

Abstract
The cytoplasmic Ca2+ clearance rate affects neuronal excitability, plasticity, and synaptic transmission. Here, we examined the modulation of the plasma membrane Ca2+ ATPase (PMCA) by tyrosine kinases. In rat sensory neurons grown in culture, the PMCA was under tonic inhibition by a member of the Src family of tyrosine kinases (SFKs). Ca2+ clearance accelerated in the presence of selective tyrosine kinase inhibitors. Tonic inhibition of the PMCA was attenuated in cells expressing a dominant-negative construct or shRNA directed to message for the SFKs Lck or Fyn, but not Src. SFKs did not appear to phosphorylate the PMCA directly but instead activated focal adhesion kinase (FAK). Expression of constitutively active FAK enhanced and dominant-negative or shRNA knockdown of FAK attenuated tonic inhibition. Antisense knockdown of PMCA isoform 4 removed tonic inhibition of Ca2+ clearance, indicating that FAK acts on PMCA4. The hyaluronan receptor CD44 activates SFK-FAK signaling cascades and is expressed in sensory neurons. Treating neurons with a CD44-blocking antibody or short hyaluronan oligosaccharides, which are produced during injury and displace macromolecular hyaluronan from CD44, attenuated tonic PMCA inhibition. Ca2+-activated K+ channels mediate a slow afterhyperpolarization in sensory neurons that was inhibited by tyrosine kinase inhibitors and enhanced by knockdown of PMCA4. Thus, we describe a novel kinase cascade in sensory neurons that enables the extracellular matrix to alter Ca2+ signals by modulating PMCA-mediated Ca2+ clearance. This signaling pathway may influence the excitability of sensory neurons following injury.
Introduction
The plasma membrane Ca2+ ATPase (PMCA) is the predominant mechanism for removing small Ca2+ loads from the cytoplasm of neurons (Werth et al., 1996). It hydrolyzes ATP to drive the exchange of intracellular Ca2+ for extracellular H+ (Di Leva et al., 2008). PMCA-mediated Ca2+ clearance regulates many Ca2+-dependent processes in neurons, including excitability (Usachev et al., 2002), plasticity (Simons et al., 2009), and neurotransmitter release (Jensen et al., 2007). Transcripts of the four PMCA genes can be alternatively spliced to create ∼30 different isoforms (Strehler and Zacharias, 2001) that are heterogeneously expressed throughout the nervous system (Filoteo et al., 1997; Burette et al., 2003). The function of the various PMCA isoforms is differentially affected by protein kinases C and A (Verma et al., 1999; Guerini et al., 2003), proteases (Pászty et al., 2002; Guerini et al., 2003), and Ca2+ calmodulin (Caride et al., 2001; Pottorf and Thayer, 2002). Thus, multiple signaling pathways converge on PMCAs to alter neuronal Ca2+ signaling.
No studies describe protein tyrosine kinase (PTK) modulation of PMCAs in neurons, although there is evidence suggesting a potential role for PTKs in the regulation of pump function in other cell types. Antigen cross-linking of the B cell receptor produces an increase in intracellular Ca2+ concentration ([Ca2+]i) that is inhibited following PMCA activation by the tyrosine phosphatase SHP-1 (Chen et al., 2004). PMCA isoform 4 is phosphorylated during platelet activation, probably by focal adhesion kinase (FAK) (Wan et al., 2003), resulting in slowed Ca2+ clearance (Bozulic et al., 2007).
CD44 is an adhesion molecule expressed on the surface of most vertebrate cells, including sensory neurons (Ikeda et al., 1996), where it functions as a receptor for extracellular matrix (ECM) components, including the following: hyaluronan (HA), collagen, laminin, fibronectin, and osteopontin (Goodison et al., 1999). CD44 plays a major role in cell adhesion and migration, in part through its activation of the Src family kinases (SFKs) Lck and Fyn (Ilangumaran et al., 1999). SFKs form complexes with and activate FAK to regulate processes ranging from development to death (Grant et al., 1995; Girault et al., 1999; Zhao and Guan, 2009). The PTK cascades activated by ECM receptors exert many effects on neurons, including changes in [Ca2+]i (Ditlevsen et al., 2007), but a role for the PMCA in this process has not been previously described.
Here we tested the hypothesis that PTKs regulate Ca2+ clearance in sensory neurons. Our results indicate that a PTK cascade regulated by CD44 is present in neurons and that it modulates PMCA-mediated Ca2+ clearance. These data suggest a novel mechanism by which changes in ECM can shape the amplitude, duration, and location of [Ca2+]i signals.
Materials and Methods
Materials.
Indo-1 acetoxymethyl ester (AM), Pluronic F-127, Ham's F12 media, and sera were purchased from Invitrogen. AG18 [tyrphostin A23; α-cyano-(3,4-dihydroxy)cinnamonitrile], pyrazolopyrimidine 2 (PP2), PP3, and 2,3-dihydro-N,N-dimethyl-2-oxo-3-[(4,5,6,7-tetrahydro-1H-indol-2-yl)methylene]-1H-indole-5-sulfonamide (SU6656) were purchased from Calbiochem-Novabiochem. Rat monoclonal antibody IM7 to CD44 was obtained from PharMingen/BD Biosciences. 4-(4′-Phenoxyanilino)-6,7-dimethoxyquinazoline (Src kinase inhibitor-1; SKI-1) and all other reagents were purchased from Sigma.
Short HA oligosaccharides (sHA-oligos) were prepared from high-molecular-weight HA by incubating HA (bovine vitreous humor) for 16 h at 37°C with testicular hyaluronidase (type 1-S) at a ratio of 320 U/mg HA in 0.1 m Na acetate buffer pH 5. This method was shown previously to yield sHA-oligos primarily composed of tetrasaccharides, hexasaccharides, and octasaccharides (Iacob and Knudson, 2006).
Cell culture.
Rat dorsal root ganglion (DRG) neurons were grown in culture as previously described (Werth et al., 1996). In brief, 1- to 3-d-old Sprague Dawley rats of both sexes were killed under a protocol approved by the University of Minnesota Institutional Animal Care and Use Committee. Ganglia were dissected from the thoracic and lumbar segments and incubated at 37°C in collagenase-dispase (0.8 and 6.4 U/ml, respectively) for 25 min. DRG neurons were dissociated by trituration through a flame-constricted pipette and then plated onto laminin-coated (50 μg/ml) glass coverslips (25 mm diameter). Cells were grown in Ham's F12 media supplemented with 5% heat-inactivated horse serum and 5% fetal bovine serum, 50 ng/ml NGF, 4.4 mm glucose, 2 mm l-glutamine, modified Eagle's medium vitamins, and penicillin–streptomycin (100 U/ml and 100 μg/ml, respectively). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2. Neurons with cell body diameters of 12–20 μm were used within 3–4 d of plating.
[Ca2+]i measurements.
Instrumentation for [Ca2+]i recording from single DRG neurons using indo-1 was similar to that previously described (Werth et al., 1996). Cells were placed in a flow-through chamber that was mounted on the stage of an inverted epi-fluorescence microscope equipped with a 70× objective (Leitz, NA = 1.15). Cells were loaded with 2 μm indo-1 AM in HEPES-buffered Hank's salt solution (HHSS) containing 0.04% Pluronic F-127 at 37°C for 30 min. HHSS had the following composition (in mm): 10 HEPES, 140 NaCl, 5 KCl, 1.3 CaCl2, 0.4 MgSO4, 0.5 MgCl2, 0.4 KH2PO4, 0.6 Na2HPO4, 3 NaHCO3, and 10 glucose, pH 7.35 with NaOH, 310 mOsm/kg with sucrose. The cells were washed for 30 min in dye-free HHSS at 37°C before initiating recording. Indo-1 was excited at 350 nm (10 nm bandpass) and emission detected at 405 (20) and 495 (20) nm. Fluorescence was monitored by a pair of photomultiplier tubes (Thorn, EMI) operating in photon-counting mode. Changes in the fluorescence of indo-1 were converted to [Ca2+]i by using the formula [Ca2+]i = Kdβ(R − Rmin)/(Rmax − R), where R is 405/495 nm fluorescent intensity ratio (Grynkiewicz et al., 1985). The dissociation constant used for indo-1 was 250 nm, and β was the ratio of fluorescence emitted at 495 nm and measured in the absence and presence of Ca2+. Rmin, Rmax, and β were determined in intact cells by applying 10 μm ionomycin in Ca2+-free buffer (1 mm EGTA) and saturating Ca2+ (5 mm Ca2+). Values for Rmin, Rmax, and β were 1.30, 10.87, and 3.16, respectively. To evoke action potentials in intact neurons, extracellular field stimulation was used (Piser et al., 1994). At 4 min intervals, a 4 s train of voltage pulses (0.1 ms) was applied across platinum electrodes at a rate of 5–8 Hz using a Grass S44 stimulator with a SIU-5 stimulus isolation unit (Astro-Med).
Perforated patch-clamp recordings.
