

Abstract
COPII vesicles transport proteins from the ER to the Golgi apparatus. Previous cryoEM structures of the COPII cage lacked the resolution necessary to determine the residues of Sec13 and Sec31 that mediate assembly and flexibility of the COPII cage. Here we present a 12Å-resolution structure of the COPII cage, where the tertiary structure of Sec13 and Sec31 is clearly identifiable. We employ this structure and a homology model of the Sec13-Sec31 complex to create a reliable pseudo-atomic model of the COPII cage. We combined this model with hydrogen/deuterium exchange mass spectrometry analysis to characterize four distinct contact regions at the vertices of the COPII cage. Furthermore, we found that the 2-fold symmetry of the Sec31 dimeric region of Sec13-31 is broken on cage formation, and that the resulting hinge is essential to form the proper edge geometry in COPII cages.
Introduction
The COPII proteins including Sec13-Sec31 (Sec13-31), Sec23-Sec24 (Sec23-24), and Sar1 are involved in transporting membrane proteins and other cargo from the endoplasmic reticulum (ER) to the Golgi apparatus 1. Sar1 plays a GTP-dependent regulatory role in the assembly and disassembly of COPII coats and has also been implicated in the process of vesicle fission 2. Sec23-24 is the cargo adaptor, in which Sec23 is the Sar1 GTPase-activating protein (GAP) and Sec24 has specific binding sites for exit sequences in the cargo molecules. Sec13-31 has an organizational role assembling the entire complex into a coherent bud through interactions with Sec23.
Human Sec13-31 has been shown to self-organize in vitro into a cage-like assembly. Cryogenic electron microscopy (cryoEM) studies of the Sec13-31 cages revealed the cage to form a cuboctahedral assembly, in which 24 Sec13-31 heterotetramers make up the edges of the cuboctahedron 3. When combined with crystal structures of fragments of the edges, models have been constructed of the COPII cage 3,4, revealing that the interactions that facilitate the assembly of the cage occur between adjacent WD40 β-propeller domains in Sec13 and Sec31. However, the resolution of the COPII cryoEM maps determined thus far has been limited to 20–40 Å, which is too low to identify the specific regions of Sec13 and Sec31 that interact with each other. 3–5
Recent studies have focused on identifying the effectors of COPII cage flexibility. Previously it was determined that though Sec13 is essential in yeast, mutations in three different membrane proteins, so called bypass-of-Sec13 (bst) mutations, could restore COPII function in cells with defective Sec136. Recently, Čopič et al. 7, demonstrated that in general, the bst mutations render the ER membrane more deformable. Furthermore they showed that one of the roles of Sec13 in the COPII cage is to impose rigidity that allows for the transport of cargos with opposing curvature. Another study recently showed that monoubiquitylation of particular residues of Sec31 drives the transport of large cargos such as procollagen 8. From these studies, it is clear that the COPII cage must be flexible enough to transport large cargos but rigid enough to impart curvature on the ER membrane.
Here we set out to determine the residues that mediate the assembly and flexibility of the COPII cage. We used cryoEM to determine the structure of the cage to 12 Å resolution by the FSC0.143 criterion. A homology model of human Sec13-31 was then flexibly fitted to the EM density to identify the residues that interact to mediate COPII cage assembly. These data were then substantiated by combining them with the results of hydrogen/deuterium exchange mass spectrometry (HDX-MS)Sec13-31. HDX-MS analysis identifies protein regions whose H/D exchange rates change in different functional states due to changes in conformation, flexibility, hydrogen bonding, and/or solvent exposure. HDX-MS measures the exchange behavior of peptide backbone amide hydrogens. For a given peptide, a folded state can have NH hydrogen exchange rates that are several orders of magnitude slower than those in an unfolded state 9. Another factor contributing to a peptide’s HDX profile is its solvent accessibility: the buried residues generally exchange hydrogens relatively slower than the surface residues. Thus, HDX-MS results directly reflect differences in solvent exposure by comparison of the same peptide segment from free and bound protein in a complex. The Sec13-31 complex studied here represents one of the largest complexes analyzed by HDX-MS to date. By combining cryoEM, modeling, and HDX-MS we were able to identify four unique contact regions at the cage vertices and a hinge region at the interface of Sec13 and Sec31, each of which is implicated in the assembly and flexibility of the COPII cage.
Results
We hypothesized that earlier Sec13-31 reconstructions were limited in resolution by a combination of a low density to volume ratio for the cage particles and conformational heterogeneity. To test this, we used cryotomography to characterize the heterogeneity of the cage sample. This revealed that the sample contained multiple intact and homogeneous cages and a significant amount of amorphous protein at the air/water interface (Supplementary Fig. 1a). We hypothesized that these were cages that had flattened and/or denatured at the air/water interface during vitrification. To overcome this problem, the COPII cages were centrifuged through a sucrose/glutaraldehyde gradient (GRAFIX) 10 that separated cages from unassembled and aggregated protein. The fraction containing pure cages was collected and prepared for cryoEM. The GRAPHIX procedure had a dramatic effect on the resolvable features of the cages, and complete cages were easily distinguishable (Supplementary Fig. 1b,c).
Given the improvement in the Sec13-31 cage preparation in vitreous ice, we collected a large dataset for single particle reconstruction. 5,052 high magnification images of the fixed cages were collected in vitreous ice, to yield 66,867 cage particles that were used in single particle reconstruction. The particles were classified with EMAN 11 and SPIDER 12 scripts using the previously published cuboctahedral Sec13-31 cage as an initial model. The refinement converged on a model at between 12 Å (FSC0.143) and 15 Å (FSC0.5) resolution, revealing new features of the COPII cage (Fig. 1a).
