

Graphical abstract

Highlights
► Genomic approaches for the detection of G-quadruplex folding sequences. ► Cellular imaging of G-quadruplex structures. ► Ligand driven alkylation and tag transfer onto G-quadruplex structures.
Keywords: G-quadruplex, Genomic mapping, Pull-down, Cross-linking, Chromatin immuno-precipitation (ChIP), Cellular imaging, Quinone methides, DNA damage response, Unbiased approaches, Sequencing
Abstract
Guanine-rich nucleic acids can fold into non-canonical DNA secondary structures called G-quadruplexes. The formation of these structures can interfere with the biology that is crucial to sustain cellular homeostases and metabolism via mechanisms that include transcription, translation, splicing, telomere maintenance and DNA recombination. Thus, due to their implication in several biological processes and possible role promoting genomic instability, G-quadruplex forming sequences have emerged as potential therapeutic targets. There has been a growing interest in the development of synthetic molecules and biomolecules for sensing G-quadruplex structures in cellular DNA. In this review, we summarise and discuss recent methods developed for cellular imaging of G-quadruplexes, and the application of experimental genomic approaches to detect G-quadruplexes throughout genomic DNA. In particular, we will discuss the use of engineered small molecules and natural proteins to enable pull-down, ChIP-Seq, ChIP-chip and fluorescence imaging of G-quadruplex structures in cellular DNA.
1. Introduction
Guanine-rich nucleic acids are well known for their ability to adopt non Watson–Crick hydrogen-bonded structures [1,2]. These structures are better known as G-quadruplexes and share the common feature of stacked guanine tetrads as basic motif [3]. The formation of such guanine based supramolecular assemblies is particularly favored under physiological conditions, with respect to pH and the presence of alkali cations (i.e. K+ and Na+). Their kinetic and thermodynamic folding parameters as well as the different topologies that they can adopt have been extensively investigated [4]. A large number of sequences capable of folding into stable G-quadruplex structures have been described in the literature. In addition, the relevance of loop sequences and G-track lengths to the formation of such structures has been systematically studied in both DNA and RNA [5–8]. Interestingly, it has been shown that such sequences often occur in functionally important regions of the genome suggesting the possibility that G-quadruplexes behave as structural switches of cellular processes and therefore provide a basis for therapeutic intervention [9–13]. Because of their importance, several biophysical assays have been developed over the past decade to characterise both the folding and the presence of these structures in nucleic acids sequences in vitro [14]. In particular, the telomeric single stranded G-overhang comprising the tandem repeat sequence (TTAGGG)n has been shown to fold into stable G-quadruplex structures in vitro [15]. Several small molecules have been synthesized with the aim to interfere with the action of telomerase enzyme via the stabilization of telomeric G-quadruplex DNA. Such molecules, including the synthetic molecule Phen–DC3 and the natural product telomestatin, showed tight stabilization of the telomeric G-quadruplex resulting in telomere shortening presumably via interaction with G-quadruplex [16,17]. Pyridostatin-based molecules (Fig. 1) also displayed similar properties towards the telomeric G-quadruplex, interfering with the hexameric protein complex shelterin [18–20]. In particular pyridostatin was shown to compete for binding with the shelterin component hPOT-1 leading to the activation of DNA damage responses at telomeres. More recently, it has been shown that the telomeric-repeat-containing RNAs (TERRA) generated by the transcription of mammalian telomeres, were also able to fold into stable G-quadruplex structures [21–23]. Although direct implications of these RNA sequences in the telomere machinery have not been yet demonstrated, it has been shown that such sequences can fold into G-quadruplex structures in cellulo [24]. G-quadruplexes may also have an impact on the transcriptional activity of several oncogenes such as c-MYC [25], c-KIT [26], k-RAS [27], VEGF [28], BCL-2 [29] and more recently SRC [30]. It has been hypothesised that G-quadruplex structures can preferentially form at the promoter regions of certain genes and that ligand-induced stabilization of the quadruplex can alter the transcriptional state of the gene [9]. This has suggested a framework for targeting G-quadruplex structures as a new anti-cancer strategy in addition to pre-established approaches based on telomere targeting. More recent studies focused on the biological aspects of the cancer related functions of G-quadruplex folding sequences have unravelled further new insights. The transcription factor SP-1 has been shown to bind to a new G-quadruplex forming sequence recently discovered in the promoter sequence of the proto-oncogene c-KIT [31]. Thus, we continue to see new perspectives for the relationship between G-quadruplexes and transcriptional regulation. Interestingly, cells deficient in the G-quadruplex resolving FANCJ helicase showed epigenetic instability, suggesting that the generation of these structures may somehow alter the stability of some histone marks [32–34]. Moreover, it has been shown that the human Pif1 (hPif1) helicase and the selective G-quadruplex stabilizing small molecule pyridostatin target overlapping genomic sites in human cells [30]. It is noteworthy that hPif1 has also been shown to bind to and unwind G-quadruplex structures in vitro [35]. Likewise, the Bloom’s (BLM) and Werner’s (WRN) syndrome helicases have also been shown to efficiently disrupt G-quadruplex DNA in vitro, and such enzymatic activity can be inhibited by treatment with G-quadruplex interacting small molecules [36–38]. Very recently, Kamath-Loeb et al. suggested the possibility that beyond its helicase activity WRN might also act as a G-quadruplex binding scaffold that recruits additional DNA processing proteins [39].
Fig. 1.

Molecular structure of pyridostatin.
Despite the amount of biophysical and biochemical data that has been collected, the field could benefit from more direct evidence for the existence and biological relevance of G-quadruplex structures in a cellular context. The design of molecular tools to probe and monitor G-quadruplex formation throughout the genome and the transcriptome is therefore a worthy challenge to pursue. Such tools need to function in the context of genomic complexity and be amenable to use on a cellular system. Herein, we discuss recent approaches that have been developed to address such goals.
