

Abstract
Rhabdomyosarcoma is a soft tissue sarcoma arising from cells of a mesenchymal or skeletal muscle lineage. Alveolar rhabdomyosarcoma (ARMS) is more aggressive than the more common embryonal (ERMS) subtype. ARMS is more prone to metastasis and carries a poorer prognosis. In contrast to ERMS, the majority of ARMS tumors carry one of several characteristic chromosomal translocations, such as t(2;13)(q35;q14), which results in the expression of a PAX3-FOXO1 fusion transcription factor. In this review we discuss the genes that cooperate with PAX3-FOXO1, as well as the target genes of the fusion transcription factor that contribute to various aspects of ARMS tumorigenesis. The characterization of these pathways will lead to a better understanding of ARMS tumorigenesis and will allow the design of novel targeted therapies that will lead to better treatment for this aggressive pediatric tumor.
Keywords: Alveolar rhabdomyosarcoma, PAX3-FOXO1, PAX7-FOXO1, FGFR4, CNR1, IRIZIO, N-MYC, IGF2, MET, CXCR4, p53, MDM2, P-Cadherin, TFAP2B, miR17-92
Introduction
According to the American Cancer Society, rhabdomyosarcoma (RMS) comprises about three percent of childhood cancers, with about 350 new cases occurring annually in the US [1], and it affects slightly more males than females [2]. RMS is a small, round, blue cell tumor usually arising in skeletal muscle tissue, and it is thought to originate from mesenchymal cells likely committed to the skeletal muscle lineage. Consistent with a myogenic origin, RMS tumors express skeletal muscle markers such as skeletal muscle actin and myosin, desmin, myoglobin, Z-band protein, MYOD and often myogenin [3-7]. RMS consists of two major histological subtypes, embryonal and alveolar RMS. The embryonal subtype (ERMS) is thought to be histologically reminiscent of embryonic developing skeletal muscle [7]. ERMS is the most prevalent of the subtypes, accounting for about 60% of RMS cases [2]. It occurs mainly in children younger than 10 years and is usually associated with a favorable prognosis, with a failure-free survival rate of 81% [8,9]. Tumors usually localize to the head and neck (including the extraocular muscles of the eye), the genitourinary tract and the retroperitoneum [7,8]. The alveolar subtype (ARMS) constitutes approximately another 20% of RMS cases [10] and occurs predominantly in adolescents. Histologically, ARMS tumors typically contain alveoli structures similar in appearance to those seen in the lung [7], though solid-variant ARMS does occur [11]. Primary ARMS tumors typically arise in the extremities and trunk [7-9,12], and they are more aggressive than their ERMS counterparts. ARMS is associated with a poorer prognosis, with a 5-year failure-free survival of 65% [8].
A characteristic of the ARMS subtype is the occurrence of recurrent chromosomal translocations. The most common of these is t(2;13)(q35;q14), which results in the expression of an oncogenic fusion protein. This fusion protein consists of the paired and homeodomains of the PAX3 transcription factor with the potent transcriptional activation domain of FOXO1 (FKHR), a member of the forkhead (FOX) family of transcription factors [13-15]. The PAX3 homeodomain is required to recapitulate PAX3-FOXO1-induced tumorigenesis, though the paired domain may play a minor role [16,17]. The PAX3-FOXO1 fusion protein can be detected in about 55% of ARMS cases [18]. A similar translocation of t(1;13)(p36;q14) fuses the PAX7 DNA-binding domains, the closest homolog of PAX3, to FOXO1 [19]. This translocation occurs in a further 22% of ARMS cases [18]. Recently, further similar translocations have been found in individual ARMS cases: t(2;X)(q35;q13), which results in PAX3-AFX fusion [20], and t(2;2)(q35;p23) and t(2;8)(q35;q13), which generate a fusion protein of PAX3-NCOA1 and PAX3-NCOA2, respectively [21,22]. These “cryptic” rare fusion variants are thought to be present in up to another 10% of ARMS tumors [7]. During normal development, PAX3 expression occurs in the neural tube and dermomyotome [23], and it is required for the normal migration of skeletal muscle precursors to the limb bud [24]. PAX7 expression is a marker of satellite cells in adult skeletal muscle [25] and is required for normal self-renewal [26]. Unlike skeletal muscle-specific PAX3 and PAX7, FOXO1A, AFX, NCOA1 and NCOA2 are widely expressed and mediate gene transcription downstream of cell signaling pathways [27-31]. All of the ARMS fusion proteins consist of the PAX3/7 DNA-binding domains fused to the transcriptional activation domains of more potent transcription factors (see Figure 1) [14,15,22]. Genome-wide transcription factor-binding studies have not yet been performed to determine whether wild-type PAX3 and PAX7-binding sites differ from these PAX3/7 fusion transcription factor-binding sites.
Figure 1.
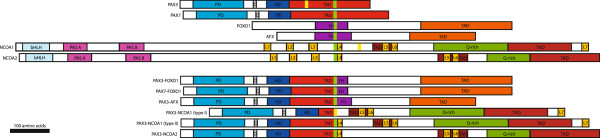
Gene translocation in alveolar rhabdomyosarcoma. Scale diagram showing the parent proteins and the resulting fusion proteins arising from chromosomal translocations occurring in ARMS. Green or yellow indicates the protein fusion sites [14,19-22]. Homologous domains are indicated in like colors. DNA-binding domains are indicated as: paired domain (PD), homeodomain (HD), fork head DNA-binding domain (FH) and basic helix-loop-helix domain (bHLH). Regions of the proteins known to act as transcriptional activation domains are indicated (TAD). Other domains include the octapeptide domain (O), PAS domains (PAS A/B), LXXLL motifs (L1-L7) and glutamine-rich region (Q-rich). Maps were derived from the following references: [14,19-22,32-34].
Expression of these ARMS fusion transcription factors is thought to abrogate normal skeletal muscle differentiation, allowing aberrant cell division and tumor development. PAX3 expression can inhibit myogenic differentiation of cultured myoblasts [35]. Although PAX3 protein is rapidly degraded during early myogenic differentiation, PAX3-FOXO1 has a significantly longer half-life than wild-type PAX3 [36]. PAX3/7-FOXO1 is capable of suppressing MyoD expression and activity [37,38]. PAX7-FOXO1 expression induces NFκB signaling, which inhibits myogenesis via activation of cyclin D1/CDK4 complexes. These complexes sequester MyoD, which would normally drive cell cycle withdrawal and myogenic differentiation [39]. In addition transcriptionally inactive MyoD can enhance PAX3-FOXO1 transcriptional activity [37]. There is also evidence that MyoD transcriptional activity is abrogated in ERMS tumors [40].
The remaining ARMS tumors are classed as fusion-negative ARMS. However, fusion-negative ARMS are indistinguishable on the levels of gene expression and in clinical outcome from ERMS tumors, leading some to argue that translocation status should be the defining factor of ARMS [41-44]. Within the ARMS subtype, prognosis can vary by disease stage at diagnosis as well as translocation status. For example, patients presenting with metastatic disease have an estimated 4-year overall survival rate of 75% for PAX7-FOXO1, while those with the PAX3-FOXO1 translocation have only 8% estimated 4-year overall survival [18]. Indeed, there is evidence that the PAX3-FOXO1 is a more potent oncogene than PAX7-FOXO1. Barr et al.[45] found that only 1/24 PAX3-FOXO1-positive ARMS tumors had amplification of the PAX3-FOXO1 gene, while PAX7-FOXO1 was amplified in 10/11 PAX7-FOXO1 ARMS, implying that genomic amplification of PAX7-FOXO1 is required for tumorigenesis, while a single copy of PAX3-FOXO1 is sufficient. However the gene expression profiles of PAX3-FOXO1- and PAX7-FOXO1-expressing tumors have not, to the author’s knowledge, been specifically compared to identify the gene set responsible for this difference in prognosis between ARMS tumors with these two fusion genes.
Review
PAX3-FOXO1 is the most common fusion gene in ARMS. This fusion transcription factor is thought to drive the gene expression that causes the worse prognosis in ARMS tumors. Many studies have sought to identify the differences in gene expression between ERMS and ARMS, as well as the genes aberrantly regulated by PAX3-FOXO1. In this review, we summarize these gene expression changes specific to PAX3-FOXO1 expression and/or ARMS. Moreover, we consolidate these data into a list of genes that may well represent the means by which ARMS tumors obtain a more aggressive phenotype than ERMS.
Cooperating mutations in ARMS tumors
It is likely that PAX3/7-FOXO1 translocation is one of the earliest events in ARMS tumorigenesis as it occurs in the majority of ARMS cases, more often than any other genetic lesion characterized in the disease. However, PAX3-FOXO1 expression in normal cells is not sufficient to induce transformation, and other genetic alterations are required [46-48]. Genomic amplification is common in ARMS tumors. The three most common amplifications seen in ARMS involve regions of chromosomes 2, 12 and 13 [49].
The region of chromosome 12 amplification spans 12q13-15 and is reported in 28% to 56% of ARMS tumors [49-52]. This 12q13-15 region includes genes such as C/EBP-homolog and transcription factor CHOP/DDIT3/GADD153, sarcoma-amplified sequence and transmembrane 4 superfamily member SAS/TSPAN31, alpha 2-macroglobulin receptor A2MR/LRP1, Sonic hedgehog (SHH) pathway effector and zinc finger transcription factor GLI1, cyclin-dependent kinase cell cycle regulator CDK4 and p53 pathway modulator MDM2. In most cases, gene amplification accompanies an increase in gene expression [50,53].
Though GLI1 is amplified genetically, the expression of this gene is not always associated with its genetic amplification. When GLI1 is overexpressed in RMS, it has been associated with an undifferentiated subtype rather than ERMS or ARMS, indicating that GLI1 may play a role in tumors that show primitive histopathological features [54]. Thus, GLI1 overexpression cannot be well associated with the ARMS pathology.
MDM2 is perhaps the best candidate oncogene in this region because of its inhibitory effect on p53 function [55]. However, MDM2 is not always included in this 12q13-15 amplification. RH30, an ARMS cell line, lacks amplification of MDM2 but shows amplification and overexpression of SAS, CHOP, GLI1 and A2MR [53]. In addition, the frequency of MDM2 gene amplification specifically may be as low as 10% in ARMS tumors [56]. One study found only 2 of 34 ARMS samples to be highly immunoreactive for MDM2 [12]. Moreover, MDM2 expression shows no association with patient prognosis or other clinicopathologic parameters [12]. Thus, it may be amplification of one of the other genes at this chromosome 12 locus that is the important cooperating mutation with PAX3-FOXO1.
