

Abstract
Cerebral edema forms in the early hours of ischemic stroke by processes involving increased transport of Na and Cl from blood into brain across an intact blood–brain barrier (BBB). Our previous studies provided evidence that the BBB Na–K–Cl cotransporter is stimulated by the ischemic factors hypoxia, aglycemia, and arginine vasopressin (AVP), and that inhibition of the cotransporter by intravenous bumetanide greatly reduces edema and infarct in rats subjected to permanent middle cerebral artery occlusion (pMCAO). More recently, we showed that BBB Na/H exchanger activity is also stimulated by hypoxia, aglycemia, and AVP. The present study was conducted to further investigate the possibility that a BBB Na/H exchanger also participates in edema formation during ischemic stroke. Sprague-Dawley rats were subjected to pMCAO and then brain edema and Na content assessed by magnetic resonance imaging diffusion-weighed imaging and magnetic resonance spectroscopy Na spectroscopy, respectively, for up to 210 minutes. We found that intravenous administration of the specific Na/H exchange inhibitor HOE-642 significantly decreased brain Na uptake and reduced cerebral edema, brain swelling, and infarct volume. These findings support the hypothesis that edema formation and brain Na uptake during the early hours of cerebral ischemia involve BBB Na/H exchanger activity as well as Na–K–Cl cotransporter activity.
Keywords: blood–brain barrier, brain edema, cerebral ischemia, Na/H exchange, stroke
Introduction
Ischemic stroke is currently the fourth leading cause of death in the United States and the brain edema that occurs during ischemia is a major cause of morbidity and mortality. Despite this, the mechanisms underlying edema formation are not well understood. In the early hours of ischemic stroke, cytotoxic cerebral edema forms by processes involving increased secretion of Na, Cl, and water from the blood into brain across an intact blood–brain barrier (BBB). At the same time, astrocytes swell as they take up the secreted ions and water. Blood–brain barrier breakdown with vasogenic edema occurs later, ∼4 hours after the onset of ischemia.1 The BBB Na transporters that participate in edema formation have not been well understood. Our previous studies provided evidence that a luminal BBB membrane Na–K–Cl cotransporter is a major contributor to ischemia-induced edema formation. In studies using cultured cerebral microvascular endothelial cells, we found that the cotransporter is quite sensitive to stimulation by hypoxia, aglycemia, and arginine vasopressin (AVP), three prominent factors present during cerebral ischemia.2, 3, 4 Using the rat permanent middle cerebral artery occlusion (pMCAO) model of ischemic stroke, we also found that inhibition of BBB Na–K–Cl cotransporter activity by intravenous bumetanide significantly reduces edema as assessed by magnetic resonance imaging (MRI) diffusion-weighted imaging (DWI), as well as brain swelling and infarct.3, 5
In more recent studies, we found evidence that a luminal BBB Na/H exchanger may also participate in secretion of Na and water from the blood into brain during ischemic stroke. Both NHE1 and NHE2 isoforms of Na/H exchange are present in cultured bovine cerebral microvascular endothelial cells and in the rat luminal BBB membrane in situ. Further, cerebral microvascular endothelial cell Na/H exchanger activity, evaluated as HOE-642-sensitive H+ flux, is rapidly stimulated by hypoxia, aglycemia, and AVP.6
Early studies showed that BBB transport of ions and water from the blood into brain accounts for up to 30% of brain interstitial fluid formation in healthy, normoxic brain, with the remainder coming from the choroid plexus.7 A role for Na/H exchange in this process was suggested by the observation that Na transport from the blood into brain of rats is inhibited by intravenous administration of the Na/H exchange inhibitors amiloride or dimethylamiloride8, 9 without affecting brain uptake of α-aminoisobutyric acid, a marker of paracellular permeability. These findings, together with our observations that NHE proteins reside in the luminal BBB membrane and are stimulated by ischemic factors, suggests a role for the exchanger in the increased Na uptake and edema that occur in the early hours of ischemic stroke.
Several studies have provided evidence that Na/H exchange inhibitors can reduce brain damage in models of cerebral ischemia–reperfusion. The exchange inhibitor SM-20220 has been shown to reduce BBB disruption and brain damage observed 1 to 7 days after transient MCAO in the rat10, 11 as well as edema and neutrophil accumulation 3 days after transient MCAO.12 Another Na/H exchange inhibitor, FR183998, was found to reduce infarct volume in spontaneously hypertensive rats following 3 days of focal cerebral ischemia.13 More recently, the highly potent Na/H exchange inhibitor HOE-642 was shown to reduce brain lesion volume in mouse transient focal ischemia induced by 30 minutes of MCAO and 1 to 72 hours of reperfusion.14 Studies of NHE1-null mice have shown reduced brain damage and neuronal injury following transient ischemia.15, 16 However, these studies all examined reperfusion injury, and did not address the mechanisms of edema formation occurring during the early stages of stroke when Na transport across the BBB is increased, before BBB breakdown and vasogenic edema. A study of spontaneously hypertensive rats subjected to 4 hours of ischemia without reperfusion demonstrated that the Na/H exchange inhibitor dimethylamiloride significantly reduced infarct volume17 although brain Na uptake and edema formation were not evaluated. Blood–brain barrier Na/H exchange protein is upregulated by ischemia17 and by decreases in shear stress,18 further suggesting a role for this Na transporter in ischemia-induced edema formation.
The present study was conducted to investigate the hypothesis that Na/H exchange contributes to brain Na uptake and cerebral edema formation during the early stages of ischemia (before reperfusion and vasogenic edema). We report here that both Na/H exchange NHE1 and NHE2 protein isoforms reside predominantly in the luminal BBB membrane of ischemic rat brain, as they do in normoxic brain. Using MRI DWI to evaluate cerebral edema formation during pMCAO in rat, we also provide evidence that intravenous HOE-642 attenuates edema and 2,3,5-triphenyltetrazolium chloride (TTC)-defined lesion volume. Further, by nuclear magnetic resonance (NMR) Na spectroscopy we show that HOE-642 and bumetanide reduce brain Na uptake during the early hours of pMCAO and also significantly improve neurologic outcome.
Materials and methods
Induction of Focal Ischemia by Permanent Middle Cerebral Artery Occlusion
This study was conducted in accordance with the Animal Use and Care Guidelines issued by the National Institutes of Health using a protocol approved by the Animal Use and Care Committee at University of California Davis. Normotensive adult male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA, USA) weighing 250 to 300 g were subjected to pMCAO using the intraluminal suture method as previously described.3, 5 Briefly, rats were anesthetized by intraperitoneal injection of sodium pentobarbital (65 mg/kg body wt). For imaging studies, nephrectomy was performed immediately before MCAO as described previously.3, 5 Body temperature was monitored and maintained at 36.8°C to 37.0°C by electric heating pad and rectal probe (Cole-Parmer Instruments, Vernon Hills, IL, USA) during surgery, and water heating pad (Gaymar, Orchard Park, NY, USA) and rectal probe during NMR data acquisition. Arterial blood pressure was continuously monitored via a left femoral artery cannula. The left femoral vein was also cannulated for injection of HOE-642 (gift from Sanofi-Aventis Pharmaceuticals, Bridgewater, NJ, USA), bumetanide (ICN Biomedicals, Costa Mesa, CA, USA) or vehicle. Immediately before euthanasia, blood samples were drawn from the descending aorta for determination of electrolytes, pH, pCO2, glucose, hemoglobin, and hematocrit (I-STAT; Sensor Devices, Waukesha, WI, USA).
