

Abstract
Recruitment of neutrophils and release of reactive oxygen species are considered to be major pathogenic components driving acute lung injury (ALI). However, NADPH oxidase, the major source of reactive oxygen species in activated phagocytes, can paradoxically limit inflammation and injury. We hypothesized that NADPH oxidase protects against ALI by limiting neutrophilic inflammation and by activating Nrf2, a transcriptional factor that induces anti-oxidative and cytoprotective pathways. Our objective was to delineate the roles of NADPH oxidase and Nrf2 in modulating acute lung inflammation and injury in clinically relevant models of acute gastric aspiration injury, a major cause of ALI. Acid aspiration caused increased ALI (as assessed by bronchoalveolar lavage fluid albumin concentration) in both NADPH oxidase-deficient (p47phox−/−) mice and in Nrf2−/− mice compared to wild-type mice. NADPH oxidase reduced airway neutrophil accumulation, but Nrf2 decreased ALI without affecting neutrophil recovery. Acid injury resulted in a 120-fold increase in mitochondrial DNA, a pro-inflammatory and injurious product of cellular necrosis, in cell-free bronchoalveolar lavage fluid. Pharmacologic activation of Nrf2 by the triterpenoid, CDDO-Im, limited aspiration-induced ALI in wild-type mice and reduced endothelial cell injury caused by mitochondrial extract-primed human neutrophils, leading to the conclusion that NADPH oxidase and Nrf2 have coordinated, but distinct, functions in modulating inflammation and injury. These results also point to Nrf2 as a therapeutic target to limit ALI by attenuating neutrophil-induced cellular injury.
Introduction
Acute lung injury (ALI) is a syndrome that results from a number of insults that damage the alveolar-capillary wall (ACW), leading to pulmonary edema and recruitment of inflammatory cells. Acute respiratory distress syndrome (ARDS), the most severe form of ALI, is associated with a high mortality rate (1, 2). Specific insults that result in direct cell injury, such as gastric aspiration, can cause a mild self-limited illness, or progress to fatal ARDS. This range of illness is likely to be modulated by both the nature of the caustic insult, underlying patient co-morbidities, and host factors governing the inflammatory response (3). Prior studies have implicated neutrophils (PMNs) and reactive oxygen species (ROS) in the pathogenesis of ALI (4–7). Interestingly, endogenous hydrogen peroxide can have anti-inflammatory, protective effects in experimental ALI (8). Thus, although ROS can cause direct cellular injury and activate inflammatory responses, they can also mediate signaling functions that are protective. The lack of demonstrated benefit for anti-oxidant strategies in improving ALI (9) may reflect ROS having both exacerbating and protective effects in ALI.
NADPH oxidase generates the oxidative “burst” in PMNs, leading to the production of ROS (e.g., superoxide anion, hydrogenperoxide, hydroxyl anion, and hypohalous acid), and activation and release of PMN granular proteases (10–13). The critical role of NADPH oxidase in antimicrobial host defense is demonstrated by chronic granulomatous disease, an inherited disorder of NADPH oxidase characterized by severe bacterial and fungal infections and by excessive inflammation, such as Crohn’s-like inflammatory bowel disease (14–16). In addition to this enzyme’s critical host defense function, NADPH oxidase also regulates inflammation. In studies of lung inflammation induced by microbial-derived products, NADPH oxidase restrained lung inflammation by activation of Nrf2, a redox-sensitive anti-oxidative and anti-inflammatory transcription factor (17).
Pathogen recognition receptors (e.g., TLRs) sample microbial motifs and initiate signaling that may result in NADPH oxidase activation. In addition to inhaled antigens and microbes, the lung is exposed to caustic irritants through inhalation and gastric acid aspiration. Common innate recognition pathways are activated by microbial products and released intracellular products of cell injury, termed damage associated molecular patterns (DAMPs) (18, 19). Seen in this light, ALI can result both from the direct insult and inflammation-induced injury primed by DAMPs.
This study uses clinically relevant models of aspiration-induced ALI to investigate mechanisms that modulate both inflammation and injury. There is a broad range of conditions that predispose to gastric aspiration-induced ALI (e.g., general anesthesia, alcohol and narcotic abuse, and neurological disorders). Gastric aspiration results in a spectrum of potential outcomes ranging from being asymptomatic, producing a rapidly resolved pneumonitis, or progression to a severe and sustained ALI. ARDS is seen in 10–25% of witnessed gastric aspiration events (20–23), and carries a mortality rate of 35–60% (22, 24, 25). Our laboratory has established gastric aspiration-induced ALI/ARDS models in mice and rats that have remarkable fidelity with human ALI in terms of histopathology, ACW permeability, severe hypoxemia, reduction in surfactant activity, lung compliance, and lung vital capacity (26–30). In these models, the ALI/ARDS picture is manifest when the contents of the aspirate contain both acidic and small gastric food particle components. When acid alone is instilled into the lungs a two-phase injury results. The initial injury phase (within 1 h of acid exposure) is primarily due to the acid’s direct caustic effects on pulmonary tissue (31), while the second injury phase (beginning at 3–4 h and peaking at 4–6 h post-exposure) results from recruited PMNs (32). When sterile small gastric particles are instilled into the lungs in a non-acidic vehicle the initial direct caustic tissue damage is not seen, but the acute PMN inflammatory phase is manifest within the same time frame as with acid-induced injury. In both cases, the inflammatory phase quickly resolves within 24 h. The combination of acid (caustic agent) and gastric particles (a non-caustic proinflammatory agent) produces a synergistic lung injury (i.e., greater than the additive effect of each single insult) that is sustained for ≥ 48 hr in mice and rats (27, 30). This synergistic lung injury is likely to be a major factor predisposing to ARDS following gastric aspiration. Together, these results point to both the aspiration insult (e.g., pH, particle concentration) and host factors that calibrate the inflammatory response influencing the severity of and recovery from ALI. Since NADPH oxidase and Nrf2 regulate inflammation and oxidative stress, it was reasoned that both pathways would have specific roles in modulating ALI following aspiration insults.
Our previous results demonstrated that NADPH oxidase-deficient (p47phox−/−) mice had increased and sustained pulmonary PMN accumulation, capillary leak, and proinflammatory cytokines compared to wild-type (WT) mice following acid aspiration (33). These were paradoxical results since NADPH oxidase-generated ROS would be expected to increase lung damage. We hypothesized that NADPH oxidase would reduce aspiration-induced ALI both by limiting neutrophilic inflammation and by activating Nrf2. In the current study, the roles of NADPH oxidase and Nrf2 in modulating acute lung inflammation and injury in response to clinically relevant models of aspiration were evaluated. NADPH oxidase and Nrf2 limited acid aspiration-induced ALI, but through distinct mechanisms. The major effect of NADPH oxidase was to reduce PMN alveolitis, whereas the major effect of Nrf2 was to decrease ALI without significantly affecting PMN airway accumulation. Together, these results challenge the notion that ROS have exclusively an injurious effect in driving ALI and point to a more complex role for NADPH oxidase-derived ROS in modulating inflammation and injury following caustic insult.
Materials and Methods
Mouse knockout models
Mice with a targeted disruption of the p47phox gene have a defective NADPH oxidase, rendering phagocytes incapable of generating measurable superoxide (34). p47phox−/− mice were derived from C57BL/6 and 129 intercrosses, and were backcrossed 14 generations in the C57BL/6 lineage. Nrf2−/− mice were generated from C57BL/6 and 129 intercrosses as described (35), and were backcrossed 9 generations in the C57BL/6 lineage. Nrf2−/− mice were kindly provided through a Material Transfer Agreement from Jefferson Chan, MD (University of Southern California School of Medicine, Irvine, CA). Knockout mice were expanded by homozygous mating at Roswell Park Cancer Institute. Male mice were used in all experiments (8–12 weeks old) and were maintained under specific pathogen free conditions. All procedures performed on animals were approved by the Institutional Animal Care and Use Committee at the Roswell Park Cancer Institute and complied with the U.S. Department of Health and Human Services’ Guide for the Care and Use of Laboratory Animals.
Gastric aspiration injury models
The gastric aspiration injury models in this study used an intratracheal (i.t.) instillation of one of the following injury vehicles: normal saline, pH ≈ 5.3 (vehicle control), saline + HCl, pH = 1.25 (acid), 7.5 mg/ml gastric particles, pH ≈ 5.3 (part.), or saline + HCl + 7.5 mg/ml gastric particles, pH = 1.25 (acid+part.), as previously described (30, 33). A ventral midline tracheostomy was performed following induction of isoflurane anesthesia and 3.6 ml/kg of injury vehicle + 0.2 ml air was instilled with a 1 cc syringe through a 22 gauge catheter inserted in the trachea (33). Mice were fluid-resuscitated with 1 ml normal saline injected subcutaneously into the ventral neck and allowed to recover in room air with food and water ad lib. Particles were derived from gastric filtrates, as previously described (28, 30). The particle preparation procedure removes gastric enzymes and bile salts and results in a mixture of gastric particles that ranged in size from 2 – 30 μm in diameter. The animals were sacrificed at 5 h post-injury vehicle instillation, the time of the peak synergistic inflammatory injury phase.