Other alterations in the p53 pathway have been found in ARMS. In ARMS tumor samples mutated p53 was reported in 0 to 22% of cases [12,56,57]. RMS cell lines show a significantly higher rate of p53 abnormalities with 60%, indicating establishment of these cell lines through xenograft and cell culture increases the proportion of cell lines with p53 alterations [56]. Looking at p53 and MDM2 expression levels, both are low in ARMS and ERMS. Metastatic ERMS tumors show significantly higher p53 protein expression, indicating that p53 gene alterations are a late event in rhabdomyosarcomagenesis. Again, p53 status did not show any correlation to prognosis [58].
Chromosome 2 has been shown to be amplified at 2p24 in 32 to 60% of ARMS tumors [49,51,59,60]. This region is known to contain the proto-oncogene N-MYC. Two independent studies have shown that a gain in the genomic copy number of the N-MYC gene is associated with an unfavorable disease outcome [59,61]. In addition, N-MYC is more highly expressed in ARMS cells lines than ERMS lines, despite the fact that it was found to only be genomically amplified in one of the five lines, indicating more than one mechanism of N-MYC overexpression in ARMS. However, in this study no clear relationship in N-MYC expression was seen with regard to primary tumor samples [62].
Another chromosomal region frequently amplified in ARMS is 13q31-32, showing amplification in between 14 and 19% of ARMS tumors [49,51]. Presence of this amplification is significantly associated with poorer failure-free survival in ARMS [63]. The minimum overlapping region of amplification at this region was originally defined as only containing two genes: GPC5 and C13ORF25. The C12ORF25 gene encodes the micro-RNA cluster miR-17-92 (MIR17HG) in an intron [64]. GPC5 overexpression can increase cell proliferation through the modulation of the growth factor activity of FGF2, HGF and WNT1a [64]. However, more thorough mapping of the genetic amplification showed that the entire GPC5 locus was only amplified in 12.5% of 13q31 amplified ARMS tumors, while the minimally amplified region contains only the peptidylprolyl isomerase pseudogene (LOC390419) and MIR17HG. This amplification is particularly prevalent in PAX7-FOXO1-positive ARMS tumors. The miR-17-92 cluster of micro-RNAs has been shown to play a role in a variety of cancer types (for review, see [65]). In PAX7-FOXO1, but not PAX3-FOXO1 expressing ARMS, overexpression of miR-17, -19a, -19b, 20a and 92a is specifically associated with an increased rate of 2-year treatment failure. This indicates a possible pro-tumorigenic interaction between PAX7-FOXO1 and miR-17-92 locus overexpression [63].
Rhabdomyosarcoma can also be associated with a loss of heterozygosity (LOH) or loss of imprinting (LOI) at 11p15.5 [66,67]. This region contains several imprinted genes such as IGF2, which is maternally imprinted (paternal allele is expressed), and H19 and p57/Kip2, which are paternally imprinted (maternal allele is expressed) [68-70]. IGF2 expression appears to be specifically upregulated by changes in imprinting or LOH at this locus in RMS. ERMS tumors are associated predominantly with a LOH at the IGF2 locus, though there is some discrepancy in the proportion of ERMS tumors showing this change: 23% according to Anderson et al. [66] and 72% according to Visser et al. [67]. Conversely, IGF2 is upregulated by LOI in 46% of fusion-positive ARMS tumors, while imprinting of H19 is conserved in 93% [66]. This indicates that an increase in IGF2 expression in RMS is important for tumorigenesis, though the mechanism of this upregulation, either LOH or LOI, varies by subtype.
A screen for PAX3-FOXO1-interacting proteins using ARF−/− primary mouse myoblasts expressing PAX3-FOXO1 and an RH30 cDNA expression library identified a gene that could induce tumor formation where ARF−/− myoblasts expressing PAX3-FOXO1 alone did not. The RH30 gene expression library expressed a truncated fragment of this novel gene dubbed IRIZIO, and expression of either this truncated form or the full-length wild-type IRIZIO were protumorigenic in this model [71]. Due to the nature of the screen, and given that abrogation of the p53 and pRb pathways are required for PAX3-FOXO1-driven cell transformation [46,47], this screen was expected to identify proteins that could abrogate the pRb pathway [71]. The mechanism of the interaction between IRIZIO and pRb, however, has yet to be identified.
PAX3-FOXO1 target genes
Many gene expression studies have been performed by various groups to try to identify genes that are either downstream of PAX3-FOXO1 gene expression in various cell types or are indicative of ARMS tumor gene expression profiles (see Table 1). Only a small proportion of these studies have gone on to further investigate the mechanism of PAX3-FOXO1 regulation of these genes and/or what role these genes may play in ARMS tumorigenesis.
Table 1.
PAX3-FOXO1 target genes in ARMS
| Gene | Description | Regulation | References | PF target |
|---|---|---|---|---|
| ABAT |
4-Aminobutyrate aminotransferase; |
Up |
[72-74] |
Yes |
| ACTC |
Actin, alpha, cardiac muscle 1 |
Up |
[5,75] |
Yes |
| ADAM10 |
A disintegrin and metalloproteinase domain 10 |
Up |
[73][72] |
Yes |
| ADRA2A |
Alpha-2 adrenergic receptor subtype C10 |
Up |
[72,74,76] |
Yes |
| ADRA2C |
Alpha-2 adrenergic receptor subtype C4 |
Up |
[22,74,76] |
Yes |
| ALK |
ALK tyrosine kinase receptor, anaplastic lymphoma kinase Ki-1 |
Up |
[22,72,74,76,77] |
Yes |
| ANK2 |
Ankyrin 2 |
Up |
[73,74,76] |
Yes |
| ASS |
Argininosuccinate synthase |
Up |
[73,74,78] |
? |
| ASTN2 |
Astrotactin 2 |
Up |
[72,76] |
Yes |
| BIN1 |
Bridging integrator 1 |
Down |
[72,79] |
Yes |
| BMP5 |
Bone morphogenic protein 5 |
Up |
[72-74,76] |
Yes |
| C10ORF6 |
Family with sequence similarity 178, member A |
Up |
[75,76] |
Yes |
| CCND1 |
Cyclin D1 |
Down |
[75][79] |
? |
| CD9 |
CD9 molecule/Tetraspanin-29 |
Up |
[73,76] |
Yes |
|
CDH3 |
Cadherin 3, type 1, P-cadherin (placental) |
Up |
[72,76,80] |
Yes |
| CHD7 |
Chromodomain helicase DNA-binding protein 7 |
Up |
[76,81] |
Yes |
| CKM |
Creatine kinase M chain, muscle |
Up |
[5,79] |
Yes |
|
CNR1 |
Cannabinoid receptor 1 |
Up |
[22,72,74,82-84] |
Yes |
| COL18A1 |
Collagen type 18 α 1 |
Up |
[73,74] |
? |
|
CXCR4 |
C-X-C chemokine receptor type 4 |
Up |
[75,76,85-87] |
Yes |
| CXCR7 |
C-X-C chemokine receptor type 7 |
Down |
[76,87] |
Yes |
| DCX |
Neuronal migration protein doublecortin |
Up |
[73,74,81] |
? |
| DES |
Desmin |
Down |
[72,79] |
Yes |
| DKFZP762M127 |
Unknown |
Up |
[73,74,81] |
Yes |
| DUSP4 |
Dual specificity phosphatase 4 |
Down |
[73,81] |
? |
| DZIP3 |
DAZ interacting protein 3, zinc finger |
Up |
[72,74,81] |
Yes |
| ELA1 |
Elastase-1 |
Up |
[22,72,74] |
Yes |
| ENC1 |
Ectodermal-neural cortex 1 (with BTB-like domain) |
Up |
[75,76] |
Yes |
| ENO3 |
Enolase 3 (beta, muscle) |
Up |
[74,79] |
? |
| EPHA4 |
Ephrin type-A receptor 4 |
Up/down |
[76,81,82] |
Yes |
| FGFR2 |
Fibroblast growth factor receptor 2 |
Up |
[72,76] |
Yes |
|
FGFR4 |
Fibroblast growth factor receptor 4 |
Up |
[22,73,76,88,89] |
Yes |
| FLNB |
Filamin B, beta |
Down |
[76,82] |
Yes |
| FNBP1 |
Formin-binding protein 1 |
Down |
[22,75,76] |
? |
| FOXF1 |
Forkhead box protein F1 |
Up |
[22,72,74,76,81] |
Yes |
| FOXO1 |
Forkhead box O1 |
Up |
[75,76] |
Yes |
| GADD45A |
Growth arrest and DNA-damage-inducible protein GADD45 alpha |
Up |
[73,76,81] |
Yes |
| GRAF |
GTPase regulator associated with FAK |
Up |
[22,72] |
Yes |
| GTF3C1 |
General transcription factor 3C |
Up |
[72,81] |
Yes |
|
H19 |
Imprinted maternally expressed gene, untranslated mRNA |
Up |
[5,79] |
Yes |
| HDAC5 |
Histone deacetylase 5 |
Up |
[73,76] |
Yes |
| HUMMLC2B |
Myosin regulatory light chain 2, skeletal muscle isoform |
Down |
[72,73] |
Yes |
|
IGF2 |
Insulin-like growth factor II |
Up |
[5,75] |
Yes |
| IGFBP3 |
IGF-binding protein 3 |
Down |
[73,81] |
Yes |
| IGFBP5 |
IGF-binding protein 5 |
Up |
[5] |
Yes |
| IL4R |
Interleukin 4 receptor |
Up |
[72,73,75,76] |
Yes |
| JAKMIP2 |
Janus kinase and microtubule-interacting protein 2 |
Up |
[72-74,76] |
Yes |
| KCNN3 |
Small conductance calcium-activated potassium channel protein 3 |
Up |
[73,74,81] |
Yes |
| KCNS3 |
Potassium voltage-gated channel subfamily S member 3 |
Up |
[73,74] |
? |
| LRRFIP2 |
Leucine-rich repeat (in FLII) interacting protein 2 |
Up |
[72,74] |
Yes |
| MARCH3 |
Membrane-associated RING finger protein 3 |
Up |
[73,81] |
Yes |
| MCAM |