Figure 1. CryoEM reconstruction of the COPII cage.
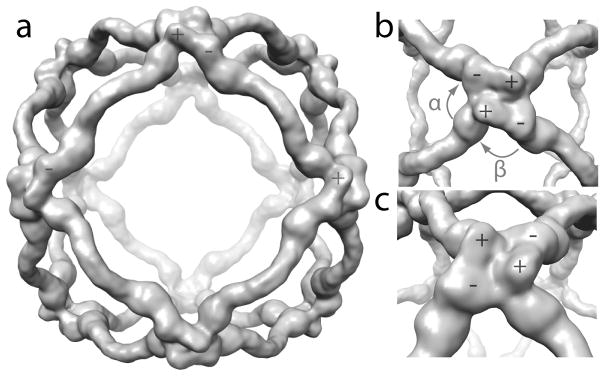
a) 12 Å resolution reconstruction of the Sec13-31 COPII cage. b) Close-up view of a vertex of the cage. The plus (+) and minus (−) ends of individual Sec13-31 heterotetramers are highlighted. The α and β angles between adjacent edges are shown. c) View of a vertex highlighting intermolecular interactions between adjacent Sec31 WD40 domains.
COPII cages are comprised of two Sec13s and two Sec31s in every edge of the cuboctahedron. The center of the Sec13-31 heterotetramer, however, is not at the center of the cuboctahedral edge; one of the Sec13 and Sec31 pairs is closer to its nearest vertex than is the other 3,13. We have dubbed the end of a given Sec13-31 edge that is closest to a vertex the plus end and the other end the minus end. The interactions at the vertices of the cages occur between the Sec31 WD40 domains at the plus and minus ends of adjacent Sec13-31 edges. Given the definitions of the ends, the angles between adjacent edges in a cuboctahedral COPII cage are named α and β. When a vertex is viewed from the outside of the cage, α is the 60° angle between the plus end of an edge and the minus end of the adjacent edge in the clockwise direction, and β is the 90° angle between a minus end and the plus end of the adjacent edge in the clockwise direction (Fig. 1b).
The most obvious improvement in resolution of the present reconstruction is in the Sec13 and Sec31 WD40 domains. The contours of the WD40 domains are clearly resolvable, and the boundaries between individual proteins can be delineated (Fig. 1c). Contacts between adjacent WD40 domains at the cage vertices are extensive, and the improved resolution of the reconstruction allowed us to create a pseudo-atomic model of the COPII cage to determine the specific regions of Sec13-31 that mediate assembly.
Model building
The first step in building a pseudo-atomic model of the cage was to create a homology model of the human Sec13-31 heterotetramer with the yeast Sec13-31 crystal structures as a template. The yeast proteins 4 were previously studied as two complexes, crystallized separately: Sec13 in complex with the Sec31 N-terminal WD40 domain, and Sec13 with the C-terminal α-solenoid domain/ACE1 motif of Sec31. The remaining 35% of Sec31 comprising its C terminus was not included. We combined Sec13-31the two structures (PDB IDs 2PM6 and 2PM9) by aligning them each relative to Sec13 to create a Sec13-31 heterotetramer. The yeast Sec13 and Sec31 sequences were then aligned with the human proteins. Given the low sequence identity between the yeast and human proteins, multi-sequence alignment was performed with 10 different Sec13 and Sec31 sequences to facilitate a more accurate alignment to the template sequence. MODELLER 14 was then used to generate an all-atom model of the human Sec13-31 edge. Several loop regions were unresolved in the yeast structure and were included in our model (Supplementary Fig. 2a).
The relative arrangements of Sec13 and Sec31 in our homology model agree with the cryoEM density, but the quaternary structure of the human Sec13-31 heterotetramer in the cage shows a greater amount of curvature. To obtain better agreement between the homology model and the EM density, the fit of the homology model was optimized by molecular dynamics flexible fitting (MDFF) 15. An edge was extracted from the COPII cage density map, to which the homology model was flexibly fitted (Supplementary Movie 1). The MDFF algorithm incorporates a term for the EM density in its force field and restraints that preserve secondary structure, thereby allowing the atomic structure of the homology model to be optimized to the density map in a physically realistic way. After MDFF, the homology model fitted well with most of the protein inside the EM density (Fig. 2a). The single edge model was then octahedrally symmetrized to generate a complete pseudo-atomic model of the COPII cage. Finally, MDFF was performed on the complete model within the complete EM map in order to relieve any steric clashes and optimize the structure of the vertices (Supplementary Movie 1, Supplementary Fig. 3). Although symmetry was not imposed during the simulation, symmetrically realigning the individual edges yielded a mean RMSD of 1.173 Å (Supplementary Fig. 2b,c).
Figure 2. Pseudo-atomic model of human Sec13-31.

a) Human homology model flexibly-fitted into a single edge of the cryoEM reconstruction of the Sec13-31 cage. The Sec13 chains are colored red and cyan and the Sec31 chains are colored yellow and blue. The α-solenoid domains of Sec31 are highlighted with a bracket. b) Superposition of the plus and minus end of an individual Sec13-31 heterotetramer. The chains were superimposed based on Sec13 (dark orange/blue) to highlight the conformational change in Sec31 (light orange/blue). c) Plus end of the fitted heterotetramer. d) Minus end of the fitted heterotetramer. The plus end α-solenoid domain is more tightly packed against Sec13 at the plus end in c) than it is at the minus end in d).