2. Structure-based methods
2.1. Theoretical mapping based on computational analysis
Biophysical studies on G-quadruplexes have provided the knowledge to enable the computational prediction of where putative G-quadruplex forming sequences (PQS) occur in genomes [40]. Several algorithms have been developed based on different criteria to identify sequences with a propensity to fold into G-quadruplex in vitro [41]. One approach is based on guanine density within a sequence window, because this correlates with the likelihood of G-quadruplex formation [42,43]. Alternatively, one can consider sequences that consist of four runs of tandem guanines or calculate the density of guanine runs rather than the density of the base itself. Programs based on these algorithms have been widely used, providing valuable insights into how predicted G-quadruplex motifs are distributed throughout genomes [44–48]. Moreover, Todd et al. demonstrated that guanine-rich sequences often display multiple folding possibilities, further complicating the analysis of data based on apparent loop sizes [49]. More recently Huppert and co-workers exploited a novel Bayesian prediction framework based on Gaussian regression process to determine the thermodynamic stability of any G-quadruplex from the sequence information alone [50]. The authors also introduced an active learning procedure useful to iteratively acquire data in an optimal fashion. Despite the limitations in the accuracy of all such computational PQS genome-wide analyses, this general approach has been valuable and has stimulated wider interest in biology by making G-quadruplex prediction and searching through sequence space readily accessible. However, there is no algorithm that takes into account how the local chromatin structure and superhelicity of the DNA may affect the G-quadruplex formation [51].
2.2. Experimental mapping based on protein immunoprecipitation
The identification of selective G-quadruplex binding proteins provides an opportunity to locate these structures in cellular DNA [52]. Specifically, the existence of such proteins allows the possibility to exploit Chromatin Immunoprecipitation (ChIP) approaches such as ChIP-Seq and ChIP-chip. ChIP experiments are widely used in biology to identify DNA–protein binding sites throughout the genome in vivo [53,54]. Typically, this procedure enables isolation of protein–DNA complexes with the aim of identifying the exact binding loci of a protein of interest. The isolation protocol can be either performed on native chromatin or chromatin that has been chemically fixed with formaldehyde prior to the immunoprecipitation step. The cells are first lysed, the chromatin is then isolated and the DNA fragmented either mechanically or enzymatically. DNA–protein complexes are then selectively precipitated using antibodies against the protein of interest. The DNA sequences recovered are released from protein by thermal denaturation, amplified by PCR and analysed on DNA-microarray (ChIP-chip) or by high throughput sequencing (ChIP-Seq). The first application of ChIP-Seq approaches has been focused on mapping the genomic binding sites of chromatin proteins and transcription factors [55–57]. However, this method is, in principle, applicable to map the existence of non B-DNA structures such as G-quadruplexes.
Recent literature has provided two examples of such techniques applied to G-quadruplexes. Paeschke et al. studied the effect of knocking down Pif1 helicase in Saccharomyces cerevisiae. Pif1 was reported to be a G-quadruplex resolvase in vitro and interestingly the authors found that yeast lacking this helicase showed replication fork stalling in the proximity of G-quadruplex forming sequence, which eventually led to DNA breakage [58]. This suggests that G-quadruplex structures are formed in vivo and that resolving helicases are crucial for DNA replication and to sustain genomic stability [59]. ChIP-chip experiments revealed 1584 Pif1 interaction sites including guanine-rich sites, most of which were predicted to be PQS by computational analysis. It is noteworthy that the authors found an enrichment of the enzyme DNA Pol II at those sites together with a good correlation with DNA breaks observed in the absence of the active helicase at the protein binding sites. These data suggested that helicase activities might sustain genomic stability during replication by interacting with or unfolding G-quadruplex motifs.
Law et al. have used a similar approach in an independent study focused on the ATR-X syndrome protein. In their study, the authors exploited ChIP-chip and ChIP-Seq technologies to map the interaction sites of the ATR-X protein throughout the human genome. Interestingly, the ATR-X protein was found to bind to tandem repeats with high guanine density, most of which were predicted to be G-quadruplexes by bioinformatics analysis [60]. Moreover, the authors demonstrated the G-quadruplex binding properties of the ATR-X protein by means of gel shift assays. Furthermore, mutations that impair ATR-X function were associated with misregulation of the associated genes in vivo. It is noteworthy that these data were obtained without prior knowledge that ATR-X was a G-quadruplex interacting protein emphasising the need of unbiased approaches such as ChIP-Seq technologies to identify relevant genomic sites where G-quadruplex DNA may occur and have a function in vivo.
Other results have been obtained in the analysis of G-quadruplex formation in the transcriptome by microarray experiments. Ashley et al. studied the FMRP protein that is involved in Fragile-X retardation syndrome [61]. In particular, the absence of FMRP that naturally binds to its RNA targets, including its own RNA FMR1, has been hypothesised to generate mistranslation of the RNAs and leading to the syndrome itself. The FMR1 associated gene harbours a (CGG)n tandem repeat in its 5′ untranslated region which undergoes an expansion in the patient affected by the syndrome leading to the eventual silencing of the gene [62]. Remarkably, the authors found that a significant proportion (≈70%) of the transcripts that were immunoprecipitated from mouse brain tissues were also predicted to contain a G-quadruplex structure [63]. This underlines how translation may be altered by G-quadruplex formation in the 5′ untranslated region of mRNA via interacting with FMRP in mouse tissues. Further use of proteins that target G-quadruplex structures will help develop deeper insights from such genome-wide experiments. Pif1, ATR-X and FMPR are naturally occurring proteins and, in principle, several other natural proteins can be used for the same purpose, such as telomere-end binding proteins [64]. The development of recombinant engineered proteins that interact with G-quadruplex structures, such as Gq1 [65,66], as well as of high affinity single chain antibodies such as HF1 [67] and scFv [68,69], and their controlled endogenous expression can also provide the means to sense G-quadruplex structures in vivo. In more details, such detection can be achieved by using ChIP-Seq analysis and immunofluorescence microscopy techniques.
