallog sect B. 1985;41:244–247. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.