Current-clamp recordings were performed using the whole-cell configuration of the perforated-patch technique. Membrane potential was amplified using an Axopatch 200B, filtered at 2 kHz, and digitized at 11 kHz with a Digidata interface controlled by pClamp software (Molecular Devices). Pipettes (Narishige) with open resistances of 2–4 MΩ were filled with solution that contained the following: 135 mm K-gluconate, 10 mm NaCl, 10 mm HEPES, and 200 μg/ml amphotericin B, pH 7.25 with KOH, 290 mOsm/kg. Recordings were performed at room temperature (22°C) in an extracellular solution that contained the following (in mm): 140 NaCl, 5 KCl, 1.3 CaCl2, 0.4 MgSO4, 0.5 MgCl2, 0.4 KH2PO4, 0.6 Na2HPO4, 3 NaHCO3, 10 HEPES, and 10 glucose, pH 7.35 with NaOH, and 310 mOsm/kg with sucrose. Solutions were applied by a gravity-fed superfusion system. Membrane potential was held at −65 mV and action potentials evoked by current injection (4 s train, 10–50 Hz; 0.1 ms pulse).
Transfection and DNA constructs.
Gene transfer into DRG neurons was performed using a biolistic particle delivery system as previously described (Usachev et al., 2000). All constructs were cotransfected with an expression plasmid for GFP (pEGFP-C1, Clontech). After 48 h, transfected cells were identified by green fluorescence [excitation = 480 (10) nm, emission = 540 (25) nm]. All constructs were propagated in Escherichia coli DH5α strain (Invitrogen), isolated using Maxiprep kits (Qiagen), and sequenced.
Modulation of kinase function was accomplished using constitutively active and dominant-negative approaches. A constitutively active Lck construct (CA-Lck) with the Y505F mutation in vector pSM (Turner et al., 1990) was kindly provided by Dan Littman (New York University Skirball Institute, New York, NY). A dominant-negative Lck construct (DN-Lck) containing the K273R mutation in vector pcDNA3 (Yousefi et al., 2003) was kindly provided by Donald Branch (University of Toronto, Toronto, ON). Dominant-negative Fyn (DN-Fyn) with the K299M mutation and dominant-negative Src (DN-Src) with the K295M mutation in pRK5 (Mariotti et al., 2001) were kindly provided by Filippo Giancotti (Memorial Sloan Kettering Cancer Center, New York, NY; Addgene plasmids 16033 and 19965). A constitutively active FAK construct (CA-FAK) in which FAK was fused in frame to the membrane-targeting region of the IL-2 receptor and a dominant-negative mutant (DN-FAK; Y397F) (Tamura et al., 1999) were kindly provided by Kenneth Yamada (National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD). An expression vector (pcDNA3.1/myc) harboring T-cell SH2 Adapter (TSAd) (Richard et al., 2006) was kindly provided by Mark Nachtigal (Dalhousie University, Halifax, NS).
To knock down PMCA4, cells were transfected with a mammalian expression plasmid (pCI-neo) harboring cDNA encoding nucleotides 71–443 of PMCA4 in the antisense orientation (Garcia et al., 2001). Effective knockdown of PMCA4 was confirmed by immunohistochemistry with PMCA4-specific antibody JA9 as previously described (Usachev et al., 2002).
Knockdown of tyrosine kinases was accomplished using shRNA expression vectors obtained from Open Biosystems/Thermo Fisher Scientific. shRNA in the pLKO.1 vector were cotransfected with a GFP expression plasmid (pEGFP-C1, Clontech) to identify transfected cells; the pGIPZ vector incorporates turbo GFP to track transfected cells. Cells were transfected with nonsilencing shRNA as a negative control (NS-pGIPZ). Effective knockdown was accomplished by transfecting with 2 or 3 shRNA constructs for Lck (pLKO.1 vector; sense sequences #1 GCTGCAAGACAACCTGGTTAT, #2 CCCTTCAACTTCGTGGCGAAA, #3 GAGAACATTGACGTGTGTGAA), Fyn (pGIPZ vector; sense sequences #1 CTGTGTGCAATGTAAGGAT, #2 GACAGAAGATGACCTGAGT, #3 GAGACCATGTCAAACATTA), Src (pGIPZ vector: sense sequences #1 GGCCTAAATGTGAAACACT, #2 TAAGGTCATGCAGGGACTC), or FAK (pGIPZ vector; sense sequences #1 TACTTGATGTTATTGATCA, #2 GACCTGGCATCTTTGATAT, #3 AGTTTGGAGATGTACATCA).
In DRG neurons expressing shRNA to Src message, neither PMCA-mediated [Ca2+]i clearance kinetics nor sensitivity to tyrosine kinase inhibitors was affected; thus, we confirmed that the constructs were effective. Because of the low efficiency of DRG neuron transfection using the gene gun, effective RNA knockdown was validated using the rat C6 glioma cell line. C6 cells were obtained from the ATCC and grown in culture as recommended. Cells were transfected with shRNA constructs and message knockdown determined by quantitative real-time reverse transcription-PCR (Q-RT-PCR). C6 cells were transfected with shRNA expression plasmids using the Amaxa Nucleofector System (Lonza) with Nucleofector Solution V per the manufacturer's protocol for this cell line (transfection efficiency ranged from 44 to 64%). After 15 h, RNA was extracted using an RNA isolation kit (Zymo Research) and Q-RT-PCR performed on 100 ng of isolated RNA using a SYBR Green Q-RT-PCR kit (Stratagene). Primers designed against Src (sense-GACAGTGGCGGATTCTACATC, antisense-ACGGTAGTGAGACGGTGAC) were purchased from Primerdesign. In addition to using equal amounts of total RNA as defined by UV spectroscopy, we also included an internal reference control (GAPDH) to account for the integrity of mRNA. QuantiTect primers (Qiagen) were used for amplification of GAPDH mRNA. PCR cycling and detection was performed on an Mx3005P PCR System and quantitative analysis performed with MxPro-Mx3005P Software v4.01 (Stratagene). The shRNA cocktail knocked down Src mRNA by 57 ± 9% (n = 3).
Data analysis.
All data are presented as mean ± SE. Significance was determined using Student's t test, ANOVA with Bonferroni post hoc test for multiple comparisons, or repeated-measures ANOVA with Bonferroni post hoc test for comparing time course data (Prism 5.0, GraphPad Software). Exponential functions were fitted to [Ca2+]i recovery traces using a nonlinear, least-squares curve-fitting algorithm (Origin 8.1 software). Experiments in which the correlation (r2) of the fitted curve was <0.8 were excluded from analysis.
Results
The extrusion of Ca2+ from the neuronal cytoplasm by PMCAs is modulated by kinases, proteases, and the duration and amplitude of [Ca2+]i increases. The effects of PTKs on the plasma membrane Ca2+ pump in neurons have not previously been investigated. To examine PMCA modulation by PTKs, small (300–400 nm) increases in [Ca2+]i were evoked by brief trains of action potentials (4 s, 5–8 Hz) in rat DRG neurons in primary culture and the rate of PMCA-mediated return to basal [Ca2+]i was quantified using indo-1-based microfluorimetry (Fig. 1A). [Ca2+]i recovered by a process well described by a single exponential (Fig. 1B). We used the rate constant (k = 1/τ) derived from fitting exponential curves to these traces as an index of PMCA function. Repeated stimuli evoked [Ca2+]i transients with reproducible recovery kinetics (Fig. 1A,C, untreated). In some cells, a slight decline in peak [Ca2+]i was compensated by increasing the frequency of the stimulus train. We have shown previously that Ca2+ recovery kinetics from these small [Ca2+]i increases are predominantly determined by PMCAs (Usachev et al., 2002). Neither mitochondrial Ca2+ uptake nor Na+/Ca2+ exchange contributed significantly to the rate of recovery to basal levels in response to this stimulus, presumably because [Ca2+]i does not reach sufficiently high levels to recruit these low-affinity, high-capacity Ca2+ clearance mechanisms (Thayer et al., 2002). The sarcoplasmic and endoplasmic reticulum Ca2+ ATPase (SERCA) will participate in clearing these small Ca2+ loads (Usachev et al., 2006), and thus, all experiments were performed in the presence of 10 μm cyclopiazonic acid to block SERCA function.
Figure 1.

Broad-spectrum PTK inhibitor AG18 accelerates Ca2+ clearance. A, Representative traces show action potential-induced [Ca2+]i transients recorded using indo-1-based microfluorimetry as described in Materials and Methods. Extracellular field stimulation (4 s, 5 Hz) was applied at the times indicated (↑). The recording was performed in the presence of 10 μm cyclopiazonic acid. One hundred micromolar AG18 was applied by superfusion at the time indicated by the horizontal bar. B, The recovery phases of the responses indicated in A (↓) were normalized to peak and fitted with an exponential curve (heavy line). C, Plot summarizes changes in [Ca2+]i recovery kinetics (normalized k) for untreated cells (open circles, n = 5) and cells treated with AGI8 (solid circles, n = 5) at the time indicated by the horizontal bar. Data are mean ± SE. **p < 0.01, ***p < 0.001 relative to untreated, repeated-measures ANOVA with Bonferroni post hoc test.
PTKs inhibit PMCA-mediated Ca2+ clearance in sensory neurons
To determine whether PTKs affected PMCA function, we applied the broad spectrum tyrosine kinase inhibitor AG18 (100 μm) and monitored changes in [Ca2+]i recovery kinetics. AG18 inhibits tyrosine kinases in the 10–100 μm range; for example, it inhibits the epidermal growth factor receptor with an IC50 of 35 μm (Levitzki and Gazit, 1995). After eliciting three control responses, drug was applied by superfusion (Fig. 1A). AG18 produced a time-dependent increase in recovery rate (Fig. 1A–C) elevating k from 3.9 ± 0.6 min−1 to 5.1 ± 0.7 min−1 (p < 0.05 ANOVA repeated measures). Thus, the PMCA appears to be under tonic inhibition by a PTK-dependent process.