The yeast Sec13-31 crystal structure has 2-fold symmetry about the dimeric interface of Sec31 in the heterotetramer. Fitting the human Sec13-31 model to the EM density, however, required breaking the 2-fold symmetry. Superimposing the resulting Sec13s of an individual heterotetramer revealed a large conformational change at the interface between Sec13 and the α-solenoid region of Sec31 (Fig. 2b). A hinge between Sec13 and the Sec31 α-solenoid motif allows the domains to flex relative to each other. The hinge has an angular range of 17° approximately in the plane normal to the 4-fold axis of the cage. At the plus end, Sec13 is packed tightly against the Sec31 α-solenoid motif (Fig. 2c), whereas at the minus end, the interaction is less extensive (Fig. 2d). By comparison, the interface between Sec13 and the WD40 domain of Sec31 is similar between the two ends (Fig. 2b), indicating that the apposition of these two proteins changes minimally whether they are at a plus or a minus end.
Solvent accessibility
We used the pseudo-atomic model to predict the residues that contact each other at the cage vertices by calculating the solvent-accessible surface area (SASA) for an isolated edge and an edge assembled in a COPII cage. Candidate contacts were identified by determining the residues whose SASA changed between the assembled and isolated states (Fig. 3). Previous studies predicted four different contact surfaces between the four Sec31 WD40 domains that form the vertex and the four Sec13 WD40 domains (cI – cIV) 4,13. The SASA calculations predict changes at contact regions cI – cIV. As shown in Figure 3, cI is predicted to occur between the two plus end Sec31 WD40 domains. cII occurs between a plus end WD40 and the minus end WD40 of an adjacent Sec13-31 in the clockwise direction (clockwise adjacent) (Fig. 3). cIII occurs between a minus end WD40 and the plus end WD40 of a clockwise adjacent Sec31. Contact cIV was previously predicted to occur between a plus end Sec13 and a clockwise adjacent minus end Sec31 WD40. In our current model, however, those proteins are too distant to permit interaction. We observe that a single loop at a plus end of Sec31 contributes to the interaction with a clockwise adjacent minus end forming cII and also makes contact with the adjacent plus end. We redefine this contact as cIV.
Figure 3. Identification of the contact regions in the COPII cage.

Contact regions were predicted by calculating the solvent-accessible surface area in isolated Sec13-31 edges, and Sec13-31 in cages (colored). Unique contact regions are labeled cI-cIV.
Identification of regions that change upon assembly
The predicted contacts from the EM reconstruction and modeling were tested by hydrogen/deuterium exchange mass spectrometry to identify residues whose hydrogen exchangeability differed between assembled and disassembled states. HDX kinetics were initially characterized for free Sec13-31 edges. Edges were soaked in a deuterium solution for time periods ranging from 30 s to 14,400 s then exchange was quenched by 1% formic acid, the proteins digested by protease XIII, and the masses of the resulting peptides determined by Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) (Fig. 4). Due to the large size of the COPII cage, new methodological approaches had to be developed in order to resolve, identify, and monitor the masses of the large number of peptides for each sample. Several recent developments applied to HDX experiments coupled with high resolution FT-ICR MS16, include automation 17,18, faster chromatographic separation 19, more efficient protein digestion 20,21, and enhanced data analysis algorithms 22–25. Application of HDX-MS to higher molecular weight targets results in increased mass spectral complexity. Analysis of the Sec13-31 complex benefited from the use of a high peak capacity 14.5 T FT-ICR mass spectrometer 26. Our HDX-MS data was analyzed by a modified custom analysis package 24, with which peptides were identified by their accurate mass and LC retention time. The detailed HDX-MS profiles for individual peptides are presented in Supplementary Figure. 4a,b.
Figure 4. HDX heat map for Sec13 and Sec31 in free Sec13-31 heterotetramers.

The deuteration level (in percentage) is calculated by dividing the observed deuterium uptake by the theoretical maximum deuterium uptake for each peptide (e.g., a peptide consisting of n amino acids can take up a maximum of n−1 observable deuteriums if no prolines are present). For each peptide, the deuteration level is calculated for each of the experimental HDX incubation periods (left, proceeding from top to bottom in each segment: 30, 60, 120, 240, 480, 900, 1,800, 3,600, 7,200, 14,400 s incubation period) and mapped onto the sequence. Residues are colored white when there is no data available for that residue at any incubation period. The predicted secondary structure elements are also labeled above the Sec31 sequence.
HDX-MS analysis revealed regions of Sec13-31 that are relatively resistant to HDX and regions that exchange more rapidly. The C-terminal region of Sec31 (residues 753–1,166) is generally more exchangeable than the rest of the protein (Fig. 4). The sequence coverage is lower in that region (75%), particularly in the so-called proline-rich region (residues 762–996) likely due to prolines in that region hindering protease digestion. Among the detected peptides, residues 771–798 and 920–936 are relatively less available to HDX, indicating that they are buried or part of a secondary structure element. Those regions flank the region of Sec31 that is predicted to interact with Sec23. Yeast residues 907–942, which map to human residues 841–858, interact with Sec23 by binding to a groove on the membrane-distal surface of that protein 27. The slowly exchanging residues that flank that region may serve to stabilize the Sec23-interacting region during COPII coat assembly. For the rest of the C-terminal domain, the increased relative HDX rates suggest greater flexibility, in agreement with secondary structure predictions that show the region to be disordered. This result may explain the observation that the C-terminal region of Sec31 is not resolved in the cryoEM reconstructions of the cage, even though the specimens used in the reconstructions used full-length protein.