2.3. Experimental mapping of DNA sites by small molecule-affinity isolation
Natural products and synthetic small molecules are powerful tools to enable the investigation of biology. They provide the means to control and modulate biological processes in reversible and dose-dependent manners with a temporal resolution that is difficult to achieve using molecular biological methods such as RNA interference. Over the last two decades, a large number of G-quadruplex interacting small molecules have emerged to help address the longstanding question of the existence of G-quadruplex nucleic acid structures in living organisms and their putative biological function(s). Moreover, small molecule ligands have the ability to recognise a pre-formed G-quadruplex structure [30] and/or induce the folding of these structures upon sequence recognition in vitro [70]. Inspired by the literature [71], we developed a G-quadruplex affinity matrix based on the potent and selective scaffold of pyridostatin [18], which contains an additional biotin tag for affinity isolation (Fig. 2). We hypothesised that synthetic small molecules could act as engineered mimics of naturally occurring proteins and antibodies. In vitro experiments demonstrated that G-quadruplex DNA could be isolated from a mixture of diverse oligonucleotides including single stranded (ss) DNA, double stranded (ds) DNA and hairpin RNA by incubating the mixture with the affinity reagent, followed by streptavidin-coated magnetic solid phase separation [72]. This particular strategy was successful in pulling-down human telomeric DNA from genomic DNA derived from cellular extracts and holds promise for the discovery of other genomic DNA and RNA G-quadruplexes that have been predicted by means of computational analysis. Several features were considered for the design of such a probe, including: (i) the presence of an affinity tag to pull down DNA fragments, (ii) the ability to recognise various G-quadruplex structures with a high level of specificity over double-stranded DNA (ds-DNA), (iii) the capacity to release intact DNA for subsequent analysis by specific PCR or sequencing (ChIP-Seq, RNA-Seq), and (iv) a detectable biological activity related to the cellular target of interest. However, we encountered some technical issues using biotin as affinity tag in cellular experiments. Therefore, we have so far limited its use to cell extracts, rather than a strictly cellular context. The fact that, telomere shortening was observed after several days exposure to the small molecule modified with the affinity tag suggests that the chemical introduction of a biotin does not preclude biological activity of the main scaffold, thus the technical issues may be related to DNA isolation. A further consideration is that specific G-quadruplex motifs isolated by this method must ideally be correlated with a biological function, and it is inevitable that many such structures, whilst present in extracts, maybe devoid of any specific biological relevance. By analogy, genome-wide studies that map transcription factor binding sites have also found that many experimentally determined binding sites are non-functional in the context of the experimental settings used by the authors [73]. Therefore, a new generation of G-quadruplex reagents must ideally be developed to resolve some of these issues and to alleviate the likelihood of experimental artifacts.
Fig. 2.

Biased approach to identify G-quadruplex structures in human genomic DNA.
Herein we discuss several types of ligands and strategies that might be considered in future work to develop new G-quadruplex probes.
-
(i)
The first approach relies on the use of a molecule containing an affinity tag capable of tight binding to G-quadruplex structures, by means of specific non-covalent interactions (i.e. π–π, hydrogen bonding and electrostatic). This approach, reminiscent of ChIP experiments based on stable protein-nucleic acids complexes, can expand the use of such molecules for cellular pull-down of G-quadruplex forming sequences. This strategy offers the possibility to disrupt the DNA-ligand complex upon thermal denaturing and/or exposure to a set of chaotropic reagents such as urea and guanidinium salts. This step is crucial to recover the pulled down nucleic acids for further sequencing and genome-wide mapping (see Fig. 2).
-
(ii)
The second approach is to use paraformaldehyde or glutaraldehyde cross-linking to strengthen the stability of the small molecule-nucleic acid adducts, an experimental protocol that resembles regular ChIP experiments. To do so, a stable covalent methylene bridge may be formed between the primary amine of the ligand-containing probe and one of the free amino guanine functions of the G-quadruplex motif. Formaldehyde cross-linking protocols have been extensively used in molecular and cell biology to freeze biological processes and are well established. The covalent link formed between the probe and the DNA motifs can also be thermally reversed in relatively mild conditions. Similarly, the bound oligonucleotides can be recovered, sequenced and mapped. However, there are two main limitations in the use of formaldehyde (i) the first is the requirement for primary amine moieties on the ligand,essential for the methylene bridge generation between ligand and DNA (ii) the second is represented by the morphological modifications and the perturbations that might be inevitably introduced in the cell by using such a non-specific cross-linking agent. In particular this last point can easily lead to the generation of artefacts especially in microscopy experiments [74]. Therefore the use of milder and more selective reversible cross-linking agents would be preferred.
-
(iii)
The third approach consists of the use of small molecules embedded with alkylating agents. In contrast to a genome-wide cross-link approach mediated by formaldehyde, this strategy offers the unique advantage of creating selective cross-links proximal to the genomic area of interest brought about by the DNA-ligand interaction at specific loci. Bertrand et al. described the first G-quadruplex hybrid ligand/alkylating agent, limited so far by the inability to recover the DNA fragment from the ligand due to the strength of the newly formed chemical covalent cross-link [75]. Similar hybrid molecules have been successively described with the aim to investigate the stabilization effect on the G-quadruplex structure induced by the covalent binding of the ligand [76,77]. However, it is important to consider that for such agents: (i) non-specific nucleophilic attack should be minimised and (ii) the alkylation process should ideally be reversible to enable the release of the alkylated sequence after the pull-down. Di Antonio et al. proposed to tether quinone methide precursors to G-quadruplex ligands in order to achieve selective cross-linking of small molecules to these structures [78]. In particular, they employed naphthalendiimide (NDI) derivative as a G-quadruplex recognition moiety following the previous report by Cuenca et al. (Fig. 3) [79]. Quinone methides are capable of reversible alkylation with properties that can be chemically tuned, and they are transient species that must be generated in situ from stable precursor [80]. Interestingly, their generation can be achieved via bioorthogonal means, such as photochemical [81,82], reductive [83], oxidative [84], thermal [85], and fluoride anion treatment [86]. More recently the conjugation of these reactive species with PNA provided the means of sequence-selective and reversible cross-linking [87,88].
Fig. 3.

Embedding quinone methide (QM) precursors to the NDI core lead to selective cross-linking of G-quadruplex folded structures (left). Schematic representation of small molecule-based sequencing and cellular imaging achievable by exploiting QM combined with G-quadruplex ligands (right).
The authors have shown cross-linking of the human telomeric G-quadruplex sequence under mild thermal activation (40 °C) with very high selectivity over ds-DNA. This system can be further embedded with affinity tags and to exploit this unique chemical reactivity for G-quadruplex in genome-wide identification or cellular imaging studies as depicted in Fig. 3. Unfortunately, the alkylation efficiency obtained with the above mentioned molecules was poor for human telomeric G-quadruplex, thus prompting the authors to improve the NDIs recognition properties by synthetizing tri- and tetra-substituted analogues as previously reported [89]. However, this strategy did not lead to notable improvement in terms of alkylation efficiency together with some other experimental limitations which still need to be tackled, such as alkylation yields and the selectivity over ds-DNA [90]. Nevertheless, the design of small molecules for covalent G-quadruplex interaction through triggerable and reversible cross-linking represents an important improvement towards genome-wide pull-down methods that requires further investigation.