To identify the kinase involved, we tested selective PTK inhibitors (Fig. 2). PP2 inhibits SFKs with IC50 values in the nanomolar range in vitro, but required a concentration of 10 μm to block Lck- and Fyn-mediated tyrosine phosphorylation in intact cells (Hanke et al., 1996). PP2 (10 μm) significantly accelerated PMCA-mediated [Ca2+]i recovery. The increase in k was not significantly different from that produced by the broad-spectrum kinase inhibitor AG18. This effect is consistent with the participation of an SFK, because the inactive analog PP3 did not affect recovery kinetics (Fig. 2A,B). SU6656 inhibits a subset of SFKs including Src, but failed to affect recovery kinetics. SU6656 is a 25-fold more potent inhibitor of Src (IC50 in vitro = 0.28 μm) than of Lck (Blake et al., 2000). In contrast, SKI-1, which inhibits Lck (IC50 = 0.011 μm) and Src (IC50 = 0.012 μm) with similar potency, was effective (Bain et al., 2007). These pharmacological experiments strongly suggested a SFK modulated PMCA function.
Figure 2.

Selective PTK inhibitors suggest that a Src family kinase inhibits PMCA in DRG neurons. A, Plot summarizes changes in [Ca2+]i recovery kinetics (normalized k) for cells treated with the Src family tyrosine kinase inhibitor PP2 (10 μm; open circles, n = 5) or its inactive analog PP3 (10 μm; solid squares, n = 5). Drug was present during the time indicated by the horizontal bar. Data are mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001 relative to PP3, repeated-measures ANOVA with Bonferroni post hoc test. B, Bar graph summarizes percentage change in recovery rate for untreated cells (control) (from Fig. 1C), or cells treated with 100 μm AG18 (AG18) (from Fig. 1A–C) (n = 5), 10 μm PP2 (n = 5), 10 μm PP3 (n = 5), 10 μm SU6656 (n = 5), or 10 μm SKI-1 (n = 4). Percentage change in recovery rate = 100[(kdrug − kcontrol)/kcontrol], where kcontrol is the mean of the three k values determined before drug application (t = 0, 4, and 8 min) and kdrug is the mean of the last three k values following drug application (t = 24, 28, and 32 min). Initial rate constants (k) for the cells before their respective treatments were 4.1 ± 0.5, 3.9 ± 0.6, 4.1 ± 0.2, 4.4 ± 0.3, 6.3 ± 0.7, and 6 ± 1 min−1. **p < 0.01, ***p < 0.001 relative to control, ANOVA with Bonferroni post hoc test.
Lck and Fyn inhibit PMCA
Based on our findings using PTK inhibitors, we used complementary genetic approaches to modulate the function or expression of specific SFKs. Inhibition of PMCA activity by Lck would be consistent with the pharmacological results. Even though Lck is best known for its effects in lymphocytes, it is expressed throughout the nervous system (Omri et al., 1996). Expression of a dominant-negative Lck construct reduced tonic inhibition of PMCA function, as indicated by reduced sensitivity to AG18 and an elevated initial recovery rate before the application of AG18, compared to control neurons (Fig. 3A,F). In GFP-expressing control neurons, AG18 accelerated Ca2+ clearance by 32 ± 3% (Fig. 3C,F), whereas in cells expressing DN-Lck, AG18 increased k by only 5 ± 2% (Fig. 3A,F). Before AG18 application, the initial [Ca2+]i clearance rate in cells expressing DN-Lck was 5.4 ± 0.2 min−1, a significant increase above the initial k (3.1 ± 0.3 min−1) in control cells (p < 0.05). In a complementary experiment in which constitutively active Lck was expressed, the [Ca2+]i clearance rate before addition of AG18 was 2.6 ± 0.3 min−1, and tonic inhibition, as indicated by sensitivity to AG18, increased to 54 ± 9% (Fig. 3A,F). We also used a knockdown approach. DRG neurons were transfected with three Lck shRNA expression vectors as described in Materials and Methods and a GFP expression plasmid to identify transfected neurons. Cells expressing Lck-shRNA no longer responded to AG18 (Fig. 3B,G), in contrast to cells expressing GFP (Fig. 3C,F), empty vector (pLKO) (Fig. 3B,G), or nonsilencing shRNA (NS-pGIPZ) (Fig. 3E,G), in which AG18 evoked responses comparable to control. In T cells, Lck binds to the adapter protein TSAd. Overexpression of TSAd binds free Lck to act in a dominant-negative manner (Sundvold-Gjerstad et al., 2005). Expression of TSAd in DRG neurons attenuated tonic PMCA inhibition as indicated by a reduction in sensitivity to AG18 (13 ± 2%) (Fig. 3C,F). In T-cells Lck forms a membrane proximal signaling complex with the SFK Fyn (Salmond et al., 2009). We found that dominant-negative Fyn (Fig. 3D) or knockdown of Fyn mRNA (Fig. 3E) attenuated tonic inhibition of Ca2+ clearance (Fig. 3F,G). The AG18-induced change in k was reduced to 4 ± 2% in DN-Fyn-expressing cells. The change in k was reduced to only 4 ± 3% in cells expressing Fyn-shRNA, which was significantly different from the 35 ± 4% (p < 0.001) inhibition observed in cells expressing NS-pGIPZ. Our pharmacological experiments suggested that Src does not regulate PMCA-mediated Ca2+ clearance in DRG neurons (Fig. 2B). Consistent with these results, neither expression of dominant-negative Src (Fig. 3D) nor knockdown of Src mRNA (Fig. 3E) affected AG18-sensitive Ca2+ clearance kinetics (Fig. 3F,G). AG18 increased the Ca2+ clearance rate by 32 ± 4% in DN-Src-expressing cells and 39 ± 3% in Src-shRNA-expressing cells, which was comparable to their respective controls (GFP and NS-pGIPZ). The Src-shRNA was confirmed effective in rat C6 glioma cells as described in Materials and Methods. Thus, two SFK kinases, Lck and Fyn, appear to be required for tonic inhibition of PMCA-mediated Ca2+ clearance. However, no precedent suggests that these kinases might directly phosphorylate the PMCA.
Figure 3.
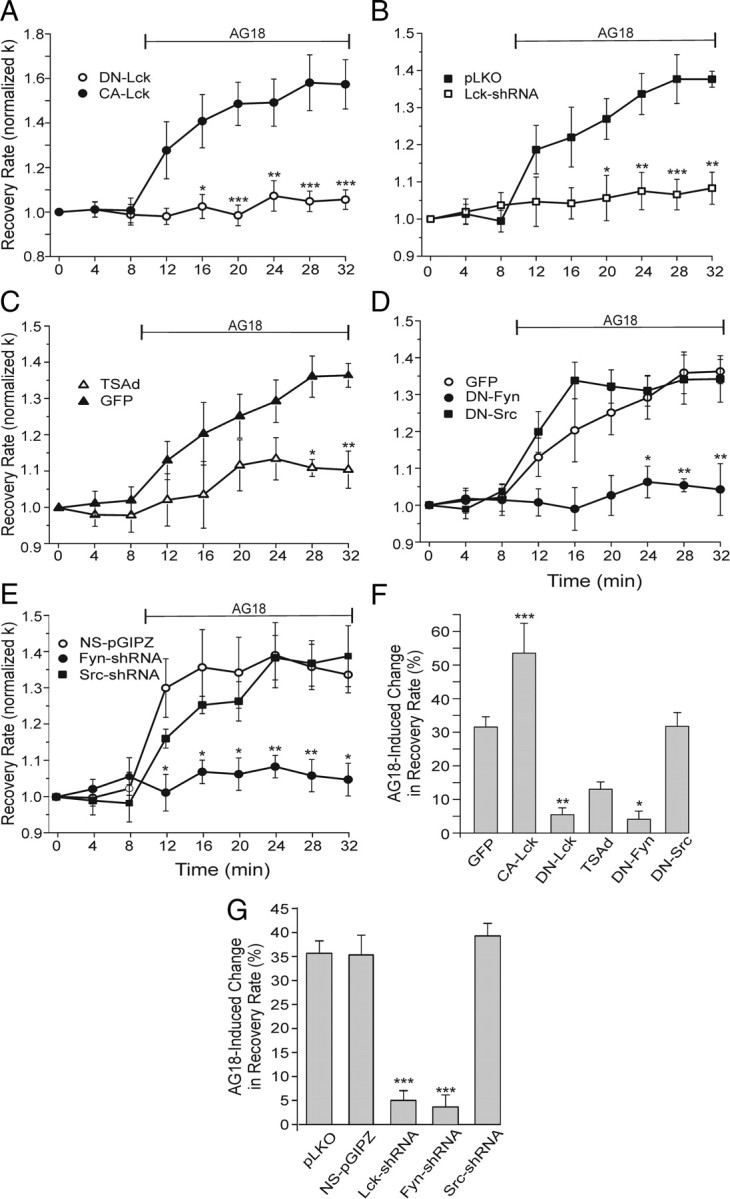
Lck and Fyn activity slow Ca2+ pump kinetics. A–E, Plots summarize changes in [Ca2+]i recovery kinetics (normalized k) for cells expressing the indicated constructs. AG18 (100 μm) was added at the time indicated by the horizontal bar. Significance was determined using repeated-measures ANOVA with Bonferroni post hoc test. A, Plot summarizes normalized k values for cells expressing DN-Lck (open circles; n = 5) or CA-Lck (closed circles; n = 7). *p < 0.05, **p < 0.01, ***p < 0.001 relative to CA-Lck. B, Plot of Ca2+ clearance rate (k) for control (pLKO empty vector; n = 4) and Lck-shRNA cells (n = 8). Before AG18 application, k values were 4.8 ± 0.6 and 4.5 ± 0.6 min−1, respectively. *p < 0.05, **p < 0.01, ***p < 0.001 relative to pLKO. C, Plot of Ca2+ clearance rate (k) for control (n = 7) and Tsad-expressing (n = 6) cells. Before AG18 application, k values were 3.1 ± 0.3 and 4.4 ± 0.4 min−1, respectively. *p < 0.05, **p < 0.01 relative to GFP. D, Plot of Ca2+ clearance rate (k) for cells expressing GFP (open circles; n = 7), DN-Fyn (solid circles; n = 3), or DN-Src (solid squares; n = 4). Before AG18 application, k values were 3.1 ± 0.3, 2.4 ± 0.5, and 3.1 ± 0.7 min−1, respectively. *p < 0.05, **p < 0.01 relative to GFP. E, Plot of Ca2+ clearance rate (k) for cells expressing nonsilencing shRNA (NS-pGIPZ; open circles; n = 6), Fyn-shRNA (solid circles n = 4), or Src-shRNA (solid squares; n = 5). Before AG18 application, k values were 3.6 ± 0.9, 3.6 ± 0.9, and 3.4 ± 0.6 min−1, respectively. *p < 0.05, **p < 0.01 relative to NS-pGIPZ. F, G, Bar graphs summarize the percentage change in Ca2+ clearance kinetics (k) induced by 100 μm AG18 in DRG neurons expressing GFP (n = 7), CA-Lck (n = 7), DN-Lck (n = 5), TSAd (n = 6), DN-Fyn (n = 3), DN-Src (n = 4), pLKO (n = 4), NS-pGIPZ (n = 6), Lck-shRNA (n = 8), Fyn-shRNA (n = 4), or Src-shRNA (n = 5). Percentage change in recovery rate = 100[(kAG18 − kcontrol)/kcontrol)], where kcontrol is the mean of the three rate constants determined before drug application (t = 0, 4, and 8 min) and kAG18 is the mean of the last three rate constants following application of AG18 (t = 24, 28, and 32 min).*p < 0.05, **p < 0.01, ***p < 0.001 relative to GFP (F) or relative to NS-pGIPZ (G), ANOVA with Bonferroni post hoc test.