Bronchoalveolar lavage (BAL) and lung processing
Mice were anesthetized with isoflurane, exsanguinated by transecting the inferior vena cava, and the lung vasculature was flushed with 5 ml saline injected into the right ventricle. A 22-gauge cannula was secured in the trachea and BAL was performed with five 1 ml saline instillations (33). The lung lobes were collected, flash frozen in liquid N2, and stored at −80°C. For histological analysis, tracheas were cannulated and lungs fixed by insufflating with 10% neutral buffered formalin for 24 h. Lungs were then paraffin-embedded, cut in 5 μm sections, and stained with hematoxylin and eosin.
The recovered BAL was centrifuged at 1,500 × g for 3 min at 4°C. The pelleted cells were counted by a Coulter counter (Beckman Coulter, Fullerton, CA), and the white cell differential was assessed using a Diff-Quik® staining kit. Cell-free BAL supernatants were stored at −80°C until further analysis. BAL albumin concentration ([albumin]) was determined by ELISA (Bethyl Laboratories, Inc., Montgomery, TX). BAL TNFα bioactivity was assessed by a cytotoxicity assay using WEHI 164 subclone 13 cells, as previously described (26). Other cytokines were measured by ELISA, using capture and detection antibodies and recombinant cytokine standards from R&D Systems (Minneapolis, MN).
NAD(P)H:quinone oxidoreductase 1 (NQO1) quantitation in lung homogenates by Western blot
Flash frozen lungs were ground into a powder by a mortar and pestle with liquid nitrogen, suspended in buffer (150 mM NaCl, 1 M HEPES pH=7.9, 1% NP-40, 0.2 M EDTA, PMSF), and subjected to dounce homogenization. Nuclear and cytoplasmic extracts were generated from lung homogenates using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific, Rockford, IL) and stored at −80°C. In prior studies using this method, protein expression of GADPH, (a cytoplasmic protein), and TBP (a nuclear protein) were almost entirely restricted to the cytoplasmic and nuclear fractions of lung homogenates, respectively (data not shown). Proteins from cytoplasmic fractions were quantified using the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA). Samples (40 μg of protein/well) were loaded onto a 10% acrylamide gel, electrophoresed, and transferred to a PVDF membrane. The membrane was incubated with goat anti-NQO1 (Imgenex, San Diego, CA) or rabbit anti-actin (Sigma-Aldrich, St. Louis, MO) followed by alkaline phosphatase-conjugated donkey anti-goat or goat anti-rabbit secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Proteins were visualized with the ECF™ system (GE Healthcare, Piscataway, NJ) according to the manufacturer’s protocol and fluorescence was detected using the Storm® fluorescence scanner (GE Healthcare). Signal intensities were measured using ImageQuant™ software (GE Healthcare), and NQO1 expression was quantitated by fluorescence normalized to β-actin expression in each sample.
Treatment with CDDO-Im (1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole)
CDDO-Im is a semi-synthetic triterpenoid that directly induces Nrf2 activity (36). CDDO-Im was dissolved in PBS + 10% DMSO + 10% cremaphor-EL. In experiments using CDDO-Im treatment, mice were injected i.p. with 100 μl of 2 mg/ml CDDO-Im (0.2 mg/mouse), or vehicle, 5–10 min post-acid+part. instillation.
Extraction and quantitation of mitochondrial DNA (mtDNA) from cell-free BAL
Total DNA was isolated from 400 μl of cell-free BAL using the QIAamp DNA Mini kit (Qiagen, Valencia, CA) according to manufacturer’s instructions. DNA was eluted in a 40 μl volume and 5 μl was used for PCR reactions. Primers for mouse mtDNA 16S rRNA (forward 5′-CTAGAAACCCCGAAACCAAA-3′ and reverse 5′-CCAGCTATCACCAAGCTCGT-3′), and mouse nuclear DNA β2-microglobulin (forward 5′-ATGGGAAGCCGAACATACTG-3′ and reverse 5′-CAGTCTCAGTGGGGGTGAAT-3′) were synthesized (Invitrogen, Carlsbad, CA). Primer sequences have no significant homology with DNA found in any bacterial species published on NCBI. Purified mtDNA was extracted from mouse liver mitochondria (Mitochondria Isolation Kit for Tissue, Pierce Chemical, Rockford, IL), which was used for generation of standard curves. No protein contamination was detected in the purified mtDNA and nuclear DNA was minimal. Quantitative real time-PCR was performed in triplicate for absolute quantification of BAL mtDNA using SYBR® Green ER master mix (Invitrogen) in a 7900HT Fast Real Time PCR system (Applied Biosystems, Bedford, MA). Samples that produced no PCR products after 40 cycles were considered “undetectable”.
Assessment of mitochondrial damage-associated molecular pattern (MTD)-activated PMNs effects on in vitro endothelial cell permeability
EA.hy926 cells, derived by the fusion of HUVECs with the continuous human lung carcinoma cell line A549, were obtained from Dr. Cora-Jean C. Edgell (University of North Carolina at Chapel Hill, NC) and maintained in DMEM medium with 10% FBS and penicillin and streptomycin in a 5% CO2 at 37°C (37). Transendothelial electrical resistance was measured in EA.hy926 monolayers by seeding 2–3 x105 cells onto cysteine and fibronectin-pretreated 8W10E+ cultureware (40 electrodes/well) and incubating at 37°C, 5% CO2 in an electric cell-substrate impedance sensing system (ECIS, Applied BioPhysics, Troy, NY) (38, 39). This system measures changes in impedance of the monolayer to AC current flow. The micro-currents used have no detectable effect on the cells. Confluence was determined by capacitance at 64 kHz coming to a plateau (~10 nF) after an overnight incubation. In this system impedance decreases and capacitance increases as permeability increases. Endothelial cell permeability change was assessed as capacitance change at 64 kHz (38, 40). Capacitance values in each well were normalized to capacitance at the start of treatments. Values from each microelectrode were pooled at discrete time points and plotted versus time as the mean ± SD (N = 2 wells).
Mitochondria were isolated from 200 mg of mouse liver (Mitochondria Isolation Kit for Tissue, Pierce Chemical), suspended in 1 ml HBSS+protease inhibitor cocktail, and sonicated on ice (VCX130-Vibra Cell, Sonics and Materials, Newtown, CT) for 10 cycles of 30 s on and 30 s off at 50% amplitude. The disrupted mitochondria were centrifuged at 12,000 × g for 10 min at 4°C, and the supernatant was used as a crude MTD solution. PMNs from healthy human donors were isolated from heparinized blood using PMN isolation medium (Thermo Fisher Scientific, Hudson, NH) (40). At the time of permeability measurement, the medium in each well was replaced with 200 μl medium containing various dilutions of MTD or 20 μg/ml CpG (Invivogen, San Diego, CA) ± 2×105 PMNs ± 100 nM CDDO-Im, and incubated at 37°C in 5% CO2. The endothelial cell layer capacitance was evaluated over 20 hr. Studies performed using human PMNs were approved by the Institutional Review Board of the Beth Israel Deaconess Medical Center and conducted in accordance with the guidelines of the World Medical Association’s Declaration of Helsinki. Written informed consent was received from participants prior to inclusion in the study.
Survival analysis
Survival analysis of the WT and p47phox−/− mouse strains as a function of the different modes of aspiration was assessed by the Kaplan-Meier method with all data censored by 48 h. The comparison between the two survival curves for WT and p47phox−/− mice at different gastric particle concentrations was made using the Mantel-Cox log-rank test.
Statistical analysis
Descriptive statistics were expressed as mean ± SEM. Comparisons between genotypes of continuous variables were assessed by Student’s t-test with Bonferroni correction for multiple comparisons after confirmation of normal distribution by Shapiro-Wilk W testing. If data was not normally distributed, the Mann-Whitney-Wilcoxon rank-sum test was used to determine statistical significance. Prism for Mac X OS version 5.0c (GraphPad Software, San Diego, CA) software was used for the statistical analyses mentioned above.
To evaluate synergy between pulmonary exposure to acid and gastric particles, a 2-way ANOVA using a generalized linear regression fit model was performed using JMP® v.7.0.1 (SAS Institute, Cary, NC) statistic software. Synergy was defined as an interaction between variables (e.g. acid + particles) resulting in an effect (e.g., albumin leak) that is significantly greater than additive. The primary analysis consisted of fitting a standard 2 × 2 factorial ANOVA model with factors for acid (no, yes) and particulate (no, yes) for each genetic mouse strain (i.e., WT, p47phox−/−, or Nrf2−/−). Each factor in the model was coded as a 0/1 indicator variable such that it could be reparameterized in a generalized linear regression fit model. The full model consisted of the main effects of the 2 factors (i.e., acid, particulate) and their interaction (i.e., acid x particulate), where the interactions within the regression model were given as products of indicator variables corresponding to the main effects. The equation fitted for each time point was as follows:
with y = measured parameter (i.e., BAL [albumin]), A = intercept, B = acid effect, b = acid indicating variable (0 = no acid, 1 = acid), C = particulate effect, c = indicating variable (0 = no particulate, 1 = particulate), and D = acid/particulate interaction effect. The fit model was performed without polynomial centering, to accentuate the analysis for interaction, as opposed to the main effects of the 2 factors. Data presented in the figures that are statistically significant for synergistic interaction (more than additive) are indicated with the “§” symbol over the appropriate group on the figure.