A common limitation shared by the design of all the above mentioned G-quadruplex selective molecular probes arose from the chemical modification of the main binding scaffold to introduce an affinity tag, such as biotin, necessary for selective DNA isolation after target recognition, or the introduction of a quinone methide precursor. The chemical linker associated to the affinity tag can adversely affect cellular uptake and binding. Similarly, the presence of a quinone methide precursor can dramatically affect the G-quadruplex binding properties of the main scaffold or its selectivity for G-quadruplex vs. ds-DNA recognition. Moreover, these modifications raise the lipophilic character of the molecule interfering with the water solubility of these analogues. Therefore we suggest exploring similar strategies based on the introduction of a smaller alkyne moiety as tag. Terminal alkynes might behave as “modifiable” affinity tags that can be pulled down by means of ‘‘click chemistry’’ onto azido functionalized beads, or fluorescently labelled in cells [91].
2.4. Cellular imaging
In cellulo sensing of G-quadruplex structure can be achieved by either using modified antibodies [68], G-quadruplex based aptamers [92] or by using small molecules that selectively light up as response to the binding [93–95]. Engineering a small molecule to achieve cellular imaging of G-quadruplex by fluorescence represents a challenging task from an experimental point of view. Indeed, a selective “turn on” fluorescence mechanism upon binding with the target is ideally required and chemically non-trivial to achieve. Moreover, single molecule resolution can be experimentally hard to achieve owing to cellular fluorescence background. In this section we will discuss recent achievements in cellular detection of G-quadruplexes structures.
Luedtke and co-workers exploited the self-assembling properties of phthalocyanines to develop G-quadruplex-specific fluorescent probes [96,97]. In particular, the authors have shown that this class of compounds exhibits a self-aggregation based fluorescence quenching in aqueous solution. The interaction with a G-quadruplex folded oligonucleotide restores the fluorescence disrupting the aggregate by means of end-stacking of a single molecule with the G-quadruplex. Phthalocyanines are suitable for G-quadruplex sensing in the nanomolar range and the authors showed cellular imaging is possible using such molecules. However, experiments that reveal insights that are explicit for particular G-quadruplexes in the cell, e.g. co-localisation with G-quadruplex binding proteins, remain to be carried out for this promising class of molecules.
Several chemical strategies have been instead developed for sensing proteins in cellulo, e.g. covalent fluorophore transfer mediated by chemical or enzymatic reactions [98–100], and such approaches can, in principle, be applied for sensing of particular nucleic acid structures. In fact, this chemistry has been successfully used to sense protein activity or expression in vivo. Three strategies can be explored taking inspiration from the protein chemistry as summarised in Figs. 4 and 5: (i) fluorophore transfer mediated by alkylation induced by proximity effects. In this approach, a recognition event mediated by a small molecule concentrates the fluorophore nearby the target and the transfer is chemically triggered either by proximity effect with nucleophilic residues on site, or enzymatically. (ii) A “clickable” tag may be first incorporated in cellulo on the target by using analogous chemical strategies. By doing so the fluorophore can be incorporated in a second step providing a temporal and spatial flexibility of the sensing process (Fig. 5). (iii) The clickable tag is directly embedded on the small molecule used as G-quadruplex binding platform. It is clear that the recent development and applications of click chemistry have allowed the covalent labelling of proteins, nucleic acids and oligosaccharides in cells and whole animals [101]. In our recent studies, we have taken advantage of click chemistry to chemically label a G-quadruplex binding small molecule in human cells [30]. In particular, we have created a variant of the G-quadruplex binding ligands pyridostatin comprising an alkyne functionality for the introduction of a tag at a late stage for G-quadruplex sensing or isolation. The introduction of a terminal alkyne to the main scaffold had no effect on the potency of the parent small molecule pyridostatin in cellular experiments making this analogue suitable for use as an affinity reagent or a surrogate antibody to detect G-quadruplex structures. This protocol enabled us to visually trace the small molecule into human cell nuclei by means of high-resolution confocal microscopy. Here, we observed a striking overlap of foci formed by accumulation of the small molecule and the GFP-labelled G-quadruplex binding helicase hPif1. In this experiment, the small molecule was incubated with cells that were pre-fixed with formaldehyde and then reacted with the fluorophore. The characteristic staining pattern observed for GFP-hPif1 in absence of small molecule treatment, which co-localised with the labelled drug upon treatment, provided evidence for G-quadruplex structure formation in unperturbed cells (see Figs. 5 and 6). This approach now paves the way to study the distribution of small molecules in a cellular environment and to further assess the localisation with other known factors. We anticipate that this general approach will enable azide-based affinity capture to isolate the bound nucleic acids (Figs. 4 and 6) for sequence determination. Additionally, this method may also enable the isolation and identification of proteins that are naturally associated with, or proximal to G-quadruplex containing genomic domains by proteomics. Such methods will facilitate the elucidation of biological function for these structures.
Fig. 4.

Chemical strategies exploitable to localise genomic area containing G-quadruplex: (A) Fluorophore transfer from the ligand to the target induced by proximity (B) Alkyne transfer from the ligand to the target induced by proximity.
Fig. 5.
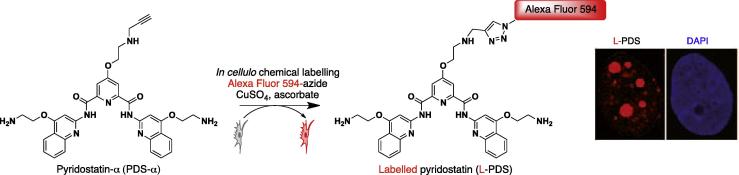
In cellulo chemical labeling strategy to localise genomic area containing G-quadruplex structures.
Fig. 6.

Unbiased approaches to identify G-quadruplex nucleic acids based on structure (small molecule pull-down) and function (ChIP-Seq).