FAK is required for inhibition of PMCA function
FAK directly phosphorylates the carboxyl tail of PMCA in platelets (Wan et al., 2003). Furthermore FAK is a known target of SFKs, including activation by Lck–Fyn complexes (Cabodi et al., 2010). Thus, we used the same dominant-negative and shRNA approach used to study the role of SFKs in PMCA regulation to determine whether FAK also participated in the regulation of Ca2+ clearance in DRG neurons. We found that dominant-negative FAK attenuated and constitutively active FAK increased tonic inhibition of Ca2+ clearance (Fig. 4A,C). The AG18-induced change in k was reduced to 9 ± 5% in DN-FAK-expressing cells and increased considerably to 95 ± 12% in CA-FAK-expressing cells. Expressing DN-FAK increased Ca2+ clearance rate before AG18 application from 3.3 ± 0.7 min−1 in control (pIL2R empty vector) cells to 4.8 ± 0.7 min−1. In cells expressing CA-FAK, k was significantly lower (1.8 ± 0.3 min−1) than in cells expressing DN-FAK (p < 0.01). Similar to the DN-FAK experiments, cells expressing shRNA targeted to FAK message displayed reduced tonic inhibition. The AG18-induced change in k was reduced to only 5 ± 2% in cells expressing FAK shRNA (Fig. 4B,C). The AG18-induced change in k in cells expressing nonsilencing shRNA (NS-pGIPZ; 35 ± 4%) was comparable to naive cells.
Figure 4.
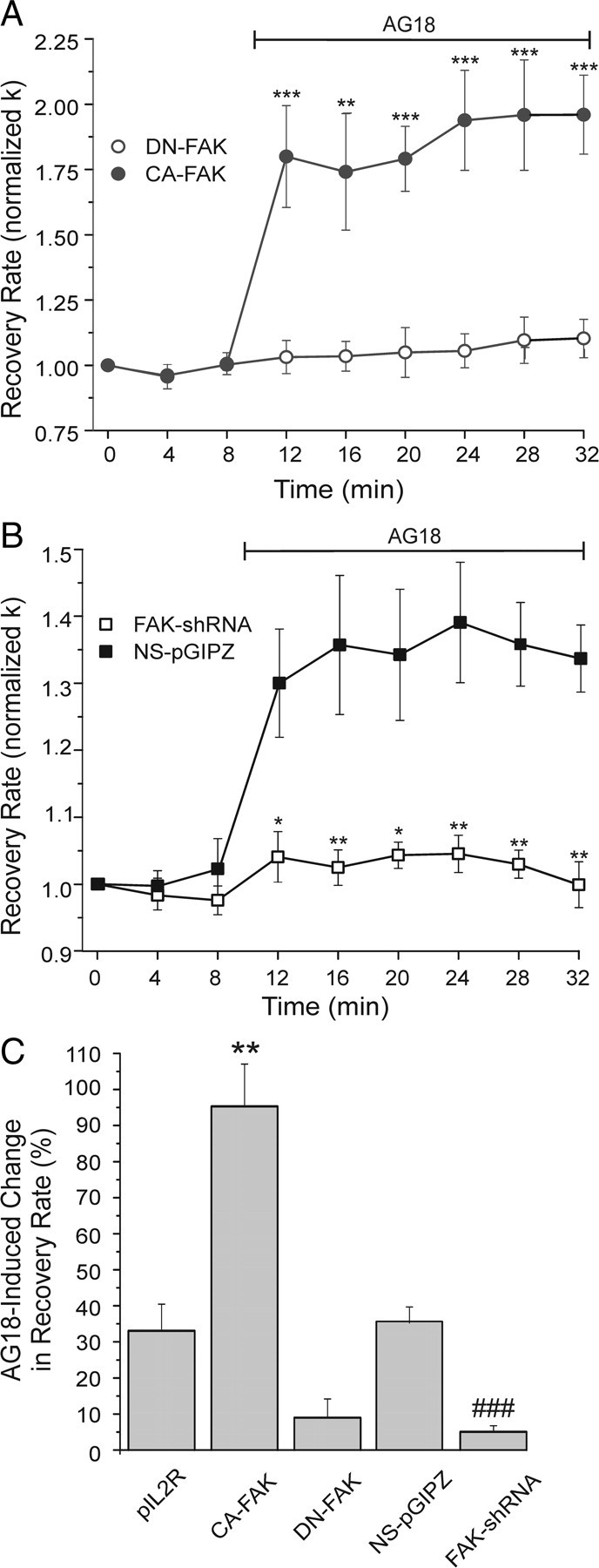
FAK inhibits Ca2+ pump kinetics. A, Plot summarizes [Ca2+]i recovery kinetics (normalized k) for cells expressing DN-FAK (open circles; n = 5) or CA-FAK (closed circles; n = 6). AG18 (100 μm) was added at the time indicated by the horizontal bar. **p < 0.01, ***p < 0.001 relative to DN-FAK, repeated-measures ANOVA with Bonferroni post hoc test. B, Plot of Ca2+ clearance rate (normalized k) for cells expressing nonsilencing shRNA (NS-pGIPZ; solid squares; n = 6) or FAK shRNA (open squares; n = 4). Before AG18 application, k values were 3.6 ± 0.9 and 3.9 ± 0.8 min−1, respectively. *p < 0.05, **p < 0.01 relative to NS-pGIPZ, repeated-measures ANOVA with Bonferroni post hoc test. C, Bar graph summarizes the percentage change in Ca2+ clearance kinetics (k) induced by 100 μm AG18 in DRG neurons expressing pIL2R (n = 4), CA-FAK (n = 6), DN-FAK (n = 5), NS-pGIPZ (n = 6), or FAK-shRNA (n = 4). Percentage change in recovery rate = 100[(kAG18 − kcontrol)/kcontrol)], where kcontrol is the mean of the three rate constants determined before drug application (t = 0, 4, and 8 min) and kAG18 is the mean of the last three rate constants following application of AG18 (t = 24, 28, and 32 min).**p < 0.01 relative to pIL2R, ANOVA with Bonferroni post hoc test. ###p < 0.001 relative to NS-pGIPZ, Student's t test.
PTKs modulate PMCA isoform 4
We next determined the PMCA isoform modulated by kinases in DRG neurons. Because tyrosine kinases inhibited PMCA4 in platelets, we focused on isoform 4. We have shown previously that expression of an antisense construct to PMCA4 (AS-PMCA4) knocks down PMCA4 immunoreactivity and inhibits PMCA-mediated Ca2+ clearance (Usachev et al., 2002). In cells expressing AS-PMCA4, tonic inhibition of Ca2+ clearance was blocked (p < 0.05), as indicated by a complete loss of AG18-induced increase in k (Fig. 5A,B). Similarly, the enhanced tonic inhibition in cells expressing CA-Lck was blocked (p < 0.05) in cells coexpressing AS-PMCA4 (Fig. 5B).