Melanoma cell adhesion molecule |
Up |
[73,81] |
Yes |
| MEG3 |
Maternally expressed 3 |
Up |
[73-75] |
? |
| MN1 |
Meningioma (disrupted in balanced translocation) 1 |
Up |
[73,76] |
Yes |
|
MET |
Hepatocyte growth factor receptor |
Up |
[73,76,81,90-92] |
Yes |
| MTUS2 |
Microtubule associated tumor suppressor candidate 2 |
Up |
[72,76] |
Yes |
| MYBPH |
Myosin-binding protein H |
Up/down |
[5,72,75] |
Yes |
|
MYCN |
N-MYC proto-oncogene protein |
Up |
[72,73,76,78,81] |
Yes |
| MYH8 |
Myosin, heavy chain 8, skeletal muscle, perinatal |
Up/down |
[5,72] |
Yes |
| MYL1 |
Myosin, light chain 1, alkali; skeletal, fast |
Up/down |
[5,79] |
Yes |
| MYL4 |
Myosin, light chain 4, alkali; atrial, embryonic |
Up/down |
[5,72] |
Yes |
| MYOD |
Myoblast determination protein 1 |
Up |
[5,73,76,81] |
Yes |
| MYOG |
Myogenin (myogenic factor 4) |
Up |
[5,90] |
Yes |
| NEBL |
Nebulette |
Up/down |
[72,73,76,81] |
Yes |
| NELL1 |
NEL-like protein 1 |
Up |
[22,72-74,76] |
Yes |
| NHLH1 |
Nescient helix loop helix 1 |
Up |
[22,76] |
Yes |
| NPTX2 |
Neuronal pentraxin II |
Down |
[76,81] |
Yes |
| NRCAM |
Neuronal cell adhesion molecule |
Up |
[72-74] |
Yes |
| OLIG2 |
Oligodendrocyte transcription factor 2 |
Up |
[22,74] |
? |
| PALMD |
Palmdelphin |
Down |
[72,76] |
Yes |
| PBK |
PDZ-binding kinase |
Up |
[73,76] |
Yes |
| PCDH7 |
Protocadherin 7 |
Up |
[76,82] |
Yes |
| PDZRN3 |
PDZ domain containing ring finger 3 |
Up |
[74,76] |
Yes |
| PGBD5 |
PiggyBac transposable element-derived protein 5 |
Up |
[22,72,74] |
Yes |
| PHF17 |
PHD finger protein 17 |
Up |
[74,76] |
Yes |
| PIPOX |
Pipecolic acid oxidase |
Up |
[22,72,74,76] |
Yes |
| PKP1 |
Plakophilin 1 (ectodermal dysplasia/skin fragility syndrome) |
Up |
[72,76] |
Yes |
| PLAG1 |
Pleiomorphic adenoma gene 1 protein |
Down |
[75,81] |
Yes |
| PLK2 |
Polo-like kinase 2 |
Down |
[73,76] |
Yes |
| PODXL |
Podocalyxin-like protein 1 |
Up |
[22,74,76] |
Yes |
| POU4F1 |
Brain-specific homeobox/POU domain protein 3A |
Up |
[72,73,76] |
Yes |
| PPARGC1A |
Peroxisome proliferator-activated receptor gamma, coactivator 1 alpha |
Up |
[22,76] |
Yes |
| PRKAR2B |
Protein kinase, cAMP-dependent, regulatory, type II, beta |
Up |
[73,81,82] |
Yes |
| PRKCA |
Protein kinase C, alpha |
Up |
[73,76] |
Yes |
| PSEN2 |
Presenilin 2 (Alzheimer’s disease 4) |
Up |
[73,74] |
? |
| PTHLT |
Parathyroid hormone-like hormone |
Up |
[76,82] |
Yes |
| QDPR |
Quinoid dihydropteridine reductase |
Up |
[74,81] |
Yes |
| RASSF4 |
Ras association (RalGDS/AF-6) domain family 4 |
Up |
[72,74,76] |
Yes |
| RRP22 |
Ras-like protein family member 10A, on chm 22 |
Up |
[73,74] |
? |
| RYR1 |
Skeletal muscle-type ryanodine receptor |
Up |
[5,74] |
Yes |
| RYR3 |
Brain-type ryanodine receptor |
Up |
[74,76,81] |
Yes |
| SLC24A3 |
Solute carrier family 24 (sodium/potassium/calcium exchanger), member 3 |
Up |
[74,76] |
Yes |
| SIX1 |
SIX homeobox 1 |
Up |
[5,75] |
Yes |
| SOX14 |
SRY (sex determining region Y)-box 14 |
Up |
[22,76] |
Yes |
| STX11 |
Syntaxin 11 |
Up |
[76,82] |
Yes |
| SULF1 |
Sulfatase 1 |
Up |
[73,76] |
Yes |
| SVIL |
Supervillin |
Down |
[72,76] |
Yes |
| TCF712 |
Transcription factor 7-like 2 (T-cell specific, HMG-box) |
Up |
[73,81] |
Yes |
| TGFB1 |
Transforming growth factor, beta 1 |
Up |
[5,76] |
Yes |
|
TFAP2B |
Transcription factor AP-2 beta |
Up |
[22,72] |
Yes |
| TIAF1 |
TGF-beta-1-induced antiapoptotic factor 1 |
Up |
[73,74] |
? |
| TM4SF10 |
Transmembrane 4 superfamily member 10 |
Up |
[73,81] |
Yes |
| TNFAIP3 |
Tumor necrosis factor, alpha-induced protein 3 |
Up |
[73,76] |
Yes |
| TNNC2 |
Troponin C type 2 (fast) |
Up/down |
[5,72,73,79] |
Yes |
| TNNI2 |
Troponin I type 2 (skeletal, fast) |
Up/down |
[5,72] |
Yes |
| TNNT2 |
Troponin T type 2 (cardiac) |
Up |
[5,79] |
Yes |
| TNNT3 |
Troponin T type 3 (skeletal, fast) |
Down |
[72,79] |
Yes |
| TRAM2 |
Translocation-associated membrane protein 2 |
Up |
[73,76] |
Yes |
| TSC22D2 |
TSC22 domain family, member 2 |
Up |
[74,76] |
Yes |
| UBE2G2 |
Ubiquitin-conjugating enzyme E2G 2 (UBC7 homolog, yeast) |
Up |
[22,76] |
Yes |
| WSCD1 |
WSC domain-containing protein 1 |
Up |
[22,74] |
? |
| WVA5A | Von Willebrand factor A domain containing 5A | Up | [72-74] | Yes |
Summary of genes found to be differentially regulated in more than one reference in ARMS tumors and/or cell lines, or by PAX3-FOXO1 overexpression, in various cell types. Gene name and description are indicated and also the relative expression in ARMS or PAX3-FOXO1. Also indicated is whether the study indicates that these genes are downstream of PAX3-FOXO1 expression. Genes indicated in bold will be further discussed.
Two of these genes have already been mentioned as cooperating mutations seen in ARMS tumors, N-MYC and IGF2. The N-MYC locus is known to be amplified in a proportion of ARMS tumors, and the IGF2 locus is known to show LOI in ARMS tumors (see cooperating mutations in ARMS tumors). However, these studies also indicate that PAX3-FOXO1 may regulate the gene expression from these loci.
N-MYC expression has been shown to be upregulated in four independent studies using PAX3-FOXO1 targeting siRNA in the ARMS cell line, RH4 [72], PAX3-FOXO1 overexpression in the RD (ERMS) cell line [73,81] and in ARMS versus non ARMS tumor cell lines [78]. A tamoxifen (4-OHT)-inducible PAX3-FOXO1-ER construct induced upregulation of both N-MYC mRNA and protein in the transduced ERMS cell line, RD, and this was not sensitive to cycloheximide treatment, indicating that N-MYC is a direct transcriptional target of PAX3-FOXO1. However the PAX3-FOXO1 regulatory region for N-MYC did not appear to be contained within −1871 to +1058 of the N-MYC gene. Consistent with a transforming role for both PAX3-FOXO1 and N-MYC, the two genes synergized in soft agar colony-forming assays [81]. In addition, knockdown of N-MYC expression identified a positive feedback loop between N-MYC and PAX3-FOXO1 [93].
IGF2 was shown to be specifically overexpressed in ARMS compared to Ewing’s sarcoma cell lines [75], which is perhaps not surprising given that LOI of the IGF2 is seen in almost half of ARMS tumors [66]. However, Khan et al. [5] have shown that, in NIH3T3 cells, PAX3-FOXO1 expression can induce the upregulation of IGF2 mRNA. Interestingly, H19 expression was also found to be upregulated in response to PAX3-FOXO1 expression in these cells. Khan et al. [5] did not investigate whether PAX3-FOXO1 can regulate imprinting of this locus, and the mechanism of PAX3-FOXO1 regulation of IGF2 mRNA expression remains unknown.
Two factors shown to be upregulated in PAX3-FOXO1-expressing cells, MET [73,81,90,91] and CXCR4 [85,86], are thought to play a role in the increased propensity for metastasis seen with ARMS.
MET is the receptor for hepatocyte growth factor/scatter factor (HGF/SF) [94]. HGF-MET signaling has been shown to play an important role in both normal skeletal muscle development and regeneration, and it is involved in regulating myogenic cell migration, survival, proliferation and differentiation [95,96]. MET appears to be a downstream target of PAX3. Splotch mice, which express a mutant PAX3 and fail to form limb muscles because of an inhibition of myogenic precursor cell migration, show a decreased expression of MET [24,97]. MET has also been found in five independent studies to be downstream of the PAX3-FOXO1 fusion protein in ARMS [73,81,90-92]. However, in the case of ARMS, it appears that HGF may play a role in chemoattraction of tumor cells to the bone marrow, which is a common site of metastasis in ARMS cases [98,99]. ARMS cell line CW9019 shows chemotaxis toward bone marrow-derived-fibroblast-conditioned media in vitro, and this migration is inhibited by the MET-blocking agent, K-252a. Moreover, both RH30 and CW9019 ARMS lines home to the bone marrow in lethally irradiated mice, while the ERMS cell lines RD, SMS-CTR, and RH18 do not [99]. Conversely, siRNA against PAX3-FOXO1 prevents migration of cells in wound-healing assays of RH30 cells in response to HGF. In addition to a migratory function for MET in ARMS, knockdown of MET by shRNA in ERMS and ARMS inhibits cell proliferation and induces apoptosis. Moreover, shRNA-mediated knockdown of MET inhibits anchorage-independent growth of ARMS and ERMS, and mutant MET-expressing MEFS prevent PAX3-FKHR transformation [91]. Consistent with MET as a PAX3-FOXO1 target, high MET expression in RMS correlates with ARMS histology, advanced disease at diagnosis and bone marrow involvement [100].