We mapped the relative deuterium uptake for the N-terminal part of Sec13-31 onto our homology model (Fig. 5). As anticipated, the residues that exchanged hydrogens the slowest are in general the most buried in the structure. The residues in the centers of the WD40 domains were particularly slow to exchange hydrogens. The α-solenoid domain exchanged hydrogens relatively quickly, suggesting that this region is relatively more conformationally variable than the rest of the protein.
Figure 5. HDX data mapped onto the Sec13-31 structure.

Identified fragments in the HDX-MS experiment are colored according to the deuterium uptake (lowest in blue and highest in red), shown for Sec13-31 soaked in D2O for 30 s, 240 s, 1,800 s, and 7,200 s before quenching. The α-solenoid region takes up deuterium relatively faster than do the WD40 domains, suggesting more flexibility than the WD40 domains.
Next, we repeated the HDX-MS experiment for assembled cages. The deuterium uptake kinetics for the cages and edges were then compared. The differential uptake was then mapped onto the Sec13-31 sequence and onto our flexibly fitted model (Fig. 6a,b, Supplementary Fig. 5). Differences in deuterium uptake were scattered all over the protein, suggesting extensive changes in conformation, flexibility, and/or solvent exposure upon assembly of Sec13-31 edges into COPII cages. We anticipated that most of the differences would be localized to the contact regions at the vertices. Indeed several peptides at the vertices that we predicted would change their exchangeability took up less deuterium in the assembled state (Fig. 6c). Peptides whose hydrogen exchangeability changed were analyzed with respect to the cage structure and were annotated based on the proposed reasons for the difference in exchangeability (Supplementary Table 1). From that analysis, it is clear that there are a few key regions for assembly. Interestingly, the same residues from different edges are involved in making multiple unique contacts. Residues in regions 202–214, 246–253, and 336–375 contact each other to form cII (when a plus end and a clockwise adjacent minus end come together) and cIV (at the interface of two plus ends and a minus end). Residues 202–214, also participate in cIII along with residues 143–145 and 162–170 at the contacts between a minus end and a clockwise adjacent plus end. Surprisingly, none of the residues predicted to form cI exhibited changes in their H/D exchangeability, however Sec31 residues 257–268 were not detected by FT-ICR MS. The lack of contacts at cI can be explained by the observation that residues that come closest to each other at cI in our model are D236 and D282. Charge/charge repulsion would disfavor the negatively charged aspartic acids from making contact. As a result, these residues at cI would not come close enough to observe changes in HDX rates.
Figure 6. Difference in deuterium uptake between Sec13-31 edge and cage.

The deuterium uptake difference for each peptide is calculated by the ARDD method 18. Briefly, for each H/D exchange period, the relative difference in D uptake between edge and cage is divided by the D uptake for edge. The relative difference for that segment averaged over all HDX incubation periods defines ARDD. a) In this figure and throughout the text, the percent relative difference in H/D exchange is positive when a fragment of the unassembled edge experiences more D uptake than the corresponding fragment in the assembled cage. Sec31 is depicted above, and Sec13 below. b) and c): Peptides that took up less deuterium in the assembled cage than as free-floating edges are shaded red according to the magnitude of their relative H/D exchange rate differences. b) A complete edge. c) An assembled vertex. The inset in c) illustrates the contours of the N-terminal Sec31 WD40 domains and identifies the locations of contacts cI-IV.
In addition to changes at the vertex peptides, there were extensive changes throughout the α-solenoid domain (Supplementary Table 1). In agreement with the fitting results in Fig. 2, there were changes in the hinge and α-helices at the interface between the α-solenoid domain and Sec13 (Fig. 6b). With sufficient signal-to-noise ratio, one would expect to see a bimodal distribution in the masses of H/D-exchanged segments in different environments at the plus and minus ends. Indeed, we have identified one peptide at residues 439–456 at the interface of the hinge region that exhibits a bimodal HDX distribution for the cage, but not for the edge, in agreement with the model for the hinge (Supplementary Fig. 6).
Changes in differential HDX were observed in the center of the α-solenoid domain near the Sec31 dimer interface. There are a few possible explanations for the changes observed in this region. It could be that there is a conformational change that occurs upon assembly. That possibility agrees with the observed difference between the yeast crystal structure, which is nearly linear in this region, and the EM structure which shows a large bend. Another possibility is that this region changes in flexibility upon assembly, as supported by the observation that the α-solenoid region has faster relative deuterium uptake in the unassembled state.
The C-terminal unstructured region of Sec31 appeared to take up less deuterium in the assembled state (Fig. 6a). That result likely indicates that the C-terminal region becomes less flexible when assembled into a cage, although it is also possible that some intermolecular contacts occur between heterotetramers on cage assembly.