Figure 5.
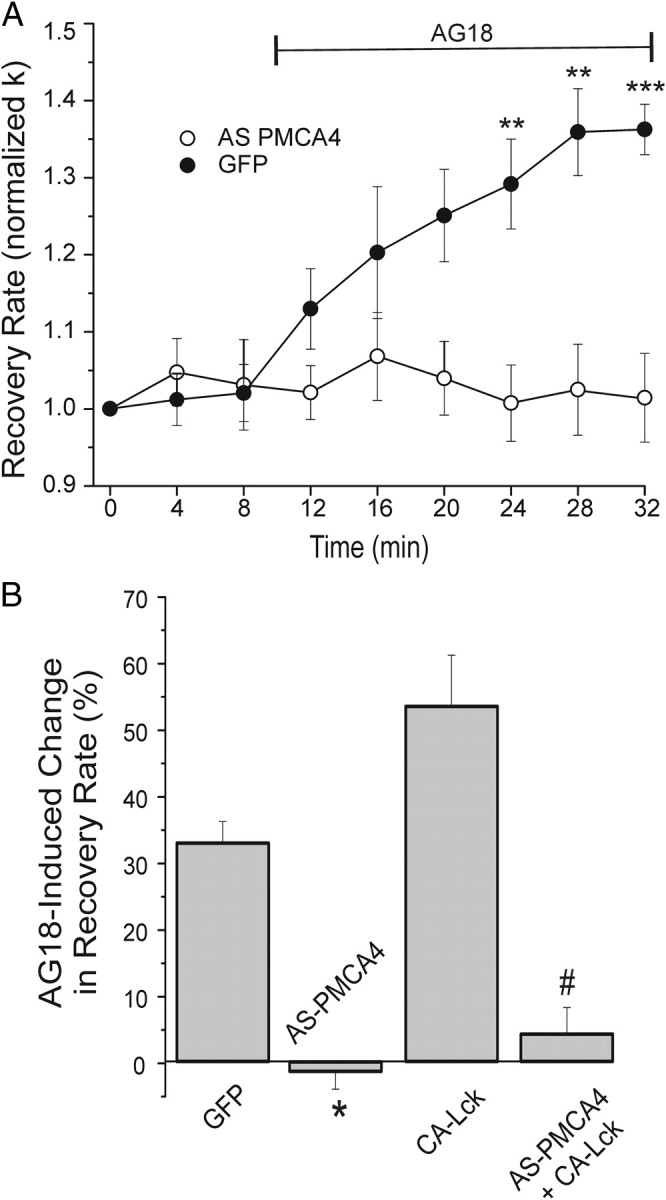
Knockdown of PMCA4 prevents PTK modulation of Ca2+ clearance. A, Plot summarizes changes in [Ca2+]i recovery kinetics (normalized k) for cells expressing AS-PMCA4 (open circles; n = 7) or GFP (closed circles; n = 6). AG18 (100 μm) was added at the time indicated by the horizontal bar. Data plotted are mean ± SE. **p < 0.01, ***p < 0.001 relative to GFP, repeated-measures ANOVA with Bonferroni post hoc test. B, Bar graph summarizes the percentage change in Ca2+ clearance kinetics (k) induced by 100 μm AG18 in DRG neurons expressing GFP (n = 7), AS-PMCA4 (n = 6), CA-Lck (n = 7), or AS-PMCA4 + CA-Lck (n = 5). Before AG18 application, k values were 3.1 ± 0.3, 3.8 ± 0.5, 2.6 ± 0.3, and 3.5 ± 0.5 min−1, respectively. Percentage change in recovery rate = 100[(kAG18 − kcontrol)/kcontrol)], where kcontrol is the mean of the three rate constants determined before drug application (t = 0, 4, and 8 min) and kAG18 is the mean of the last three rate constants following application of AG18 (t = 24, 28, and 32 min). *p < 0.05 relative to GFP; #p < 0.05 relative to CA-Lck, Student's t test.
Hyaluronic acid receptor modulates PTK-dependent inhibition of PMCA4
CD44 is a cell surface receptor for ECM components such as hyaluronic acid that is expressed in virtually all cell types, including neurons (Glezer et al., 2009). Because CD44 activates Lck in T lymphocytes (Taher et al., 1996), we examined the possibility that CD44 was activating the PTK cascade responsible for tonic inhibition of PMCA4 in DRG neurons. Treatment with a CD44-blocking antibody for 24 h completely blocked tonic inhibition of Ca2+ clearance; AG18 had no effect in antibody-treated cells (p < 0.05) (Fig. 6A,B). HA is a high-molecular-weight polymer composed of disaccharide repeats that is a component of the ECM. Following tissue injury and in certain disease states, HA is broken down into short oligosaccharides (Jiang et al., 2007). We treated high-molecular-weight HA with hyaluronidase to produce a mixture of short tetrasaccharides, hexasaccharides, and octasaccharides (sHA-oligos). These sHA-oligos have been previously shown to act as antagonists at CD44 (Hua et al., 1993; Lesley et al., 2000). Acute treatment of DRG neurons with sHA-oligos (40 μg/ml) did not affect the [Ca2+]i clearance rate (n = 3). However, treatment with sHA-oligos for 24 h attenuated tonic inhibition of PMCA-mediated Ca2+ clearance (p < 0.05) (Fig. 6A,B). In contrast, 24 h treatment with high-molecular-weight HA had no effect on PMCA function (Fig. 6A,B).
Figure 6.

CD44 activates PTK-dependent inhibition of PMCA4. A, Plot summarizes changes in [Ca2+]i recovery kinetics (normalized k) for cells treated for 24 h with 5 μg/ml CD44-blocking antibody (open circles; n = 5), 40 μg/ml sHA-oligos (filled squares; n = 3), or 40 μg/ml full-length HA (filled triangles; n = 3). Before AG18 application, k values were 8 ± 2, 2.9 ± 0.7, and 5 ± 1 min−1, respectively. AG18 (100 μm) was added at the time indicated by the horizontal bar. Data are mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001 relative to full-length HA, repeated-measures ANOVA with Bonferroni post hoc test. B, Bar graph summarizes the percentage change in Ca2+ clearance kinetics (k) induced by 100 μm AG18 in DRG neurons treated for 24 h with CD44-blocking antibody (CD44 Ab; n = 5), high-molecular-weight HA (full-length HA; n = 3), and sHA-oligos (n = 3). Percentage change in recovery rate = 100[(kAG18 − kcontrol)/kcontrol)], where kcontrol is the mean of the three rate constants determined before drug application (t = 0, 4, and 8 min) and kAG18 is the mean of the last three rate constants following application of AG18 (t = 24, 28, and 32 min). *p < 0.05 relative to full-length HA, ANOVA with Bonferroni post hoc test.
PTKs modulate afterhyperpolarization by altering Ca2+ clearance rate
Altered Ca2+ clearance might affect the duration and amplitude of transient increases in [Ca2+]i to modulate the function of Ca2+-dependent processes. We examined one such process. Sensory neurons exhibit a pronounced slow afterhyperpolarization (AHP) following trains of action potentials. The slow AHP is mediated by Ca2+-activated K+ channels and thus would be expected to be reduced in amplitude in cells in which PMCA-mediated Ca2+ clearance was stimulated. We used the perforated-patch technique to record the AHP in DRG neurons held in current clamp. Trains of action potentials were evoked by current injection. Naive cells and cells expressing GFP were stimulated with a 50 Hz train (4 s train) of depolarizing stimuli (0.1 ms pulses) to evoke a burst of action potentials. The stimulus frequency was increased relative to that used to evoke [Ca2+]i increases for studying Ca2+ clearance kinetics to increase the amplitude of the AHP and, thus, enable a more accurate measurement. The amplitude of the AHP at the end of the burst was measured before and after application of the SFK inhibitor PP2. PP2 reduced the amplitude of the AHP to 62 ± 5 and 74 ± 4% of control, for naive and GFP-expressing cells, respectively (Fig. 7A). In cells expressing AS-PMCA4, the 50 Hz stimulus produced a greatly increased AHP, possibly resulting from excess Ca2+ accumulation due to reduced PMCA-mediated Ca2+ clearance. To evoke AHPs of comparable amplitude to those observed in control cells, the stimulus frequency was decreased to either 10 or 20 Hz, which produced AHPs with amplitudes that were slightly smaller or larger than control. PP2 had no affect in cells expressing AS-PMCA4, regardless of stimulus strength (Fig. 7A,B). These results are consistent with the idea that SFKs modulate the AHP indirectly by tonic inhibition of PMCA4-mediated Ca2+ clearance.
Figure 7.