CXCR4 is normally expressed in satellite cells within skeletal muscle and is used as a marker of mononucleated cells capable of differentiating into myofibers [101]. CXCR4 is a cell surface receptor; it binds and mediates the signaling of stromal-derived factor-1 (SDF-1) and induces cell chemotaxis [102]. SDF-1 can induce migration and chemotactic invasion in ARMS cell lines, but not ERMS cell lines [85], and this migration can be inhibited by SDF-1-neutralizing antibody or the CXCR4 inhibitor AMD3100 [103]. Moreover, expression of CXCR4 and MET in ARMS lines appears to synergize to induce the migration of cells toward bone marrow-derived fibroblast conditioned media in vitro. Inhibition of each receptor reduces migration, and combined inhibition reveals synergism between these receptors [99]. SDF-1 can also induce proliferation of the ARMS cell line, RH30 [103]. Consistent with CXCR4 expression being downstream of PAX3-FOXO1 transgene expression, CXCR4 expression in RMS correlates with the ARMS histology, unfavorable primary site, advanced disease at diagnosis and bone marrow involvement [100].
Other genes have been further confirmed as downstream genes of PAX3-FOXO1. Cannabinoid receptor 1 (CNR1) is specifically upregulated at both the mRNA and protein level in fusion-positive ARMS cells [74]. CNR1 is normally highly expressed in brain [104] but is also expressed in skeletal muscle at levels detectable by RT-PCR [105]. CNR1 has been confirmed, using ChIP analysis, as a direct target of both PAX3 and PAX3-FOXO1 transcriptional activity [82]. Furthermore, the homeodomain of PAX3-FOXO1 appears to be the important domain for the regulation of CNR1 expression [72]. CNR1 has been proposed to be a potential drug target in ARMS. Treatment with CNR1 agonists can induce apoptosis in some ARMS cell lines [83]. In addition, CNR1 expression has been linked with an increase propensity for PAX3-FOXO1 expressing mouse myoblast invasiveness and lung metastasis formation. Moreover, treatment with an inverse agonist to CNR1 can abrogate in vitro invasion and in vivo lung metastasis formation [84]. Thus, CNR1 may represent a viable therapeutic target specific for the increased metastatic capacity of PAX3-FOXO1 expressing ARMS.
Transcription factor AP2β (TFAP2B) has been shown to be a downstream target of PAX3-FOXO1 and appears to require the paired domain of PAX3-FOXO1 to be induced. TFAP2B promoter expression is induced by PAX3, which has been shown to bind to the TFAP2B promoter by ChIP analysis. siRNA targeting TFAP2B introduced into PAX3-FKHR-positive ARMS induces apoptosis, indicating that TFAP2B can mediate cell survival in ARMS, downstream of PAX3-FOXO1 [72].
FGFR4 has been identified as a direct transcriptional target of PAX3 and PAX3-FOXO1, which bind to a downstream enhancer region [76,106]. Accordingly, FGFR4 is significantly upregulated by PAX3-FOXO1 expression [22,73,76,88,89,107]. However, upregulation of FGFR4 downstream of PAX3-FOXO1 in primary myoblasts does not appear to act as an effector of PAX3-FOXO1-mediated myoblast transformation given that wild-type FGFR4 upregulation is not required for PAX3-FOXO1-induced proliferation, transformation, invasion or inhibition of myogenic differentiation [89]. However, knockdown of FGFR4 in RMS cell lines does show a reduction in cell proliferation and an increase in apoptosis, suggesting that at later stages of ARMS tumorigenesis FGFR4 overexpression may interact with other unknown genetic lesions within these cell lines to induce pro-survival and proliferation effects [107]. It is interesting to note however that kinase domain-activating mutations in FGFR4 have been identified in 7.5% of RMS, including fusion-positive ARMS [108], and can contribute to myoblast growth advantage and transformation [89]. Thus, FGFR4-activating mutations likely represent cooperating mutations in RMS and upregulation of FGFR4 in fusion-positive ARMS would enhance this effect.
The CDH3/P-cadherin gene has been identified as a direct transcriptional target of PAX3/7-FOXO1 [72,76,80]. P-cadherin expression in the C2C12 myoblast cell line inhibits myogenic differentiation and maintains a proliferative state through maintaining cyclin D1 expression. This in turn results in transformation of C2C12 cells, allowing colony formation in soft agar. Additionally, P-cadherin expression resulted in enhanced cell motility, as well as cadherin switching, a hallmark of epithelial to mesenchymal transition and metastatic progression [80].
In vitro models of ARMS
Many different cell lines have been derived from human ARMS tumors; these are regularly used to investigate the biology of ARMS. These include ARMS cell lines derived from human tumors in the laboratory of Dr. Peter Houghton: RH3, RH4, RH10, RH28, RH30 and RH41, all of which express the PAX3-FOXO1 fusion protein [13,109,110] and have been widely used in the field. In addition, the NCI-supported Pediatric Preclinical Testing Program (PPTP) uses RH10, RH28, RH30, RH30R, RH41 and RH65 subcutaneous xenograft tumors to test drug efficacy in a well-characterized preclinical model of many pediatric cancers [111-113].
Other in vitro models that have been used involve the introduction of the PAX3/7-FOXO1 fusion proteins into both myogenic and non-myogenic cell lines including fibroblast cell lines, ERMS cell lines, MEFs, mesenchymal stem cells (MSC), normal or immortalized human or mouse myogenic cells and even an osteosarcoma cell line [5,81,82,91,114-119]. Given that the cell of origin for ARMS has yet to be identified, perhaps this variation in model cell lines is prudent. However, it is likely that ARMS and ERMS tumors are derived from a mesenchymal cell likely of the the myogenic lineage because of skeletal muscle lineage-specific gene expression seen in these tumors [7].
Conversely, some studies have used endogenous PAX3-FOXO1 in ARMS cell lines to determine the transcriptional targets of this fusion protein within the ARMS tumor cell context. Both Kikuchi et al. [90] and Ebauer et al. [72] used siRNA specifically targeting PAX3-FOXO1 or both PAX3 and PAX3-FOXO1 sequences, respectively. Inhibition of PAX3-FOXO1 expression reduced cell proliferation and motility and allowed some myogenic differentiation [90]. In addition, comparative gene expression studies were performed identifying over 100 PAX3-FOXO1 gene targets. Cao et al. [76] performed ChIP sequencing studies using a PAX3-FOXO1-specific antibody and were able to identify 1,463 putative PAX3-FOXO1-binding sites in the human genome. Furthermore, PAX3-FOXO1-binding sites adjacent to MyoD, FGFR4 and IGF1R were verified as transcriptionally regulated by PAX3-FOXO1.
In vivo models of ARMS
Many different transgenic and knock-in animal models have been attempted to recapitulate ARMS tumor formation in vivo. Several of these models have attempted to constitutively express PAX3/7-FOXO1 fusion proteins in the skeletal muscle lineage during development, only to result in developmental defects and not tumor formation [120-123]. Transgenic mice expressing PAX3-FOXO1 under the control of the PAX3 promoter and enhancer regions resulted in expression of PAX3-FOXO1 in the dorsal neural tube and lateral dermomyotome. PAX3-FOXO1 expression in this context appeared to interfere with normal PAX3 developmental functions including neural tube and neural crest abnormalities similar to those seen in PAX3 mutant Splotch mice. The majority of defects appeared to be in neural development, though defects were seen in hind limb skeletal muscle; however, no tumors developed [121,122].
Lagutina et al. [120] developed a model where PAX3-FOXO1 was knocked into the PAX3 locus. This knock-in locus expressed low amounts of PAX3-FOXO1, which in heterozygous pups was sufficient to result in developmental defects in the heart and diaphragm, leading to congestive heart failure and perinatal death, as well as malformations of some hypaxial muscles. However, neither chimeric adults nor their newborn heterozygous pups developed malignancies. It was hypothesized that PAX3-FOXO1 expression from the PAX3 control sequences was insufficient to cause ARMS formation, and downstream regions of the FOXO1 locus may be required to induce sufficient PAX3-FOXO1 expression to induce tumor development.
A PAX7-FOXO1 model of ARMS was also attempted in Drosophila [123]. Expression of UAS-hPAX7-FOXO1, under control of myosin heavy-chain Gal4, also resulted in developmental defects in the fly, evidenced by disorganized myogenic patterning. Though nothing resembling tumor formation was seen, this group did note dissemination and infiltration of non-native tissue by PAX7-FOXO1 expressing mononucleated cells, indicating an increase in invasive capacity of these cells.
Keller et al. [47] used a conditional PAX3-FOXO1 knock-in into the PAX3 locus, and Myf6-driven Cre expression. This allowed, upon Cre recombination, expression of PAX3-FOXO1 driven by the PAX3 promoter and 3’ FOXO1 genomic sequences that potentially contain cis-regulatory elements, a region absent from previous PAX3 knock-in strategies. This was the first animal model that successfully recapitulated the formation of ARMS, though at the low frequency of approximately 0.4% (1/228) and with latency of over 1 year (383 days). However, this frequency was greatly enhanced, and latency greatly reduced, in homozygote PAX3P3Fa/P3Fa mice also lacking Trp53 or Ink4a/Arf. Subsequently, ARMS tumors have developed in this conditional PAX3-FOXO1 knock-in model with a Pax7CreER and MCre (Pax3 hypaxial muscle enhancer) also lacking functional Trp53 [124]. Moreover, histologically diagnosed fusion-negative ARMS tumors have been found to develop in conditional Ptch1+/−Trp53−/− mice when Cre is expressed from Pax7CreER. The latency and incidence of ARMS tumor development in these different models have yet to be compared.