3. Function-based methods
Over the past 10 years, several biological processes have been linked to G-quadruplex structures providing the means to detect functional markers to identify biologically relevant DNA secondary structures. Examples of such processes include (i) replication of DNA sequences containing PQS that is modulated by the Dog1 helicase in C. elegans [102], Pif1 in yeast and Rev1 in DT40 cells [103]; (ii) transcription of certain genes that may be modulated by the stalling of RNA Pol II at sites containing G-quadruplex motifs and by the helicase ATR-X in human cells [103]; (iii) genomic instability promoted by selective G-quadruplex interacting small molecules [16,18,30,104–106]; (iv) DNA recombination near sites containing a G-quadruplex structure in the pathogen Neisseria gonorrhoeae [107]; (v) telomere maintenance that is impaired in the presence of G-quadruplex binding small molecules [108]; (vi) RNA splicing that may be affected by the presence of G-quadruplex structure in the pre-mRNA of hTERT [109]; (vii) RNA translation that may be modulated by proteins such as FMRP known to bind to G-quadruplex motifs [110]. Whilst the perturbation of these biological processes induced by a molecule can be detected, the identification of a protein specifically linked to a particular phenotype or a mechanism is non-trivial. We have developed and applied an approach based on the immunoprecipitation of a surrogate marker of DNA damage, namely the phosphorylated form of histone H2AX (γH2AX), to identify by sequencing and computational analysis the nature and location of sequences and genomic locations of sites associated with breaks promoted by pyridostatin. This unbiased approach has ultimately led to the discovery of genomic domains that contain clusters of G-quadruplex motifs including therapeutically relevant oncogenes such as SRC and MYC. In this study, we observed that the G-quadruplex binding small molecule pyridostatin targets non-telomeric human genomic loci. DNA double-strand breaks are promoted at these sites upon binding of the small molecule to the DNA by a mechanism that may be linked to the stalling of polymerases during transcription and replication. The phosphorylation of H2AX is, in fact, one of the earliest events of the DNA damage response (DDR), thus acts as a read-out of DNA double strand breaks. A direct comparison of the immunoprecipitated sequences with the map of human PQS obtained by computational analysis revealed a set of G-quadruplex containing domains that are functional in the presence of the small molecule. Further analysis also suggested that some of these loci still exhibit residual genomic instability in the absence of drug treatment and may be considered as natural fragile sites. The power of such a method lies in the fact that it is unbiased and focused on the study of biologically relevant PQS since it is a direct readout of a biological function, associated to the G-quadruplex structure.
4. Correlation of G-quadruplex structure with function
The existence and function of each individual G-quadruplex structure mapped in the human genome is still questionable. To provide a greater degree of certainty, one can carry out a careful cross-comparison of motifs revealed by structural isolation with those identified by means of functional detection. This can be illustrated by the finding that SRC is a potential target for G-quadruplex interacting small molecules. The occurrence of DNA damage within the coding region of certain genes upon treatment with pyridostatin revealed a set of genes that contain multiple sequences with a propensity to adopt a G-quadruplex conformation as defined by biophysical experiments and computational mapping. A first validation of the genes targeted was performed by RT-PCR gene expression measurement. This revealed a clear down-regulation of the genes studied as seen by a decrease in their respective mRNA levels upon pyridostatin treatment. Following gene-target identification by ChIP-Seq and a first RT-PCR validation, it was then possible to cross-validate the gene set of interest by regular specific ChIP-q-PCR. This protocol consist in precipitating the protein (i.e. γH2AX) linked to the phenotype investigated (i.e. DNA damage) and quantifying the level of bound DNA by using PCR primers specific for each genes previously identified by ChIP-Seq. This method is well established in the study of DNA damage phenotype [111]. Moreover, in both cases the chaotropic treatment that follows the immunoprecipitation step ensures the release of the small molecule from the targeted DNA sequences, thus avoiding artefacts that G-quadruplex binding small molecule can generate during PCR experiments [112].
To complete this analysis, the development of an efficient protocol such as the one described in Fig. 6 should be performed to isolate G-quadruplex motifs that are directly involved in promoting this phenotype. Indeed, over 25 PQS have been computationally mapped in SRC based on the Quadparser algorithm. Furthermore, these motifs are spread throughout the gene and their individual contribution to the phenotype observed (i.e. DNA damage induction, gene expression alteration) remains to be elucidated. A deep-sequencing method based on small molecule-DNA pull-down protocols, reminiscent of traditional ChIP-Seq, is a worthy technical challenge that will undoubtedly shed further light on these mechanisms.
5. Discussions and Conclusion
The invention and development of general methods for mapping G-quadruplex forming sequences in the genome and the transcriptome of cells remains a major challenge in biology. It will be equally valuable to develop techniques for identifying G-quadruplexes that are functional and/or suitable targets for small molecule intervention. The recent advances in ligands appended with bio-orthogonal groups or affinity tags provide practical approaches to follow for visualising or isolating nucleic acid sequences involved in G-quadruplex formation in cellular DNA. The major advances in high-throughput sequencing of nucleic acids allows such probes to be used to address such questions throughout the entire genome (or transcriptome), as has been shown [30]. Further development and exploitation of the types of methods we have described will enable us and others to address the central questions in relation to the existence, location, functions and druggability of G-quadruplex nucleic acids in an explicit fashion within the context of living systems.
