

Abstract
The plant virus, Cowpea mosaic virus (CPMV), is developed as a carrier of the chemotherapeutic drug doxorubicin (DOX). CPMV-DOX conjugate, in which eighty DOX molecules are covalently bound to external surface carboxylates of the viral nanoparticle (VNP), shows greater cytotoxicity than free DOX toward HeLa cells when administered at low dosage. At higher concentrations CPMV-DOX cytotoxicity is time-delayed. The CPMV-conjugate is targeted to the endolysosomal compartment of the cells, in which the proteinaceous drug carrier is degraded and the drug released. This study is the first demonstrating the utility of CPMV as a drug delivery vehicle.
Keywords: Cowpea mosaic virus; doxorubicin, drug delivery; viral nanoparticle; cancer
Introduction
Doxorubicin (DOX) is a highly potent drug used in the treatment of various cancers, but its clinical utility is limited by low solubility and severe adverse effects such as congestive heart failure. It is clear that a delivery vehicle is required to increase efficacy while reducing off-target effects. Doxil is the first clinically approved nanoparticle-based drug delivery system,1 and diverse classes of nanomaterials,2 including dendrimers, polymers, and metallic nanoparticles, are currently undergoing clinical trials.3 The optimum drug delivery system provides targeted delivery with controlled release so as to improve the efficacy of the therapeutic agent and minimise systemic toxicity.
Viruses provide an ideal basis for the development of targeted drug delivery vehicles as they have evolved naturally to deliver cargos to host cells with high efficiency.4 Several viral nanoparticle (VNP)-based nanomedicines are under development. VNPs from plant viruses and bacteriophages are favoured because these platforms are non-infectious toward mammals.5–7 Genome-free viral capsids of the bacteriophage MS2 have been modified to covalently bind taxol on their interior surface. The taxol-MS2, when incubated with MCF-7 breast cancer cells, released taxol and the resulting cell viability levels were similar to those observed for free taxol in solution.8 MS2 has also been loaded internally with a cocktail of drugs (DOX, cisplatin and 5-fluorouracil) and, after external surface modification, used as a targeted carrier.9 Further, phage Qβ,10 plant viruses Red clover necrotic mosaic virus (RCNMV)11 and Hibiscus chlorotic ringspot virus,12 and animal virus-like particles of the VP6 major structural protein of rotavirus13 have been used as drug carriers for DOX delivery.
Our research focuses on the plant virus Cowpea mosaic virus (CPMV). CPMV is an icosahedral virus with approximate diameter of 30 nm, the structure of which is known to near-atomic resolution.14 The virions comprise 60 copies each of two different types of coat protein that together form the asymmetric unit. The multiple copies of the asymmetric unit provide regularly spaced attachment units; facilitating the coupling and presentation of a wide range of different moieties on the exterior surface.15–18 Here we describe the covalent modification of CPMV with DOX. Two chemical ligation strategies were evaluated: i) covalent modification via a stable amide bond and ii) a labile disulfide bridge. When DOX is conjugated via stable amide bond to the CPMV carrier system, the formulation induces time-delayed, but enhanced, toxicity to HeLa cells compared to free drug.
Experimental Section
Materials and Instrumentation
1-ethyl-3-(3-dimethlyaminopropyl)carbodiimide hydrochloride (EDC) was purchased from Novabiochem, N-hydroxysuccinimide (NHS) from Fluka, 3,3'-dithiobis(sulfosuccinimidylpropionate) (DTSSP) from Thermo Scientific and doxorubicin hydrochloride (DOX·HCl) from Sigma. UV-visible spectra were recorded on a Perkin Elmer Lambda 25 spectrophotometer using UVWINLab software. Zeta potential was measured on a Malvern Instruments Zetasizer-Nano ZS: 1 mL of 0.5 mg/mL CPMV particles dispersed in 10 mM sodium phosphate buffer pH 7.2. Zeta cells were equilibrated at 21 °C for two minutes before recording three measurements each of ten runs. Data was collected with automatic attenuation selected and analysed using the Smoluchowski module. Transmission electron microscopy (TEM) images were obtained on a FEI Tecnai 20 TEM, FEI UK Ltd, using carbon-coated copper EM grids (400 mesh, Agar Scientific). Dynamic light scattering (DLS) was measured on a DynaPro Titan, Wyatt Technology Corporation (laser wavelength 830 nm, scattering angle 20 °).