Results
NADPH oxidase limits aspiration-induced ALI
Different models of gastric aspiration that mimic what is observed in the clinic were utilized to understand mechanisms of aspiration-induced ALI. Aspiration of acid alone results in direct caustic injury, whereas aspiration of particles in a non-acidic vehicle causes foreign-body-induced inflammation. Challenge with acid plus gastric particles resulted in inflammation-mediated synergistic lung injury results (i.e., an interaction that produces injury greater than the injuries produced by acid or particles alone), as assessed by extravasation of albumin into the airspaces, as well as blood oxygenation (i.e., PaO2/FiO2) (27–30). Histopathological analysis at 5 h after challenge demonstrated distinct patterns of lung inflammation and injury in WT mice as a function of the aspiration vehicle contents. Acid alone resulted in direct epithelial injury with denuded bronchiolar epithelium and necrotic cells and debris within the bronchial and bronchiolar airspaces with moderate PMN infiltrate; gastric particles alone provoked generalized PMN infiltration as well as focal areas of dense PMN inflammation without evidence of necrotic cells; and combined acid and gastric particles led to a similar PMN infiltration pattern as seen in part.-injury, but also resulted in focal areas of bronchial epithelial cell necrosis (Figure 1). These results establish that the nature of the aspirate is linked to the extent of acute lung inflammation and injury.
Figure 1. Pulmonary histopathology of WT mice 5 h following acid and gastric particulate aspiration.

Normal saline (saline) + HCl, pH = 1.25 (acid), 7.5 mg/ml small, non-acidified gastric particles (part.), or 7.5 mg/ml part. + HCl, pH = 1.25 (acid+part.) was instilled into the lungs, intratracheally (i.t.), of WT mice. The mice were sacrificed 5 h later, and lung sections were analyzed by H&E. Boxes in the left column images (bars = 100 μm) indicate areas that are magnified in their respective right column images (bars = 25 μm). A & B) uninjured; C & D) In acid-injured mice, necrotic cells and debris within the bronchial and bronchiolar airspaces (arrows) with moderate PMN infiltrate and denuded bronchiolar epithelium (arrow heads) were observed; E & F) Administration of particles alone resulted in generalized PMN infiltration and focal areas of dense PMN accumulation in the alveolar spaces and in some of the bronchioles without evidence of necrotic cells; G & H) In mice administered acid+part., both PMN infiltration similar to mice administered particles alone and necrotic bronchial epithelial cells (arrows) were observed. Images shown are representative of lungs from 2 – 3 mice from 3 independent experiments.
Using these distinct clinically relevant modes of aspiration, this study sought to understand host factors that modulate acute lung inflammation and injury. Our previous results demonstrated that NADPH oxidase-deficient (p47phox−/−) mice developed significantly greater alveolar PMN leukocytosis and BAL [albumin] (a marker of loss of ACW integrity) compared with WT mice (33). Based on these findings, a comprehensive evaluation of the role NADPH oxidase plays in modulating direct caustic injury (acid) versus foreign body-induced PMN inflammation (particles) and combined insult (acid + particles) was conducted. The analysis was focused on an early time point (5 h) after aspiration based on previous findings that PMN-dependent ALI begins at 3–4 h and peaks at 4–6 h following the initial caustic insult (32).
The p47phox−/− mice were more susceptible to acid+part.-induced mortality compared to similarly treated WT mice (Figure 2 and Supplement Figure 1), strongly supporting a beneficial role of NADPH oxidase in mitigating injury. Administration of acid alone or particles alone did not result in mortality in either genotype. To test the hypothesis that NADPH oxidase plays a role in the synergistic lung injury induced by gastric acid and particle aspiration, BAL [albumin] was measured from WT and p47phox−/− mice that were injured by pulmonary instillation of saline, acid, part., or acid+part. and sacrificed 5 h later. The particle concentration in the part., and acid+part. injury models was limited to 7.5 mg/ml so a non-lethal lung injury could be studied.
Figure 2. Effect of acid+part. particle concentration on survival of WT and p47phox−/− mice.
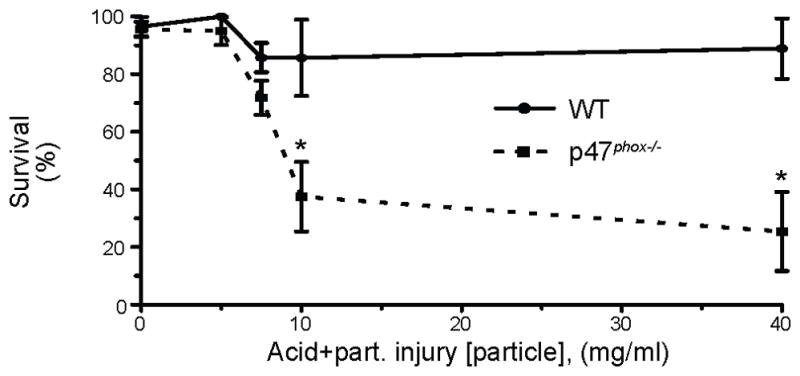
Acid+part. injury vehicle was instilled, i.t., into the lungs of WT and p47phox−/− mice at the indicated [particle]. Experiments were designed to sacrifice the mice at 5, 24, or 48 h post-injury. The data was censored at the designated sacrifice time point, therefore, 48 h was the maximum time of censor. WT (filled circle with solid line) and p47phox−/− (filled square with dotted line) survival displayed as mean ± SEM. For WT mice: N = 59 (0 mg/ml), 13 (5 mg/ml), 68 (7.5 mg/ml), 8 (10 mg/ml), 10 (40 mg/ml). For p47phox−/− mice: N = 92 (0 mg/ml), 20 (5 mg/ml), 97 (7.5 mg/ml), 16 (10 mg/ml), 40 (40 mg/ml). Data was assembled from 50 independent experiments. In addition, forty-five p47phox−/− mice had 40 mg/ml part.-alone instilled, i.t., into the lungs and none died. * P < 0.05 for comparison between genotypes at the same particle concentration by Mantel-Cox log-rank test. Individual Kaplan-Meier plots are displayed in Supplemental Figure 1.
Consistent with a previous study (33), the acid pulmonary insult produced an increase (>2.5-fold) in albumin leakage in the p47phox−/− genotype compared to the WT mice (12,317±3,581 μg/ml compared to 3,929±994 μg/ml, P<0.025), pointing to a protective role for NADPH oxidase in acid aspiration. The p47phox−/− genotype did not have a significant effect on BAL [albumin] in the saline (vehicle control), part., or acid+part. injury groups. Also consistent with previous results (30), WT mice demonstrated a synergistic increase in lung injury (assessed by albumin leakage into the airspaces) that resulted from the combined injuries, as determined by 2-way ANOVA, P<0.03 for interaction (Figure 3A). However, the synergistic increase in lung injury did not occur in the p47phox−/− mice (P>0.96). The synergistic increase in BAL [albumin] in the WT mice was 165% more than what would be expected if the vehicle instillation, acid, and particulate main effects were additive. Using the same calculation, the aspiration injury in p47phox−/− mice resulted in only a 2% increase over the additive effects, showing that NADPH oxidase is required for synergistic ALI. Together, these results demonstrate a protective role for NADPH oxidase in enhancing survival following acid+part. challenge and in limiting acid aspiration-induced ALI.
Figure 3. Effect of p47phox genotype on acid and gastric particulate aspiration-induced synergistic lung injury and inflammation.

Saline, acid, part., or acid+part. was instilled into the lungs, i.t., of WT and p47phox−/− mice. BAL was performed 5 h later and the recovered BAL was assessed for: A) albumin concentration ([albumin]), as an indicator of lung injury (white inset bars within the acid+part. data represent predicted values if injury was additive); B) PMN alveolitis; and C) ratio of BAL [albumin] to # of recovered PMN, an indicator of the degree of lung injury/PMN. N = 6 – 9 for each group from 7 independent experiments. * P < 0.05 for comparisons indicated by the brackets, § P < 0.05 for synergistic interaction of acid and particulate components within the indicated genotype by 2-way ANOVA (i.e., WT or p47phox−/− mice).
NADPH oxidase limits PMN accumulation into the airspaces following aspiration
There was substantial discordance in the accumulation of PMNs into the airspaces compared to the BAL [albumin] (Figure 3B). When injured with acid-alone, the p47phox−/− mice had a 2.7-fold increase in the number of PMNs in the BAL compared to WT mice (2.2±0.4×105 PMNs compared to 8.0±2.4×104 PMNs, P < 0.013). The p47phox−/− mice experienced a >5.8-fold increase in BAL PMNs, compared to WT mice after part. injury (2.7±0.5×106 PMNs compared to 4.7±1.2×105 PMNs, P < 0.004), and a >3.8-fold increase over WT mice after acid+part. injury (2.4±0.4×106 compared to 6.2±1.1×105 PMNs, P < 0.0008).
Paradoxically, accumulation of PMNs into the airspaces did not correlate with the BAL [albumin] caused by the different injury models in either mouse genotype. To assess injury relative to inflammation, the ratio of BAL [albumin]:PMN recovery was analyzed as a function of genotype and aspiration contents; a higher ratio indicating increased ACW dysfunction per recovered PMN. The BAL [albumin]:PMN ratio was greatest for acid and lowest for part. aspiration in both genotypes (Figure 3C). These results are consistent with similar aspiration experiments in rats (data not shown). The p47phox−/− mice also exhibited a lower BAL [albumin]:PMN ratio following part. and acid+part. aspirations compared to similarly treated WT mice (Figure 3C). These findings point to NADPH oxidase limiting the degree of PMN alveolitis while increasing the injury per recovered PMN.