Focal cerebral ischemia was induced by occlusion of the left MCA as described previously.3, 5, 19 Briefly, the left common carotid artery was exposed and occipital and thyroid artery branches of the external carotid artery and pterygopalatine artery were ligated. The external carotid artery was ligated ∼3 to 5 mm distal to its origin and a 3-0 dermalon filament (3 cm length) with blunted tip was inserted into the ICA and advanced to the origin of the MCA. Middle cerebral artery occlusion was confirmed by Laser Doppler (Moor Instruments, Wilmington, DE, USA) and cerebral blood flow evaluated immediately before and after initiation of pMCAO.3 In these studies, cerebral blood flow was reduced to 24.1%±6.6%, 22.0%±8.7%, 20.5%±7.3%, and 19.7%±8.9% of pre-MCAO values for rats treated with vehicle, HOE-642, bumetanide and HOE-642 plus bumetanide, respectively (mean value±s.d., of 26, 17, 21, and 18 rats, respectively). For some experiments, rats were subjected to MCAO surgery without insertion of the filament (Sham). HOE-642 and/or bumetanide were administered intravenously (15 or 30 mg/kg in 2 to 4 doses, respectively, of 7.5 mg/kg) starting at 20 minutes before initiation of pMCAO. For neurologic outcome experiments, some rats were given HOE-642 and/or bumetanide by a single intraperitoneal injection. Upon initiation of pMCAO, rats were placed supine on a Plexiglas stage with bite bar and ear clamps, then the stage was positioned in the magnet bore of a 7-T Bruker Biospec MRS/MRI system (Bruker Biospin MRI, Billerica, MA, USA). Rats were then subjected to DWI or to 23Na chemical shift imaging (CSI) spectroscopy for up to 210 minutes to determine cerebral edema and Na uptake, respectively, as described in the following sections. HOE-642 and bumetanide were prepared as fresh stock solutions for each experiment. HOE-642 was dissolved in water. Bumetanide was prepared in saline solution containing 0.5% albumin.3
Immunoelectron Microscopy Methods
Rat brains subjected to left pMCAO for 90 minutes underwent cardiac perfusion fixation for 60 minutes using 4% paraformaldehyde plus 0.05% glutaraldehyde in 0.1 mol/L phosphate buffer (pH 7.4) as described previously.3, 6 The brains were postfixed in 4% paraformaldehyde overnight then subjected to freeze substitution.6, 20 Tissues were embedded in lowiacryl resin, sectioned onto carbon-coated grids, and immunolabeled with monoclonal NHE1 antibody (4E9, Millipore, Bedford, MA, USA) or polyclonal NHE2 antibody (AB3038, Millipore) with 15 nm gold particle-conjugated anti-mouse IgG or anti-rabbit IgG, respectively, as described previously.3, 6 After staining sections with uranyl acetate and lead citrate, images were collected using a Philips 410 electron microscope (F.E.I. Company, Hillsboro, OR, USA). Gold particles in both luminal and abluminal BBB membranes were counted using a double-blind method. The percentage of plasma membrane gold particles found in the luminal membrane was determined for each of 111 and 115 electron micrographs examined for NHE1 and NHE2, respectively.
Magnetic Resonance Diffusion Weighted Imaging Analysis of Apparent Diffusion Coefficient Values
Diffusion-weighted imaging was performed using a 7-T Bruker Biospec MRS/MRI system as described previously.3, 5 Briefly, rats were placed into a 72-mm radio frequency probe inside the 7-T magnet and spin echo images (2 mm slices) were then acquired at 50 to 210 minutes following occlusion of the MCA or Sham surgery. Apparent diffusion coefficient (ADC) values were determined for selected brain regions of interest (ROIs) using four gradient strengths21 and Paravision 2.1 software (Bruker Biospin GmbH, Rheinstetten, Germany). Apparent diffusion coefficient values for anatomically corresponding ipsilateral (left) and contralateral (right) hemisphere ROIs were compared and ratios of L/R ADC values were calculated.
Magnetic Resonance Chemical Shift Imaging of Brain Na
Magnetic resonance CSI was used to determine brain Na content of rats, either total brain Na (vascular plus extravascular) or extravascular Na. In experiments quantifying extravascular Na, the NMR chemical shift/relaxation reagent Dysprosium triethylenetetraminehexaacetic acid (DyTTHA) was prepared and infused as previously described.22, 23 Briefly, 250 mmol/L DyTTHA was infused intravenously at 0.3 mL/min to achieve a final dose of 1.5 mmol per liter per kg and allowed to equilibrate across the various body compartments for 20 minutes. Since MCAO does not cause complete occlusion, this will include brain vasculature in both hemispheres. For the first ∼4 hours after pMCAO, BBB permeability remains low so DyTTHA remains within the vascular space of the sampled region and shifts the frequency of intravascular sodium.24, 25
For Na CSI, rats with the Plexiglas stage were inserted into the bore of a 7-T Bruker Biospec MRS/MRI system (Bruker) such that the head was centered in a double tuned 1 H/23Na probe (Doty Scientific, Columbia, SC, USA). For experiments using DyTTHA, rats were placed in the 7-T bore after the start of DyTTHA infusion. Using the 1H channel, shimming was optimized and scout images were acquired for locating Na CSI voxels. One-pulse 23Na experiments were performed at 30 minute intervals before, between, and after CSI experiments to confirm plasma [DyTTHA] was stable as measured by maintenance of vascular/extracellular Na shift of 1.8±0.1 p.p.m.
Two-dimensional Na CSI images were acquired via the standard Bruker CSI protocol using a 2-millisecond three-lobe sinc pulse for slice selection with ∼2.75 milliseconds from the center of excitation pulse to start of data acquisition. Slices were 4 mm thick and acquired in a 32 × 32 matrix with a FOV of 64 mm. Each 23Na CSI data set was acquired in 21 minutes using a repetition time of 250 milliseconds. Data were zero filled and Fourier transformed in all three dimensions using MATLAB to generate a spectrum for each pixel corresponding to 1 mm × 1 mm × 4 mm voxels. Na CSI spectra were further analyzed using MATLAB to integrate over the unshifted extravascular Na peak and that of an external standard to calculate brain [Na] using the following equation:
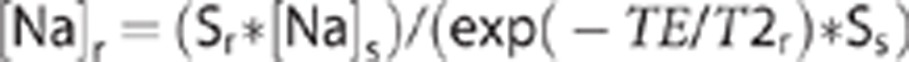 |
where S is the integrated signal intensity measured for any voxel, TE is the delay between the excitation pulse and the beginning of data acquisition, and the subscripts r and s indicate rat brain and external standard, respectively. This equation is derived from the standard equation for T1 and T2 dependent signal decay assuming TR is >5 T1 for Na in both rat and external standard and that TE is much smaller than T2 for Na in the external standard.
2,3,5-Triphenyltetrazolium Chloride Assessment of Lesion Size
At the conclusion of DWI experiments (210 minutes), rats were euthanized and the brain quickly removed and sectioned into 2 mm thick slices starting at the frontal pole using a Brain Matrix Slicer (Vibratome, St Louis, MO, USA). Slices were then immersed in 2% TTC (Sigma Aldrich, St Louis, MO, USA) in a petri dish and incubated at 37oC for 20 minutes. Slices were then scanned (Epson Perfection 1200U scanner, Epson America Inc., Long Beach, CA, USA and Adobe Photoshop software, Adobe Systems Inc., San Jose, CA, USA) and brain slices then analyzed for infarct volume using Image-J analysis software (public domain software developed at NIH and available on the internet at http://rsb.info.nih.gov/nih-image). Percent infarct was calculated as described by Swanson et al:26
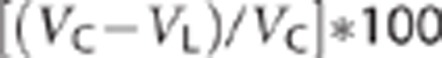 |
where VC is the volume of control hemisphere and VL is the volume of noninfarcted tissue in the lesioned hemisphere.