Electrophoresis Methods
Agarose gel electrophoresis of 5–10 μg of CPMV particles suspended in 10 mM sodium phosphate buffer pH 7.0 with 2 μL of loading dye (Coomassie staining solution or MBI Fermentas dye) were analysed on 1.2 % (w/v) agarose gel in an electric field of 60 V for 1–2 hours. For ethidium bromide staining (of nucleic acid), 0.1 μg/ mL (4–5 L) in 1× TBE buffer was added to the gel. Particles were visualized on a UV transilluminator at 302 nm using Gene Genius Bio Imaging System with software Gene Snap (Syngene). For coat protein staining, gels were treated with Coomassie staining solution (50 % (v/v) methanol; 10 % (v/v) acetic acid; 0.25 % (w/v) Coomassie Brilliant Blue G-250) for 1h followed by destaining solution (50 % (v/v) methanol; 20 % (v/v) acetic acid in Milli-Q water) overnight. Sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed on 5–10 μg of virus in 10 mM sodium phosphate buffer pH 7.0 mixed with 3 μL of 4× RunBlue LDS Sample Buffer (Expedeon), samples were heated for 5–10 minutes at 100 °C in a thermoblock in order to denature the protein. The samples were analysed on 12 % TEO-CI SDS RunBlue precast gels (Expedeon) under non-reducing conditions at 180 V for 30–70 minutes using 600–700 mL of 20:1 dilution of 20× RunBlue SDS Running Buffer (Expedeon). The bands when stained with 20 mL of InstantBlue (Expedeon) were visible after a few minutes. Gels were washed with Milli-Q water prior to imaging. Gel images were recorded using camera or scanner.
CPMV-DOX
Carboxylic acid groups on the capsid surface were first activated by the carbodiimide method using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS). CPMV in either 10 mM or 0.1 M sodium phosphate buffer pH 7.4 was incubated with a freshly prepared Milli-Q water solution of EDC (1000 molar excess) for 5 minutes followed by the addition of a freshly prepared dimethylsulfoxide (DMSO) solution of NHS (4000 molar excess). The DMSO concentration was, if necessary, adjusted to be less than or equal to 20% (v/v). The reaction was allowed to proceed for 2h at room temperature with gentle stirring. The O-succinimide esterified CPMV particles were purified by methods described below. The activated particles, after purification, were incubated with a 2000 molar excess of DOX·HCl in 20% (v/v) DMSO. The reaction mixture was stirred overnight (16h) at 4 °C. Modified particles (CPMV-DOX) were eluted through a PD-10 column, fractions collected and concentrated on 100 kDa cut-off columns before being layered onto 5 ml 10–50% sucrose gradients. Fractions were collected and dialysed against 10 mM sodium phosphate buffer pH 7.0 for 2–4 days using 100 kDa molecular weight cut-off membranes. The yield of CPMV-DOX conjugate was between 60–80% based on the initial virus concentration. The number of DOX molecules was determined by measuring the absorbance at 490 nm (extinction coefficient, ε: 13500 M−1 cm−1; Supporting Information).
CPMV-SS-DOX
CPMV suspended in 0.1 M sodium phosphate, 0.15 M NaCl, pH 7.2 was incubated with a 2000 molar excess of aqueous 3,3'-dithiobis(sulfosuccinimidylpropionate) (DTSSP) solution. The reaction was left to proceed for 2h at ambient temperature. CPMV-dithiouni(sulfosuccinimidylpropionate) (CPMV-DTUSSP) particles were purified on a PD-10 gel filtration column. Freshly prepared CPMV-DTUSSP particles were incubated with 2000 molar excess of DOX·HCl dissolved in DMSO. The DMSO level was adjusted to 20% (v/v) of the reaction volume. The reaction was left overnight while gently stirring at 4 °C. CPMV-SS-DOX particles were purified on 5 ml 10–50% sucrose gradients. The fractions containing CPMV-SS-DOX were collected and dialysed against 10 mM sodium phosphate buffer for 3–5 days using 100 kDa molecular weight cut-off membranes. The yield was 40–50% relative to the initial CPMV concentration as determined by UV-visible spectrophotometry.
Virus Particle Purification
Sucrose gradients
Sucrose solutions of 50%, 40%, 30%, 20%, and 10% (w/v) in 10 mM sodium phosphate buffer pH 7.4 were prepared and used within 2 weeks. Gradients were prepared by underlying sucrose solutions of decreasing density (175 or 500 μL) in a centrifuge tube of (2.1 mL or 5 mL, respectively) and carefully overlaying the sample to fill the tube (normally 300–500 μL). Gradients were centrifuged at 137000 g for 1.5–2.5h at 4 °C in a swing-out rotor (AH-650); 175–300 μL fractions containing modified particles were collected and buffer exchanged for 10 mM sodium phosphate buffer pH 7.4.