Discussion
The present results suggest that a few key regions of COPII are important for assembly. Several loop regions on opposite sites of the Sec31 WD40 domain appear to contribute to multiple contacts depending on whether they are at the plus end or the minus end. Sec31 residues 343–372 appear to participate in contacts cII and cIV. These residues were not resolved in the Sec31 crystal structure, and we modeled them as an extended loop structure. They exchanged deuterium rapidly in the disassembled state, suggesting that they are quite flexible, and modeling suggests that they become more ordered during assembly. Moreover, these residues exchange less deuterium when cages are formed. The other peptides that form the cII and cIV contacts include residues 204–215 and 246–253. Interestingly, residues 204–215 participate in cII at the plus end of an edge and cIV at the minus end of an edge, reminiscent of quasi-equivalence in viruses for which the same residues make different contacts in a molecular assembly depending upon their particular structural environment. Moreover, our structure suggests that at the plus end, each edge in the cage makes contact with all three of its adjacent edges at the cage vertex; a given plus end Sec31 WD40 domain participates in contacts cI with the opposite plus end, cII with the clockwise minus end, cIII with the counterclockwise minus end, and cIV with the clockwise minus end and the opposite plus end, likely lending stability to the otherwise flexible COPII cage structures.
The bend at the center of Sec13-31 appears to be quite variable, and this variability in bend angle appears to be a characteristic of the ancestral coatamer element (ACE1) motif comprising the N-terminal 2/3 of Sec31. ACE1 motifs are found in several of the proteins involved in trafficking and forming the nuclear pore complex 28 and often mediate homo-dimerization via the so called crown region where dimerization occurs via a domain swap. For instance, Sec16 contains an ACE1 domain and forms a dimer through interactions at the crown region. Like Sec31, Sec16 binds Sec13 via a β-propeller insertion into the WD40 domain of Sec13 29. The structure of the Sec13-16 complex is remarkably similar to Sec13-31; it dimerizes via a domain swap at the crown region of the ACE1 motif and also forms a bend at the center of the Sec13-16 heterotetramer. However, Sec13-16 forms a 90° angle at the dimeric interface whereas Sec13-31 in the yeast crystal structure forms a 165° angle, and human Sec13-31 in the cage structure forms a 135° angle and a 145° angle in the presence of Sec23 30. The flexibility suggested by these structures is corroborated by our HDX data on free Sec13-31 in which the α-solenoid region exchanges hydrogens relatively more rapidly than the N-terminal domains (Fig. 5). The flexibility is also supported by early EM studies of Sec13-31 that showed the protein to be highly conformationally variable 31.
It has been shown that the COPII cage can form a variety of geometries depending on the conditions of the cage assembly 13,30,32. Two key angles were identified; α that occurs between a plus end and a clockwise adjacent minus end, and β that occurs between a minus end and a clockwise adjacent plus end. α is always 60° in the cage structures determined so far, whereas β varies between 90° and 108° 13. It was previously predicted that contacts cII and cIV mediate formation of the α angle, and the generation of cages of different diameters is facilitated by changing the β angle by flexing about cI and cIII. A paradox set up by this prediction is that cI and cIII are required to change dramatically to allow changes in β, which would not be likely if cI and cIII involved extensive contacts. Our results with HDX show no contacts at cI and relatively few contacts at cIII. The relative sparsity of contacts at this region likely allows the cage to flex to form different geometries in response to the needs of cargo.
Recent studies indicate that monoubiquitylation of Sec31 in vivo leads to the formation of large COPII coated vesicles that allow for the secretion of large cargos such as procollagen 8. These studies showed that CUL3–KLHL12 preferentially monoubiquitylates mouse Sec31 residues Lys 647 or Lys 1217 (human residues 608 and 1,155), located in the α-solenoid domain and C terminal unstructured region. Based on our results, it is unlikely that ubiquitylation at those residues plays a structural role in promoting the formation of larger assemblies. Instead, it is likely that ubiquitylation recruits additional factors or somehow delays vesiculation allowing the formation of larger COPII coats as Jin et al suggested 8.
Our structure demonstrates that cage assembly is accompanied by a large conformational change at the interface between Sec13 and the Sec31 α-solenoid domain. A hinge between Sec13 and Sec 31 was predicted by by Čopič et al. 7 who showed that yeast Sec13 knockouts were not able to transport cargos that were likely to confer opposing curvature to the COPII coats. They concluded that Sec13 serves to rigidify the COPII cage. We extend their analysis by proposing a structural rationale for their observations. We propose that the function of the Sec13-31 hinge is to allow the Sec31 WD40 domains at the plus and minus ends of an edge to assume different conformations, allowing for the structural convergence of the plus and minus ends at the vertices, apparently required for cage formation. Sec13, then, plays a key role in rigidifying the hinge by providing the structural integrity needed to bud off cargo from the ER while maintaining the flexibility required to form the structurally differentiated plus and minus ends of Sec13-31 edges that we observe in the COPII cage.
An increase in the resolution of our COPII cage reconstruction over previous reconstructions has allowed for the generation of a reliable pseudo-atomic model of the Sec13-31 heterotetramer in the COPII cage. This result, combined with HDX-MS, has allowed us to identify residues contributing to four distinct contact regions at the vertices of the COPII cage. Furthermore, our investigation shows that the 2-fold symmetry of Sec13-31 is broken during assembly, and this observation helps explain the structural role of Sec13 in cage assembly. Sec13 contributes to a hinge that is an essential feature of Sec13-31 in the formation of edges into COPII cages. The confluence of techniques used in this investigation exemplifies the viability of our approach, which may prove to be applicable to the determination of more complex interactions such as the Sec13-31 – Sec23-24 complex. Our results demonstrate the power of combining cryoEM, modeling, and HDX-MS to derive conclusions that would not be attainable from the individual techniques alone.