Neurologic Assessment
Rats treated with vehicle, HOE-642 and/or bumetanide and subjected to pMCAO were allowed to recover from anesthesia, then assessed for neurologic function at 4 hours, 1 day, and 2 days after the start of pMCAO. Sensorimotor deficits were assessed by the 14-score protocol.27 Briefly, rats were hand-held in an immobilizing grip then visual and tactile forelimb and hindlimb placing as well as proprioceptive hindlimb placing were tested. Rats were also evaluated by Rotarod test28 Here, rats were allowed to run for up to 300 seconds on a rod rotating at constant speed (16 r.p.m.). Rats were subjected to baseline/training trials the day before pMCAO or Sham surgery and then reevaluated at days 1 and 2 following induction of pMCAO to determine the length of time they could run on the apparatus without falling. Trials were repeated three times and the highest score was recorded.
Statistical Analyses
Values are presented as mean±s.d. Data shown were analyzed for significance using analysis of variance or by Student's t-test. P values <0.05 were considered to indicate significant difference.
Results
In Situ Distribution of NHE1 and NHE2 Na/H Exchange Isoform Proteins in Cerebral Microvascular Endothelial Cells of Ischemic Rat Brain
If NHE1 and/or NHE2 of BBB endothelial cells participates in edema formation during cerebral ischemia by transporting Na from the blood into brain, then one or both of the NHE proteins should be present in the luminal BBB membrane in situ in ischemic brain. To test this, we used rat brains perfusion fixed after 90 minutes of pMCAO and immunoelectron microscopy with antibodies that specifically recognize NHE1 or NHE2 proteins. Representative immunoEM micrographs reveal that both NHE1 (Figure 1A) and NHE2 (Figure 1B) reside predominantly in the luminal BBB membrane of ipsilateral (ischemic) cortical brain sections, whether core or penumbra, as well as in contralateral (normoxic) cortical sections. Quantitation of the in situ BBB gold particle distribution observed in multiple micrographs shows that NHE1 luminal distribution was 66% to 75% of total plasma membrane NHE1 in contralateral cortex and 83% to 86% in the ipsilateral cortex (Figure 1C). The apparent increase in luminal NHE1 of ipsilateral versus contralateral cortex reached statistical significance for ipsilateral penumbra but not core. We also found that 75% to 80% of NHE2 resides in the luminal BBB membrane, with no significant differences observed between ipsilateral core and penumbra compared with contralateral cortical sections (Figure 1D).
Figure 1.

Immunoelectron microscopy localization of NHE1 and NHE2 proteins in microvascular endothelial cell membranes of ischemic rat brain. (A, B) Rats were subjected to 90 minutes of permanent middle cerebral artery occlusion (pMCAO), then brains perfusion fixed and prepared for immunoelectron microscopy as described in Materials and Methods. The brain sections were labeled with NHE1 or NHE2 antibodies at dilutions of 1:2,000 and 1:1,000 (NHE1 and NHE2, respectively). Sections were then labeled with gold particle-conjugated secondary antibody. ImmunoEM images shown are representative micrographs. Vessel lumens are at the top of each image with the basal lamina underlying the endothelium and separating the cells from perivascular astrocytes and neurons. Locations of gold particles are indicated with arrowheads. (C, D) NHE1 and NHE2 distribution between luminal (L) and abluminal (A) microvascular endothelial cell membranes of ischemic (ipsilateral core and penumbra) and control (contralateral) frontoparietal cortex of perfusion-fixed rat brains as determined by quantitation of gold particles in immunoelectron micrographs. Values shown are mean values±s.d. of 23 to 32 microvessels for NHE1 core and penumbra 1:1,000 and 1:2,000 antibody dilutions and 22 to 37 microvessels for NHE2 core and penumbra 1:500 and 1:1,000 antibody dilutions. *Significantly different from contralateral penumbra by analysis of variance (ANOVA) with Bonferroni Dunn post hoc test (P<0.0001) p value for NHE2 ANOVA comparing all eight groups is 0.947. It should be noted that the fixation methods used here, required to retain good antigenicity of the tissue, sacrifice some structural detail in the images. Also, although NHE transporter proteins are abundant in the tissue, the section thickness and size of the field of view result in few gold particles visible in each image. Quantitation of microvessels from >20 micrographs for each antibody and dilution revealed that NHE1 and NHE2 are distributed 66% to 75% and 75% to 80%, respectively, in the luminal membrane. Qualitatively, the immunoEM images reveal that NHE1 and NHE2 proteins are found predominantly in the luminal blood–brain barrier (BBB) membrane.
Intravenous HOE-642 Reduces Cerebral Edema and Infarct in Permanent Middle Cerebral Artery Occlusion
Our previous finding that BBB Na/H exchange activity is rapidly stimulated by hypoxia, aglycemia, and AVP suggests that, like BBB Na–K–Cl cotransporter activity, the Na/H exchanger participates in early ischemia-induced cerebral edema formation. To evaluate the contribution of Na/H exchange activity to edema formation occurring during the initial stages of pMCAO before BBB breakdown, we examined the effects of intravenous HOE-642 on pMCAO-induced edema in rats using MRI DWI. Rats were administered intravenous HOE-642 or vehicle, then pMCAO immediately induced and edema formation evaluated over a 210-minute time course. From the DWI data, we calculated ADC values for four brain ROI in both ipsilateral and contralateral hemispheres (regions L1–L4 and R1–R4, respectively) as depicted in Figure 2A, representative MR proton images captured 210 minutes after the start of left pMCAO in rats given vehicle, HOE-642 or HOE-642+Bumetanide. In these images, hyperintensity of the left hemisphere corresponds to edema formation. Figures 2B and 2C (ROIs 1 and 2) show that in rats given vehicle and subjected to pMCAO, the left/right ADC ratios fell below the Sham ratio of 1.0 (depicted with the dotted line). However, in rats given HOE-642 (15 mg/kg), the fall in ADC ratios was attenuated, indicating reduction of edema formation. Specifically, the fall in ADC values was reduced by 24% to 73% and 29% to 63% for RO1s 1 and 2, respectively, over the 210-minute time course. This attenuation of edema formation was sustained throughout the 210-minute experiment. Because our previous studies showed significant reduction of edema formation during pMCAO in rats given intravenous bumetanide, in the present study we also tested whether treating rats with intravenous HOE-642 plus bumetanide (15 mg/kg each) provides a greater reduction of edema. Here, the fall in ADC values was reduced by 71% to 90% and 67% to 98% for ROIs 1 and 2, respectively, over the 210-minute time course. While there is a trend for greater edema reduction with HOE-642 plus bumetanide compared with HOE-642 alone, the differences reached statistical significance only at 50 minutes for L1/R1 values and at 70, 90, and 110 minutes for L2/R2 values (by analysis of variance with Neuwman–Keuls post hoc test). Similar results were obtained for ROI 4 (data not shown).
Figure 2.