Size-exclusion chromatography
Gel filtration (AKTA Purifier-900) on a Sephacryl S-500 column (GE Healthcare) and PD-10 desalting columns (Amersham) were according to the manufacturer's instructions. The columns were pre-equilibrated with 10 mM sodium phosphate buffer.
Ultrafiltration
Ultrafiltration was used as a method for purification and concentration of virus particles. Samples were applied to centrifugal filter units of various volumes (500 μL-15 mL) with 100, 300, and 10000 kDa molecular–weight (Microcon, Amicon Millipore, and Sartorius Stedim) and centrifuged at 14000 g for 5–15 minutes depending on the volume. The particles were retained on the filter of these tubes while the buffer and small impurities passed through.
Dialysis
Slide-A-Lyzer cassettes (Pierce), Float-A-Lyze tubing and Snakeskin tubing were used for dialysis.
Conjugate stability
CPMV-DOX and CPMV-SS-DOX conjugates (100 μg and 300 μg) in a 96 well plate were incubated with 100 μL of Minimum Essential Media (MEM) supplemented with 10% (v/v) fetal bovine serum, 1% (w/v) penicillin-streptomycin, 1% (w/v) glutamine at 37 °C and 5% CO2. The plate was incubated for seven days and samples taken and run on agarose gels at set time points.
Tissue Culture
HeLa cells were obtained from ATCC and cultured and maintained on MEM supplemented with 10% (v/v) fetal bovine serum, 1% (w/v) penicillin-streptomycin, 1% (w/v) glutamine at 37 °C and 5% CO2. All culture media reagents were purchased from Invitrogen.
Cell Viability Assay
XTT Cell Proliferation Assay Kit (ATCC) and CellTiter-Glo Luminescent Cell Viability Assay (Promega) were used to determine cell viability. For XTT assay, HeLa cells were seeded on a 96-well plate (15,000 cells/well; 100 μL MEM/well) and incubated for 24h at 37 °C, 5% CO2, washed, and then incubated in MEM containing varying concentrations of CPMV-DOX and CPMV-SS-DOX (2 × 107 − 1 × 108 CPMV/cell, i.e. 0.5–15 M DOX) and corresponding amounts of CPMV and free DOX. All assays were analyzed in triplicates and repeated at least twice. CPMV-conjugated DOX, CPMV and free DOX formulations were incubated with the cells for 3h, washed with saline to remove any unbound particles and drug. Fresh medium was added and time course studies were conducted: cells were incubated for a further 24h, 72h and 7 days prior to measuring cell viability. At the end of each incubation period, 50 μL of XTT reagent (reconstituted as per instructions in the kit) was added to each well and the plates were incubated for another 2–3 h for color development. The absorbance at 450 nm and 630 nm was then recorded on TECAN Infinite 200 PRO multimode plate reader; data analyzed as recommended by the supplier. For the CellTiter-Glo Luminescent assay, equivalent numbers of cells on opaque walled plates were treated with similar concentrations of CPMV-DOX, CPMV-SS-DOX, CPMV and free DOX for 3h, washed and incubated for a further 24h, 48h, 72h and 5 days. At the end of each incubation period, 100 μL of reconstituted CellTiter-Glo reagent was added to the plates, mixed for 2 min on orbital shaker and then luminescence was measured on a TECAN Infinite 200 PRO multimode plate reader. Average of triplicates for each concentration were then plotted with respect to control cells which were considered hundred percent viable.