NADPH oxidase modulates cytokine responses in gastric aspiration
BAL was analyzed for concentrations of the acute pro-inflammatory cytokines, TNFα and IL-1β (Figures 4A and 4B). Part. injury led to a robust TNFα response that was blunted when the gastric particulate injury vehicle was acidified (i.e., acid+part.) in the WT mice (525±95 pg/ml and 194±50 pg/ml, respectively, P<0.008), and, to a much greater degree, in p47phox−/− mice (1,059±150 pg/ml and 57±15 pg/ml, respectively, P<0.0001). BAL [TNFα] was two-fold greater in p47phox−/− compared to WT mice in response to part. (P<0.0085), but was lower in p47phox−/− compared to WT mice following acid injury (p=0.022) and acid+part. injury (p=0.018). Thus, paradoxically, there was an inverse relationship between BAL [TNFα] (Figure 4A) and BAL [albumin] per recovered BAL PMN in both genotypes (Figure 3C).
Figure 4. Effect of p47phox genotype on acid and gastric particulate aspiration-induced pulmonary proinflammatory cytokine and CXC chemokine production.

Cell-free BAL recovered from WT and p47phox−/− mice 5 h following pulmonary injury, as described in Figure 3, were analyzed for: A) TNFα, by WEHI bioassay; B) IL-1β, by ELISA; C) KC, by ELISA; and D) MIP-2, by ELISA. N = 6 – 9 for each group from 7 independent experiments. * P < 0.05 and † 0.05 < P < 0.1 for comparisons indicated by the brackets.
In contrast to BAL [TNFα] data, BAL IL-1β levels followed the expected paradigm of pro-inflammatory cytokines with regard to the loss of ACW integrity (Figure 4B). Acid+part. aspiration resulted in an increase in BAL [IL-1β] relative to part.-induced albumin BAL levels in both WT mice (6,932±1,232 pg/ml and 201±51 pg/ml, respectively, P<0.0001) and in p47phox−/− mice (7,993±1,673 pg/ml and 450±104 pg/ml, respectively, P<0.0005). There was no difference in BAL [IL-1β] between the two genotypes for any of the three aspiration injuries.
Based on the increased BAL PMN leukocytosis in p47phox−/− following aspiration injury (Figure 3B), BAL [keratinocyte chemoattractant (KC)] and [MIP-2], the major PMN chemoattractant chemokines, were evaluated (Figures 4C and 4D). There was a small increase in BAL [MIP-2] in p47phox−/− compared to WT mice injured with acid+part. (51.7±7.0 pg/ml and 31.1±0.5 pg/ml, respectively; P<0.015). As with the BAL [TNFα], part. caused greater BAL [KC] compared to acid+part. in both WT mice (406±44 pg/ml and 81±16 pg/ml, respectively, P<0.0001) and in p47phox−/− mice (477±88 pg/ml and 134±43 pg/ml, respectively, P<0.003). A similar effect was observed with BAL [MIP-2] in WT mice (166±28 pg/ml and 31±0.5 pg/ml, respectively, P<0.0001) and in p47phox−/− mice (244±42 pg/ml and 52±7 pg/ml, respectively, P<0.0003). No significant differences in BAL concentrations occurred between the genotypes with regards to other inflammation-associated cytokines tested (IFNγ, IL-6, IL-10, IL-12, IL-17, MCP-1, MIP-1α). These results point to NADPH oxidase modulating airway levels of pro-inflammatory cytokines as a function of the type of aspiration insult.
Nrf2−/− mice have increased lung injury compared to WT mice following acid aspiration, but have a distinct phenotype from NADPH oxidase-deficient mice
Nrf2 is a transcription factor that is activated by intracellular oxidants to upregulate expression of numerous antioxidant and cytoprotective genes (41). Since NADPH oxidase had a protective role following acid aspiration, we hypothesized that oxidant-induced activation of Nrf2 might be a mechanism by which NADPH oxidase limits ALI. To test this hypothesis, saline, acid, part., or acid+part., was instilled i.t., in WT and Nrf2−/− mice, followed by sacrifice at 5 h. Similar to p47phox−/− mice, acid produced an increase (>2-fold) in BAL albumin levels in Nrf2−/− compared to WT mice (8,227±761 μg/ml and 3,929±994 μg/ml, respectively, P<0.005) (Figure 5A). Interestingly, there was no difference in BAL [albumin] between the Nrf2−/− and WT mice following the part. or acid+part. insults. In contrast to p47phox−/− mice, synergistic lung injury occurred in Nrf2−/− mice subjected to acid+part., with the increase in BAL [albumin] being 126% more than what would be expected if the vehicle instillation, acid, and particulate main effects were additive.
Figure 5. Effect of Nrf2 genotype on acid and gastric particulate aspiration-induced synergistic lung injury and inflammation.

BAL recovered from WT and Nrf2−/− mice 5 h following pulmonary injury, as described in Figure 3, were analyzed for: A) [albumin] (white inset bars within the acid+part. data represent predicted values if injury was additive); B) PMN alveolitis; and C) ratio of BAL [albumin] to # of recovered PMN, an indicator of the degree of lung injury/PMN. N = 6 – 9 for each group from 7 independent experiments. * P < 0.05 for comparisons indicated by the brackets, § P < 0.05 for synergistic interaction of acid and particulate components within the indicated genotype by 2-way ANOVA (i.e., WT or Nrf2−/− mice).
Nrf2 has no effect on PMN accumulation into the airspaces following gastric aspiration
Nrf2−/− mice had levels of PMN inflammation that were similar to WT mice following all gastric aspiration insults (Figure 5B). These findings are in contrast to the increased BAL PMN leukocytosis observed in p47phox−/− mice after acid, part., and acid+part. administration (Figure 3B). In addition, Nrf2−/− mice had greater BAL [albumin]:PMN ratio (a reflection of injury per recovered PMN) compared to WT mice following acid aspiration (0.24±0.06 μg/ml/PMN and 0.11±0.03 μg/ml/PMN, respectively, P = 0.02), reflecting greater injury per recovered PMN. (Figure 5C). Together, these results demonstrate that while both NADPH oxidase and Nrf2 limit acid aspiration-induced ALI, they do so through distinct mechanisms. The major effect of NADPH oxidase is to reduce BAL PMN alveolitis, whereas the major effect of Nrf2 is to decrease lung injury without significantly affecting airway PMN accumulation.
NADPH oxidase is required for full Nrf2 activation following acid aspiration
Our prior studies showed that NADPH oxidase limited zymosan-induced acute lung inflammation, in part, through activation of Nrf2 (17). We, therefore, postulated that NADPH oxidase would also be required for Nrf2 activation following acid insult. To assess Nrf2 activation in injured lungs, protein levels of NQO1 were examined in cytoplasmic extracts of lung homogenates of saline- and acid-injured WT, p47phox−/− and Nrf2−/− mice, the latter serving as a specificity control for Nrf2-independent NQO1 expression (Figure 6). There was a small increase in NQO1 levels in WT lungs following acid injury compared to the saline-injured group (3.54±0.19×104 NQO1/β-actin density units and 2.82±0.27×104 NQO1/β-actin density units, respectively, P<0.025). NQO1 levels were reduced in lungs of p47phox−/− compared to WT mice following both saline and acid administration, and, in contrast to WT mice, acid challenge did not increase NQO1 levels in the lungs of p47phox−/− mice. Lung NQO1 protein expression was lowest in Nrf2−/− mice. Although NADPH oxidase and Nrf2 have distinct functions in modulating inflammation, these results support a model in which NADPH oxidase-induced Nrf2 activation is one of a number of pathways that protects against inflammation-mediated injury.
Figure 6. NADPH oxidase stimulates the expression of NQO1, a Nrf2-inducible gene.
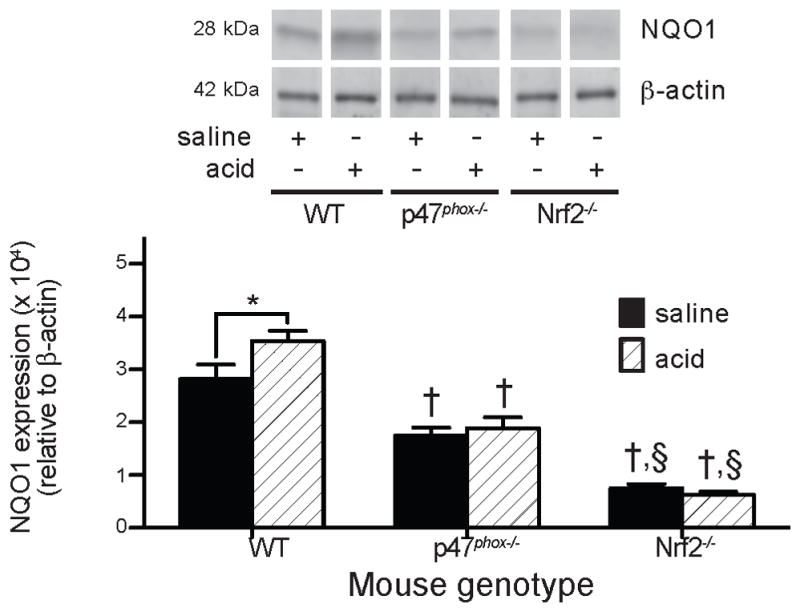
NQO1 protein levels were assessed in cytoplasmic extracts of lungs harvested from WT, p47phox−/−, and Nrf2−/− mice at 5 h after saline or acid injury by quantitated Western blot and normalized to β-actin expression. Group replicates were run on the same gel and bands from representative lanes are displayed. N = 7 – 9 for each group from 7 independent experiments. * P < 0.025 for comparison indicated by bracket, † P < 0.004 compared to WT mice of the same injury, § P < 0.0001 compared to p47phox−/− mice of the same injury.