Clearly the problems that have arisen during the development of an animal model for ARMS indicate that the timing and the cell lineage targeted for PAX3-FOXO1 expression are very important for the development of ARMS tumor formation and for avoiding developmental defects. In a review [125] following the publication of the animal model [47], Keller et al. discuss the possibilities for the cell of origin for ARMS; because Keller et al. achieved the formation of ARMS tumors in their mouse model using Myf6-Cre-driven conditional PAX3-FOXO1, and Myf6 is usually expressed in differentiating skeletal muscle myotubes, they propose a potential dedifferentiation mechanism for ARMS development. However, the formation of a fusion gene such as PAX3-FOXO1 suggests that the cell of origin for ARMS should express both PAX3 and FOXO1 at the time that the translocation occurs, given that open chromatin is likely required for these two genomically distinct regions to translocate. Anecdotal evidence for this includes that the genome translocations that occur in many different cancer types occur between genes that are expressed in the cell type of origin. A recent study by Osborne et al. [126] showed that the MYC and IGH genes, which are involved in a chromosomal translocation common in Burkitt lymphoma, are colocalized at the same transcription factories more often in activated B-cells, the originating cell of Burkitt lymphoma, than resting B cells. This colocalization at the same transcription factory allows for close proximity of these gene loci in euchromatin, providing the circumstances where these genes would be in close association, facilitating the specific translocation event. PAX3 is rapidly downregulated upon myoblast differentiation, so it would be unlikely that the PAX3 loci would be expressed in a nascent myotube expressing Myf6, making it difficult to understand how translocation could occur in nascent myotubes and therefore cast some doubt on whether the dedifferentiation model is feasible. However, it is possible that Myf6 expression does rarely occur in a small subset of undifferentiated myogenic cells in conjunction with PAX3. This could allow for this model to produce ARMS tumors and account for the low frequency at which these tumors are seen as well as the requirement for homozygous PAX3-FOXO1 knock-in alleles [47].
From these animal models it is apparent that the timing of PAX3-FOXO1 expression is critical for ARMS development. Too early and widespread expression of PAX3-FOXO1 expression can result in developmental defects and no apparent tumor development [120-123], whereas later expression of PAX3-FOXO1, via a Myf6-driven Cre recombinase, does cause disease, though at a low frequency [47]. Perhaps inducible expression, driven by various myogenic genes with carefully characterized expression profiling, would result in an increased frequency of disease and help to narrow down the exact stage in which PAX3-FOXO1 expression drives ARMS tumorigenesis. Nevertheless, the cell of origin for ARMS is yet to be identified, and animal models of ARMS will no doubt play an important role in its identification.
Conclusion
To date, numerous factors (outlined in Figure 2) have been identified that contribute to ARMS tumor development and its aggressive clinical phenotype. These consist of both PAX3/7-FOXO1 target genes, such as N-MYC, IGF2, MET, CXCR4, CNR1, TFAP2B, FGFR4 and P-cadherin, and PAX3/7-FOXO1 cooperating factors, such as the abrogation of the p53 pathway, IGF2 deregulation, N-MYC and miR17-92 amplification, and IRIZIO expression. Future ARMS research will continue to discover the mechanisms by which ARMS tumorigenesis occurs. This will involve the identification of more PAX3-FOXO1 target and cooperating genes; more importantly, the mechanisms by which these genes contribute to tumorigenesis will be elucidated. It is critical that we develop a mechanistic understanding of how these factors contribute and interact to perpetrate ARMS tumorigenesis. This will allow new opportunities to develop specifically targeted therapies for this aggressive pediatric disease.
Figure 2.

Review summary: Fusion gene regulated genes contributing to alveolar rhabdomyosarcoma. Rhabdomyosarcoma develops from an unknown cell of origin from the mesodermal lineage that may be skeletal muscle specified. This cell likely expresses both PAX3/7 and FOXO1 and may also express Myf6. A gene fusion event resulting in a PAX3/7 DNA-binding domain fused to a more potent transcriptional activation domain occurs. This fusion transcription factor is capable of inducing a group of PAX3-FOXO1-regulated genes that contribute to ARMS development in conjunction with other genetic lesions.
Abbreviations
ARMS: Alveolar rhabdomyosarcoma; bHLH: Basic helix loop helix domain; CDK: Cyclin-dependent kinase; CNR1: Cannabinoid receptor 1; ERMS: Embryonal rhabdomyosarcoma; FH: Forkhead DNA-binding domain; FKHR: Forkhead in rhabdomyosarcoma (now known as FOXO1); HD: Homeodomain DNA-binding domain; HGF/SF: Hepatocyte growth factor/scatter factor; LOH: Loss of heterozygosity; LOI: Loss of imprinting; MEF: Mouse embryonic fibroblast; miR: Micro RNA; MSC: Mesenchymal stem cells; PD: Paired box DNA-binding domain; PPTP: Pediatric Preclinical Testing Program; RMS: Rhabdomyosarcoma; SDF-1: Stromal-derived factor-1; SHH: Sonic hedgehog; siRNA: Short interfering RNA; TFAP2B: Transcription factor AP2 b.
Competing interests
The authors have no competing interests to declare.
Authors’ contributions
AM was responsible for the drafting of the manuscript. GG was responsible for critical revision of the content and approved the final version of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Amy D Marshall, Email: a.marshall@centerary.org.au.
Gerard C Grosveld, Email: gerard.grosveld@stjude.org.
References
- American Cancer Society. Rhabdomyosarcoma. http://www.cancer.org/Cancer/Rhabdomyosarcoma/DetailedGuide/rhabdomyosarcoma.
- Punyko JA, Mertens AC, Baker KS, Ness KK, Robison LL, Gurney JG. Long-term survival probabilities for childhood rhabdomyosarcoma. A population-based evaluation. Cancer. 2005;103:1475–1483. doi: 10.1002/cncr.20929. [DOI] [PubMed] [Google Scholar]
- Tonin PN, Scrable H, Shimada H, Cavenee WK. Muscle-specific gene expression in rhabdomyosarcomas and stages of human fetal skeletal muscle development. Cancer Res. 1991;51:5100–5106. [PubMed] [Google Scholar]
- Dias P, Chen B, Dilday B, Palmer H, Hosoi H, Singh S, Wu C, Li X, Thompson J, Parham D. et al. Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol. 2000;156:399–408. doi: 10.1016/S0002-9440(10)64743-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J, Bittner ML, Saal LH, Teichmann U, Azorsa DO, Gooden GC, Pavan WJ, Trent JM, Meltzer PS. cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci USA. 1999;96:13264–13269. doi: 10.1073/pnas.96.23.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerema-McKenney A, Wijnaendts LC, Pulliam JF, Lopez-Terrada D, McKenney JK, Zhu S, Montgomery K, Mitchell J, Marinelli RJ, Hart AA. et al. Diffuse myogenin expression by immunohistochemistry is an independent marker of poor survival in pediatric rhabdomyosarcoma: a tissue microarray study of 71 primary tumors including correlation with molecular phenotype. Am J Surg Pathol. 2008;32:1513–1522. doi: 10.1097/PAS.0b013e31817a909a. [DOI] [PubMed] [Google Scholar]
- Wexler L, Meyer W, Helman L. In: Principles and Practice of Pediatric Oncology. Fifthth edition. Pizzo PA, Poplack D, editor. Philidelphia: Lippincott Williams and Wilkins; 2006. Rhabdomyosarcoma and the undifferentiated sarcomas; pp. 971–1001. [Google Scholar]
- Meza JL, Anderson J, Pappo AS, Meyer WH. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children's Oncology Group. J Clin Oncol. 2006;24:3844–3851. doi: 10.1200/JCO.2005.05.3801. [DOI] [PubMed] [Google Scholar]
- Xia SJ, Pressey JG, Barr FG. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol Ther. 2002;1:97–104. doi: 10.4161/cbt.51. [DOI] [PubMed] [Google Scholar]
- Punyko JA, Mertens AC, Gurney JG, Yasui Y, Donaldson SS, Rodeberg DA, Raney RB, Stovall M, Sklar CA, Robison LL, Baker KS. Long-term medical effects of childhood and adolescent rhabdomyosarcoma: a report from the childhood cancer survivor study. Pediatr Blood Cancer. 2005;44:643–653. doi: 10.1002/pbc.20310. [DOI] [PubMed] [Google Scholar]
- Cerveira N, Torres L, Ribeiro FR, Henrique R, Pinto A, Bizarro S, Ferreira AM, Lopes C, Teixeira MR. Multimodal genetic diagnosis of solid variant alveolar rhabdomyosarcoma. Cancer Genet Cytogenet. 2005;163:138–143. doi: 10.1016/j.cancergencyto.2005.06.020. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Oda Y, Kawaguchi K, Tamiya S, Yamamoto H, Suita S, Tsuneyoshi M. Altered expression and molecular abnormalities of cell-cycle-regulatory proteins in rhabdomyosarcoma. Mod Pathol. 2004;17:660–669. doi: 10.1038/modpathol.3800101. [DOI] [PubMed] [Google Scholar]
- Douglass EC, Valentine M, Etcubanas E, Parham D, Webber BL, Houghton PJ, Houghton JA, Green AA. A specific chromosomal abnormality in rhabdomyosarcoma. Cytogenet Cell Genet. 1987;45:148–155. doi: 10.1159/000132446. [DOI] [PubMed] [Google Scholar]
- Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ 3rd, Emanuel BS, Rovera G, Barr FG. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;5:230–235. doi: 10.1038/ng1193-230. [DOI] [PubMed] [Google Scholar]
- Fredericks WJ, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J, Barr FG, Rauscher FJ 3rd. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol. 1995;15:1522–1535. doi: 10.1128/mcb.15.3.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam PY, Sublett JE, Hollenbach AD, Roussel MF. The oncogenic potential of the Pax3-FKHR fusion protein requires the Pax3 homeodomain recognition helix but not the Pax3 paired-box DNA binding domain. Mol Cell Biol. 1999;19:594–601. doi: 10.1128/mcb.19.1.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Schwartz J, Wang C. Comparative analysis of paired- and homeodomain-specific roles in PAX3-FKHR oncogenesis. Int J Clin Exp Pathol. 2009;2:370–383. [PMC free article] [PubMed] [Google Scholar]
- Sorensen PH, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM, Bridge JA, Crist WM, Triche TJ, Barr FG. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children's oncology group. J Clin Oncol. 2002;20:2672–2679. doi: 10.1200/JCO.2002.03.137. [DOI] [PubMed] [Google Scholar]
- Davis RJ, D'Cruz CM, Lovell MA, Biegel JA, Barr FG. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994;54:2869–2872. [PubMed] [Google Scholar]
- Barr FG, Qualman SJ, Macris MH, Melnyk N, Lawlor ER, Strzelecki DM, Triche TJ, Bridge JA, Sorensen PH. Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res. 2002;62:4704–4710. [PubMed] [Google Scholar]
- Sumegi J, Streblow R, Frayer RW, Dal Cin P, Rosenberg A, Meloni-Ehrig A, Bridge JA. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes Chromosomes Cancer. 2010;49:224–236. doi: 10.1002/gcc.20731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachtel M, Dettling M, Koscielniak E, Stegmaier S, Treuner J, Simon-Klingenstein K, Buhlmann P, Niggli FK, Schafer BW. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res. 2004;64:5539–5545. doi: 10.1158/0008-5472.CAN-04-0844. [DOI] [PubMed] [Google Scholar]
- Goulding MD, Chalepakis G, Deutsch U, Erselius JR, Gruss P. Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO J. 1991;10:1135–1147. doi: 10.1002/j.1460-2075.1991.tb08054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daston G, Lamar E, Olivier M, Goulding M. Pax-3 is necessary for migration but not differentiation of limb muscle precursors in the mouse. Development. 1996;122:1017–1027. doi: 10.1242/dev.122.3.1017. [DOI] [PubMed] [Google Scholar]
- Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–786. doi: 10.1016/S0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Oustanina S, Hause G, Braun T. Pax7 directs postnatal renewal and propagation of myogenic satellite cells but not their specification. EMBO J. 2004;23:3430–3439. doi: 10.1038/sj.emboj.7600346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J. 2000;349:629–634. doi: 10.1042/0264-6021:3490629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misiti S, Koibuchi N, Bei M, Farsetti A, Chin WW. Expression of steroid receptor coactivator-1 mRNA in the developing mouse embryo: a possible role in olfactory epithelium development. Endocrinology. 1999;140:1957–1960. doi: 10.1210/en.140.4.1957. [DOI] [PubMed] [Google Scholar]
- Voegel JJ, Heine MJ, Zechel C, Chambon P, Gronemeyer H. TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J. 1996;15:3667–3675. [PMC free article] [PubMed] [Google Scholar]
- Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27:352–360. doi: 10.1016/S0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- Xu J, Li Q. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol. 2003;17:1681–1692. doi: 10.1210/me.2003-0116. [DOI] [PubMed] [Google Scholar]
- Kempf BE, Vogt PK. A genetic analysis of PAX3-FKHR, the oncogene of alveolar rhabdomyosarcoma. Cell Growth Differ. 1999;10:813–818. [PubMed] [Google Scholar]
- Li H, Gomes PJ, Chen JD. RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc Natl Acad Sci USA. 1997;94:8479–8484. doi: 10.1073/pnas.94.16.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belandia B, Parker MG. Functional interaction between the p160 coactivator proteins and the transcriptional enhancer factor family of transcription factors. J Biol Chem. 2000;275:30801–30805. doi: 10.1074/jbc.C000484200. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Lam P, Jepeal L, Maas RL, Shapiro DN. Pax3 inhibits myogenic differentiation of cultured myoblast cells. J Biol Chem. 1995;270:11719–11722. doi: 10.1074/jbc.270.20.11719. [DOI] [PubMed] [Google Scholar]
- Miller PJ, Hollenbach AD. The oncogenic fusion protein Pax3-FKHR has a greater post-translational stability relative to Pax3 during early myogenesis. Biochim Biophys Acta. 2007;1770:1450–1458. doi: 10.1016/j.bbagen.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olguin HC, Patzlaff NE, Olwin BB. Pax7-FKHR transcriptional activity is enhanced by transcriptionally repressed MyoD. J Cell Biochem. 2011;112:1410–1417. doi: 10.1002/jcb.23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhabeu F, Hayashi S, Morgan JE, Relaix F, Zammit PS. Alveolar rhabdomyosarcoma-associated proteins PAX3/FOXO1A and PAX7/FOXO1A suppress the transcriptional activity of MyoD-target genes in muscle stem cells. Oncogene. 2012. Epub ahead of print. [DOI] [PubMed]
- Charytonowicz E, Matushansky I, Domenech JD, Castillo-Martin M, Ladanyi M, Cordon-Cardo C, Ziman M. PAX7-FKHR fusion gene inhibits myogenic differentiation via NF-kappaB upregulation. Clin Transl Oncol. 2012;14:197–206. doi: 10.1007/s12094-012-0784-4. [DOI] [PubMed] [Google Scholar]
- Tapscott SJ, Thayer MJ, Weintraub H. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science. 1993;259:1450–1453. doi: 10.1126/science.8383879. [DOI] [PubMed] [Google Scholar]
- Wexler LH, Ladanyi M. Diagnosing alveolar rhabdomyosarcoma: morphology must be coupled with fusion confirmation. J Clin Oncol. 2010;28:2126–2128. doi: 10.1200/JCO.2009.27.5339. [DOI] [PubMed] [Google Scholar]
- Anderson JR, Barr FG, Hawkins DS, Parham DM, Skapek SX, Triche TJ. Fusion-negative alveolar rhabdomyosarcoma: modification of risk stratification is premature. J Clin Oncol. 2010;28:e587–588. doi: 10.1200/JCO.2010.30.5466. author reply e589-590. [DOI] [PubMed] [Google Scholar]
- Davicioni E, Anderson MJ, Finckenstein FG, Lynch JC, Qualman SJ, Shimada H, Schofield DE, Buckley JD, Meyer WH, Sorensen PH, Triche TJ. Molecular classification of rhabdomyosarcoma–genotypic and phenotypic determinants of diagnosis: a report from the Children's Oncology Group. Am J Pathol. 2009;174:550–564. doi: 10.2353/ajpath.2009.080631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson D, Missiaglia E, de Reynies A, Pierron G, Thuille B, Palenzuela G, Thway K, Orbach D, Lae M, Freneaux P. et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. 2010;28:2151–2158. doi: 10.1200/JCO.2009.26.3814. [DOI] [PubMed] [Google Scholar]
- Barr FG, Nauta LE, Davis RJ, Schafer BW, Nycum LM, Biegel JA. In vivo amplification of the PAX3-FKHR and PAX7-FKHR fusion genes in alveolar rhabdomyosarcoma. Hum Mol Genet. 1996;5:15–21. doi: 10.1093/hmg/5.1.15. [DOI] [PubMed] [Google Scholar]
- Naini S, Etheridge KT, Adam SJ, Qualman SJ, Bentley RC, Counter CM, Linardic CM. Defining the cooperative genetic changes that temporally drive alveolar rhabdomyosarcoma. Cancer Res. 2008;68:9583–9588. doi: 10.1158/0008-5472.CAN-07-6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004;18:2614–2626. doi: 10.1101/gad.1244004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp R, Recio JA, Jhappan C, Otsuka T, Liu S, Yu Y, Liu W, Anver M, Navid F, Helman LJ. et al. Synergism between INK4a/ARF inactivation and aberrant HGF/SF signaling in rhabdomyosarcomagenesis. Nat Med. 2002;8:1276–1280. doi: 10.1038/nm787. [DOI] [PubMed] [Google Scholar]
- Weber-Hall S, Anderson J, McManus A, Abe S, Nojima T, Pinkerton R, Pritchard-Jones K, Shipley J. Gains, losses, and amplification of genomic material in rhabdomyosarcoma analyzed by comparative genomic hybridization. Cancer Res. 1996;56:3220–3224. [PubMed] [Google Scholar]
- Ragazzini P, Gamberi G, Pazzaglia L, Serra M, Magagnoli G, Ponticelli F, Ferrari C, Ghinelli C, Alberghini M, Bertoni F. et al. Amplification of CDK4, MDM2, SAS and GLI genes in leiomyosarcoma, alveolar and embryonal rhabdomyosarcoma. Histol Histopathol. 2004;19:401–411. doi: 10.14670/HH-19.401. [DOI] [PubMed] [Google Scholar]
- Gordon AT, Brinkschmidt C, Anderson J, Coleman N, Dockhorn-Dworniczak B, Pritchard-Jones K, Shipley J. A novel and consistent amplicon at 13q31 associated with alveolar rhabdomyosarcoma. Genes Chromosomes Cancer. 2000;28:220–226. doi: 10.1002/(SICI)1098-2264(200006)28:2<220::AID-GCC11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Bridge JA, Liu J, Qualman SJ, Suijkerbuijk R, Wenger G, Zhang J, Wan X, Baker KS, Sorensen P, Barr FG. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes Chromosomes Cancer. 2002;33:310–321. doi: 10.1002/gcc.10026. [DOI] [PubMed] [Google Scholar]
- Forus A, Florenes VA, Maelandsmo GM, Meltzer PS, Fodstad O, Myklebost O. Mapping of amplification units in the q13-14 region of chromosome 12 in human sarcomas: some amplica do not include MDM2. Cell Growth Differ. 1993;4:1065–1070. [PubMed] [Google Scholar]