Confocal Microscopy
Cellular uptake of CPMV and CPMV-DOX was evaluated using confocal laser scanning microscopy. HeLa cells (25,000 cells/well) were grown for 24 h on glass coverslips placed in an untreated 24-well plate in MEM at 37 °C, 5% CO2. Cells were washed and CPMV (1 × 107 CPMV/cell) introduced, cells were incubated for 3h, and then washed to remove any unbound CPMV. Time course studies were conducted, and cells were incubated for 24h, 48h, 72h and 7 days. At the end of the incubation period, cells were washed with saline solution and fixed for 5 min at room temperature using DPBS containing 4% (v/v) paraformaldehyde and 0.3% (v/v) glutaraldehyde. Cell membranes were stained using 1:1000 wheat germ agglutinin (WGA) conjugated with AlexaFluor-647 (WGA-A647; Invitrogen) in 5% (v/v) goat serum (GS) for 45 min at room temperature in the dark followed by subsequent washing with DPBS (Invitrogen). Cells were permeabilized using 0.2% (v/v) Triton-X 100 for 2 min, washed, and blocked using 5% (v/v) GS for 45 min. CPMV was stained using specific antibodies: cells were incubated with anti-CPMV antibodies (rabbit IgG, Pacific Immunology) at 1:200 dilution for 60 min at room temperature. Endolysosomes were stained using a mouse anti-human LAMP-1 antibody (Biolegend, 1:200; 5% GS) for 60 min. Secondary antibodies, goat anti-mouse-AlexaFluor 488 (secondary to LAMP-1 Ab) and goat anti-rabbit-AlexaFluor 555 (secondary to anti-CPMV Ab) at 1:500 dilutions (mixed together) were then used to label the primary antibodies for 60 min. Nuclei were stained with DAPI (MP Biomedicals, 1:7500) for 5 min. Cells were washed with DPBS in between each staining step. The coverslips were then mounted using a mounting media (Permount, Fisher Chemicals) on glass slides and sealed using nail polish. Confocal images were captured on Olympus FluoView FV1000 LSCM and data processed using Image J 1.44o software (http://imagej.nih.gov/ij).
Results and Discussion
DOX was either directly covalently coupled to carboxylate groups on the external surface of native CPMV utilising the EDC/NHS method,19 Figure 1A, to give CPMV-DOX or to amine groups on the surface with an eight-atom spacer arm crosslinker containing a central disulfide bond, CPMV-SS-DOX, Figure 1B. CPMV doxorubicin conjugates were recovered in yields, relative to starting virus concentration, of 60–80% and 40–50%, respectively. After rigorous purification, the number of DOX molecules per virus was determined from the absorbance at 490 nm to be 80 ± 8 for CPMV-DOX and 270 ± 14 for CPMV-SS-DOX. There are 180 chemically addressable carboxylates on the surface of CPMV,19,20 in the case of CPMV-DOX the loading of DOX is approximately 45% compared to the maximum. This level of loading was the highest achievable under the reaction conditions used and may be a consequence of steric crowding at the surface. In CPMV-SS-DOX the DOX molecules are coordinated to surface amine groups with a long linker. The coverage is within the range that can be achieved under forcing conditions (300 addressable lysines per virus capsid21). A control experiment, in which CPMV is incubated with DOX under identical reaction conditions but in the absence of coupling reagent, provided no evidence for non-specific binding as shown by the absence of fluorescence in an unstained agarose gel of the recovered virus (data not shown). Aqueous suspensions of DOX-modified CPMV are colored bright red, Figure 1C. Successful DOX coupling was monitored using native agarose gel electrophoresis. In the electric field, free DOX migrates toward the cathode, while CPMV particles move toward the anode, Figure 2. When conjugated to CPMV, DOX migrates with its carrier toward the anode. Free DOX and DOX-conjugated CPMV-DOX and CPMV-SS-DOX can be visualized in the gels under UV light based on the intrinsic fluorescence of the drug. When stained with ethidium bromide (EtBr stains the encapsulated CPMV genome) or Coomassie Blue (CB stains the coat proteins) the labelled and native CPMV can be detected. The band pattern is fully consistent with DOX being bound to the external surface of the VNP. For CPMV-DOX, Figure 2A, mobility of CPMV toward the anode slows down upon conjugation of NHS-linkers to external carboxylates and further slows down upon DOX conjugation. The sequential loading can be followed step-by-step. For CPMV-SS-DOX, Figure 2D, the mobility slows on coupling of DSSTP and is then enhanced on DOX conjugation, likely as a consequence of a combination of charge and mass effects, consistent with modification of the virus surface. The zeta potential for the conjugates was also different, being considerably more negative than that of native CPMV (CPMV, −12.3 mV; CPMV-DOX, −38.1 mV; CPMV-SS-DOX −36.7 mV. Supporting Information). Uranyl acetate stained transmission electron micrographs show that the virus particles retain their integrity when DOX is bound and are monodisperse with an average diameter of 32 nm; this is consistent with dynamic light scattering measurements in solution, Figure 3. Within 10 days of preparation the modified particles show a tendency to aggregate.
Figure 1.