References
- 1.Gellert M., Lipsett M.N., Davies D.R. Proc. Nat. Acad. Sci. USA. 1962;48:2013–2018. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sen D., Gilbert W. Nature. 1988;334:364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- 3.Davis J.T. Angew. Chem. Int. Ed. Engl. 2004;43:668–698. doi: 10.1002/anie.200300589. [DOI] [PubMed] [Google Scholar]
- 4.Burge S., Parkinson G.N., Hazel P., Todd A.K., Neidle S. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bugaut A., Balasubramanian S. Biochemistry. 2008;47:689–697. doi: 10.1021/bi701873c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang A.Y., Bugaut A., Balasubramanian S. Biochemistry. 2011;50:7251–7258. doi: 10.1021/bi200805j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hazel P., Huppert J., Balasubramanian S., Neidle S. J. Am. Chem. Soc. 2004;126:16405–16415. doi: 10.1021/ja045154j. [DOI] [PubMed] [Google Scholar]
- 8.Rachwal P.A., Brown T., Fox K.R. Biochemistry. 2007;46:3036–3044. doi: 10.1021/bi062118j. [DOI] [PubMed] [Google Scholar]
- 9.Balasubramanian S., Hurley L.H., Neidle S. Nat. Rev. Drug Disc. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Folini M., Venturini L., Cimino-Reale G., Zaffaroni N. Expert Opin. Ther. Targets. 2011;15:579–593. doi: 10.1517/14728222.2011.556621. [DOI] [PubMed] [Google Scholar]
- 11.Neidle S. FEBS J. 2010;277:1118–1125. doi: 10.1111/j.1742-4658.2009.07463.x. [DOI] [PubMed] [Google Scholar]
- 12.Miller K.M., Rodriguez R. Expert Rev. Clin. Pharmacol. 2011;4:139–142. doi: 10.1586/ecp.11.4. [DOI] [PubMed] [Google Scholar]
- 13.Bugaut A., Rodriguez R., Kumari S., Hsu S.T., Balasubramanian S. Org. Biomol. Chem. 2010;8:2771–2776. doi: 10.1039/c002418j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murat P., Singh Y., Defrancq E. Chem. Soc. Rev. 2011;40:5293–5307. doi: 10.1039/c1cs15117g. [DOI] [PubMed] [Google Scholar]
- 15.Parkinson G.N., Lee M.P., Neidle S. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 16.De Cian A., DeLemos E., Mergny J.L., Teulade-Fichou M.P., Monchaud D. J. Am. Chem. Soc. 2007;129:1856–1857. doi: 10.1021/ja067352b. [DOI] [PubMed] [Google Scholar]
- 17.Kim M.Y., Vankayalapati H., Shin-Ya K., Wierzba K., Hurley L.H. J. Am. Chem. Soc. 2002;124:2098–2099. doi: 10.1021/ja017308q. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez R., Müller S., Yeoman J.A., Trentesaux C., Riou J.F., Balasubramanian S. J. Am. Chem. Soc. 2008;130:15758–15759. doi: 10.1021/ja805615w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Lange T. Gene. Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 20.Koirala D., Dhakal S., Ashbridge B., Sannohe Y., Rodriguez R., Sugiyama H., Balasubramanian S., Mao H. Nat. Chem. 2011;3:782–787. doi: 10.1038/nchem.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martadinata H., Phan A.T. J. Am. Chem. Soc. 2009;131:2570–2578. doi: 10.1021/ja806592z. [DOI] [PubMed] [Google Scholar]
- 22.Collie G.W., Parkinson G.N., Neidle S., Rosu F., De Pauw E., Gabelica V. J. Am. Chem. Soc. 2010;132:9328–9334. doi: 10.1021/ja100345z. [DOI] [PubMed] [Google Scholar]
- 23.Collie G.W., Haider S.M., Neidle S., Parkinson G.N. Nucleic Acids Res. 2010;38:5569–5580. doi: 10.1093/nar/gkq259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y., Suzuki Y., Ito K., Komiyama M. Telomeric repeat-containing RNA structure in living cells. Proc. Nat. Acad. Sci. USA. 2010;107:14579–14584. doi: 10.1073/pnas.1001177107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siddiqui-Jain A., Grand C.L., Bearss D.J., Hurley L.H. Proc. Nat. Acad. Sci. USA. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernando H., Reszka A.P., Huppert J., Ladame S., Rankin S., Venkitaraman A.R., Neidle S., Balasubramanian S. Biochemistry. 2006;45:7854–7860. doi: 10.1021/bi0601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cogoi S., Xodo L.E. Nucleic Acids Res. 2006;34:2536–2549. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun D., Guo K., Rusche J.J., Hurley L.H. Nucleic Acids Res. 2005;33:6070–6080. doi: 10.1093/nar/gki917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai J., Dexheimer T.S., Chen D., Carver M., Ambrus A., Jones R.A., Yang D. J. Am. Chem. Soc. 2006;128:1096–1098. doi: 10.1021/ja055636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez R., Miller K.M., Forment J.V., Bradshaw C.R., Nikan M., Britton S., Oelschlaegel T., Xhemalce B., Balasubramanian S., Jackson S.P. Nat. Chem. Bio. 2012;8:301–310. doi: 10.1038/nchembio.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.E.A. Raiber, R. Kranaster, E. Lam, M. Nikan, S. Balasubramanian, Nucleic Acids Res. (2011), doi:10.1093/nar/gkr882. [DOI] [PMC free article] [PubMed]
- 32.P. Sarkies, P. Murat, L.G. Phillips, K.J. Patel, S. Balasubramanian, J.E. Sale, Nucleic Acids Res. (2011), doi:10.1093/nar/gkr868. [DOI] [PMC free article] [PubMed]
- 33.Wu Y., Brosh R.M., Jr. Cell Cycle. 2010;9:4080–4090. doi: 10.4161/cc.9.20.13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.London T.B., Barber L.J., Mosedale G., Kelly G.P., Balasubramanian S., Hickson I.D., Boulton S.J., Hiom K. J. Biol. Chem. 2008;283:36132–36139. doi: 10.1074/jbc.M808152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanders C.M. Biochem. J. 2010;430:119–128. doi: 10.1042/BJ20100612. [DOI] [PubMed] [Google Scholar]
- 36.Huber M.D., Lee D.C., Maizels N. Nucleic Acids Res. 2002;30:3954–3961. doi: 10.1093/nar/gkf530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson J.E., Cao K., Ryvkin P., Wang L.S., Johnson F.B. Nucleic Acids Res. 2010;38:1114–1122. doi: 10.1093/nar/gkp1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J.L., Harrison R.J., Reszka A.P., Brosh R.M., Jr., Bohr V.A., Neidle S., Hickson I.D. Biochemistry. 2001;40:15194–15202. doi: 10.1021/bi011067h. [DOI] [PubMed] [Google Scholar]