PTKs alter Ca2+-activated K channels by regulating Ca2+ clearance. DRG neurons were held at −65 mV in current clamp using the perforated patch technique as described in Materials and Methods. Bursts of action potentials (4 s trains) were evoked by injecting depolarizing current (0.1 ms pulses) every 2 min using a protocol similar to that used to elicit increases in [Ca2+]i to study Ca2+ clearance. The stimulus frequency was 50 Hz for naive and GFP-expressing cells and was reduced to 10 or 20 Hz for AS-PMCA4-expressing cells as indicated. A, Recordings show the mean of three sweeps recorded before (control) and 8 min after superfusion of 10 μm PP2. Action potentials were truncated to better display the AHP. Representative recordings are shown from a naive cell, a cell expressing GFP, and cells expressing AS-PMCA4 stimulated with 10 or 20 Hz as indicated. B, Bar graph displays mean amplitude of AHP before (open bar) and after application of PP2 (solid bars) recorded from naive (n = 5) and GFP- (n = 5) and AS-PMCA4- (10 Hz n = 4; 20 Hz n = 5) expressing cells. Stimulus frequency is indicated in parentheses. *p < 0.05 relative to control, paired Student's t test.
Discussion
The present study demonstrates that a tyrosine kinase cascade regulates the plasma membrane Ca2+ pump in sensory neurons. The PMCA is a point of cross talk that enables the interaction of the ECM with CD44 to regulate Ca2+ signaling via activation of a Lck, Fyn, FAK cascade. A scheme summarizing this novel CD44 to PMCA signaling pathway is displayed in Figure 8. All of the proteins included in this model are present in neurons, and many of the proposed protein–protein interactions have been previously described for non-neuronal cells. However, this is the first description of the full pathway. Changes in Ca2+ clearance rate can potentially modulate many neuronal processes; we provided the specific example of PTKs controlling the action potential afterhyperpolarization through the regulation of a Ca2+-dependent K+ current.
Figure 8.

Schematic diagram of proposed CD44-regulated signaling pathway.
CD44 is a cell surface glycoprotein receptor that participates in adhesion and migration by binding to HA and other molecules in the ECM (Goodison et al., 1999). It is expressed throughout the nervous system (Glezer et al., 2009), although relatively little is known about its role in neuronal function. We found that a CD44-blocking antibody completely abolished PTK-dependent modulation of the PMCA in sensory neurons (Fig. 6). CD44 participates in the extension of axons from retinal ganglion cells onto a laminin substrate (Ries et al., 2007), consistent with its role in cell adhesion and migration. The DRG neurons studied here were plated on laminin-coated coverslips, suggesting that laminin is the ligand responsible for the constitutive activation of CD44. The modulation of L-type Ca2+ channels by HA with subsequent changes in synaptic plasticity supports the idea that ECM regulation of Ca2+ signaling might alter neuronal function (Kochlamazashvili et al., 2010). It is plausible that increases in Ca2+ influx would be accompanied by inhibition of Ca2+ efflux to amplify a Ca2+ response as described previously for lymphocytes (Habib et al., 2007). The level of PMCA expression has a profound effect on synaptic plasticity (Simons et al., 2009), so it is reasonable to expect that the signaling pathway described here might also regulate synaptic strength. We found that sHA-oligos attenuated tonic PMCA inhibition. This observation is consistent with the known antagonist-like actions of sHA-oligos acting on CD44 (Hua et al., 1993; Lesley et al., 2000). The dissociation of high-molecular-weight hyaluronic acid and other high-molecular-weight ligands from CD44-expressing cells is slow (t1/2 ≈ 0.5–2 h) (Lesley et al., 2000), consistent with our observation that sHA-oligos required prolonged exposure to reverse CD44-mediated tonic inhibition of the PMCA. Because HA is degraded to sHA-oligos at sites of injury and in arthritic joints (Jiang et al., 2007) and sHA-oligos result in increased Ca2+ clearance rate (Fig. 6), we speculate that sHA-oligos might increase the excitability of sensory neurons and contribute to the hyperalgesia associated with these conditions. Indeed, injection of high-molecular-weight hyaluronic acid into the knee joint relieves osteoarthritic pain, although the mechanism has been attributed to the elastoviscosity of the HA solution (Gomis et al., 2004). CD44 can also act as a coreceptor for receptor tyrosine kinases and serve as a platform to assemble signaling molecules. For example, CD44 mediates the ERBB2–ERBB3 heterodimerization in response to neuregulins, a function essential for Schwann-cell differentiation, survival, and proliferation in the peripheral nervous system (Ponta et al., 2003).
The Lck–Fyn complex proposed in this report is based on the results of pharmacological and genetic approaches. The excellent agreement between complementary dominant-negative, constitutively active, and shRNA strategies provides compelling evidence that Lck and Fyn are required for the tonic PTK activity that suppressed PMCA-mediated Ca2+ clearance rate. While Lck and Fyn are expressed in the nervous system (Omri et al., 1996; Thomas and Brugge, 1997), the presence of this complex in neurons has not previously been described. The activation of Lck and Fyn by CD44 is well described in T lymphocytes (Taher et al., 1996; Ilangumaran et al., 1999). In some cells, these kinases activate parallel, redundant pathways; in others, the pathways diverge; and in some cases, as described here, Lck and Fyn appear to act in series (Filipp and Julius, 2004). Clearly our data demonstrate that Lck, Fyn, and FAK participate in the CD44 signaling pathway, and there is precedent for these kinases assembling into a membrane proximal signaling complex (Li et al., 2001); the serial activation of these kinases is plausible, but this interpretation was not explicitly tested in this study. Little is known about the role of Lck in neurons. Fyn plays a major role in the development (Maness, 1992) and plasticity (Grant et al., 1992) of the nervous system.
Among the well known targets activated by SFK signaling complexes is FAK (Cabodi et al., 2010). Indeed, in fyn knock-out animals, FAK is hypophosphorylated and long-term potentiation of CA1 synapses in the hippocampus impaired (Grant et al., 1995). CD44-dependent activation of FAK initiates an anti-apoptotic effect in malignant cells (Fujita et al., 2002) and promotes cell motility (Kim et al., 2008). We speculate that CD44 to FAK signaling may participate in axon guidance, an idea supported by misrouting of axons following enzymatic removal of HA (Chan et al., 2007). Localized increases in the [Ca2+]i enhance the residency of FAK at focal adhesions and trigger the disassembly of focal adhesions required for directed movement (Giannone et al., 2004). Coordinated modulation of PMCA-mediated Ca2+ clearance might help to shape [Ca2+]i microdomains needed for localized adhesion. FAK phosphorylation of PMCA4 in platelets contributes to thrombin-induced platelet adhesion (Bozulic et al., 2007), consistent with the idea that FAK regulation of Ca2+ clearance modulates cell motility.
The modulation of PMCA4 by PTKs in platelets and B cells (Chen et al., 2004; Bozulic et al., 2007) provides a precedent for the Ca2+ pump modulation described here. The loss of tonic inhibition of Ca2+ clearance in PMCA4 knockdown cells supports this contention (Fig. 5). DRG neurons predominantly express PMCA isoforms 2 and 4 (Usachev et al., 2002). Thus, knockdown of PMCA4 is, in part, compensated by PMCA2. However, PMCA2 does not appear to be inhibited by PTKs, and thus AS-PMCA4-expressing cells retain the ability to pump Ca2+ across the plasma membrane but lose sensitivity to PTKs. This interpretation is supported by the unique sensitivities to kinases displayed by the various PMCA isoforms. For example, PMCA4b is stimulated by protein kinase C (Enyedi et al., 1996), while isoforms 2a and 2b are inhibited (Enyedi et al., 1997). It will be interesting to examine the sensitivity of Ca2+ clearance in various central neurons to PTK activity as PMCAs are heterogeneously distributed in the brain (Stahl et al., 1992; Zacharias et al., 1995; Filoteo et al., 1997).
The physiological consequences for PMCA modulation in neurons are becoming increasingly established. We showed here that increases in PMCA-mediated Ca2+ clearance reduce the amplitude of the slow AHP that is generated by a Ca2+-activated K+ current (Fig. 7). The regulation of the AHP by SFKs was completely absent in AS-PMCA4-expressing cells. The pronounced AHP in DRG neurons contributes to spike frequency adaptation in these cells (Cordoba-Rodriguez et al., 1999). Thus, PMCA activation would be anticipated to increase burst duration and frequency. This is the first suggestion that HA might control the excitability of sensory neurons, although enzymatic breakdown of the ECM increases the excitability of interneurons (Dityatev et al., 2007). Other Ca2+-dependent processes that might be particularly sensitive to the rate of PMCA-mediated Ca2+ clearance include cell adhesion and migration (Bozulic et al., 2007), neurotransmitter release (Jensen et al., 2007), synaptic plasticity (Simons et al., 2009), and survival (Fernandes et al., 2007).
In summary, the present report delineates a CD44-activated kinase cascade not previously described for neurons and shows for the first time that PTKs modulate PMCA-mediated Ca2+ clearance in neurons. Future experiments to determine the additional physiological roles for this pathway in sensory neurons might provide insight into mechanisms of pain transmission following wounding and the mechanism by which the ECM regulates [Ca2+]i gradients during migration, axon guidance, and neurotransmission. In the broader context of the CNS, it will be important to determine whether this CD44-activated kinase cascade is present in central neurons, how PTKs influence the unique compliments of PMCA isoforms present in central neurons, and whether this signaling pathway is altered in neurodegenerative disease. Finally, this study supports the idea that plasma membrane Ca2+ pumps are a heterogeneous and dynamically regulated family of proteins that act in coordination with Ca2+ influx pathways to determine the amplitude, duration, and spatial localization of Ca2+ signals.