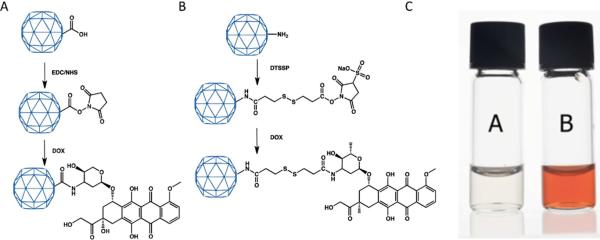
Schematic of the conjugation method for preparation of (A) CPMV-DOX and (B) CPMV-SS-DOX. (C) Photograph of aqueous suspensions of wild-type CPMV, left, and CPMV-DOX, right.
Figure 2.

Agarose gel electrophoresis of CPMV-DOX unstained (A) and Coomassie Blue stained (B). Lane 1, CPMV; lane 2, CPMV-NHS-ester; lane 3, CPMV-DOX; lane 4, free DOX; Agarose gel electrophoresis of CPMV-SS-DOX unstained (C) and ethidium bromide stained (D). Lane 1, CPMV, lane 2, CPMV-DTUSSP, lane 3, CPMV-SS-DOX; Stability of CPMV-DOX and CPMV-SS-DOX on incubation with cell culture media as monitored by DOX fluorescene on agarose electrophoresis gels (E) and after staining with ethidium bromide (F). Lane 1, CPMV; lane 2, CPMV-DOX after 24h incubation; lane 3, CPMV-DOX after 7 days; lane 4, CPMV-SS-DOX before incubation; lane 5, CPMV-SS-DOX after 24h incubation; lane 6, CPMV-SS-DOX after 7 days incubation.
Figure 3.

Transmission electron micrographs of uranyl acetate stained CPMV-DOX (A) and CPMV-SS-DOX (B). Dynamic light scattering and correlation plot for CPMV (blue-line), CPMV-DOX (red-line) and CPMV-SS-DOX (green-line).
The cell viability and drug efficacy of CPMV-DOX and CPMV-SS-DOX was evaluated using tissue culture techniques. Native CPMV nanoparticles are biocompatible and do not show any apparent cytotoxicity, Figure 4. CPMV-DOX conjugates showed effective cell killing, Figure 4. We observed significant differences in the cellular toxicity patterns comparing CPMV-DOX and CPMV-SS-DOX. The efficacy of CPMV-SS-DOX was essentially that of the free drug. Data indicate that the effective dose of DOX remains unaltered when conjugated to CPMV via a disulfide bridge. This can be attributed to a reduction-induced dissociation of the disulfide-bridges in the culture media, resulting in release of doxorubicin from CPMV prior to cellular uptake. Thus, CPMV-SS-DOX essentially resembles free DOX in its activity. Nevertheless, targeting strategies could be applied to further improve either formulation. Free drug induces cardiotoxicity thus limiting its clinical utility. Conjugation via disulfide or covalent amide bond to, for example, a receptor-targeted CPMV nanoparticle22 is expected to provide advantages through tissue-specific delivery.
Figure 4.

Cytotoxicity analysis of CPMV-SS-DOX (A) and CPMV-DOX (B) using HeLa cells. Cells were treated with varying concentrations of CPMV conjugated with DOX and equivalent amounts of free DOX and CPMV carrier. Time course studies were conducted over seven days and cell viability was measured and compared to untreated control cells (100% viability). The following drug and carrier concentrations were studied: A) C-SS-D1 = 1.7 nM CPMV coupled with 0.48 μM DOX, C-SS-D2 = 8.5 nM CPMV loaded with 2.4 μM DOX, C-SS-D3 = 17 nM CPMV loaded with 4.8 μM DOX, and C-SS-D4 = 51 nM CPMV loaded with 14.4 μM DOX. Equivalent amounts of free DOX (D1–4) and CPMV (C1–4) were used. B) CD1 = 17 nM CPMV loaded with 1.45 μM DOX, CD2 = 51 nM CPMV loaded with 4.35 μM DOX, and CD3 = 102 nM CPMV loaded with 14.4 μM DOX. Equivalent amounts of free DOX (D1–3) and CPMV (C1–3) were used.