- Roberts WM, Douglass EC, Peiper SC, Houghton PJ, Look AT. Amplification of the gli gene in childhood sarcomas. Cancer Res. 1989;49:5407–5413. [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-R. [DOI] [PubMed] [Google Scholar]
- Taylor AC, Shu L, Danks MK, Poquette CA, Shetty S, Thayer MJ, Houghton PJ, Harris LC. P53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumors and cell lines. Med Pediatr Oncol. 2000;35:96–103. doi: 10.1002/1096-911X(200008)35:2<96::AID-MPO2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Felix CA, Kappel CC, Mitsudomi T, Nau MM, Tsokos M, Crouch GD, Nisen PD, Winick NJ, Helman LJ. Frequency and diversity of p53 mutations in childhood rhabdomyosarcoma. Cancer Res. 1992;52:2243–2247. [PubMed] [Google Scholar]
- Leuschner I, Langhans I, Schmitz R, Harms D, Mattke A, Treuner J. p53 and mdm-2 expression in Rhabdomyosarcoma of childhood and adolescence: clinicopathologic study by the Kiel Pediatric Tumor Registry and the German Cooperative Soft Tissue Sarcoma Study. Pediatr Dev Pathol. 2003;6:128–136. doi: 10.1007/s10024-001-0097-z. [DOI] [PubMed] [Google Scholar]
- Hachitanda Y, Toyoshima S, Akazawa K, Tsuneyoshi M. N-myc gene amplification in rhabdomyosarcoma detected by fluorescence in situ hybridization: its correlation with histologic features. Mod Pathol. 1998;11:1222–1227. [PubMed] [Google Scholar]
- Driman D, Thorner PS, Greenberg ML, Chilton-MacNeill S, Squire J. MYCN gene amplification in rhabdomyosarcoma. Cancer. 1994;73:2231–2237. doi: 10.1002/1097-0142(19940415)73:8<2231::AID-CNCR2820730832>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Williamson D, Lu YJ, Gordon T, Sciot R, Kelsey A, Fisher C, Poremba C, Anderson J, Pritchard-Jones K, Shipley J. Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J Clin Oncol. 2005;23:880–888. doi: 10.1200/JCO.2005.11.078. [DOI] [PubMed] [Google Scholar]
- Toffolatti L, Frascella E, Ninfo V, Gambini C, Forni M, Carli M, Rosolen A. MYCN expression in human rhabdomyosarcoma cell lines and tumour samples. J Pathol. 2002;196:450–458. doi: 10.1002/path.1068. [DOI] [PubMed] [Google Scholar]
- Reichek JL, Duan F, Smith LM, Gustafson DM, O'Connor RS, Zhang C, Dunlevy MJ, Gastier-Foster JM, Barr FG. Genomic and clinical analysis of amplification of the 13q31 chromosomal region in alveolar rhabdomyosarcoma: a report from the Children's Oncology Group. Clin Cancer Res. 2011;17:1463–1473. doi: 10.1158/1078-0432.CCR-10-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson D, Selfe J, Gordon T, Lu YJ, Pritchard-Jones K, Murai K, Jones P, Workman P, Shipley J. Role for amplification and expression of glypican-5 in rhabdomyosarcoma. Cancer Res. 2007;67:57–65. doi: 10.1158/0008-5472.CAN-06-1650. [DOI] [PubMed] [Google Scholar]
- Cho WC. OncomiRs: the discovery and progress of microRNAs in cancers. Mol Cancer. 2007;6:60. doi: 10.1186/1476-4598-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J, Gordon A, McManus A, Shipley J, Pritchard-Jones K. Disruption of imprinted genes at chromosome region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia. 1999;1:340–348. doi: 10.1038/sj.neo.7900052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser M, Sijmons C, Bras J, Arceci RJ, Godfried M, Valentijn LJ, Voute PA, Baas F. Allelotype of pediatric rhabdomyosarcoma. Oncogene. 1997;15:1309–1314. doi: 10.1038/sj.onc.1201302. [DOI] [PubMed] [Google Scholar]
- Bartolomei MS, Zemel S, Tilghman SM. Parental imprinting of the mouse H19 gene. Nature. 1991;351:153–155. doi: 10.1038/351153a0. [DOI] [PubMed] [Google Scholar]
- Hatada I, Inazawa J, Abe T, Nakayama M, Kaneko Y, Jinno Y, Niikawa N, Ohashi H, Fukushima Y, Iida K. et al. Genomic imprinting of human p57KIP2 and its reduced expression in Wilms' tumors. Hum Mol Genet. 1996;5:783–788. doi: 10.1093/hmg/5.6.783. [DOI] [PubMed] [Google Scholar]
- Rachmilewitz J, Goshen R, Ariel I, Schneider T, de Groot N, Hochberg A. Parental imprinting of the human H19 gene. FEBS Lett. 1992;309:25–28. doi: 10.1016/0014-5793(92)80731-U. [DOI] [PubMed] [Google Scholar]
- Picchione F, Pritchard C, Lagutina I, Janke L, Grosveld GC. IRIZIO: a novel gene cooperating with PAX3-FOXO1 in alveolar rhabdomyosarcoma (ARMS) Carcinogenesis. 2011;32:452–461. doi: 10.1093/carcin/bgq273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebauer M, Wachtel M, Niggli FK, Schafer BW. Comparative expression profiling identifies an in vivo target gene signature with TFAP2B as a mediator of the survival function of PAX3/FKHR. Oncogene. 2007;26:7267–7281. doi: 10.1038/sj.onc.1210525. [DOI] [PubMed] [Google Scholar]
- Davicioni E, Finckenstein FG, Shahbazian V, Buckley JD, Triche TJ, Anderson MJ. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006;66:6936–6946. doi: 10.1158/0008-5472.CAN-05-4578. [DOI] [PubMed] [Google Scholar]
- Lae M, Ahn EH, Mercado GE, Chuai S, Edgar M, Pawel BR, Olshen A, Barr FG, Ladanyi M. Global gene expression profiling of PAX-FKHR fusion-positive alveolar and PAX-FKHR fusion-negative embryonal rhabdomyosarcomas. J Pathol. 2007;212:143–151. doi: 10.1002/path.2170. [DOI] [PubMed] [Google Scholar]
- Baer C, Nees M, Breit S, Selle B, Kulozik AE, Schaefer KL, Braun Y, Wai D, Poremba C. Profiling and functional annotation of mRNA gene expression in pediatric rhabdomyosarcoma and Ewing's sarcoma. Int J Cancer. 2004;110:687–694. doi: 10.1002/ijc.20171. [DOI] [PubMed] [Google Scholar]
- Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, Yang F, Pineda M, Helman LJ, Meltzer PS. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010;70:6497–6508. doi: 10.1158/0008-5472.CAN-10-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corao DA, Biegel JA, Coffin CM, Barr FG, Wainwright LM, Ernst LM, Choi JK, Zhang PJ, Pawel BR. ALK expression in rhabdomyosarcomas: correlation with histologic subtype and fusion status. Pediatr Dev Pathol. 2009;12:275–283. doi: 10.2350/08-03-0434.1. [DOI] [PubMed] [Google Scholar]
- Khan J, Simon R, Bittner M, Chen Y, Leighton SB, Pohida T, Smith PD, Jiang Y, Gooden GC, Trent JM, Meltzer PS. Gene expression profiling of alveolar rhabdomyosarcoma with cDNA microarrays. Cancer Res. 1998;58:5009–5013. [PubMed] [Google Scholar]
- De Pitta C, Tombolan L, Albiero G, Sartori F, Romualdi C, Jurman G, Carli M, Furlanello C, Lanfranchi G, Rosolen A. Gene expression profiling identifies potential relevant genes in alveolar rhabdomyosarcoma pathogenesis and discriminates PAX3-FKHR positive and negative tumors. Int J Cancer. 2006;118:2772–2781. doi: 10.1002/ijc.21698. [DOI] [PubMed] [Google Scholar]
- Thuault S, Hayashi S, Lagirand-Cantaloube J, Plutoni C, Comunale F, Delattre O, Relaix F, Gauthier-Rouviere C. P-cadherin is a direct PAX3-FOXO1A target involved in alveolar rhabdomyosarcoma aggressiveness. Oncogene. 2012. Epub ahead of print. [DOI] [PubMed]
- Mercado GE, Xia SJ, Zhang C, Ahn EH, Gustafson DM, Lae M, Ladanyi M, Barr FG. Identification of PAX3-FKHR-regulated genes differentially expressed between alveolar and embryonal rhabdomyosarcoma: focus on MYCN as a biologically relevant target. Genes Chromosomes Cancer. 2008;47:510–520. doi: 10.1002/gcc.20554. [DOI] [PubMed] [Google Scholar]
- Begum S, Emami N, Cheung A, Wilkins O, Der S, Hamel PA. Cell-type-specific regulation of distinct sets of gene targets by Pax3 and Pax3/FKHR. Oncogene. 2005;24:1860–1872. doi: 10.1038/sj.onc.1208315. [DOI] [PubMed] [Google Scholar]
- Oesch S, Walter D, Wachtel M, Pretre K, Salazar M, Guzman M, Velasco G, Schafer BW. Cannabinoid receptor 1 is a potential drug target for treatment of translocation-positive rhabdomyosarcoma. Mol Cancer Ther. 2009;8:1838–1845. doi: 10.1158/1535-7163.MCT-08-1147. [DOI] [PubMed] [Google Scholar]
- Marshall AD, Lagutina I, Grosveld GC. PAX3-FOXO1 induces cannabinoid receptor 1 to enhance cell invasion and metastasis. Cancer Res. 2011;71(24):7471–7480. doi: 10.1158/0008-5472.CAN-11-0924. [DOI] [PubMed] [Google Scholar]
- Libura J, Drukala J, Majka M, Tomescu O, Navenot JM, Kucia M, Marquez L, Peiper SC, Barr FG, Janowska-Wieczorek A, Ratajczak MZ. CXCR4-SDF-1 signaling is active in rhabdomyosarcoma cells and regulates locomotion, chemotaxis, and adhesion. Blood. 2002;100:2597–2606. doi: 10.1182/blood-2002-01-0031. [DOI] [PubMed] [Google Scholar]
- Tomescu O, Xia SJ, Strezlecki D, Bennicelli JL, Ginsberg J, Pawel B, Barr FG. Inducible short-term and stable long-term cell culture systems reveal that the PAX3-FKHR fusion oncoprotein regulates CXCR4, PAX3, and PAX7 expression. Lab Invest. 2004;84:1060–1070. doi: 10.1038/labinvest.3700125. [DOI] [PubMed] [Google Scholar]
- Tarnowski M, Grymula K, Reca R, Jankowski K, Maksym R, Tarnowska J, Przybylski G, Barr FG, Kucia M, Ratajczak MZ. Regulation of expression of stromal-derived factor-1 receptors: CXCR4 and CXCR7 in human rhabdomyosarcomas. Mol Cancer Res. 2010;8:1–14. doi: 10.1158/1541-7786.MCR-09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu CR, Peterson C, Meltzer PS. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–679. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall AD, van der Ent MA, Grosveld GC. PAX3-FOXO1 and FGFR4 in alveolar rhabdomyosarcoma. Mol Carcinog. 2012;51(10):807–815. doi: 10.1002/mc.20848. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Tsuchiya K, Otabe O, Gotoh T, Tamura S, Katsumi Y, Yagyu S, Tsubai-Shimizu S, Miyachi M, Iehara T, Hosoi H. Effects of PAX3-FKHR on malignant phenotypes in alveolar rhabdomyosarcoma. Biochem Biophys Res Commun. 2008;365:568–574. doi: 10.1016/j.bbrc.2007.11.017. [DOI] [PubMed] [Google Scholar]