Online Methods
Expression and purification
Human Sec13R, and Sec31L1 (GenBank accession NM_183352 and AAI43493.1 respectively) were expressed in Sf9 and Hi5 insect cells by use of baculovirus and purified as described 3. An expression construct encoding Sec13R and Sec31L1 genes was kindly provided by the Laboratory of William Balch. Multiple chromatographic steps were used in the purification process. The first step was purification with immobilized metal affinity chromatography (IMAC) by use of an N-terminal hexa-histidine tag located on Sec13R. This was followed by anion exchange, size exclusion chromatography, and dialysis of the Sec13-31-rich pool in low salt buffer (20 mM Tris–Cl, pH 7.5, 300 mM NaCl, 1 mM MgOAc, 10 mM dithiothreitol (DTT)) against assembly buffer (20 mM Tris–Cl, pH 7.5, 700 mM KOAc, 1 mM MgOAc, 10 mM DTT) overnight.
GRAFIX
After dialysis against assembly buffer, Sec13-31 cages were purified from unassembled Sec13-31 heterotetramers by Gradient Fixation, GRAFIX 10, to obtain a more homogenous sample. A dual gradient was prepared with 5–45% sucrose together with 0–0.015% glutaraldehyde all in assembly buffer. The gradient was prepared by adding 25% glutaraldehyde to a 45% sucrose solution to obtain a final glutaraldehyde concentration of 0.015%. 5.75 mL of this sucrose-glutaraldehyde solution was added to the bottom of an ultraclear thin-walled centrifuge tube. An equal volume of 5% sucrose solution with 0% glutaraldehyde was carefully layered on top, and the gradient was prepared by tilted tube rotation 33. Sample containing Sec13-31 was layered on top and spun at 28,000 × g for 4 hours in a SW41 swinging bucket rotor. Gradient fractions that corresponded to purified Sec13-31 cages were pooled and dialyzed against assembly buffer to remove sucrose and glutaraldehyde. Sample was then dialyzed into a low salt buffer solution buffer (20 mM Tris–Cl, pH 7.5, 125 mM KOAc, 1 mM MgOAc, 10 mM DTT) overnight and prepared for cryoEM.
CryoEM
Vitrified, thin-layered samples were prepared by adding 3 μL of sample onto Quantifoil R2/1 grids (Quantifoil Micro Tools) that had been plasma cleaned for 8 s with a Gatan Solarus plasma cleaner (Gatan, Pleasanton,CA). An FEI Vitrobot (FEI Company) was used to freeze plunge sample into liquid ethane. Samples were transferred to liquid nitrogen for long term storage. Data were collected automatically with Leginon 34 on an FEI Titan Krios at 120 keV at a magnification of 37,000X with an under-focus of −1.5 – 3.5 μm and an electron dose of 15 Å−2. Images were recorded on a CCD camera with effective pixel size of 1.52 Å.
Image processing
Images were processed with the Appion software package35. Particles were picked by template matching, which resulted in 81,334 particles. CTF was estimated by use of ACE as implemented in Appion 36. Phases were corrected for individual particles based on the defocus reported from ACE for the particular micrograph from which the particle was picked. After making a stack, the stack was hand-edited to remove 14,467 particles that were poorly picked. Particles were initially reconstructed by means of EMAN refinement 11, combined with multivariate statistical analysis of each class of projections between iterations of refinement. Correspondence analysis with hierarchical ascendant classification was performed generating 2–5 subclasses for each projection. Only the subclass that correlated best with the projection of the model for that iteration was to contribute to the reconstruction for a given iteration. The resolution of the final reconstruction was 12 Å as measured by the FSC0,143 criterion and 15 Å by the FSC0.5 criterion. A total of 23,404 particles went into the final reconstruction.
The pixel size for the final reconstruction was matched to previously published COPII cage by use of xmipp_align_volumes 37 script to find the pixel size that produced the maximum cross correlation value between the final 3D reconstruction and the published COPII cage. The cross correlation value between the two density maps was found to be at a maximum at a pixel size of 2.78 Å/pixel.
Homology modeling
The homology model for the human Sec13-31 heterotetramer was created with MODELLER 14 with the yeast Sec13-31 crystal structures (PDB IDs 2PM6 and 2PM9) as templates. A complete heterotetramer model was created by superposing the Sec13 that was common to both models and joining the Sec31 molecules to create a continuous Sec13-31 heterotetramer. Human Sec31 has only 21% identity to yeast Sec31, so multi-sequence alignment was performed with Sec13 and Sec31 A and B sequences from Homo sapiens (AAI43493.1, CAH73561.1), Drosophila melanogaster (NP_724716.1, NP_610414.1), Giardia lamblia (EFO62997.1), Xenopus laevis (NP_001085250.1), Arabidopsis thaliana (NP_191905.3), Bos taurus (DAA28510.1, DAA14888.1), and Saccharomyces cerevisiae (CAA98772.1) to facilitate a more accurate alignment to the template sequence. Clustal Omega was used for the alignment 38. The aligned sequences were passed to MODELLER for structure building. Five candidate structures were constructed that differed mostly in the loop regions. The top scoring structure was then optimized to the cryoEM density by means of flexible fitting.
Flexible Fitting
The flexible fitting processes were performed by use of Molecular Dynamics Flexible Fitting (MDFF) 15 as described in Supplementary Note 1.
H/D exchange
The entire HDX experiment was optimized and automated with an HTC Pal autosampler (Eksigent Technologies, Dublin, CA). In contrast to the EM sample, the HDX samples were prepared without any fixation or sucrose gradient separation. A 5 μL stock of Sec13-31 edge or cage (~10 μM in monomer) was mixed with 45 μL of HEPES buffer in D2O, pH meter reading 7.5, to initiate each H/D exchange period. The HDX incubation periods were 0, 0.5, 1, 2, 4, 8, 15, 30, 60, 120 and 240 min, each performed in triplicate. Each HDX reaction was quenched by rapid mixing with 25 μL of 200 mM TCEP, 6 M urea solution in 1.0% formic acid and 25 μL 5-fold dilution of saturated protease type XIII in 1.0% formic acid. The final pH was ~2.3 and the protease digestion was performed for 3 min at ~1° C. The digested sample was then injected for LC-MS analysis.