Brain edema formation in permanent middle cerebral artery occlusion (MCAO): inhibition by intravenous HOE-642. (A) Magnetic resonance diffusion-weighted brain images of rats subjected to left pMCAO illustrating relative hyperintensity (edema) on left side. Regions of interest for ipsilateral and contralateral cortex (regions of interest (ROIs) L1–L3 and R1–R3, respectively) and striatum (ROI L4 and R4, respectively) are shown. Left to right panels show images of rats treated with vehicle, HOE-642 and HOE-642+Bumetanide, respectively. (B, C) Rats were administered vehicle, HOE-642 (15 mg/kg) or HOE-642 plus bumetanide (15 mg/kg each) as described in Materials and Methods and then apparent diffusion coefficient (ADC) values were determined for ROIs 1 to 4 for up to 210 minutes following induction of pMCAO. L/R ADC ratios are shown for ROIs 1 and 2 in (B, C), respectively. Values shown are mean values±s.d. for 4, 6, and 3 rats subjected to pMCAO with vehicle, HOE-642, and HOE-642+Bumet, respectively. ADC ratios for HOE-642 and HOE-642+Bumet are significantly different than Vehicle by analysis of variance (ANOVA) with Newman–Keuls post hoc test; *P<0.05, **P<0.01, ***P<0.001.
Table 1 shows that mean arterial pressure (MAP) in rats subjected to pMCAO was not significantly different among rats given vehicle, HOE-642 (15 mg/kg), or HOE-642 plus bumetanide (15 mg/kg each). Other physiological parameters, including plasma electrolytes, BUN (blood urea nitrogen), glucose, hemoglobin, and hematocrit were also not significantly altered by HOE-642 or HOE-642 plus bumetanide. We have shown previously that bumetanide alone does not alter these parameters.3 In these studies, we also evaluated edema by gravimetric methods following 180 minutes of pMCAO (Table 2). We found that the percent of water in ipsilateral brain hemisphere was significantly elevated above that in contralateral hemisphere for rats subjected to 180 minutes of pMCAO and vehicle only, as we have reported previously.3 However, in rats treated with HOE-642 or with HOE-642 plus bumetanide, the percent brain water following 180 minutes of pMCAO was not significantly different between ipsilateral and contralateral hemispheres nor was it significantly different than contralateral hemisphere of rats treated with MCAO and vehicle only. In previous studies, we found similar results for effects of bumetanide alone on percent brain water following MCAO.3
Table 1. Physiological parameters of rats subjected to MCAO ± HOE-642 ± Bumetanide.
| Vehicle | HOE-642 | HOE-642+Bumetanide | |
|---|---|---|---|
| Na+ (mmol/L) | 134.8±1.7 | 133.6±0.9 | 139.7±3.8 |
| K+ (mmol/L) | 5.7±0.7 | 5.5±0.7 | 6.1±2.9 |
| Cl− (mmol/L) | 103.8±2.4 | 101.6±1.6 | 105.3±4.2 |
| pH | 7.30±0.50 | 7.37±0.04 | 7.3±1.23 |
| pCO2 (mm Hg) | 47.6±7.8 | 42.1±4.5 | 44.5±4.2 |
| HCO3− (mmol/L) | 23.8±1.2 | 24.4±2.2 | 23.0±1.0 |
| BUN (mg/dL) | 41.8±3.7 | 43.4±2.5 | 44.3±1.2 |
| Glucose (mg/dL) | 192.0±46.8 | 204.0±26.4 | 164.3±28.2 |
| Hemoglobin (g/dL) | 13.8±1.5 | 14.2±0.9 | 13.0±0.0 |
| Hematocrit (%PCV) | 40.0±4.2 | 41.2±2.5 | 38.7±0.5 |
| MAP (mm Hg) | 87.6±6.00 | 84.8±2.80 | 90.1±20.06 |
BUN, blood urea nitrogen; MAP, mean arterial pressure; PCV, packed cell volume; pMCAO, permanent middle cerebral artery occlusion.
Before MCAO, rats were nephrectomized, treated with vehicle or HOE-642 (15 mg/kg) or HOE-642 plus bumetanide (15 mg/kg each). All physiological parameters shown except MAP were determined by iStat after 3 hours of permanent MCAO as described in Materials and Methods. Values are mean±s.d. for 6, 5, and 3 experiments with Vehicle, HOE-642, and HOE-642 + Bumetanide, respectively. MAP measurements were taken every 30 minutes over imaging period and a single average MAP value obtained for each animal. Variation of MAP within each experiment was 15.1%, 13.1%, and 17.2% of mean (s.d. as % of mean) for Sham, MCAO + Vehicle and MCAO + Bumetanide, respectively. No significant differences were found among any of the groups for any of the parameters by analysis of variance or two-tailed unpaired t-tests. P values for analysis of variance analyses were 0.1806, 0.1566, 0.1689, 0.2014, 0.3495, 0.5694, 0.4700, 0.3792, 0.3672, 0.5744, and 0.8885 for Na, K, Cl, pH, pCO2, HCO3, BUN, Glucose, Hemoglobin, Hematocrit, and MAP, respectively.
Table 2. Effects of HOE-642 and bumetanide on brain water content after 180 minutes of permanent MCAO.
| Percent brain water | ||
|---|---|---|
| |
Ipsilateral |
Contralateral |
| MCAO+Vehicle | 80.61±0.63a,b | 78.69±0.36 |
| MCAO+HOE | 79.61±0.44 | 78.75±0.24 |
| MCAO+HOE+Bumet | 79.22±0.59 | 78.44±0.24 |
MCAO, middle cerebral artery occlusion.
Nephrectomized rats were treated with either vehicle, HOE-642 (15 mg/kg) or HOE-642 + bumetanide (15 mg/kg each), and then subjected to permanent MCAO. After 180 minutes, the rats were killed and hemispheric brain water determined gravimetrically as described in Materials and Methods. Values are represented as mean±s.d. for 5, 4, and 3 rats for vehicle, HOE- and HOE+Bumetanide-treated rats, respectively.
Significantly different from contralateral values for MCAO + Vehicle, MCAO + HOE and MCAO + HOE + Bumet (by analysis of variance, P<0.001 for all three comparisons).
Significantly different from ipsilateral values for MCAO + HOE and MCAO + HOE + Bumet (by analysis of variance, P<0.01 for both comparisons).
At the end of the imaging, the rat brains were subjected to TTC staining to assess infarct volume, as described previously3 and in Materials and Methods. Figure 3A shows representative TTC images of rats subjected to pMCAO and treated with vehicle, HOE-642 or HOE-642 plus bumetanide. A characteristically large MCAO-induced lesion was observed in the left hemisphere of vehicle-treated rats, while HOE-642 (15 mg/kg)-treated rats exhibited an attenuated TTC-defined lesion. Little to no infarct was observed in rats given HOE-642 plus bumetanide (15 mg/kg). Figure 3B shows mean infarct volumes following 210 minutes of pMCAO. HOE-642 treatment markedly reduced percent infarct compared with MCAO with vehicle, as did HOE-642 plus bumetanide treatment. Here total percent infarct was 41.5%±8.7% for vehicle-treated rats compared with 10.3%±5.6% for HOE-642-treated rats (mean values±s.d.). Rats treated with HOE-642+bumetanide exhibited an apparent greater reduction in total hemispheric infarct compared with rats treated with HOE-642 alone although the difference did not reach statistical significance. Further analyses of these data revealed that HOE-642 appears to have a greater protective effect in cortex than subcortex. The subcortical infarct is 32.0%±4.5% of the total infarct in vehicle-treated rats but 66.1%±13.3% of the total infarct in HOE-642-treated rats.
Figure 3.