Footnotes
National Science Foundation Grant IOS0814549 and National Institutes of Health Grants DA07304 and DA11806 supported this work. We thank Kristen Lawson, Jessica Reinardy, and Nicole Wydeven for contributing preliminary photometry data and Elizabeth Salm for assisting with cell culture. We are grateful to the following investigators for providing expression plasmids: Donald Branch (University of Toronto) for DN-Lck, Dan Littman (New York University Skirball Institute) for CA-Lck, Filippo Giancotti (Memorial Sloan Kettering Cancer Center) for DN-Src and DN-Fyn, Mark Nachtigal (Dalhousie University) for TSAd, and Kenneth Yamada (National Institute of Dental and Craniofacial Research, National Institutes of Health) for CA-FAK and DN-FAK.
References
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, Courtneidge SA. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozulic LD, Malik MT, Dean WL. Effects of plasma membrane Ca(2+) -ATPase tyrosine phosphorylation on human platelet function. J Thromb Haemost. 2007;5:1041–1046. doi: 10.1111/j.1538-7836.2007.02508.x. [DOI] [PubMed] [Google Scholar]
- Burette A, Rockwood JM, Strehler EE, Weinberg RJ. Isoform-specific distribution of the plasma membrane Ca2+ ATPase in the rat brain. J Comp Neurol. 2003;467:464–476. doi: 10.1002/cne.10933. [DOI] [PubMed] [Google Scholar]
- Cabodi S, Di Stefano P, Leal Mdel P, Tinnirello A, Bisaro B, Morello V, Damiano L, Aramu S, Repetto D, Tornillo G, Defilippi P. Integrins and signal transduction. Adv Exp Med Biol. 2010;674:43–54. doi: 10.1007/978-1-4419-6066-5_5. [DOI] [PubMed] [Google Scholar]
- Caride AJ, Penheiter AR, Filoteo AG, Bajzer Z, Enyedi A, Penniston JT. The plasma membrane calcium pump displays memory of past calcium spikes. Differences between isoforms 2b and 4b. J Biol Chem. 2001;276:39797–39804. doi: 10.1074/jbc.M104380200. [DOI] [PubMed] [Google Scholar]
- Chan CK, Wang J, Lin L, Hao Y, Chan SO. Enzymatic removal of hyaluronan affects routing of axons in the mouse optic chiasm. Neuroreport. 2007;18:1533–1538. doi: 10.1097/WNR.0b013e3282efa065. [DOI] [PubMed] [Google Scholar]
- Chen J, McLean PA, Neel BG, Okunade G, Shull GE, Wortis HH. CD22 attenuates calcium signaling by potentiating plasma membrane calcium-ATPase activity. Nat Immunol. 2004;5:651–657. doi: 10.1038/ni1072. [DOI] [PubMed] [Google Scholar]
- Cordoba-Rodriguez R, Moore KA, Kao JPY, Weinreich D. Calcium regulation of a slow post-spike hyperpolarization in vagal afferent neurons. Proc Natl Acad Sci U S A. 1999;96:7650–7657. doi: 10.1073/pnas.96.14.7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leva F, Domi T, Fedrizzi L, Lim D, Carafoli E. The plasma membrane Ca2+ ATPase of animal cells: structure, function and regulation. Arch Biochem Biophys. 2008;476:65–74. doi: 10.1016/j.abb.2008.02.026. [DOI] [PubMed] [Google Scholar]
- Ditlevsen DK, Berezin V, Bock E. Signalling pathways underlying neural cell adhesion molecule-mediated survival of dopaminergic neurons. Eur J Neurosci. 2007;25:1678–1684. doi: 10.1111/j.1460-9568.2007.05436.x. [DOI] [PubMed] [Google Scholar]
- Dityatev A, Brückner G, Dityateva G, Grosche J, Kleene R, Schachner M. Activity-dependent formation and functions of chondroitin sulfate-rich extracellular matrix of perineuronal nets. Dev Neurobiol. 2007;67:570–588. doi: 10.1002/dneu.20361. [DOI] [PubMed] [Google Scholar]
- Enyedi A, Verma AK, Filoteo AG, Penniston JT. Protein kinase C activates the plasma membrane Ca2+ pump isoform 4b by phosphorylation of an inhibitory region downstream of the calmodulin-binding domain. J Biol Chem. 1996;271:32461–32467. doi: 10.1074/jbc.271.50.32461. [DOI] [PubMed] [Google Scholar]
- Enyedi A, Elwess NL, Filoteo AG, Verma AK, Paszty K, Penniston JT. Protein kinase C phosphorylates the a forms of plasma membrane Ca2+ pump isoforms 2 and 3 and prevents binding of calmodulin. J Biol Chem. 1997;272:27525–27528. doi: 10.1074/jbc.272.44.27525. [DOI] [PubMed] [Google Scholar]
- Fernandes D, Zaidi A, Bean J, Hui D, Michaelis ML. RNA(i)- induced silencing of the plasma membrane Ca(2+)- ATPase 2 in neuronal cells: effects on Ca(2+) homeostasis and cell viability. J Neurochem. 2007;102:454–465. doi: 10.1111/j.1471-4159.2007.04592.x. [DOI] [PubMed] [Google Scholar]
- Filipp D, Julius M. Lipid rafts: resolution of the “fyn problem?”. Mol Immunol. 2004;41:645–656. doi: 10.1016/j.molimm.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Filoteo AG, Elwess NL, Enyedi A, Caride A, Aung HH, Penniston JT. Plasma membrane Ca2+ pump in rat brain - patterns of alternative splices seen by isoform-specific antibodies. J Biol Chem. 1997;272:23741–23747. doi: 10.1074/jbc.272.38.23741. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Kitagawa M, Nakamura S, Azuma K, Ishii G, Higashi M, Kishi H, Hiwasa T, Koda K, Nakajima N, Harigaya K. CD44 signaling through focal adhesion kinase and its anti-apoptotic effect. FEBS Lett. 2002;528:101–108. doi: 10.1016/s0014-5793(02)03262-3. [DOI] [PubMed] [Google Scholar]
- Garcia ML, Usachev YM, Thayer SA, Strehler EE, Windebank AJ. Plasma membrane calcium ATPase plays a role in reducing Ca(2+)-mediated cytotoxicity in PC12 cells. J Neurosci Res. 2001;64:661–669. doi: 10.1002/jnr.1120. [DOI] [PubMed] [Google Scholar]
- Giannone G, Rondé P, Gaire M, Beaudouin J, Haiech J, Ellenberg J, Takeda K. Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J Biol Chem. 2004;279:28715–28723. doi: 10.1074/jbc.M404054200. [DOI] [PubMed] [Google Scholar]
- Girault J-A, Costa A, Derkinderen P, Studler J-M, Toutant M. FAK and PYK2/CAK[beta] in the nervous system: a link between neuronal activity, plasticity and survival? Trends Neurosci. 1999;22:257–263. doi: 10.1016/s0166-2236(98)01358-7. [DOI] [PubMed] [Google Scholar]
- Glezer I, Bittencourt JC, Rivest S. Neuronal expression of Cd36, Cd44, and Cd83 antigen transcripts maps to distinct and specific murine brain circuits. J Comp Neurol. 2009;517:906–924. doi: 10.1002/cne.22185. [DOI] [PubMed] [Google Scholar]
- Gomis A, Pawlak M, Balazs EA, Schmidt RF, Belmonte C. Effects of different molecular weight elastoviscous hyaluronan solutions on articular nociceptive afferents. Arthritis Rheum. 2004;50:314–326. doi: 10.1002/art.11421. [DOI] [PubMed] [Google Scholar]
- Goodison S, Urquidi V, Tarin D. CD44 cell adhesion molecules. Mol Pathol. 1999;52:189–196. doi: 10.1136/mp.52.4.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SG, O'Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- Grant SG, Karl KA, Kiebler MA, Kandel ER. Focal adhesion kinase in the brain: novel subcellular localization and specific regulation by Fyn tyrosine kinase in mutant mice. Genes Dev. 1995;9:1909–1921. doi: 10.1101/gad.9.15.1909. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Guerini D, Pan B, Carafoli E. Expression, purification, and characterization of isoform 1 of the plasma membrane Ca2+ pump: focus on calpain sensitivity. J Biol Chem. 2003;278:38141–38148. doi: 10.1074/jbc.M302400200. [DOI] [PubMed] [Google Scholar]
- Habib T, Park H, Tsang M, de Alborán IM, Nicks A, Wilson L, Knoepfler PS, Andrews S, Rawlings DJ, Eisenman RN, Iritani BM. Myc stimulates B lymphocyte differentiation and amplifies calcium signaling. J Cell Biol. 2007;179:717–731. doi: 10.1083/jcb.200704173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Hua Q, Knudson CB, Knudson W. Internalization of hyaluronan by chondrocytes occurs via receptor-mediated endocytosis. J Cell Sci. 1993;106:365–375. doi: 10.1242/jcs.106.1.365. [DOI] [PubMed] [Google Scholar]
- Iacob S, Knudson CB. Hyaluronan fragments activate nitric oxide synthase and the production of nitric oxide by articular chondrocytes. Int J Biochem Cell Biol. 2006;38:123–133. doi: 10.1016/j.biocel.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Nakao J, Asou H, Toya S, Shinoda J, Uyemura K. Expression of CD44H in the cells of neural crest origin in peripheral nervous system. Neuroreport. 1996;7:1713–1716. doi: 10.1097/00001756-199607290-00003. [DOI] [PubMed] [Google Scholar]