Online LC–ESI FTICR MS
After proteolysis, the peptides were separated and desalted with a Jasco high-performance liquid chromatography/supercitical fluid chromatography (HPLC/SFC) instrument (Jasco, Easton, MD) interfaced with the HTC Pal autosampler (Eksigent Technologies, Dublin, CA). For LC, 45 μL of the protein digest was injected from a 50 μL loop to a Pro-Zap Expedite MS C18 column (Grace Davidson, Deerfield, IL), HR 1.5 μm particle size, 500 Å pore size, 2.1 × 10 mm2. A rapid gradient from 2% B to 95% B in 2 min (A, acetonitrile/H2O/formic acid, 5/94.5/0.5; B, acetonitrile/H2O/formic acid, 95/4.5/0.5) was performed for eluting peptides at a flow rate of 300 μL/min. The LC eluent flow rate is reduced by ~1/1000 by a post-column splitter for efficient electrospray ionization.
The ionized LC eluent was directed to a custom-built hybrid LTQ 14.5 T FTICR mass spectrometer (ThermoFisher, San Jose, CA) 26. Mass spectra were collected from 380 < m/z < 1300 at high mass resolving power (m/Δm50% = 100, 000 at m/z 400, in which Δm50% is the mass spectral peak full width at half-maximum peak height). External ion accumulation was performed in the linear ion trap with a target ion population of three million charges collected for each FTICR measurement. LTQ-accumulated ions were transferred (1 ms transfer period) 39 through three octopole ion guides (2.2 MHz, 250 Vp–p) to a capacitively coupled 40 closed cylindrical ICR cell (55 mm i.d.) for analysis. The ion accumulation period was typically less than 100 ms during peptide elution, and the ICR time-domain signal acquisition period was 767 ms (i.e., an overall duty cycle of 1 Hz per acquisition). Automatic gain control41 and high magnetic field 42 provided excellent external calibration mass accuracy (normally less than 500 ppb root mean square mass error).
Supplementary Material
Acknowledgments
This work was supported by the NIH through R01GM086892, the AHA through #0835300N, the NSF Division of Materials Research through DMR-06-54118 and the State of Florida. We thank Dr. Christopher Hendrickson and Dr. Feng Xian for helpful discussions.
Footnotes
Accession codes
The Sec13-31 EM density map has been deposited in the Electron Microscopy Data Bank, with accession code EMD-5524.
References
- 1.Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R. COPII and the regulation of protein sorting in mammals. Nat Cell Biol. 2011;14:20–28. doi: 10.1038/ncb2390. [DOI] [PubMed] [Google Scholar]
- 2.Lee MC, Orci L, Hamamoto S, Futai E, et al. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122:605–617. doi: 10.1016/j.cell.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 3.Stagg SM, Gurkan C, Fowler DM, LaPointe P, et al. Structure of the Sec13/31 COPII coat cage. Nature. 2006;439:234–238. doi: 10.1038/nature04339. [DOI] [PubMed] [Google Scholar]
- 4.Fath S, Mancias JD, Bi X, Goldberg J. Structure and organization of coat proteins in the COPII cage. Cell. 2007;129:1325–1336. doi: 10.1016/j.cell.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 5.Bacia K, Futai E, Prinz S, Meister A, et al. Multibudded tubules formed by COPII on artificial liposomes. Sci Rep. 2011;1:17. doi: 10.1038/srep00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elrod-Erickson MJ, Kaiser CA. Genes that control the fidelity of endoplasmic reticulum to Golgi transport identified as suppressors of vesicle budding mutations. Mol Biol Cell. 1996;7:1043–1058. doi: 10.1091/mbc.7.7.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Copic A, Latham CF, Horlbeck MA, D’Arcangelo JG, Miller EA. ER Cargo Properties Specify a Requirement for COPII Coat Rigidity Mediated by Sec13p. Science. 2012 doi: 10.1126/science.1215909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin L, Pahuja KB, Wickliffe KE, Gorur A, et al. Ubiquitin-dependent regulation of COPII coat size and function. Nature. 2012;482:495–500. doi: 10.1038/nature10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engen JR. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal Chem. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kastner B, Fischer N, Golas MM, Sander B, et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat Methods. 2008;5:53–55. doi: 10.1038/nmeth1139. [DOI] [PubMed] [Google Scholar]
- 11.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 12.Frank J, Radermacher M, Penczek P, Zhu J, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 13.Stagg SM, LaPointe P, Razvi A, Gurkan C, et al. Structural basis for cargo regulation of COPII coat assembly. Cell. 2008;134:474–484. doi: 10.1016/j.cell.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;Chapter 2(Unit 2.9) doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 15.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marshall AG, Hendrickson CL, Jackson GS. Fourier transform ion cyclotron resonance mass spectrometry: a primer. Mass Spectrom Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 17.Chalmers MJ, Busby SA, Pascal BD, He Y, et al. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2006;78:1005–1014. doi: 10.1021/ac051294f. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Q, Willison LN, Tripathi P, Sathe SK, et al. Epitope mapping of a 95 kDa antigen in complex with antibody by solution-phase amide backbone hydrogen/deuterium exchange monitored by Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 2011;83:7129–7136. doi: 10.1021/ac201501z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang HM, Bou-Assaf GM, Emmett MR, Marshall AG. Fast reversed-phase liquid chromatography to reduce back exchange and increase throughput in H/D exchange monitored by FT-ICR mass spectrometry. J Am Soc Mass Spectrom. 