- 39.Kamath-Loeb A., Loeb L.A., Fry M. PloS one. 2012;7:e30189. doi: 10.1371/journal.pone.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Todd A.K. Methods. 2007;43:246–251. doi: 10.1016/j.ymeth.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 41.Huppert J.L. Biochimie. 2008;90:1140–1148. doi: 10.1016/j.biochi.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 42.Kikin O., D’Antonio L., Bagga P.S. QGRS mapper: A web-based server for predicting g-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006;34(Web Server issue):W676–W682. doi: 10.1093/nar/gkl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kostadinov R., Malhotra N., Viotti M., Shine R., D’Antonio L., Bagga P. GRSDB: A database of quadruplex forming G-rich sequences in alternatively processed mammalian pre-mRNA sequences. Nucleic Acids Res. 2006;34(Database issue):D119–D124. doi: 10.1093/nar/gkj073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eddy J., Maizels N. Nucleic Acids Res. 2006;34:3887–3896. doi: 10.1093/nar/gkl529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huppert J.L., Balasubramanian S. Nucleic Acids Res. 2005;33:2908–2916. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huppert J.L., Balasubramanian S. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rawal P., Kummarasetti V.B., Ravindran J., Kumar N., Halder K., Sharma R., Mukerji M., Das S.K., Chowdhury S. Genome Res. 2006;16:644–655. doi: 10.1101/gr.4508806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huppert J.L., Bugaut A., Kumari S., Balasubramanian S. Nucleic Acids Res. 2008;36:6260–6268. doi: 10.1093/nar/gkn511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Todd A.K., Haider S.M., Parkinson G.N., Neidle S. Nucleic Acids Res. 2007;35:5799–5808. doi: 10.1093/nar/gkm609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stegle O., Payet L., Mergny J.L., MacKay D.J., Huppert J.L. Bioinformatics. 2009;25:374–382. doi: 10.1093/bioinformatics/btp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun D., Hurley L.H. J. Med. Chem. 2009;52:2863–2874. doi: 10.1021/jm900055s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sissi C., Gatto B., Palumbo M. Biochimie. 2011;93:1219–1230. doi: 10.1016/j.biochi.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gilchrist D.A., Fargo D.C., Adelman K. Methods. 2009;48:398–408. doi: 10.1016/j.ymeth.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie Z., Hu S., Qian J., Blackshaw S., Zhu H. Cell. Mol. Life Sci. 2011;68:1657–1668. doi: 10.1007/s00018-010-0617-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson D.S., Mortazavi A., Myers R.M., Wold B. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 56.Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P., Lee W. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 58.Paeschke K., Capra J.A., Zakian V.A. Cell. 2011;145:678–691. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lopes J., Piazza A., Bermejo R., Kriegsman B., Colosio A., Teulade-Fichou M.P., Foiani M., Nicolas A. EMBO J. 2011;30:4033–4046. doi: 10.1038/emboj.2011.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Law M.J., Lower K.M., Voon H.P., Hughes J.R., Garrick D., Viprakasit V., Mitson M., De Gobbi M., Marra M., Morris A., Abbott A. Cell. 2010;143:367–378. doi: 10.1016/j.cell.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 61.Ashley C.T., Jr., Wilkinson K.D., Reines D., Warren S.T. Science. 1993;262:563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- 62.Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 63.Brown V., Jin P., Ceman S., Darnell J.C., O’Donnell W.T., Tenenbaum S.A., Jin X., Feng Y., Wilkinson K.D., Keene J.D., Darnell R.B. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 64.Paeschke K., Simonsson T., Postberg J., Rhodes D., Lipps H.J. Nat. Struct. Mol. Bio. 2005;12:847–854. doi: 10.1038/nsmb982. [DOI] [PubMed] [Google Scholar]
- 65.Ladame S., Schouten J.A., Roldan J., Redman J.E., Neidle S., Balasubramanian S. Biochemistry. 2006;45:1393–1399. doi: 10.1021/bi050229x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patel S.D., Isalan M., Gavory G., Ladame S., Choo Y., Balasubramanian S. Biochemistry. 2004;43:13452–13458. doi: 10.1021/bi048892t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fernando H., Sewitz S., Darot J., Tavare S., Huppert J.L., Balasubramanian S. Nucleic Acids Res. 2009;37:6716–6722. doi: 10.1093/nar/gkp740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaffitzel C., Postberg J., Paeschke K., Lipps H.J. Methods Mol. Biol. 2010;608:159–181. doi: 10.1007/978-1-59745-363-9_11. [DOI] [PubMed] [Google Scholar]
- 69.Schaffitzel C., Berger I., Postberg J., Hanes J., Lipps H.J., Pluckthun A. Proc. Nat. Acad. Sci. USA. 2001;98:8572–8577. doi: 10.1073/pnas.141229498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rodriguez R., Pantos G.D., Goncalves D.P., Sanders J.K., Balasubramanian S. Angew. Chem. Int. Ed. Engl. 2007;46:5405–5407. doi: 10.1002/anie.200605075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Taunton J., Hassig C.A., Schreiber S.L. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 72.Müller S., Kumari S., Rodriguez R., Balasubramanian S. Nat. Chem. 2010;2:1095–1098. doi: 10.1038/nchem.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carroll J.S., Meyer C.A., Song J., Li W., Geistlinger T.R., Eeckhoute J., Brodsky A.S., Keeton E.K., Fertuck K.C., Hall G.F., Wang Q. Nat. Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 74.Belitsky J.M., Leslie S.J., Arora P.S., Beerman T.A., Dervan P.B. Bioorg. Med. Chem. 2002;10:3313–3318. doi: 10.1016/s0968-0896(02)00204-3. [DOI] [PubMed] [Google Scholar]
- 75.Bertrand H., Bombard S., Monchaud D., Teulade-Fichou M.P. J. Biol. Inorg. Chem. 2007;12:1003–1014. doi: 10.1007/s00775-007-0273-3. [DOI] [PubMed] [Google Scholar]
- 76.Bertrand H., Bombard S., Monchaud D., Talbot E., Guedin A., Mergny J.L., Grunert R., Bednarski P.J., Teulade-Fichou M.P. Org. Biomol. Chem. 2009;7:2864–2871. doi: 10.1039/b904599f. [DOI] [PubMed] [Google Scholar]