The dissociation of DOX from CPMV-SS-DOX was confirmed by incubating CPMV-DOX and CPMV-SS-DOX with culture media under the same conditions as for the cell viability and drug efficacy studies. On taking samples at various time points, and monitoring the presence of conjugated DOX by fluorescence on unstained agarose electrophoresis gel, it was found that while CPMV-DOX was unchanged after seven days, CPMV-SS-DOX had released most of the bound DOX to the medium after 24h and had released all bound DOX after seven days, Figure 2. On staining the gel with ethidium bromide, bands typical of CPMV were observed indicating that the capsid shell remained intact under the incubation conditions.
Covalent bound CPMV-DOX conjugates offer an advantage over free DOX. At low dosage (1.45 μM DOX), CPMV-DOX was found to be more effective in conferring cytotoxicity over free DOX. At this concentration, after five days cells showed full cell viability when treated with free DOX but were effectively killed when treated with the CPMV-DOX conjugate. It is interesting that the trend of CPMV-DOX induced cytotoxicity is concentration-independent and time-delayed for the higher concentrations tested. The concentration-independence may be explained by the uptake mechanism of CPMV into HeLa cells. It has been shown that CPMV is taken up by HeLa cells via surface-expressed vimentin.23,24 It appears that cell uptake is saturated and that increasing the carrier/drug concentration does not have a net benefit. This delayed uptake may be explained by the varying fate of free DOX versus CPMV-DOX conjugates in cells. Free DOX diffuses through the cell membranes and targets the nucleus. This is in contrast to the CPMV-DOX conjugate: CPMV enter the cells via endocytosis and are targeted to the endolysosomes, in which the carrier is degraded and the drug released, Figure 5. We hypothesize that the CPMV carrier enhances cellular uptake and may distribute the drug more evenly, resulting in increased efficacy at low drug concentration.
Figure 5.

A. Imaging of CPMV-DOX in HeLa cells at 3h post incubation. CPMV is shown in red (immunostained using rabbit anti-CPMV antibody and a secondary goat anti-rabbit antibody conjugated with AlexaFluor 555), nuclei are shown in blue, DIC overlay is shown in grey. B. Time course study showing cellular uptake of CPMV using confocal scanning microscopy. CPMV was incubated with HeLa cells for 3h, cells were washed to remove excess CPMV and then incubated for 3, 24, 48, 72h and 7 days. CPMV was immunostained (pseudo-colored in green). Endolysosomes were stained with a mouse anti-human Lamp-1 antibody and a secondary goat anti-mouse antibody conjugated with AlexaFluor 488 (pseudo-colored in red). Nuclei were stained with DAPI (in blue). Scale bar is 50 μm. C. Co-localization analysis using Co-localization Highlight plug in and ImageJ software. White = co-localization of CPMV and Lamp-1 staining. Scale bar is 50 μm.
We have previously shown that native CPMV is endocytosed and targeted to the endolysosomal compartment.25 Here we analyzed this process over long-time periods. Co-localization with Lamp-1 marker (a marker of the endolyosomes) is highest at 24h and 48h post-incubation, Figure 5. At later time points (72h and 7 days) the CPMV signal fades; at this time point cell killing is induced. We hypothesize that the CPMV protein cage is digested within the protease-rich and acidic endolysosomal compartment, DOX is released and cytotoxicity induced. The observation that at low dosage (1.45 μM DOX) CPMV-DOX shows improved efficacy over free DOX may be explained by the protein cages distributing the drug more evenly and efficiently.
In this study we lay the foundation for the use of CPMV as a carrier for drug delivery. One of the major hurdles in cancer treatment is the development of drug resistance, which can lead to reoccurrence of the disease in a more aggressive form. Studies using nanoparticle-drug conjugates have shown that multidrug resistance can be overcome by efficient cellular entry and intracellular drug release in acidic organelles.26,27 The goal of this study was to evaluate uptake and drug efficacy using a surface-vimentin expressing cell line. However, further studies will include the evaluation of our drug delivery system using drug-resistant cells. Also, we will develop and test targeted devices by including targeting ligands. However, the efficacy has to be carefully evaluated when surface-modifications are introduced. For example, RCNMV loaded with up to 600 DOX moieties and decorated with cancer cell specific targeting peptides showed peptide-dependent uptake by the HeLa cells but only similar efficacy to CPMV-DOX at low concentration after 72 hours.11 A few VNP drug conjugates have been developed in recent years; in all cases the reported drug efficacy of VNP-drug was comparable to the free drug.2–13 It is possible that the vimentin-specific uptake renders CPMV advantageous over other VNP systems. We have previously shown that native and PEGylated CPMV is homed to vimentin-expressing tumours via the enhanced permeability and retention effect and targets cancer cells via surface-expressed vimentin.24
Conclusions
In conclusion, CPMV-DOX conjugate is a more effective cytotoxic agent toward HeLa cells than free DOX at low dosage and, at higher concentrations, the cytotoxic effect is time-delayed. The CPMV nanoparticles are targeted to the endolysosomal compartment of the cells without the need for modification of the capsid with cell specific targeting agents. However, the potential exists to further develop the platform technology to introduce targeting ligands to match a patient-specific profile, toward the development of personalized cancer medicines.