2,3,5-Triphenyltetrazolium chloride (TTC) assessment of infarct size: effects of HOE-642 and bumetanide. (A) At the conclusion of diffusion-weighted imaging experiments, TTC staining of brain slices was done as described in Materials and Methods. Images are from three representative experiments and show brain slices from rats given intravenous injection of vehicle, HOE-642 (15 mg/kg) or HOE-642 plus bumetanide (15 mg/kg each) then subjected to 210 minutes of permanent middle cerebral artery occlusion (pMCAO). In all three panels, the brain slices start at 2 mm from the frontal pole and continue in 2 mm increments to 16 mm from the frontal pole. (B) The TTC-defined total hemispheric infarct volume was determined by summing infarct volumes of all 2 mm brain slices for each rat (2- to 16-mm slices from the frontal pole). Data represent infarct volume as percent of the total ipsilateral hemisphere volume, corrected for brain swelling, and are mean values±s.d. of 4, 6, and 3 rats subjected to MCAO+Vehicle, MCAO+HOE-642, and MCAO+HOE-642+Bumet, respectively. Values for HOE-642 and HOE-642+Bumet are significantly different from Vehicle by analysis of variance (ANOVA) with Newman–Keuls post hoc test; *P<0.05, **P<0.01.
Intravenous HOE-642 and Bumetanide Reduce Brain Na Uptake in Permanent Middle Cerebral Artery Occlusion
The hypothesis that a luminal BBB Na/H exchanger and/or Na–K–Cl cotransporter works with the abluminal Na/K ATPase to increase Na transport from blood into brain during ischemic stroke predicts that intravenous HOE-642 and/or bumetanide will reduce not only edema but also brain Na uptake, the driving force for water entry into the brain during cerebral ischemia. To examine the contribution of Na/H exchange and Na–K–Cl cotransporter activities to brain Na uptake occurring during the initial stages of pMCAO, we employed NMR Na CSI methods, as described in Materials and Methods. Here, we evaluated brain Na content following induction of pMCAO, determining ipsilateral/contralateral ROI brain Na content ratios. Figure 4A shows that in vehicle-treated rats subjected to pMCAO, the Na content ratio rose linearly, reaching a 1.79-fold increase by 192 minutes. For rats given intravenous bumetanide or HOE-642 (30 mg/kg each) and then subjected to pMCAO, the increase in brain Na content ratio was significantly attenuated, reaching only 1.45- and 1.37-fold increases, respectively, by 192 minutes. Our hypothesis predicts that the MCAO-induced increase in brain Na is mediated by BBB secretion of Na from the blood into brain and thus the increase in total brain Na should be accounted for specifically by an increase in brain extravascular Na. To test this, we used magnetic resonance Na CSI to evaluate changes in brain extravascular Na (Figures 4B and 4C). Here, vascular and extravascular Na spectral peaks were separated through use of the chemical shift regent DyTTHA, as described in Materials and Methods. We found that brain extravascular Na increased as expected and that the rise was greater than that for total brain Na. By linear regression analysis, the rate of total brain [Na] increase determined for MCAO+Vehicle (Figure 4A) was 20.0%±6.7%/h while the rate of extravascular brain [Na] increase determined for MCAO+Vehicle was 48.1%±3.6% and 39.3%±4.4% for Figures 4B and 4C, respectively, and 43.5%±4.2%/h for combined analysis of Figures 4B and 4C. Also, as expected the MCAO-induced rise in extravascular Na was significantly attenuated by HOE-642 or bumetanide.
Figure 4.

HOE-642 and bumetanide reduction of brain Na uptake in middle cerebral artery occlusion (MCAO). (A) Rats were administered intravenous HOE-642 (30 mg/kg), bumetanide (30 mg/kg), or vehicle immediately before initiation of pMCAO. Localized 23Na chemical shift imaging (CSI) magnetic resonance spectroscopy (MRS) was used to determine brain [Na] of specified regions of interest (ROI) as described in Materials and Methods. ROIs are as depicted in Figure 2A. Values are mean values±s.d. for 6, 9, and 6 rats for vehicle, bumetanide, and HOE-642, respectively. Sham-operated rat Na L/R ratios, depicted in the figure as the dashed line, averaged 1.0±0.02 (mean±s.d., n=6) over the 192 minute time course following induction of pMCAO. Brain [Na] values for bumetanide and HOE-642-treated rats are significantly different than vehicle-treated rats by analysis of variance (ANOVA) with Newman–Keuls post hoc test at 104, 126, 170, and 192 minutes for bumetanide (P<0.01, P<0.05, P<0.05, and P<0.01, respectively) and at 126, 148, 170, and 192 minutes for HOE-642 (P<0.05, P<0.05, P<0.01, and P<0.001, respectively). Asterisks indicate values that are significantly different from MCAO+Vehicle by two-tailed t-test; *P<0.05, **P<0.01). (B, C) Rats were administered intravenous HOE-642 (30 mg/kg) or bumetanide (30 mg/kg) immediately before pMCAO. Localized 23Na CSI MRS of DyTTHA-infused rats was used to determine extravascular brain [Na] of specified ROIs as described in Materials and Methods. Values are mean values±s.d. of eight rats each for vehicle and HOE-642 (B) five and four rats for vehicle and bumetanide, respectively (C). Extravascular brain [Na] values for HOE are significantly different than vehicle by ANOVA with Newman–Keuls post hoc test at 126 and 148 minutes (P<0.05 for both). Extravascular brain [Na] values for bumetanide are significantly different than vehicle by ANOVA with Newman–Keuls post hoc test at 120, 150, and 180 minutes (P<0.05, P<0.001, and P<0.001, respectively). For (B, C), asterisks indicate values that are different from MCAO+Vehicle by two-tailed t-test; *P<0.05, **P<0.01, ***P<0.001.
Intraperitoneal and Intravenous HOE-642 and Bumetanide Improve Neurologic Outcome Following Permanent Middle Cerebral Artery Occlusion
In the present study, we also determined whether the reduction of cerebral edema and brain Na uptake observed with HOE-642 and/or bumetanide is associated with significant improvement in neurologic outcome following pMCAO. Rats were given a single intraperitoneal injection of HOE-642, bumetanide or HOE-642 plus bumetanide immediately before pMCAO, then subjected to a 14-point neurologic assessment, as described in Materials and Methods. As shown in Figure 5A, rats subjected to pMCAO and vehicle exhibited poor neurologic outcome compared with Sham-operated rats at 4 hours, 1 day, and 2 days, respectively, after the start of pMCAO. For rats given bumetanide, significant increases in neurologic scores were observed at days 1 and 2 (but not 4 hours), while rats treated with HOE-642 showed significant increases in neurologic score at all three times, as did rats treated with HOE-642+bumetanide. Scores were not significantly different among HOE-642-, bumetanide- and HOE-642+bumetanide-treated rats at days 1 and 2, nor were scores for HOE-642 and HOE-642+bumetanide different at 4 hours, suggesting that the effects of HOE-642 and bumetanide by this neurologic test are not additive. We further evaluated neurologic function using the Rotarod test, as described in Materials and Methods. Rats treated with a single injection of HOE-642, bumetanide or HOE-642+bumetanide all showed significant improvement in Rotarod scores at day 2 compared with rats treated with vehicle (Figure 5B). Similar findings were obtained at day 1 (not shown). Rats were not tested at 4 hours after the start of pMCAO. Rotarod scores for rats treated with HOE-642+bumetanide were not significantly different than Sham-operated rat values.
Figure 5.