- Ilangumaran S, Borisch B, Hoessli DC. Signal transduction via CD44: role of plasma membrane microdomains. Leuk Lymphoma. 1999;35:455–469. doi: 10.1080/10428199909169610. [DOI] [PubMed] [Google Scholar]
- Jensen TP, Filoteo AG, Knopfel T, Empson RM. Pre-synaptic plasma membrane Ca2+ ATPase isoform 2a regulates excitatory synaptic transmission in rat hippocampal CA3. J Physiol. 2007;579:85–99. doi: 10.1113/jphysiol.2006.123901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol. 2007;23:435–461. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- Kim Y, Lee YS, Choe J, Lee H, Kim YM, Jeoung D. CD44-epidermal growth factor receptor interaction mediates hyaluronic acid-promoted cell motility by activating protein kinase C signaling involving Akt, Rac1, Phox, reactive oxygen species, focal adhesion kinase, and MMP-2. J Biol Chem. 2008;283:22513–22528. doi: 10.1074/jbc.M708319200. [DOI] [PubMed] [Google Scholar]
- Kochlamazashvili G, Henneberger C, Bukalo O, Dvoretskova E, Senkov O, Lievens PM, Westenbroek R, Engel AK, Catterall WA, Rusakov DA, Schachner M, Dityatev A. The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca(2+) channels. Neuron. 2010;67:116–128. doi: 10.1016/j.neuron.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesley J, Hascall VC, Tammi M, Hyman R. Hyaluronan binding by cell surface CD44. J Biol Chem. 2000;275:26967–26975. doi: 10.1074/jbc.M002527200. [DOI] [PubMed] [Google Scholar]
- Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- Li R, Wong N, Jabali MD, Johnson P. CD44-initiated cell spreading induces Pyk2 phosphorylation, is mediated by Src family kinases, and is negatively regulated by CD45. J Biol Chem. 2001;276:28767–28773. doi: 10.1074/jbc.M100158200. [DOI] [PubMed] [Google Scholar]
- Maness PF. Nonreceptor protein tyrosine kinases associated with neuronal development. Dev Neurosci. 1992;14:257–270. doi: 10.1159/000111670. [DOI] [PubMed] [Google Scholar]
- Mariotti A, Kedeshian PA, Dans M, Curatola AM, Gagnoux-Palacios L, Giancotti FG. EGF-R signaling through Fyn kinase disrupts the function of integrin alpha6beta4 at hemidesmosomes: role in epithelial cell migration and carcinoma invasion. J Cell Biol. 2001;155:447–458. doi: 10.1083/jcb.200105017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omri B, Crisanti P, Marty MC, Alliot F, Fagard R, Molina T, Pessac B. The Lck tyrosine kinase is expressed in brain neurons. J Neurochem. 1996;67:1360–1364. doi: 10.1046/j.1471-4159.1996.67041360.x. [DOI] [PubMed] [Google Scholar]
- Pászty K, Verma AK, Padányi R, Filoteo AG, Penniston JT, Enyedi A. Plasma membrane Ca2+ ATPase isoform 4b is cleaved and activated by caspase-3 during the early phase of apoptosis. J Biol Chem. 2002;277:6822–6829. doi: 10.1074/jbc.M109548200. [DOI] [PubMed] [Google Scholar]
- Piser TM, Lampe RA, Keith RA, Thayer SA. ω-Grammotoxin blocks action-potential-induced Ca2+ influx and whole-cell Ca2+ current in rat dorsal root ganglion neurons. Pflugers Arch. 1994;426:214–220. doi: 10.1007/BF00374774. [DOI] [PubMed] [Google Scholar]
- Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- Pottorf WJ, Thayer SA. Transient rise in intracellular calcium produces a long-lasting increase in plasma membrane calcium pump activity in rat sensory neurons. J Neurochem. 2002;83:1002–1008. doi: 10.1046/j.1471-4159.2002.01221.x. [DOI] [PubMed] [Google Scholar]
- Richard KC, Bertolesi GE, Dunfield LD, McMaster CR, Nachtigal MW. TSAd interacts with Smad2 and Smad3. Biochem Biophys Res Commun. 2006;347:266–272. doi: 10.1016/j.bbrc.2006.06.068. [DOI] [PubMed] [Google Scholar]
- Ries A, Goldberg JL, Grimpe B. A novel biological function for CD44 in axon growth of retinal ganglion cells identified by a bioinformatics approach. J Neurochem. 2007;103:1491–1505. doi: 10.1111/j.1471-4159.2007.04858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R. T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol Rev. 2009;228:9–22. doi: 10.1111/j.1600-065X.2008.00745.x. [DOI] [PubMed] [Google Scholar]
- Simons SB, Escobedo Y, Yasuda R, Dudek SM. Regional differences in hippocampal calcium handling provide a cellular mechanism for limiting plasticity. Proc Natl Acad Sci U S A. 2009;106:14080–14084. doi: 10.1073/pnas.0904775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl WL, Eakin TJ, Owens JW, Jr, Breininger JF, Filuk PE, Anderson WR. Plasma membrane Ca2+-ATPase isoforms: distribution of mRNAs in rat brain by in situ hybridization. Brain Res Mol Brain Res. 1992;16:223–231. doi: 10.1016/0169-328x(92)90229-5. [DOI] [PubMed] [Google Scholar]
- Strehler EE, Zacharias DA. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol Rev. 2001;81:21–50. doi: 10.1152/physrev.2001.81.1.21. [DOI] [PubMed] [Google Scholar]
- Sundvold-Gjerstad V, Granum S, Mustelin T, Andersen TC, Berge T, Shapiro MJ, Shapiro VS, Spurkland A, Lea T. The C terminus of T cell-specific adapter protein (TSAd) is necessary for TSAd-mediated inhibition of Lck activity. Eur J Immunol. 2005;35:1612–1620. doi: 10.1002/eji.200425638. [DOI] [PubMed] [Google Scholar]
- Taher TE, Smit L, Griffioen AW, Schilder-Tol EJ, Borst J, Pals ST. Signaling through CD44 is mediated by tyrosine kinases. Association with p56lck in T lymphocytes. J Biol Chem. 1996;271:2863–2867. doi: 10.1074/jbc.271.5.2863. [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Danen EH, Takino T, Miyamoto S, Yamada KM. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem. 1999;274:20693–20703. doi: 10.1074/jbc.274.29.20693. [DOI] [PubMed] [Google Scholar]
- Thayer SA, Usachev YM, Pottorf WJ. Modulating Ca2+ clearance from neurons. Front Biosci. 2002;7:d1255–d1279. doi: 10.2741/A838. [DOI] [PubMed] [Google Scholar]
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Turner JM, Brodsky MH, Irving BA, Levin SD, Perlmutter RM, Littman DR. Interaction of the unique N-terminal region of tyrosine kinase p56lck with cytoplasmic domains of CD4 and CD8 is mediated by cysteine motifs. Cell. 1990;60:755–765. doi: 10.1016/0092-8674(90)90090-2. [DOI] [PubMed] [Google Scholar]
- Usachev YM, Khammanivong A, Campbell C, Thayer SA. Particle-mediated gene transfer to rat neurons in primary culture. Pflugers Arch. 2000;439:730–738. doi: 10.1007/s004249900240. [DOI] [PubMed] [Google Scholar]
- Usachev YM, DeMarco SJ, Campbell C, Strehler EE, Thayer SA. Bradykinin and ATP accelerate Ca2+ efflux from rat sensory neurons via protein kinase C and the plasma membrane Ca2+ pump isoform 4. Neuron. 2002;33:113–122. doi: 10.1016/s0896-6273(01)00557-8. [DOI] [PubMed] [Google Scholar]
- Usachev YM, Marsh AJ, Johanns TM, Lemke MM, Thayer SA. Activation of protein kinase C in sensory neurons accelerates Ca2+ uptake into the endoplasmic reticulum. J Neurosci. 2006;26:311–318. doi: 10.1523/JNEUROSCI.2920-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma AK, Paszty K, Filoteo AG, Penniston JT, Enyedi A. Protein kinase C phosphorylates plasma membrane Ca2+ pump isoform 4a at its calmodulin binding domain. J Biol Chem. 1999;274:527–531. doi: 10.1074/jbc.274.1.527. [DOI] [PubMed] [Google Scholar]
- Wan TC, Zabe M, Dean WL. Plasma membrane Ca2+-ATPase isoform 4b is phosphorylated on tyrosine 1176 in activated human platelets. Thrombosis and Haemostasis. 2003;89:122–131. [PubMed] [Google Scholar]
- Werth JL, Usachev YM, Thayer SA. Modulation of calcium efflux from cultured rat dorsal root ganglion neurons. J Neurosci. 1996;16:1008–1015. doi: 10.1523/JNEUROSCI.16-03-01008.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi S, Ma XZ, Singla R, Zhou YC, Sakac D, Bali M, Liu Y, Sahai BM, Branch DR. HIV-1 infection is facilitated in T cells by decreasing p56lck protein tyrosine kinase activity. Clin Exp Immunol. 2003;133:78–90. doi: 10.1046/j.1365-2249.2003.02187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias DA, Dalrymple SJ, Strehler EE. Transcript distribution of plasma membrane Ca2+ pump isoforms and splice variants in the human brain. Brain Res Mol Brain Res. 1995;28:263–272. doi: 10.1016/0169-328x(94)00215-z. [DOI] [PubMed] [Google Scholar]
- Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]