Supporting Information
Acknowledgement
This work was supported by NIH/NIBIB grants R00 EB009105 (to N.F.S.), P30 EB011317 (to N.F.S.), and the Biotechnology and Biological Sciences Research Council, UK (Core Strategic Grant to the John Innes Centre (JIC) and Industrial Partnership Award, D.J.E. and G.P.L., JIC DTG to A. A. A. A.). Mrs J. Elaine Barclay and Mr Andrew Davis of JIC are thanked for technical assistance and photography, respectively.
Footnotes
Publisher's Disclaimer: “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a free service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are accessible to all readers and citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.
Supporting Information UV-visible spectra and zeta potential measurements are presented. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Shi J, Xiao Z, Kamaly N, Farokhzad OC. Self-Assembled Targeted Nanoparticles: Evolution of Technologies and Bench to Bedside Translation. Acc. Chem. Res. 2011;44:1123–1134. doi: 10.1021/ar200054n. [DOI] [PubMed] [Google Scholar]
- 2.Yu MK, Park P, Jon S. Targeting Strategies for Multifunctional Nanoparticles in Cancer Imaging and Therapy. Theranostics. 2012;2:3–44. doi: 10.7150/thno.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. http://clinicaltrials.gov.
- 4.Yildiz I, Shukla S, Steinmetz NF. Applications of Viral Nanoparticles in Medicine. Curr. Opin. Biotech. 2011;22:901–908. doi: 10.1016/j.copbio.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franzen F, Lommel SA. Targeting Cancer with `Smart Bombs': Equipping Plant Virus Nanoparticles for a `Seek and Destroy' Mission. Nanomedicine. 2009;4:575–588. doi: 10.2217/nnm.09.23. [DOI] [PubMed] [Google Scholar]
- 6.Ren Y, Wong SM, Lim L-Y. Application of Plant Viruses as Nano Drug Delivery Systems. Pharm. Res. 2010;27:2509–2513. doi: 10.1007/s11095-010-0251-2. [DOI] [PubMed] [Google Scholar]
- 7.Steinmetz NF, Manchester M. Viral Nanoparticles: Tools for Materials Science and Biomedicine. Pan Stanford Publishing; Singapore: 2011. pp. 207–245. [Google Scholar]
- 8.Wu W, Hsiao SC, Carrico ZM, Francis MB. Genome-Free Viral Capsids as Multivalent Carriers for Taxol Delivery. Angew. Chem. Int. Ed. 2009;48:9493–9497. doi: 10.1002/anie.200902426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashley CE, Carnes EC, Phillips GK, Durfee PN, Buley MD, Lino CA, Padilla DP, Phillips B, Carter MB, Willman CL, Brinker CJ, Caldeira JC, Chackerian B, Wharton W, Peabody DS. Cell-Specific Delivery of Diverse Cargos by Bacteriophage MS2 Virus-Like Particles. ACS Nano. 2011;5:5729–5745. doi: 10.1021/nn201397z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pokorski JK, Hovlid ML, Finn MG. Cell Targeting with Hybrid Qβ Virus-Like Particles Displaying Epidermal Growth Factor. ChemBioChem. 2011;12:2441–2447. doi: 10.1002/cbic.201100469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lockney DM, Guenther RN, Loo L, Overton W, Antonelli R, Clark J, Hu M, Luft C, Lommel SA, Franzen S. The Red Clover Necrotic Mosaic Virus Capsid as a Multifunctional Cell Targeting Plant Viral Nanoparticle. Bioconjugate Chem. 2011;22:67–73. doi: 10.1021/bc100361z. [DOI] [PubMed] [Google Scholar]
- 12.Ren Y, Wong SM, Lim L-Y. Folic Acid-Conjugated Protein Cages of a Plant Virus: A Novel Delivery Platform for Doxorubicin. Bioconjugate Chem. 2007;18:836–843. doi: 10.1021/bc060361p. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Q, Chen W, Chen Y, Zhang L, Zhang J, Zhang Z. Self-Assembled Virus-Like Particles from Rotavirus Structural Protein VP6 for Targeted Drug Delivery. Bioconjugate Chem. 2011;22:346–352. doi: 10.1021/bc1002532. [DOI] [PubMed] [Google Scholar]