- 77.Georgiades S.N., Abd Karim N.H., Suntharalingam K., Vilar R. Angew. Chem. Int. Ed. Engl. 2010;49:4020–4034. doi: 10.1002/anie.200906363. [DOI] [PubMed] [Google Scholar]
- 78.Di Antonio M., Doria F., Richter S.N., Bertipaglia C., Mella M., Sissi C., Palumbo M., Freccero M. J. Am. Chem. Soc. 2009;131:13132–13141. doi: 10.1021/ja904876q. [DOI] [PubMed] [Google Scholar]
- 79.Cuenca F., Greciano O., Gunaratnam M., Haider S., Munnur D., Nanjunda R., Wilson W.D., Neidle S. Bioorg. Med. Chem. Lett. 2008;18:1668–1673. doi: 10.1016/j.bmcl.2008.01.050. [DOI] [PubMed] [Google Scholar]
- 80.Weinert E.E., Dondi R., Colloredo-Melz S., Frankenfield K.N., Mitchell C.H., Freccero M., Rokita S.E. J. Am. Chem. Soc. 2006;128:11940–11947. doi: 10.1021/ja062948k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Percivalle C., La Rosa A., Verga D., Doria F., Mella M., Palumbo M., Di Antonio M., Freccero M. J. Org. Chem. 2011;76:3096–3106. doi: 10.1021/jo102531f. [DOI] [PubMed] [Google Scholar]
- 82.Arumugam S., Popik V.V. J. Am. Chem. Soc. 2011;133:5573–5579. doi: 10.1021/ja200356f. [DOI] [PubMed] [Google Scholar]
- 83.Di Antonio M., Doria F., Mella M., Merli D., Profumo A., Freccero M. J. Org. Chem. 2007;72:8354–8360. doi: 10.1021/jo7014328. [DOI] [PubMed] [Google Scholar]
- 84.Kuang Y., Balakrishnan K., Gandhi V., Peng X. J. Am. Chem. Soc. 2011;48:19278–19281. doi: 10.1021/ja2073824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rodriguez R., Adlington R.M., Moses J.E., Cowley A., Baldwin J.E. Org. Lett. 2004;6:3617–3619. doi: 10.1021/ol048479d. [DOI] [PubMed] [Google Scholar]
- 86.Rokita S.E., Yang J., Pande P., Greenberg W.A. J. Org. Chem. 1997;62:3010–3012. doi: 10.1021/jo9700336. [DOI] [PubMed] [Google Scholar]
- 87.Liu Y., Rokita S.E. Biochemistry. 2012;51:1020–1027. doi: 10.1021/bi201492b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang H., Rokita S.E. Angew. Chem. Int. Ed. Engl. 2010;49:5957–5960. doi: 10.1002/anie.201001597. [DOI] [PubMed] [Google Scholar]
- 89.Hampel S.M., Sidibe A., Gunaratnam M., Riou J.F., Neidle S. Bioorg. Med. Chem. Lett. 2010;20:6459–6463. doi: 10.1016/j.bmcl.2010.09.066. [DOI] [PubMed] [Google Scholar]
- 90.Nadai M., Doria F., Di Antonio M., Sattin G., Germani L., Percivalle C., Palumbo M., Richter S.N., Freccero M. Biochimie. 2011;93:1328–1340. doi: 10.1016/j.biochi.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 91.Kolb H.C., Finn M.G., Sharpless K.B. Angew. Chem. Int. Ed. Engl. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 92.Gatto B., Palumbo M., Sissi C. Curr. Med. Chem. 2009;16:1248–1265. doi: 10.2174/092986709787846640. [DOI] [PubMed] [Google Scholar]
- 93.Meguellati K., Koripelly G., Ladame S. Angew. Chem. Int. Ed. Engl. 2010;49:2738–2742. doi: 10.1002/anie.201000291. [DOI] [PubMed] [Google Scholar]
- 94.Lu Y.J., Yan S.C., Chan F.Y., Zou L., Chung W.H., Wong W.L., Qiu B., Sun N., Chan P.H., Huang Z.S., Gu L.Q. Chem. Commun. 2011;47:4971–4973. doi: 10.1039/c1cc00020a. [DOI] [PubMed] [Google Scholar]
- 95.Ren J., Qin H., Wang J., Luedtke N.W., Wang E., Wang J. Anal. Bioanal. Chem. 2011;399:2763–2770. doi: 10.1007/s00216-011-4669-0. [DOI] [PubMed] [Google Scholar]
- 96.Alzeer J., Luedtke N.W. Biochemistry. 2010;49:4339–4348. doi: 10.1021/bi9020583. [DOI] [PubMed] [Google Scholar]
- 97.Alzeer J., Vummidi B.R., Roth P.J., Luedtke N.W. Angew. Chem. Int. Ed. Engl. 2009;48:9362–9365. doi: 10.1002/anie.200903685. [DOI] [PubMed] [Google Scholar]
- 98.Sellars J.D., Landrum M., Congreve A., Dixon D.P., Mosely J.A., Beeby A., Edwards R., Stee P.G. Org. Biomol. Chem. 2010;8:1610–1618. doi: 10.1039/b920443a. [DOI] [PubMed] [Google Scholar]
- 99.Li X., Kapoor T.M. J. Am. Chem. Soc. 2010;132:2504–2505. doi: 10.1021/ja909741q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tsukiji S., Miyagawa M., Takaoka Y., Tamura T., Hamachi I. Nat. Chem. Biol. 2009;5:341–343. doi: 10.1038/nchembio.157. [DOI] [PubMed] [Google Scholar]
- 101.Moses J.E., Moorhouse A.D. Chem. Soc. Rev. 2007;36:1249–1262. doi: 10.1039/b613014n. [DOI] [PubMed] [Google Scholar]
- 102.Cheung I., Schertzer M., Rose A., Lansdorp P.M. Nat. Gen. 2002;31:405–409. doi: 10.1038/ng928. [DOI] [PubMed] [Google Scholar]
- 103.Edmunds C.E., Simpson L.J., Sale J.E. Mol. Cell. 2008;30:519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 104.Rizzo A., Salvati E., Porru M., D’Angelo C., Stevens M.F., D’Incalci M., Leonetti C., Gilson E., Zupi G., Biroccio A. Nucleic Acids Res. 2009;37:5353–5364. doi: 10.1093/nar/gkp582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Salvati E., Leonetti C., Rizzo A., Scarsella M., Mottolese M., Galati R., Sperduti I., Stevens M.F., D’Incalci M., Blasco M., Chiorino G. J. Clin. Invest. 2007;117:3236–3247. doi: 10.1172/JCI32461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gomez D., Aouali N., Londono-Vallejo A., Lacroix L., Megnin-Chanet F., Lemarteleur T., Douarre C., Shin-ya K., Mailliet P., Trentesaux C., Morjani H. J. Biol. Chem. 2003;278(50):50554–50562. doi: 10.1074/jbc.M308440200. [DOI] [PubMed] [Google Scholar]
- 107.Cahoon L.A., Seifert H.S. Science. 2009;325:764–767. doi: 10.1126/science.1175653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Salvati E., Scarsella M., Porru M., Rizzo A., Iachettini S., Tentori L., Graziani G., D’Incalci M., Stevens M.F., Orlandi A., Passeri D. Oncogene. 2010;29:6280–6293. doi: 10.1038/onc.2010.344. [DOI] [PubMed] [Google Scholar]
- 109.Gomez D., Lemarteleur T., Lacroix L., Mailliet P., Mergny J.L., Riou J.F. Nucleic Acids Res. 2004;32:371–379. doi: 10.1093/nar/gkh181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Phan A.T., Kuryavyi V., Darnell J.C., Serganov A., Majumdar A., Ilin S., Raslin T., Polonskaia A., Chen C., Clain D., Darnell R.B. Nat. Struct. Mol. Biol. 2011;18:796–804. doi: 10.1038/nsmb.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shanbhag N.M., Rafalska-Metcalf I.U., Balane-Bolivar C., Janicki S.M., A Greenberg R. Cell. 2010;141:970–981. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reed J., Gunaratnam M., Beltran M., Resza A.P., Vilar R., Neidle S. Anal. Biochem. 2008;380:99–105. doi: 10.1016/j.ab.2008.05.013. [DOI] [PubMed] [Google Scholar]