Neurologic assessment of rats subjected to middle cerebral artery occlusion (MCAO) and treated with HOE-642 and/or bumetanide. (A) Rats were administered intraperitoneal bumetanide (30 mg/kg), HOE-642 (30 mg/kg), Bumetanide+HOE-642 (30 m/kg each) or Vehicle then subjected to pMCAO. The rats were allowed to awaken from anesthesia and then tested for sensorimotor function at 4 hours, 1 day, and 2 days after induction of MCAO by the 14-score test, as described in Materials and Methods. Values are mean±s.d. with n values of 5, 11, 7, 5, and 7 for Sham, MCAO+Vehicle, MCAO+Bumet, MCAO+HOE, and MCAO+Bumet+HOE, respectively. Values are significantly different than MCAO vehicle scores by analysis of variance (ANOVA) with Newman–Keuls post hoc test; **P<0.01, ***P<0.001. MCAO+HOE score at 4 hours is significantly different from MCAO+vehicle score by two-tailed t-test. (B) Rotarod test. Values are mean±s.d. with n values of 5, 24, 20, 10, and 18 for Sham, MCAO+Vehicle, MCAO+Bumet, MCAO+HOE, and MCAO+Bumet+HOE, respectively. Values are significantly different than MCAO vehicle scores by ANOVA with Newman–Keuls post hoc test; *P<0.05, **P<0.01, ***P<0.001. In some experiments, rats were given bumetanide and/or HOE-642 by intravenous rather than intraperitoneal injection. No differences in outcome between the two routes of administration were observed (data not shown).
Discussion
Previous studies from our group provided evidence that BBB Na–K–Cl cotransport is a major contributor to ischemia-induced cerebral edema formation and that intravenous bumetanide reduces edema and infarct following pMCAO. However, additional BBB Na transporters likely also contribute to edema formation. We recently demonstrated that Na/H exchange is present in the luminal BBB membrane in situ and that its activity is stimulated by the ischemic factors hypoxia, aglycemia, and AVP.6 In the present study, we now provide additional evidence supporting a role for BBB Na/H exchange in ischemia-induced cerebral edema formation. Here, we show that NHE1 and NHE2 isoform proteins are present in the luminal BBB in situ during ischemia as well as normoxia. Using NMR DWI and Na spectroscopy, we also show that edema formation and brain Na uptake occurring in the rat pMCAO model of ischemic stroke are significantly attenuated by intravenous administration of HOE-642. Further, we provide evidence that HOE-642 reduces the TTC-defined lesion and improves neurologic outcome in rats subjected to pMCAO.
Our immunoelectron microscopy studies reveal that both NHE1 and NHE2 protein isoforms are present predominantly in the luminal BBB membrane in situ of ischemic cortex, both core and penumbral areas, as well as normoxic cortex, with 66% to 75% and 75% to 80% of plasma membrane NHE1 and NHE2, respectively, found in the luminal membrane. These values are in very good agreement with our earlier finding that in normoxic rat brains (not subjected to MCAO) NHE1 and NHE2 proteins are distributed 65% to 70% and 75% to 80%, respectively, in the luminal BBB membrane,6 indicating that ischemic conditions do not alter the plasma membrane distribution of NHE proteins. Our present study also found that in contralateral brain sections anatomically corresponding to the ipsilateral penumbra, luminal membrane NHE1 protein is 83% to 86% of total, higher than that found in control brain. The reasons for this apparent increase and whether it is of physiological significance will require further study. These findings support the hypothesis that a luminal BBB Na/H exchanger participates in vectorial transport of Na from the blood into brain not only in normoxic brain but also in ischemic brain when the secretion of Na into the brain is increased, contributing to edema formation as obligatory water follows.
The results of our MRI DWI studies reveal for the first time that intravenous HOE-642 reduces edema formation occurring in the early hours of pMCAO-induced cerebral ischemia in the rat. The finding that the fall in ADC values was attenuated over the entire 210 minutes experiment indicates that HOE-642 causes a sustained reduction in edema formation rather than simply slowing edema formation. Further, the combination of HOE-642 and bumetanide appears to provide a greater attenuation of edema that is also sustained throughout the experiment. Together with our previous studies of bumetanide effects on edema during MCAO, these findings are consistent with the hypothesis that BBB Na/H exchanger and Na–K–Cl cotransporter activities both contribute to ischemia-induced cerebral edema formation. These MRI findings are confirmed by our gravimetry experiments that reveal significant reduction of brain water after 3 hours of pMCAO in rats treated with HOE-642 or HOE-642+bumetanide compared with vehicle. Our studies also show that the MCAO-induced infarct, assessed as the TTC-defined lesion volume, is significantly reduced in rats treated with HOE-642 and that there is a trend for an even greater reduction of lesion volume with HOE-642 and bumetanide in combination. It should be noted that the doses of HOE-642 and bumetanide (15 to 30 mg/kg) used in this study are estimated to result in plasma concentrations sufficiently high to maximally inhibit the readily accessible luminal BBB membrane Na/H exchanger and Na–K–Cl cotransporter. In ongoing high performance liquid chromatography tandem mass spectrometry studies, rats given intravenous HOE-642 (7 mg/kg) or bumetanide (30 mg/kg) had plasma [HOE-642] and [bumetanide] of∼2 and ∼40 μmol/L, respectively, 60 minutes after injection (data not shown), maximally effective doses for both inhibitors.2, 6 It should also be noted that based on their chemical structures,29, 30, 31, 32 neither HOE-642 nor bumetanide are predicted to significantly enter the brain when the BBB is intact. Thus, only after BBB breakdown, as occurs for example, with ischemia/reperfusion, should these agents be readily accessible to brain parenchyma and inhibit Na transporter activities of astrocytes and neurons. Neither HOE-642 nor bumetanide, alone or in combination, was found to alter physiological parameters of the rats, including plasma electrolytes and MAP as shown here and in our previous study of bumetanide3 nor did they alter cerebral blood flow (data not shown). Thus, the effects of HOE-642 and bumetanide on edema and infarct during pMCAO are unlikely due to changes in these parameters. A recent study of ischemia/reperfusion brain injury in mice showed that HOE-642 effectively reduced brain lesion volume assessed by DWI T2 imaging.14 Together with our present finding, this suggests that Na/H exchange participates in ischemia/reperfusion injury as well as in the detrimental formation of edema that occurs early during ischemia before any reperfusion event.
An important finding of the present study is that both intravenous HOE-642 and bumetanide reduce brain Na uptake during ischemia. Here, we demonstrate that brain [Na] increases linearly over the course of our pMCAO experiments with the rate of brain [Na] increase significantly reduced by a single injection of either HOE-642 or bumetanide. This finding is consistent with our hypothesis that luminal BBB Na/H exchange and Na–K–Cl cotransport, coupled with the abluminal Na/K pump, perform net transport of Na from blood into brain, providing the driving force for osmotically obliged water to follow. It is important to note that although MRI DWI is valuable for determining whether a potential therapy causes a sustained reduction of edema formation or simply delays edema formation, this method has the limitation that ADC values do not decrease in proportion to the increase in brain water occurring as edema progresses (as assessed by gravimetry). That is, the methodology limits the ADC values from falling below a minimum ‘floor' value. Gravimetry studies have documented that during cerebral ischemia, total brain water continues to increase through at least 12 hours after the start of ischemia, as does brain Na, evaluated by flame photometry.33, 34 The advantage of the 23Na MRS (magnetic resonance spectroscopy) methods used in this study is that changes in brain [Na] can be monitored in real-time in vivo during MCAO. Our findings regarding linear Na uptake during ischemia are in agreement with previous 23Na MRI studies of Jones et al.35 showing that rat brain Na increases linearly for up to 7 hours during ischemia induced by MCA surgical transection. Linear brain Na uptake has also been observed during 5 hours of ischemia induced by MCAO in the rat36 and macaque.37 The rate of total brain [Na] increase observed in our pMCAO experiments (20.0%±6.7%/h) agrees quite well with values reported previously for brain [Na] increases of 25.0%±4.7% and 22%±4%/h as determined by 23Na MRI for MCA transection35 and intraluminal MCAO,36 respectively.