2009;20:520–524. doi: 10.1016/j.jasms.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cravello L, Lascoux D, Forest E. Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Communications in Mass Spectrometry. 2003;17:2387–2393. doi: 10.1002/rcm.1207. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Settembre E, Xu C, Dormitzer PR, et al. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc Natl Acad Sci U S A. 2008;105:1867–1872. doi: 10.1073/pnas.0711623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weis D, Engen J, Kass I. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. Journal of The American Society for Mass Spectrometry. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 23.Pascal BD, Chalmers MJ, Busby SA, Griffin PR. HD Desktop: An Integrated Platform for the Analysis and Visualization of H/D Exchange Data. Journal of The American Society for Mass Spectrometry. 2009;20:601–610. doi: 10.1016/j.jasms.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kazazic S, Zhang HM, Schaub TM, Emmett MR, et al. Automated data reduction for hydrogen/deuterium exchange experiments, enabled by high-resolution Fourier transform ion cyclotron resonance mass spectrometry. J Am Soc Mass Spectrom. 2010;21:550–558. doi: 10.1016/j.jasms.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z, Zhang A, Xiao G. Improved Protein Hydrogen/Deuterium Exchange Mass Spectrometry Platform with Fully Automated Data Processing. Analytical Chemistry. 2012;84:4942–4949. doi: 10.1021/ac300535r. [DOI] [PubMed] [Google Scholar]
- 26.Schaub TM, Hendrickson CL, Horning S, Quinn JP, et al. High-performance mass spectrometry: Fourier transform ion cyclotron resonance at 14.5 Tesla. Anal Chem. 2008;80:3985–3990. doi: 10.1021/ac800386h. [DOI] [PubMed] [Google Scholar]
- 27.Bi X, Mancias JD, Goldberg J. Insights into COPII coat nucleation from the structure of Sec23. Sar1 complexed with the active fragment of Sec31. Dev Cell. 2007;13:635–645. doi: 10.1016/j.devcel.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brohawn SG, Leksa NC, Spear ED, Rajashankar KR, Schwartz TU. Structural evidence for common ancestry of the nuclear pore complex and vesicle coats. Science. 2008;322:1369–1373. doi: 10.1126/science.1165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whittle JRR, Schwartz TU. Structure of the Sec13-Sec16 edge element, a template for assembly of the COPII vesicle coat. J Cell Biol. 2010;190:347–361. doi: 10.1083/jcb.201003092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhattacharya N, O Donnell J, Stagg SM. The Structure of the Sec13/31 COPII Cage Bound to Sec23. J Mol Biol. 2012;420:324–334. doi: 10.1016/j.jmb.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lederkremer GZ, Cheng Y, Petre BM, Vogan E, et al. Structure of the Sec23p/24p and Sec13p/31p complexes of COPII. Proc Natl Acad Sci U S A. 2001;98:10704–10709. doi: 10.1073/pnas.191359398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Donnell J, Maddox K, Stagg S. The structure of a COPII tubule. J Struct Biol. 2011;173:358–364. doi: 10.1016/j.jsb.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 33.Coombs DH, Watts NR. Generating sucrose gradients in three minutes by tilted tube rotation. Anal Biochem. 1985;148:254–259. doi: 10.1016/0003-2697(85)90654-2. [DOI] [PubMed] [Google Scholar]
- 34.Suloway C, Pulokas J, Fellmann D, Cheng A, et al. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 35.Lander GC, Stagg SM, Voss NR, Cheng A, et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mallick SP, Carragher B, Potter CS, Kriegman DJ. ACE: automated CTF estimation. Ultramicroscopy. 2005;104:8–29. doi: 10.1016/j.ultramic.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 37.Scheres SHW, Núñez-Ramírez R, Sorzano COS, Carazo JM, Marabini R. Image processing for electron microscopy single-particle analysis using XMIPP. Nat Protoc. 2008;3:977–990. doi: 10.1038/nprot.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sievers F, Wilm A, Dineen D, Gibson TJ, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilcox B, Hendrickson C, Marshall A. Improved ion extraction from a linear octopole ion trap: SIMION analysis and experimental demonstration. Journal of The American Society for Mass Spectrometry. 2002;13:1304–1312. doi: 10.1016/S1044-0305(02)00622-0. [DOI] [PubMed] [Google Scholar]
- 40.Beu SC, Laude DA. Elimination of axial ejection during excitation with a capacitively coupled open trapped-ion cell for Fourier transform ion cyclotron resonance mass spectrometry. Analytical Chemistry. 1992;64:177–180. [Google Scholar]
- 41.Schwartz J, Senko M, Syka J. A two-dimensional quadrupole ion trap mass spectrometer. Journal of The American Society for Mass Spectrometry. 2002;13:659–669. doi: 10.1016/S1044-0305(02)00384-7. [DOI] [PubMed] [Google Scholar]
- 42.Marshall AG, Guan S. Advantages of High Magnetic Field for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Rapid Communications in Mass Spectrometry. 1996;10:1819–1823. doi: 10.1002/(SICI)1097-0231(199611)10:14<1829::AID-RCM697>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.