- Taulli R, Scuoppo C, Bersani F, Accornero P, Forni PE, Miretti S, Grinza A, Allegra P, Schmitt-Ney M, Crepaldi T, Ponzetto C. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res. 2006;66:4742–4749. doi: 10.1158/0008-5472.CAN-05-4292. [DOI] [PubMed] [Google Scholar]
- Ginsberg JP, Davis RJ, Bennicelli JL, Nauta LE, Barr FG. Up-regulation of MET but not neural cell adhesion molecule expression by the PAX3-FKHR fusion protein in alveolar rhabdomyosarcoma. Cancer Res. 1998;58:3542–3546. [PubMed] [Google Scholar]
- Tonelli R, McIntyre A, Camerin C, Walters ZS, Di Leo K, Selfe J, Purgato S, Missiaglia E, Tortori A, Astolfi A, Antitumor activity of sustained N-Myc reduction in Rhabdomyosarcomas and transcriptional block by antigene therapy. Clin Cancer Res. 2011. [DOI] [PubMed]
- Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande-Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- Brand-Saberi B, Muller TS, Wilting J, Christ B, Birchmeier C. Scatter factor/hepatocyte growth factor (SF/HGF) induces emigration of myogenic cells at interlimb level in vivo. Dev Biol. 1996;179:303–308. doi: 10.1006/dbio.1996.0260. [DOI] [PubMed] [Google Scholar]
- Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376:768–771. doi: 10.1038/376768a0. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Shapiro DN, Cheng J, Lam PY, Maas RL. Pax3 modulates expression of the c-Met receptor during limb muscle development. Proc Natl Acad Sci USA. 1996;93:4213–4218. doi: 10.1073/pnas.93.9.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasiewicz E, Miekus K, Kijowski J, Drabik G, Wilusz M, Bobis-Wozowicz S, Majka M. Inhibition of rhabdomyosarcoma's metastatic behavior through downregulation of MET receptor signaling. Folia Histochem Cytobiol. 2009;47:485–489. doi: 10.2478/v10042-009-0108-x. [DOI] [PubMed] [Google Scholar]
- Jankowski K, Kucia M, Wysoczynski M, Reca R, Zhao D, Trzyna E, Trent J, Peiper S, Zembala M, Ratajczak J. et al. Both hepatocyte growth factor (HGF) and stromal-derived factor-1 regulate the metastatic behavior of human rhabdomyosarcoma cells, but only HGF enhances their resistance to radiochemotherapy. Cancer Res. 2003;63:7926–7935. [PubMed] [Google Scholar]
- Diomedi-Camassei F, McDowell HP, De Ioris MA, Uccini S, Altavista P, Raschella G, Vitali R, Mannarino O, De Sio L, Cozzi DA. et al. Clinical significance of CXC chemokine receptor-4 and c-Met in childhood rhabdomyosarcoma. Clin Cancer Res. 2008;14:4119–4127. doi: 10.1158/1078-0432.CCR-07-4446. [DOI] [PubMed] [Google Scholar]
- Sherwood RI, Christensen JL, Conboy IM, Conboy MJ, Rando TA, Weissman IL, Wagers AJ. Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 2004;119:543–554. doi: 10.1016/j.cell.2004.10.021. [DOI] [PubMed] [Google Scholar]
- D'Apuzzo M, Rolink A, Loetscher M, Hoxie JA, Clark-Lewis I, Melchers F, Baggiolini M, Moser B. The chemokine SDF-1, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur J Immunol. 1997;27:1788–1793. doi: 10.1002/eji.1830270729. [DOI] [PubMed] [Google Scholar]
- Strahm B, Durbin AD, Sexsmith E, Malkin D. The CXCR4-SDF1alpha axis is a critical mediator of rhabdomyosarcoma metastatic signaling induced by bone marrow stroma. Clin Exp Metastasis. 2008;25:1–10. doi: 10.1007/s10585-007-9094-6. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- Cavuoto P, McAinch AJ, Hatzinikolas G, Janovska A, Game P, Wittert GA. The expression of receptors for endocannabinoids in human and rodent skeletal muscle. Biochem Biophys Res Commun. 2007;364:105–110. doi: 10.1016/j.bbrc.2007.09.099. [DOI] [PubMed] [Google Scholar]
- Lagha M, Sato T, Bajard L, Daubas P, Esner M, Montarras D, Relaix F, Buckingham M. Regulation of skeletal muscle stem cell behavior by Pax3 and Pax7. Cold Spring Harb Symp Quant Biol. 2008;73:307–315. doi: 10.1101/sqb.2008.73.006. [DOI] [PubMed] [Google Scholar]
- Crose LE, Etheridge KT, Chen C, Belyea B, Talbot LJ, Bentley RC, Linardic CM. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin Cancer Res. 2012;18(14):3780–3790. doi: 10.1158/1078-0432.CCR-10-3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JG, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, Yu Y, Chen QR, Shah K, Youngblood V. et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest. 2009;119:3395–3407. doi: 10.1172/JCI39703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelton BJ, Houghton JA, Parham DM, Douglass EC, Torrance PM, Holt H, Houghton PJ. Characterization of cell lines derived from xenografts of childhood rhabdomyosarcoma. Cancer Res. 1987;47:4501–4507. [PubMed] [Google Scholar]
- Croci S, Landuzzi L, Astolfi A, Nicoletti G, Rosolen A, Sartori F, Follo MY, Oliver N, De Giovanni C, Nanni P, Lollini PL. Inhibition of connective tissue growth factor (CTGF/CCN2) expression decreases the survival and myogenic differentiation of human rhabdomyosarcoma cells. Cancer Res. 2004;64:1730–1736. doi: 10.1158/0008-5472.CAN-3502-02. [DOI] [PubMed] [Google Scholar]
- Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, Gorlick R, Kolb EA, Zhang W, Lock R. et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007;49:928–940. doi: 10.1002/pbc.21078. [DOI] [PubMed] [Google Scholar]
- Kang MH, Smith MA, Morton CL, Keshelava N, Houghton PJ, Reynolds CP. National Cancer Institute pediatric preclinical testing program: model description for in vitro cytotoxicity testing. Pediatr Blood Cancer. 2011;56:239–249. doi: 10.1002/pbc.22801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale G, Su X, Morton CL, Phelps D, Gorlick R, Lock RB, Reynolds CP, Maris JM, Friedman HS, Dome J. et al. Molecular characterization of the pediatric preclinical testing panel. Clin Cancer Res. 2008;14:4572–4583. doi: 10.1158/1078-0432.CCR-07-5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia SJ, Holder DD, Pawel BR, Zhang C, Barr FG. High expression of the PAX3-FKHR oncoprotein is required to promote tumorigenesis of human myoblasts. Am J Pathol. 2009;175:2600–2608. doi: 10.2353/ajpath.2009.090192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuoppo C, Riess I, Schmitt-Ney M, Allegra P, Forni PE, Bersani F, Taulli R, Accornero P, Crepaldi T, Ponzetto C. The oncogenic transcription factor PAX3-FKHR can convert fibroblasts into contractile myotubes. Exp Cell Res. 2007;313:2308–2317. doi: 10.1016/j.yexcr.2007.02.037. [DOI] [PubMed] [Google Scholar]
- Xia SJ, Barr FG. Analysis of the transforming and growth suppressive activities of the PAX3-FKHR oncoprotein. Oncogene. 2004;23:6864–6871. doi: 10.1038/sj.onc.1207850. [DOI] [PubMed] [Google Scholar]
- Ren YX, Finckenstein FG, Abdueva DA, Shahbazian V, Chung B, Weinberg KI, Triche TJ, Shimada H, Anderson MJ. Mouse mesenchymal stem cells expressing PAX-FKHR form alveolar rhabdomyosarcomas by cooperating with secondary mutations. Cancer Res. 2008;68:6587–6597. doi: 10.1158/0008-5472.CAN-08-0859. [DOI] [PubMed] [Google Scholar]
- Roeb W, Boyer A, Cavenee WK, Arden KC. PAX3-FOXO1 controls expression of the p57Kip2 cell-cycle regulator through degradation of EGR1. Proc Natl Acad Sci USA. 2007;104:18085–18090. doi: 10.1073/pnas.0708910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressey JG, Pressey CS, Robinson G, Herring R, Wilson L, Kelly DR, Kim H. 2D-difference gel electrophoretic proteomic analysis of a cell culture model of alveolar rhabdomyosarcoma. J Proteome Res. 2011;10:624–636. doi: 10.1021/pr1008493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagutina I, Conway SJ, Sublett J, Grosveld GC. Pax3-FKHR knock-in mice show developmental aberrations but do not develop tumors. Mol Cell Biol. 2002;22:7204–7216. doi: 10.1128/MCB.22.20.7204-7216.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finckenstein FG, Davicioni E, Osborn KG, Cavenee WK, Arden KC, Anderson MJ. Transgenic mice expressing PAX3-FKHR have multiple defects in muscle development, including ectopic skeletal myogenesis in the developing neural tube. Transgenic Res. 2006;15:595–614. doi: 10.1007/s11248-006-9011-9. [DOI] [PubMed] [Google Scholar]
- Anderson MJ, Shelton GD, Cavenee WK, Arden KC. Embryonic expression of the tumor-associated PAX3-FKHR fusion protein interferes with the developmental functions of Pax3. Proc Natl Acad Sci USA. 2001;98:1589–1594. doi: 10.1073/pnas.98.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo RL, Allport JA, Olson EN. A Drosophila model of the rhabdomyosarcoma initiator PAX7-FKHR. Proc Natl Acad Sci USA. 2006;103:13439–13444. doi: 10.1073/pnas.0605926103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijo K, Hosoyama T, Bjornson CR, Schaffer BS, Prajapati SI, Bahadur AN, Hansen MS, Blandford MC, McCleish AT, Rubin BP. et al. Biomarker system for studying muscle, stem cells, and cancer in vivo. FASEB J. 2009;23:2681–2690. doi: 10.1096/fj.08-128116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Capecchi MR. New genetic tactics to model alveolar rhabdomyosarcoma in the mouse. Cancer Res. 2005;65:7530–7532. doi: 10.1158/0008-5472.CAN-05-0477. [DOI] [PubMed] [Google Scholar]
- Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ, Corcoran AE, Fraser P. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5:e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]