CDDO-Im, a Nrf2 activator, is protective in aspiration-induced ALI
CDDO-Im is a triterpenoid that potently activates Nrf2 (36). Triterpenoids directly interact with thiol groups of the Keap1 redox sensor, leading to its dissociation from Nrf2 (42). We, therefore, asked whether CDDO-Im could be used therapeutically in aspiration-induced ALI. WT mice had acid+part. instilled into the lungs, i.t., followed by i.p. CDDO-Im (0.2 mg/mouse) or vehicle 15 min later. Acid+part. was selected because it results in the greatest increase in injury and PMN recruitment. The 15 min time point was chosen to simulate administering a therapeutic agent early following a witnessed aspiration event. CDDO-Im treatment had no effect on BAL albumin levels at 5 h post-acid+part., but led to a 4-fold decrease in albumin leak at 24 h post-injury (4.2±1.1 mg/ml and 16.8±6.6 mg/ml, respectively, P<0.02) (Figure 7A). Macrophage BAL recovery was similar between treatment groups. Paradoxically, although CDDO-Im did not affect BAL PMN numbers at 5 h, it led to ≈60% increase in BAL PMN recovery at 24 h (8.4±0.8×105 and 5.3±0.7×105 PMNs in CDDO-Im and placebo-treated mice, respectively, P<0.03) (Figure 7B). At 24 h post-acid+part., CDDO-Im led to a lower BAL [albumin]:PMN ratio compared to placebo (0.007±0.001 μg/ml/PMN and 0.041±0.021 μg/ml/PMN, respectively, P<0.016) (Figure 7C). NQO1 protein expression was increased in lung homogenates of mice that received CDDO-Im at both 5 h and 24 h (Figure 7D). Thus, despite leading to an increase in airway PMN accumulation, CDDO-Im reduced ALI following acid+part. aspiration.
Figure 7. Effect of CDDO-Im treatment on acid and gastric particulate aspiration-induced lung injury, inflammation, and Nrf2-activated NQO1 expression.

WT mice were injured by i.t. instillation of acid+part. then treated 15 min later by i.p. injection of 100 μl of 2 mg/ml CDDO-Im in treatment vehicle (PBS + 10% DMSO + 10% cremaphor-EL) or vehicle alone. Mice were sacrificed and BAL performed at 5 and 24 h post-injury and recovered BAL analyzed for: A) [albumin]; B) PMN alveolitis; and C) ratio of BAL [albumin] to # of recovered PMN, an indicator of the degree of lung injury/PMN. D) In addition, the lungs were removed, homogenized, and separated into cytosolic and nuclear fractions. The cytosolic fraction was analyzed by Western blot, probed for NQO1 (a gene product under the control of the transcription factor Nrf2), and quantitated by fluorescence normalized to β-actin expression in each sample. N = 4 – 6 for each group from 1 experiment. * P < 0.05 for comparisons indicated by the brackets.
Acid aspiration causes rapid release of MTDs
Histopathological assessment of the pulmonary inflammation 5 h after instillation of the gastric aspirate indicated that the acid-containing aspirates caused necrosis of pulmonary epithelial cells, whereas, the part.-only injury did not (Figure 1). Zhang et al. (19) showed that mitochondrial DAMPs (MTDs) released systemically following traumatic injury activate PMNs and induce PMN-mediated lung injury. We hypothesized that MTDs released following acid insult may prime PMN-mediated ALI. Instillation of acid, i.t., in WT mice resulted in a 120-fold increase in recovery of mtDNA (an MTD) (measured by qPCR of the mitochondrial 16S rRNA gene) in cell-free BAL (Figure 8A). At 5 and 60 min post-aspiration, the mean percentage of PMNs in the BAL was <5% of the total cells recovered (data not shown). Thus, the recovered mtDNA at these early time points likely represents direct acid-induced cell disruption rather than inflammation-induced injury. MtDNA was not detected in serum (data not shown), suggesting restriction of the direct cell injury to the air space tissues of the lungs.
Figure 8. Acid aspiration results in release of mitochondrial DAMPs (damage-associated molecular patterns) (MTD) into the pulmonary airspaces. CDDO-Im limits endothelial cell permeability induced by MTDs and by TLR-9-primed PMNs.

A) WT mice were injured by i.t. instillation of acid then sacrificed 5, 30, 60, or 300 min later (0 min represents data from uninjured mice), and BAL performed. Cells and debris recovered in the BAL were removed by centrifugation and the supernatant analyzed for mtDNA, an MTD, by qPCR. N = 3 – 5 for each group from 2 independent experiments. * P < 0.05 compared to uninjured control. B) EA.hy926 cells (an endothelial cell-derived cell line) were cultured to confluence in wells of an ECIS electrode array and freshly isolated human PMNs (2×105/well) + various dilutions of MTDs derived from rat liver mitochondria added. Capacitance, an indicator of endothelial cell permeability, was monitored in real-time. Unstimulated PMNs did not produce an appreciable change in endothelial cell permeability, but the combination of MTDs and PMNs caused an increase that was dependent on the MTD dose. C) Addition of CDDO-Im (100 nM) reduced MTD+PMN-mediated endothelial cell injury. D) Exposure of endothelial cells to MTDs in the absence of PMNs resulted in an increase in endothelial cell permeability that was reduced by CDDO-Im. E) To investigate the effect of CDDO-Im on PMN-mediated injury separate from direct endothelial injury caused by MTDs, TLR-9-primed PMNs were used. CDDO-Im attenuated endothelial cell permeability caused by PMNs activated by CpG sequences. Together, these results show that CDDO-Im reduces endothelial cell injury induced directly by MTDs and by TLR-9 activated PMNs. Data are from continuous averaging of the capacitance measured by each of the 40 electrodes in the well’s electrode array. Error bars represent mean ± SD of 2 wells at the indicated discrete time point from 1 experiment.
CDDO-Im limits endothelial cell permeability induced by MTDs and by activated PMNs
Based on our in vivo findings that aspiration injury leads to the rapid release of mtDNA and that CDDO-Im reduced ALI, an in vitro approach was used to dissect the roles of MTDs and unstimulated versus activated PMNs in causing endothelial cell injury and the protective effect of CDDO-Im. A confluent endothelial cell monolayer was exposed to isolated human PMNs and an MTD extract derived from lysed mitochondria isolated from rat liver, and a biocapacitance system was used to monitor real-time changes in endothelial permeability changes as previously described (43). The combination of PMNs and MTDs increased endothelial cell permeability, and the injury was dependent on the MTD concentration (Figure 8B). Addition of CDDO-Im reduced endothelial cell injury caused by MTDs and PMNs, while unstimulated PMNs did not result in increased endothelial cell permeability compared to medium (Figure 8C). MTDs in the absence of PMNs resulted in increased endothelial cell permeability, which was reduced by CDDO-Im (Figure 8D). The MTD extract contains mitochondrial DNA (a ligand of TLR-9), formylated peptides, and likely other products that can prime innate immune responses and cause cellular injury (19). To investigate the effect of CDDO-Im on PMN-mediated injury as distinct from direct endothelial injury caused by MTDs, we used TLR-9-primed PMNs. In prior studies, CpG sequences (a pure TLR-9 ligand) did not directly affect endothelial cell permeability, but stimulated neutrophil injury to endothelial cells (43). Addition of CDDO-Im attenuated endothelial cell injury caused by CpG-primed PMNs (Figure 8E). Together, these results show that CDDO-Im can mitigate endothelial cell injury induced directly by MTDs and by TLR-9-activated PMNs.
Discussion
These studies demonstrate important interactions between NADPH oxidase and Nrf2 as host factors that modulate inflammatory stress and different components involved in aspiration pneumonitis and injury. Acid aspiration is a caustic insult that leads to damage and necrosis of alveolar cells and release of DAMPs that activate innate immune responses. DAMP-primed PMNs are recruited to the lung and amplify injury; this inflammation-induced injury that follows the initial insult can lead to sustained ALI and ARDS. In PMNs, NADPH oxidase is rapidly activated by infectious threat and other stimuli, and is the major source of ROS generation that targets microbes. The immediate effect of ROS generation from NADPH oxidase is expected to augment injury to cells and extracellular matrix. NADPH oxidase activation in neutrophils is also linked to generation of neutrophil extracellular traps (NETs) (13, 44, 45), which contain serine proteases and other anti-microbial products that are potentially injurious. While NETotic neutrophils can exacerbate ALI (46), studies on the specific role of proteases in aspiration-induced ALI have produced different results (32, 47, 48). Paradoxically, NADPH oxidase-deficient mice have augmented lung inflammation and capillary leak after acid challenge, pointing to NADPH oxidase also mediating protective responses that limit injury. Our results support a model in which NADPH oxidase limits injury through a number of pathways. First, NADPH oxidase limits the extent of PMN alveolitis. Second, NADPH oxidase-derived ROS are associated with activation of Nrf2, a redox-sensitive transcriptional factor that induces anti-oxidant and cytoprotective responses. Our proposed model for the interaction of NADPH oxidase and Nrf2 in modulating inflammation and injury following gastric acid challenge is summarized in Figure 9.