In the present study, we also conducted 23Na MRS experiments with the shift reagent DyTTHA to specifically evaluate changes in extravascular brain [Na], as described in previous studies.25, 38 Here, we report a steeper rise in extravascular brain [Na] compared with total brain [Na] during MCAO. This is as expected if the increase in brain Na is due to BBB secretion of Na from blood into brain, with vascular [Na] remaining constant during MCAO. Further, HOE-642 and bumetanide both significantly reduce the increase in brain extravascular [Na], providing further support for our hypothesis that BBB Na/H exchange and Na–K–Cl cotransport participate in increased vectorial transport of Na from the blood into brain during ischemia. We did not test the combined effects of bumetanide and HOE-642 in these experiments. Whether these Na transporter inhibitors have additive effects on reducing brain Na uptake will require further investigation. It should also be noted that HOE-642 is a more potent inhibitor of NHE1 than NHE2, with reported IC50 values of 0.03 to 3.4 μmol/L and 4.3 to 62 mmol/L, respectively.39 Despite this, our study was designed to ensure maximal inhibition of both NHE isoforms and thus additional studies are needed to determine the relative contributions of NHE1 and NHE2 to edema formation and brain Na uptake in pMCAO.
The present study also demonstrates that HOE-642 and bumetanide significantly improve neurologic outcome of rats through at least 2 days following onset of pMCAO. This suggests that inhibiting Na/H exchange and/or Na–K–Cl cotransport to reduce early edema formation is associated with an effective improvement in neurologic function. Additional studies will be needed to determine whether this improved outcome is sustained for longer periods.
Our studies do not allow us to conclude that the observed effects of bumetanide and HOE-642 are exclusively due to their inhibition of BBB Na/H exchanger and Na–K–Cl cotransporter activities. While preliminary studies suggest that these inhibitors do not readily penetrate into the brain under normoxic conditions, it is possible that some inhibition of astrocyte and/or neuronal Na/H exchange and Na–K–Cl cotransport may occur during the early states of ischemia and, as BBB breakdown occurs, one would predict robust inhibition of all Na/H exchange and Na–K–Cl cotransport, both at the BBB and in brain parenchymal cells. In this regard, it is important to note that the majority of previous studies investigating effects of these drugs in stroke used ischemia/reperfusion models in which the BBB breaks down and likely allows the drugs to reach their targets on astrocytes and neurons. The question of to what degree bumetanide and HOE-642 effects are due to BBB, astrocyte and/or neuronal targets is an important one that will require further investigation. It should also be noted that other BBB ion transporters and channels likely contribute to edema formation in stroke. One example is the finding that de novo synthesis of a BBB SUR1-regulated NCCa-ATP channel several hours after onset of ischemia contributes to continued edema formation during ischemic stroke.40
In summary, the results of the present study demonstrate for the first time that intravenous administration of HOE-642 and bumetanide significantly reduces cerebral edema, brain Na uptake and lesion volume and also improves neurologic outcome in rats subjected to pMCAO. These findings support the hypothesis that luminal BBB membrane Na/H exchanger and Na–K–Cl cotransporter, readily inhibited by intravenous HOE-642 and bumetanide, provide effective therapeutic targets for reduction of edema and infarct in ischemic stroke. Together with previous ischemia/reperfusion studies, these findings suggest that HOE-642 and bumetanide reduce the brain Na uptake and edema formation that occur during early hours of ischemia when the BBB is still intact and, with the onset of BBB breakdown occurring during more prolonged ischemia or with reperfusion, states in which HOE-642 and bumetanide are predicted to more readily permeate into the brain, also reduce the damaging effects of increased astrocyte and neuronal Na/H exchange and Na–K–Cl cotransporter activities.
The authors declare no conflict of interest.
Footnotes
This study was supported by NIH RO1 NS039953 (MEO) and by American Heart Association Western States Affiliate Predoctoral Fellowship (TIL). NMR spectrometer expense was funded in part by NIH RR02511 and NSF PCM-8417289. This investigation was conducted in part in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR17348-01 from the National Center for Research Resources, National Institutes of Health.
References
- Betz AL. Alterations in cerebral endothelial cell function in ischemia. Adv Neurol. 1996;71:301–313. [PubMed] [Google Scholar]
- Foroutan S, Brillault J, Forbush B, O'Donnell ME. Moderate-to-severe ischemic conditions increase activity and phosphorylation of the cerebral microvascular endothelial cell Na+-K+-Cl- cotransporter. Am J Physiol Cell Physiol. 2005;289:C1492–C1501. doi: 10.1152/ajpcell.00257.2005. [DOI] [PubMed] [Google Scholar]
- O'Donnell ME, Tran L, Lam TI, Liu XB, Anderson SE. Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab. 2004;24:1046–1056. doi: 10.1097/01.WCB.0000130867.32663.90. [DOI] [PubMed] [Google Scholar]
- Brillault J, Lam TI, Rutkowsky JM, Foroutan S, O'Donnell ME. Hypoxia effects on cell volume and ion uptake of cerebral microvascular endothelial cells. Am J Physiol Cell Physiol. 2008;294:C88–C96. doi: 10.1152/ajpcell.00148.2007. [DOI] [PubMed] [Google Scholar]
- O'Donnell ME, Lam TI, Tran LQ, Foroutan S, Anderson SE. Estradiol reduces activity of the blood-brain barrier Na-K-Cl cotransporter and decreases edema formation in permanent middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2006;26:1234–1249. doi: 10.1038/sj.jcbfm.9600278. [DOI] [PubMed] [Google Scholar]
- Lam TI, Wise PM, O'Donnell ME. Cerebral microvascular endothelial cell Na/H exchange: evidence for the presence of NHE1 and NHE2 isoforms and regulation by arginine vasopressin. Am J Physiol Cell Physiol. 2009;297:C278–C289. doi: 10.1152/ajpcell.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserr HF, Cooper DN, Milhorat TH. Flow of cerebral interstitial fluid as indicated by the removal of extracellular markers from rat caudate nucleus. Exp Eye Res. 1977;25 (Suppl:461–473. doi: 10.1016/s0014-4835(77)80041-9. [DOI] [PubMed] [Google Scholar]
- Ennis SR, Ren XD, Betz AL. Mechanisms of sodium transport at the blood-brain barrier studied with in situ perfusion of rat brain. J Neurochem. 1996;66:756–763. doi: 10.1046/j.1471-4159.1996.66020756.x. [DOI] [PubMed] [Google Scholar]
- Betz AL. Sodium transport from blood to brain: inhibition by furosemide and amiloride. J Neurochem. 1983;41:1158–1164. doi: 10.1111/j.1471-4159.1983.tb09066.x. [DOI] [PubMed] [Google Scholar]
- Horikawa N, Kuribayashi Y, Itoh N, Nishioka M, Matsui K, Kawamura N, et al. Na+/H+ exchange inhibitor SM-20220 improves endothelial dysfunction induced by ischemia-reperfusion. Jpn J Pharmacol. 2001;85:271–277. doi: 10.1254/jjp.85.271. [DOI] [PubMed] [Google Scholar]