- 14.Lin T, Johnson JE. Structures of Picorna-Like Plant Viruses: Implications and Applications. Adv. Virus Res. 2003;62:167–239. doi: 10.1016/s0065-3527(03)62004-x. [DOI] [PubMed] [Google Scholar]
- 15.Evans DJ, Steinmetz NF. Utilisation of Plant Viruses in Bionanotechnology. Org. Biomol. Chem. 2007;5:2891–2902. doi: 10.1039/b708175h. [DOI] [PubMed] [Google Scholar]
- 16.Strable E, Finn MG. Chemical Modification of Viruses and Virus-Like Particles. In: Manchester M, Steinmetz NF, editors. Curr. Top. in Microbiol. Immunol. Springer-Verlag; Berlin: 2009. pp. 1–21. [DOI] [PubMed] [Google Scholar]
- 17.Evans DJ. The Bionanoscience of Plant Viruses: Templates and Synthons for New Materials. J. Mater. Chem. 2010;363:1070–1076. [Google Scholar]
- 18.Evans DJ, Lomonossoff GP. Applications of Plant Viruses in Bionanotechnology. In: Palmer K, Gleba Y, editors. Curr. Top. in Microbiol. Immunol. Springer-Verlag; Berlin: 2012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinmetz NF, Lomonossoff GP, Evans DJ. Cowpea Mosaic Virus for Material Fabrication: Addressable Carboxylate Groups on a Programmable Nanoscaffold. Langmuir. 2006;22:3488–3490. doi: 10.1021/la060078e. [DOI] [PubMed] [Google Scholar]
- 20.Aljabali AAA, Barclay JE, Butt JN, Lomonossof GP, Evans DJ. Redox-Active Ferrocene-Modified Cowpea Mosaic Virus Nanoparticles. Dalton Trans. 2010;39:7569–7574. doi: 10.1039/c0dt00495b. [DOI] [PubMed] [Google Scholar]
- 21.Chatterji A, Ochoa WF, Paine M, Ratna BR, Johnson JE, Lin T. New Addresses on an Addressable Virus Nanoblock: Uniquely Reactive Lys Residues on Cowpea Mosaic Virus. Chem. Bio. 2004;11:855–863. doi: 10.1016/j.chembiol.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 22.Steinmetz NF, Ablack AL, Hickey JL, Ablack J, Manocha B, Mymryk JS, Luyt LG, Lewis JD. Intravital Imaging of Human Prostate Cancer Using Viral Nanoparticles Targeted to Gastrin-Releasing Peptide Receptors. Small. 2011;7:1664–1672. doi: 10.1002/smll.201000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koudelka KJ, Destito G, Plummer EM, Trauger SA, Siuzdak G, Manchester M. Endothelial Targeting of Cowpea Mosaic Virus (CPMV) via Surface Vimentin. PloS Pathog. 2009;5:e1000417. doi: 10.1371/journal.ppat.1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinmetz NF, Cho C-F, Ablack A, Lewis JD, Manchester M. Cowpea Mosaic Virus Nanoparticles Target Surface Vimentin on Cancer Cells. Nanomedicine. 2011;6:351–364. doi: 10.2217/nnm.10.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu Z, Chen K, Yildiz I, Dirksen A, Fischer R, Dawson P, Steinmetz NF. Development of Viral Nanoparticles for Efficient Intracellular Delivery. Nanoscale. 2012 doi: 10.1039/c2nr30366c. in press, DOI: 10.1039/C2NR30366C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang F, Wang YC, Dou S, Xiong MH, Sun TM, Wang J. Doxorubicin-Tethered Responsive Gold Nanoparticles Facilitate Intracellular Drug Delivery for Overcoming Multidrug Resistance in Cancer Cells. ACS Nano. 2011;5:3679–92. doi: 10.1021/nn200007z. [DOI] [PubMed] [Google Scholar]
- 27.Shapira A, Livney YD, Broxterman HJ, Assaraf JG. Nanomedicine for Targeted Cancer Therapy: Towards the Overcoming of Drug Resistance. Drug Resist. Updat. 2011;14:150–163. doi: 10.1016/j.drup.2011.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.