Figure 9. Putative model of interaction of NADPH oxidase and Nrf2 in modulating inflammation and injury following gastric aspiration.
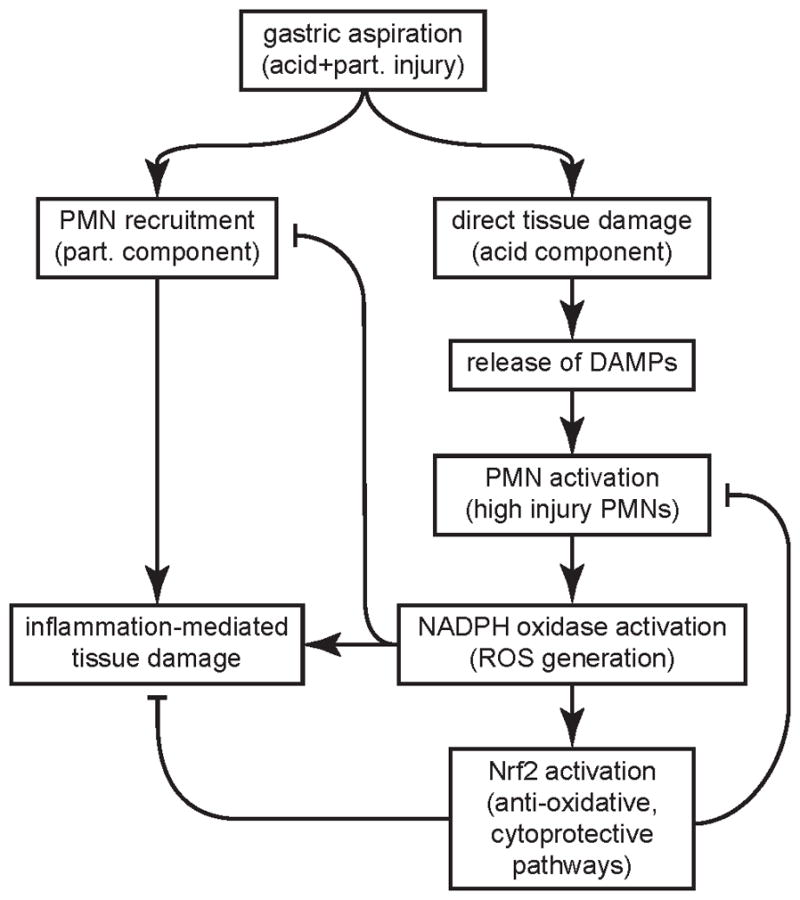
The acid component of the gastric aspirate leads to direct damage and necrosis of alveolar cells releasing DAMPs that activate PMNs producing a highly injurious PMN phenotype. Acid and particle aspiration results in synergistic ALI due to robust recruitment of PMNs and DAMP-induced priming of PMN-mediated injury. NADPH oxidase activation produces the PMN “respiratory burst” (generation of superoxide anion and downstream ROS) and release of sequestered PMN granular proteases that augment injury to cells and extracellular matrix. Despite these early pro-injurious effects, the results from this study point to NADPH oxidase limiting ALI through two mechanisms. NADPH oxidase reduces neutrophilic alveolitis and activates Nrf2, which induces anti-oxidative and cytoprotective pathways that counteract the ROS-mediated tissue damage and downmodulate the high injury PMN phenotype. They also support Nrf2 as a therapeutic target to limit ALI.
BAL PMN numbers were reduced in WT compared to p47phox−/− mice following all of the tested aspiration models. NADPH oxidase stimulation of PMN apoptosis (49) and macrophage-mediated clearance of apoptotic PMNs (efferocytosis) (50, 51), may be pathways that dampen PMN inflammation. In contrast to NADPH oxidase, the major effect of Nrf2 was to limit ALI without modulating PMN alveolitis. Nrf2 is activated by oxidative stress, and induces numerous cytoprotective pathways, such as phase 2 anti-oxidant enzymes and PPARγ (52, 53), that can limit organ injury. Together, these results point to distinct roles for NADPH oxidase and Nrf2 in modulating inflammation and injury as a function of the nature of the insult. Since NADPH oxidase and Nrf2 are expressed in several lineages, a limitation of these experiments is that they do not provide knowledge regarding in which specific cell lineages NADPH oxidase and Nrf2 activity modulate inflammation and injury.
Circulating mitochondrial DAMPs that are released following cellular injury can activate PMNs by ligation of specific pathogen recognition receptors and elicit PMN-mediated lung capillary leak in a rat model that mimics sepsis-induced ARDS (19). Mitochondrial DNA was isolated in cell-free BAL from acid-injured mice leading to the speculation that mitochondrial DAMPs released following acid-induced cellular necrosis may activate phagocytes and augment lung injury beyond the immediate cellular damage caused by acid instillation. The Nrf2 electrophile activator, CDDO-Im, was protective in acid+part.-induced ALI. The CDDO-Im was administered 15 minutes after aspiration to simulate administering a therapeutic following witnessed aspiration. The reduction in ALI was not observed at 5 h after aspiration, but was apparent at 24 h. These results suggest that Nrf2 activation induces cytoprotective pathways that do not affect the immediate injury secondary to gastric aspiration, but likely modulate inflammation-induced ALI. Paradoxically, CDDO-Im limited aspiration-induced ALI while increasing BAL PMN recovery (Figure 7B), a finding consistent with CDDO-Im reducing the capacity of recruited PMNs to cause tissue injury (Figure 7C). CDDO-Im could act at the level of recruited PMNs, lung stromal cells (e.g. endothelial and epithelial cells), or both. A limitation of these studies is that CDDO-Im can affect a number of pathways, and therefore its protective effect may be Nrf2-independent. Consistent with these in vivo results, CDDO-Im attenuated endothelial cell injury caused by MTDs and by activated PMNs (Figure 8). These in vivo and in vitro results suggest that targeting Nrf2 can limit inflammation-induced injury.
Nrf2 regulates the expression of numerous genes in vivo, most of which have cytoprotective and antioxidant functions that mitigate cellular stress induced by electrophiles and free radicals (41). The reactive oxidants, H2O2 and hypochlorous acid, activate Nrf2 in both macrophages and airway epithelial cells, in vitro (54–57). In prior studies, both acid and acid+part. aspiration result in reduced pulmonary antioxidant reserve capacity (58). These findings, along with the present study results, lead us to postulate that activation of Nrf2 may limit ALI, in part, by mitigating inflammation-induced oxidative stress.
While both p47phox−/− mice and Nrf2−/− mice had ≈2-fold greater ALI following acid aspiration compared to similarly treated WT mice (Figures 3A and 5A), there were important phenotypic differences between the two knockout mice. NADPH oxidase limited BAL PMN inflammation (Figure 3B), but Nrf2 did not affect PMN recovery (Figure 5B). Indeed, NADPH oxidase activation had either a neutral or aggravating effect on ALI when ALI was normalized to BAL PMN recovery (Figure 3C). In contrast, Nrf2−/− mice had a greater BAL [albumin]:PMN ratio compared to WT mice following acid aspiration, supporting the conclusion that Nrf2 is involved in decreasing the injury produced by recruited PMNs. This finding may reflect decreases in ROS scavenging or other cytoprotective mechanisms in Nrf2-deficient PMNs (41, 59). A change in antioxidant capacity would be predicted to produce a greater injury with decreased PMN numbers. Additionally, NADPH oxidase, but not Nrf2, was required for the synergistic lung injury resulting from the combination of acid and gastric particle aspiration. These results clearly indicate that NADPH oxidase and Nrf2 activation have distinct roles in modulating aspiration-induced lung inflammation and ALI.
Airway cytokine and chemokine levels were most strongly related to the pathogenic model of aspiration (Figure 4). BAL IL-1β levels correlated with the degree of lung injury, and were generally similar between WT and p47phox−/− mice (Figure 4B). Prior studies have produced different results regarding the role of NADPH oxidase in inflammasome activation required for caspase-regulated IL-1β production (60–62). BAL TNFα levels inversely correlated with injury, with the highest levels observed after part. (the least injurious aspiration insult). TNFα can enhance PMN recruitment by activation of adhesion molecules (63), however, in the part. model, these recruited PMNs appeared to have a lower injury potential compared to other aspiration insults (Figure 3C). These results are consistent with our prior studies showing reduced BAL TNFα levels following acid + part. compared to part. alone (29, 30), and suggest that caustic cellular injury may blunt certain cytokine responses.