- Horikawa N, Kurobayashi Y, Matsui K, Ohashi N. Relationship between the neuroprotective effect of Na+/H+ exchanger inhibitor SM-20220 and the timing of its administration in a transient middle cerebral artery occlusion model of rats. Biol Pharm Bull. 2001;24:767–771. doi: 10.1248/bpb.24.767. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Matsumoto Y, Ikeda Y, Kondo K, Ohashi N, Umemura K. SM-20220, a Na+/H+ exchanger inhibitor: effects on ischemic brain damage through edema and neutrophil accumulation in a rat middle cerebral artery occlusion model. Brain Res. 2002;945:242–248. doi: 10.1016/s0006-8993(02)02806-8. [DOI] [PubMed] [Google Scholar]
- Kitayama J, Kitazono T, Yao J, Ooboshi H, Takaba H, Ago T, et al. Inhibition of Na+/H+ exchanger reduces infarct volume of focal cerebral ischemia in rats. Brain Res. 2001;922:223–228. doi: 10.1016/s0006-8993(01)03175-4. [DOI] [PubMed] [Google Scholar]
- Ferrazzano P, Shi Y, Manhas N, Wang Y, Hutchinson B, Chen X, et al. Inhibiting the Na+/H+ exchanger reduces reperfusion injury: a small animal MRI study. Front Biosci. 2011;3:81–88. doi: 10.2741/e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Luo J, Kintner DB, Shull GE, Sun D. Na(+)-dependent chloride transporter (NKCC1)-null mice exhibit less gray and white matter damage after focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:54–66. doi: 10.1038/sj.jcbfm.9600006. [DOI] [PubMed] [Google Scholar]
- Luo J, Chen H, Kintner DB, Shull GE, Sun D. Decreased neuronal death in Na+/H+ exchanger isoform 1-null mice after in vitro and in vivo ischemia. J Neurosci. 2005;25:11256–11268. doi: 10.1523/JNEUROSCI.3271-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hom S, Fleegal MA, Egleton RD, Campos CR, Hawkins BT, Davis TP. Comparative changes in the blood-brain barrier and cerebral infarction of SHR and WKY rats. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1881–R1892. doi: 10.1152/ajpregu.00761.2005. [DOI] [PubMed] [Google Scholar]
- Chang E, O'Donnell ME, Barakat AI. Shear stress and 17beta-estradiol modulate cerebral microvascular endothelial Na-K-Cl cotransporter and Na/H exchanger protein levels. Am J Physiol Cell Physiol. 2008;294:C363–C371. doi: 10.1152/ajpcell.00045.2007. [DOI] [PubMed] [Google Scholar]
- Zea Longa E, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Golshani P, Liu XB, Jones EG. Differences in quantal amplitude reflect GluR4- subunit number at corticothalamic synapses on two populations of thalamic neurons. Proc Natl Acad Sci USA. 2001;98:4172–4177. doi: 10.1073/pnas.061013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatlisumak T, Takano K, Carano RAD, Fisher M. Effect of basic fibroblast growth factor on experimental focal ischemia studied by diffusion-weighted and perfusion imaging. Stroke. 1996;27:2292–2298. doi: 10.1161/01.str.27.12.2292. [DOI] [PubMed] [Google Scholar]
- Anderson SE, Murphy E, Steenbergen C, London RE, Cala PM.Na/H exchange in myocardium: effects of hypoxia and acidification on Na and Ca. Am J Physiol Cell Physiol 1990259(Cell Physiology 28)C940–C948. [DOI] [PubMed] [Google Scholar]
- Balschi JA. 23Na NMR demonstrates prolonged increase of intracellular sodium following transient regional ischemia in the in situ pig heart. Basic Res Cardiol. 1999;94:60–69. doi: 10.1007/s003950050127. [DOI] [PubMed] [Google Scholar]
- Bansal N, Germann MJ, Lazar I, Malloy CR, Sherry AD. In vivo Na-23 MR imaging and spectrosocpy of rat brain during TmDOTP5- infusion. J Magn Reson Imaging. 1992;2:385–391. doi: 10.1002/jmri.1880020405. [DOI] [PubMed] [Google Scholar]
- Preston E, Foster DO. Diffusion into rat brain of contrast and shift reagents for magnetic resonance imaging and spectroscopy. NMR Biomed. 1993;6:339–344. doi: 10.1002/nbm.1940060510. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- De Ryck B, Van Reempts J, Borgers M, Wauquier A, Janssen PA. Photochemical stroke model: flunarizine prevents sensorimotor deficits after neocortical infarcts in rats. Stroke. 1989;20:1383–1390. doi: 10.1161/01.str.20.10.1383. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, Pike BR, O'Dell DM, Lyeth BG, Jenkins LW. The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J Neurotrauma. 1994;11:187–196. doi: 10.1089/neu.1994.11.187. [DOI] [PubMed] [Google Scholar]
- Scholz W, Albus U, Counillon L, Gögelein H, Lang H-J, Linz W, et al. Protective effects of HOE642, a selective sodium-hydrogen exchange subtype 1 inhibitor, on cardiac ischaemia and reperfusion. Cardiovasc Res. 1995;29:260–268. [PubMed] [Google Scholar]
- Chen BP. Loop diuretics: comparison of torasemide, furosemide, and bumetanide. Drug Inform Update. 1996;60:343–354. [PubMed] [Google Scholar]
- Fischer H, Gottschlich R, Seelig A. Blood-brain barrier permeation: molecular parameters governing passive diffusion. J Membr Biol. 1998;165:201–211. doi: 10.1007/s002329900434. [DOI] [PubMed] [Google Scholar]
- Seelig A, Gottschlich R, Devant RM. A method to determine the ability of drugs to diffuse through the blood-brain barrier. Proc Natl Acad Sci. 1994;91:68–72. doi: 10.1073/pnas.91.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz AL, Keep RF, Beer ME, Ren XD. Blood-brain barrier permeability and brain concentration of sodium, potassium, and chloride during focal ischemia. J Cereb Blood Flow Metab. 1994;14:29–37. doi: 10.1038/jcbfm.1994.5. [DOI] [PubMed] [Google Scholar]
- Menzies SA, Betz AL, Hoff JT. Contributions of ions and albumin to the formation and resolution of ischemic brain edema. J Neurosurg. 1993;78:257–266. doi: 10.3171/jns.1993.78.2.0257. [DOI] [PubMed] [Google Scholar]
- Jones SC, Kharlamov A, Yanovski B, Kim DK, Easley KA, Yushmanov VE, et al. Stroke onset time using sodium MRI in rat focal cerebral ischemia. Stroke. 2006;37:883–888. doi: 10.1161/01.STR.0000198845.79254.0f. [DOI] [PubMed] [Google Scholar]
- Yushmanov VE, Kharlamov A, Yanovski B, LaVerde GC, Boada FE, Jones SC. Inhomogeneous sodium accumulation in the ischemic core in rat focal cerebral ischemia by 23Na MRI. J Magn Reson Imaging. 2009;30:18–24. doi: 10.1002/jmri.21816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVerde GC, Jungreis CA, Nemoto E, Boada FE. Sodium time course using 23Na MRI in reversible focal brain ischemia in the monkey. J Magn Reson Imaging. 2009;30:219–223. doi: 10.1002/jmri.21723. [DOI] [PubMed] [Google Scholar]
- Eleff SM, Mclennan IJ, Hart GK, Maruki Y, Traystman RJ, Koehler RC. Shift reagent enhanced concurrent 23Na and 1H magnetic resonance spectroscopic studies of transcellular sodium distribution in the dog brain in vivo. Magn Reson Imaging. 1993;30:11–17. doi: 10.1002/mrm.1910300104. [DOI] [PubMed] [Google Scholar]
- Masereel B, Pochet L, Laeckmann D. An overview of inhibitors of Na+/H+ exchanger. Eur J Med Chem. 2003;38:547–554. doi: 10.1016/s0223-5234(03)00100-4. [DOI] [PubMed] [Google Scholar]
- Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, et al. Newly expressed SUR1-regulated NCCa-ATP channel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12:433–440. doi: 10.1038/nm1390. [DOI] [PMC free article] [PubMed] [Google Scholar]