Although NADPH oxidase is the major source of ROS in activated PMNs, other sources of ROS may also alter inflammatory responses following gastric aspiration. Superoxide anion generated by xanthine oxidase can mediate both antibacterial host defense (64) and ALI (65) in NADPH oxidase-deficient mice. In addition, the p47phox subunit may also have NADPH oxidase-independent effects on inflammation and injury. Prior studies have demonstrated similar phenotypic expression of lung inflammation and injury in both p47phox−/− mice and X-linked gp91phox-deficient mice (17, 66). These consistent findings from two different mouse models support a critical role of NADPH oxidase in control of inflammation as opposed to an individual NADPH oxidase protein subunit functioning independently of NADPH oxidase or an artifact introduced during generation of one of the knockout colonies.
ROS can exacerbate ALI through several mechanisms, including direct cellular injury, NF-κB activation, and activation of injurious inflammatory responses (67–69). NADPH oxidase activation leads to rapid generation of ROS and activation of PMN granular proteases responsible for killing invading pathogens (10). While initial actions are clearly injurious, this study demonstrates that NADPH oxidase can also counterbalance these early pro-inflammatory and injurious events. This outcome may also explain the numerous unsuccessful studies using anti-oxidant approaches to limit inflammation in several diseases, including ALI and sepsis (9). We anticipate that recognition of the mechanisms involved in NADPH oxidase activation leading to the stimulation of protective pathways may result in the development of novel therapeutic approaches in gastric aspiration, as well as other ALI etiologies.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health: AI079253 (BHS), CA016056 (Center Support Grant to Roswell Park Cancer Institute), HL048889 (PRK, BAD), AI084410 (PRK, BAD), CA116430 (KKS), and NIGMS 5 R01 GM089711-03 (CJH), and DoD Hypothesis Grant award W81XWH-09-1-0472 (CJH).
Abbreviations used in this article
- acid+part
gastric particles in saline + HCl, pH=1.25
- ACW
alveolar-capillary wall
- ALI
acute lung injury
- ARDS
acute respiratory distress syndrome
- BAL
bronchoalveolar lavage
- DAMP
damage-associated molecular patterns
- i.t
intratracheal
- KC
keratinocyte chemoattractant
- MTD
mitochondrial DAMP
- mtDNA
mitochondrial DNA
- NQO1
NAD(P)H:quinone oxidoreductase 1
- p47phox−/−
NADPH oxidase-deficient mouse; part., gastric particles in saline
- PMN
polymorphonuclear leukocyte (neutrophil)
- PPARγ
peroxisome proliferator-activated receptor-γ
- ROS
reactive oxygen species
- WT
wild-type
References
- 1.Sheu CC, Gong MN, Zhai R, Chen F, Bajwa EK, Clardy PF, Gallagher DC, Thompson BT, Christiani DC. Clinical characteristics and outcomes of sepsis-related vs non-sepsis-related ARDS. Chest. 2010;138:559–567. doi: 10.1378/chest.09-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cooke CR, Shah CV, Gallop R, Bellamy S, Ancukiewicz M, Eisner MD, Lanken PN, Localio AR, Christie JD. A simple clinical predictive index for objective estimates of mortality in acute lung injury. Crit Care Med. 2009;37:1913–1920. doi: 10.1097/CCM.0b013e3181a009b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ware LB, Koyama T, Billheimer DD, Wu W, Bernard GR, Thompson BT, Brower RG, Standiford TJ, Martin TR, Matthay MA. Prognostic and pathogenetic value of combining clinical and biochemical indices in patients with acute lung injury. Chest. 2010;137:288–296. doi: 10.1378/chest.09-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinlan GJ, Lamb NJ, Tilley R, Evans TW, Gutteridge JM. Plasma hypoxanthine levels in ARDS: implications for oxidative stress, morbidity, and mortality. Am J Respir Crit Care Med. 1997;155:479–484. doi: 10.1164/ajrccm.155.2.9032182. [DOI] [PubMed] [Google Scholar]
- 5.Ruchaud-Sparagano MH, Drost EM, Donnelly SC, Bird MI, Haslett C, Dransfield I. Potential pro-inflammatory effects of soluble E-selectin upon neutrophil function. European journal of immunology. 1998;28:80–89. doi: 10.1002/(SICI)1521-4141(199801)28:01<80::AID-IMMU80>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 6.Inci I, Zhai W, Arni S, Hillinger S, Vogt P, Weder W. N-acetylcysteine attenuates lung ischemia-reperfusion injury after lung transplantation. Ann Thorac Surg. 2007;84:240–246. doi: 10.1016/j.athoracsur.2007.03.082. discussion 246. [DOI] [PubMed] [Google Scholar]
- 7.Fialkow L, Wang Y, Downey GP. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic Biol Med. 2007;42:153–164. doi: 10.1016/j.freeradbiomed.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 8.Zmijewski JW, Lorne E, Zhao X, Tsuruta Y, Sha Y, Liu G, Abraham E. Antiinflammatory effects of hydrogen peroxide in neutrophil activation and acute lung injury. Am J Respir Crit Care Med. 2009;179:694–704. doi: 10.1164/rccm.200806-851OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adhikari N, Burns KE, Meade MO. Pharmacologic therapies for adults with acute lung injury and acute respiratory distress syndrome. Cochrane Database Syst Rev. 2004:CD004477. doi: 10.1002/14651858.CD004477.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 11.Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. 2007;5:577–582. doi: 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]
- 12.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 13.Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, Reichenbach J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114:2619–2622. doi: 10.1182/blood-2009-05-221606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya-O’Brien S, Hilligoss DM, Malech HL, Gallin JI, Holland SM. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004;114:462–468. doi: 10.1542/peds.114.2.462. [DOI] [PubMed] [Google Scholar]
- 15.Siddiqui S, V, Anderson L, Hilligoss DM, Abinun M, Kuijpers TW, Masur H, Witebsky FG, Shea YR, Gallin JI, Malech HL, Holland SM. Fulminant mulch pneumonitis: an emergency presentation of chronic granulomatous disease. Clin Infect Dis. 2007;45:673–681. doi: 10.1086/520985. [DOI] [PubMed] [Google Scholar]
- 16.Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000;79:170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, Vethanayagam RR, Yull FE, Capitano M, Wallace PK, Minderman H, Christman JW, Sporn MB, Chan J, Vinh DC, Holland SM, Romani LR, Gaffen SL, Freeman ML, Blackwell TS. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS ONE. 2010;5:e9631. doi: 10.1371/journal.pone.0009631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kono H, Rock KL. How dying cells alert the immune system to danger. Nature reviews Immunology. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.ARDS_Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 21.Brower RG, Lanken PN, MacIntyre N, Matthay MA, Morris A, Ancukiewicz M, Schoenfeld D, Thompson BT. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351:327–336. doi: 10.1056/NEJMoa032193. [DOI] [PubMed] [Google Scholar]
- 22.Estenssoro E, Dubin A, Laffaire E, Canales H, Saenz G, Moseinco M, Pozo M, Gomez A, Baredes N, Jannello G, Osatnik J. Incidence, clinical course, and outcome in 217 patients with acute respiratory distress syndrome. Crit Care Med. 2002;30:2450–2456. doi: 10.1097/00003246-200211000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Fowler AA, Hamman RF, Good JT, Benson KN, Baird M, Eberle DJ, Petty TL, Hyers TM. Adult respiratory distress syndrome: risk with common predispositions. Ann Intern Med. 1983;98:593–597. doi: 10.7326/0003-4819-98-5-593. [DOI] [PubMed] [Google Scholar]
- 24.Eisner MD, Thompson T, Hudson LD, Luce JM, Hayden D, Schoenfeld D, Matthay MA. Efficacy of low tidal volume ventilation in patients with different clinical risk factors for acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;164:231–236. doi: 10.1164/ajrccm.164.2.2011093. [DOI] [PubMed] [Google Scholar]
- 25.Hudson LD, Milberg JA, Anardi D, Maunder RJ. Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1995;151:293–301. doi: 10.1164/ajrccm.151.2.7842182. [DOI] [PubMed] [Google Scholar]
- 26.Davidson BA, Knight PR, Helinski JD, Nader ND, Shanley TP, Johnson KJ. The role of tumor necrosis factor-alpha in the pathogenesis of aspiration pneumonitis in rats. Anesthesiology. 1999;91:486–499. doi: 10.1097/00000542-199908000-00024. [DOI] [PubMed] [Google Scholar]
- 27.Davidson BA, Knight PR, Wang Z, Chess PR, Holm BA, Russo TA, Hutson A, Notter RH. Surfactant alterations in acute inflammatory lung injury from aspiration of acid and gastric particulates. Am J Physiol Lung Cell Mol Physiol. 2005;288:L699–708. doi: 10.1152/ajplung.00229.2004. [DOI] [PubMed] [Google Scholar]
- 28.Knight PR, Rutter T, Tait AR, Coleman E, Johnson K. Pathogenesis of gastric particulate lung injury: a comparison and interaction with acidic pneumonitis. Anesthesia and analgesia. 1993;77:754–760. doi: 10.1213/00000539-199310000-00017. [DOI] [PubMed] [Google Scholar]
- 29.Knight PR, Davidson BA, Nader ND, Helinski JD, Marschke CJ, Russo TA, Hutson AD, Notter RH, Holm BA. Progressive, severe lung injury secondary to the interaction of insults in gastric aspiration. Exp Lung Res. 2004;30:535–557. doi: 10.1080/01902140490489162. [DOI] [PubMed] [Google Scholar]
- 30.Raghavendran K, Davidson BA, Mullan BA, Hutson AD, Russo TA, Manderscheid PA, Woytash JA, Holm BA, Notter RH, Knight PR. Acid and particulate-induced aspiration lung injury in mice: importance of MCP-1. Am J Physiol Lung Cell Mol Physiol. 2005;289:L134–143. doi: 10.1152/ajplung.00390.2004. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy TP, Johnson KJ, Kunkel RG, Ward PA, Knight PR, Finch JS. Acute acid aspiration lung injury in the rat: biphasic pathogenesis. Anesthesia and analgesia. 1989;69:87–92. [PubMed] [Google Scholar]
- 32.Knight PR, Druskovich G, Tait AR, Johnson KJ. The role of neutrophils, oxidants, and proteases in the pathogenesis of acid pulmonary injury. Anesthesiology. 1992;77:772–778. doi: 10.1097/00000542-199210000-00023. [DOI] [PubMed] [Google Scholar]
- 33.Segal BH, Davidson BA, Hutson AD, Russo TA, Holm BA, Mullan B, Habitzruther M, Holland SM, Knight PR., 3rd Acid aspiration-induced lung inflammation and injury are exacerbated in NADPH oxidase-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2007;292:L760–768. doi: 10.1152/ajplung.00281.2006. [DOI] [PubMed] [Google Scholar]
- 34.Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan JY, Kwong M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim Biophys Acta. 2000;1517:19–26. doi: 10.1016/s0167-4781(00)00238-4. [DOI] [PubMed] [Google Scholar]
- 36.Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, Gribble GW, Hill-Kapturczak N, Agarwal A, Sporn MB. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res. 2005;65:4789–4798. doi: 10.1158/0008-5472.CAN-04-4539. [DOI] [PubMed] [Google Scholar]
- 37.Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci U S A. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giaever I, Keese CR. Micromotion of mammalian cells measured electrically. Proc Natl Acad Sci U S A. 1991;88:7896–7900. doi: 10.1073/pnas.88.17.7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wegener J, Keese CR, Giaever I. Electric cell-substrate impedance sensing (ECIS) as a noninvasive means to monitor the kinetics of cell spreading to artificial surfaces. Exp Cell Res. 2000;259:158–166. doi: 10.1006/excr.2000.4919. [DOI] [PubMed] [Google Scholar]
- 40.Itagaki K, Zhang Q, Hauser CJ. Sphingosine kinase inhibition alleviates endothelial permeability induced by thrombin and activated neutrophils. Shock. 2010;33:381–386. doi: 10.1097/SHK.0b013e3181c6bb75. [DOI] [PubMed] [Google Scholar]
- 41.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 42.Dinkova-Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao X, Suh N, Williams C, Risingsong R, Honda T, Gribble GW, Sporn MB, Talalay P. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci U S A. 2005;102:4584–4589. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itagaki K, Adibnia Y, Sun S, Zhao C, Sursal T, Chen Y, Junger W, Hauser CJ. Bacterial DNA induces pulmonary damage via TLR-9 through cross-talk with neutrophils. Shock. 2011;36:548–552. doi: 10.1097/SHK.0b013e3182369fb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamada M, Gomez JC, Chugh PE, Lowell CA, Dinauer MC, Dittmer DP, Doerschuk CM. Interferon-gamma production by neutrophils during bacterial pneumonia in mice. Am J Respir Crit Care Med. 2011;183:1391–1401. doi: 10.1164/rccm.201004-0592OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, Looney MR. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. The Journal of clinical investigation. 2012;122:2661–2671. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldman G, Welbourn R, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Reactive oxygen species and elastase mediate lung permeability after acid aspiration. Journal of applied physiology. 1992;73:571–575. doi: 10.1152/jappl.1992.73.2.571. [DOI] [PubMed] [Google Scholar]
- 48.Nader ND, Davidson BA, Tait AR, Holm BA, Knight PR. Serine antiproteinase administration preserves innate superoxide dismutase levels after acid aspiration and hyperoxia but does not decrease lung injury. Anesthesia and analgesia. 2005;101:213–219. doi: 10.1213/01.ANE.0000152188.65226.FE. table of contents. [DOI] [PubMed] [Google Scholar]
- 49.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 50.Fernandez-Boyanapalli R, Frasch SC, Riches DW, Vandivier RW, Henson PM, Bratton DL. PPARgamma activation normalizes resolution of acute sterile inflammation in murine chronic granulomatous disease. Blood. 2010;116:4512–4522. doi: 10.1182/blood-2010-02-272005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernandez-Boyanapalli RF, Frasch SC, McPhillips K, Vandivier RW, Harry BL, Riches DW, Henson PM, Bratton DL. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood. 2009;113:2047–2055. doi: 10.1182/blood-2008-05-160564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cho HY, Gladwell W, Wang X, Chorley B, Bell D, Reddy SP, Kleeberger SR. Nrf2-regulated PPARgamma expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med. 2010;182:170–182. doi: 10.1164/rccm.200907-1047OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26:175–182. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 54.Zhu L, Pi J, Wachi S, Andersen ME, Wu R, Chen Y. Identification of Nrf2-dependent airway epithelial adaptive response to proinflammatory oxidant-hypochlorous acid challenge by transcription profiling. Am J Physiol Lung Cell Mol Physiol. 2008;294:L469–477. doi: 10.1152/ajplung.00310.2007. [DOI] [PubMed] [Google Scholar]
- 55.Woods CG, Fu J, Xue P, Hou Y, Pluta LJ, Yang L, Zhang Q, Thomas RS, Andersen ME, Pi J. Dose-dependent transitions in Nrf2-mediated adaptive response and related stress responses to hypochlorous acid in mouse macrophages. Toxicol Appl Pharmacol. 2009;238:27–36. doi: 10.1016/j.taap.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun Jang J, Piao S, Cha YN, Kim C. Taurine chloramine activates Nrf2, increases HO-1 expression and protects cells from death caused by hydrogen peroxide. J Clin Biochem Nutr. 2009;45:37–43. doi: 10.3164/jcbn.08-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agata N, Ahmad R, Kawano T, Raina D, Kharbanda S, Kufe D. MUC1 oncoprotein blocks death receptor-mediated apoptosis by inhibiting recruitment of caspase-8. Cancer Res. 2008;68:6136–6144. doi: 10.1158/0008-5472.CAN-08-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nader-Djalal N, Knight PR, 3rd, Thusu K, Davidson BA, Holm BA, Johnson KJ, Dandona P. Reactive oxygen species contribute to oxygen-related lung injury after acid aspiration. Anesthesia and analgesia. 1998;87:127–133. doi: 10.1097/00000539-199807000-00028. [DOI] [PubMed] [Google Scholar]
- 59.Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW, Biswal S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochemical and biophysical research communications. 2006;351:883–889. doi: 10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Bruggen R, Koker MY, Jansen M, van Houdt M, Roos D, Kuijpers TW, van den Berg TK. Human NLRP3 inflammasome activation is Nox1–4 independent. Blood. 2010;115:5398–5400. doi: 10.1182/blood-2009-10-250803. [DOI] [PubMed] [Google Scholar]
- 61.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, Suva ML, Stehle JC, Kopf M, Stamenkovic I, Corradin G, Tschopp J. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS ONE. 2009;4:e6510. doi: 10.1371/journal.pone.0006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Phadke AP, Mehrad B. Cytokines in host defense against Aspergillus: recent advances. Med Mycol. 2005;43(Suppl 1):S173–176. doi: 10.1080/13693780500052099. [DOI] [PubMed] [Google Scholar]
- 64.Segal BH, Sakamoto N, Patel M, Maemura K, Klein AS, Holland SM, Bulkley GB. Xanthine oxidase contributes to host defense against Burkholderia cepacia in the p47(phox−/−) mouse model of chronic granulomatous disease. Infect Immun. 2000;68:2374–2378. doi: 10.1128/iai.68.4.2374-2378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kubo H, Morgenstern D, Quinian WM, Ward PA, Dinauer MC, Doerschuk CM. Preservation of complement-induced lung injury in mice with deficiency of NADPH oxidase. The Journal of clinical investigation. 1996;97:2680–2684. doi: 10.1172/JCI118718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao XP, Standiford TJ, Rahman A, Newstead M, Holland SM, Dinauer MC, Liu QH, Malik AB. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: studies in p47phox−/− and gp91phox−/− mice. Journal of immunology. 2002;168:3974–3982. doi: 10.4049/jimmunol.168.8.3974. [DOI] [PubMed] [Google Scholar]
- 67.Tasaka S, Amaya F, Hashimoto S, Ishizaka A. Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxid Redox Signal. 2008;10:739–753. doi: 10.1089/ars.2007.1940. [DOI] [PubMed] [Google Scholar]
- 68.Rahman I, Kilty I. Antioxidant therapeutic targets in COPD. Curr Drug Targets. 2006;7:707–720. doi: 10.2174/138945006777435254. [DOI] [PubMed] [Google Scholar]
- 69.Zhang X, Shan P, Sasidhar M, Chupp GL, Flavell RA, Choi AM, Lee PJ. Reactive oxygen species and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase mediate hyperoxia-induced cell death in lung epithelium. Am J Respir Cell Mol Biol. 2003;28:305–315. doi: 10.1165/rcmb.2002-0156OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.