

Abstract
Nasopharyngeal carcinoma (NPC) is an Epstein-Barr virus-associated malignancy most common in East Asia and Africa. Radiotherapy and cisplatin-based chemotherapy are the main treatment options. Unfortunately, disease response to concurrent chemoradiotherapy varies among patients with NPC, and many cases are resistant to cisplatin. Increased DNA damage repair is one of the mechanisms contributing to this resistance. Jab1/CSN5 is a multifunctional protein that participates in controlling cell proliferation and the stability of multiple proteins. Jab1 overexpression has been found to correlate with poor prognosis in several tumor types. However, the biological significance of Jab1 activity in response to cancer treatment is unclear. In this study, we used three NPC cell lines (CNE1, CNE2, and HONE1) to investigate the hypothesis that Jab1 positively regulates the DNA repair protein Rad51 and, in turn, cellular response to treatment with DNA-damaging agents such as cisplatin, ionizing radiation (IR) and ultraviolet (UV). We found that Jab1 was overexpressed in two relatively cisplatin-, IR- and UV-resistant NPC cell lines, and knocking down its expression conferred sensitivity to cisplatin, IR and UV. By contrast, exogenous Jab1 expression enhanced the resistance of NPC cells to cisplatin, IR and UV Moreover, we provide a mechanism by which Jab1 positively regulated Rad51 through p53-dependent pathway, and increased ectopic expression of Rad51 conferred cellular resistance to cisplatin, IR and UV in Jab1-deficient cells. Taken together, our findings suggest that Jab1 plays an important role in the cellular response to cisplatin and irradiation by regulating DNA damage and repair pathways. Therefore, Jab1 is a novel biomarker for predicting the outcome of patients with NPC who are treated with DNA-damaging agents.
Keywords: nasopharyngeal carcinoma, Epstein-Barr virus-associated malignancy, cisplatin, ultraviolet radiation, ionizing radiation, RPPA
Introduction
Nasopharyngeal carcinoma (NPC) arises from the epithelial lining of the nasopharynx (1) and is one of the most poorly understood types of cancer. NPC has a remarkable ethnic and geographic distribution: it is highly prevalent in southern China, Southeast Asia, northern Africa, and Alaska (2). The annual incidence peaks at 50 cases per 100,000 people in endemic regions, but it is rare in the Western world (1 per 100,000 people) (3). Etiologic studies have indicated that Epstein-Barr virus (EBV) infection, environmental factors, and genetic susceptibility are associated with NPC (1, 2, 4).
Cisplatin chemotherapy and radiotherapy have become the main treatments for NPC (5). Unfortunately, many NPC patients do not benefit much from concurrent chemoradiotherapy; 30% to 40% of patients develop distant metastases within 4 years (6, 7), and once metastasis occurs, the prognosis is very poor. One of the primary causes of treatment failure is the development of resistance to antitumor drugs and radiation (8). Therefore, understanding the molecular mechanisms of radio- and chemo-sensitivity/resistance in NPC will improve the development of novel therapeutic approaches and result in better clinical outcomes.
Jab1/CSN5 (Jab1 hereafter) as we initially identified as a c-Jun coactivator, is also known as the fifth component of the COP9 signalosome (CSN) complex (CSN5) (9). Jab1/CSN5 plays an essential role in positively regulating cellular proliferation by functionally inactivating several key negative regulatory proteins and tumor suppressors through their subcellular localization and degradation, including p53, Smad 4/7, and the cyclin-dependent kinase inhibitor p27Kip1 (p27) (10). Abnormal overexpression of Jab1 is correlated with a lower rate of survival in patients with breast cancer (11), anaplastic large cell lymphoma (12), pancreatic adenocarcinomas (13), and other types of cancer (10). Recently, we have found that Jab1 overexpression is also correlated with a lower survival rate in patients with NPC (14). Because DNA-damaging strategies, such as radiotherapy and the majority of chemotherapeutic therapies, are the most frequently used non-surgical anti-cancer therapies for human cancers, it is conceivable that Jab1 overexpression contributes to the reduced antitumor effects of these therapies. Indeed, our previous studies in mice indicate that Jab1 is essential for DNA repair and linked to the maintenance of genome integrity and cell survival (15). Loss of Jab1 sensitized cells to gamma radiation–induced apoptosis and increased spontaneous DNA damage and homologous recombination defects (15). This suggests that inhibiting Jab1 in tumor cells may enhance the sensitivity of these cells to DNA-damaging therapy. Sources of DNA damage include ultraviolet (UV) light, ionizing radiation (IR), cisplatin and a variety of industrial and environmental chemical compounds (16); thus, in our study we use cisplatin, IR and UV to represent the major DNA damage agents. On the basis of our previous findings, we hypothesized that Jab1 contributes to cisplatin, IR and UV resistance.
To test this hypothesis, we analyzed the effects of Jab1 on the response of three NPC cell lines to cisplatin, IR and UV radiation. We demonstrated that knocking down Jab1 expression in these NPC cells sensitized them to cisplatin, IR and UV radiation, and conversely, overexpression of Jab1 contributes to cisplatin and irradiation resistance in NPC cells by positively regulating the levels of the DNA repair gene Rad51. These observations suggest that Jab1 is a major contributor to the cisplatin, IR and UV radiation resistance of NPC. Jab1 therefore represents a novel therapeutic target in patients with NPC and a biomarker in predicting treatment outcomes of these patients treated with cisplatin- and irradiation-based therapy.
Results
Cisplatin, IR and UV sensitivity/resistance patterns in NPC cell lines
As adjuvant cisplatin chemotherapy and radiotherapy are the main treatments for NPC, we measured the sensitivity of a panel of NPC cell lines to these genotoxic agents. We exposed three NPC cell lines to various doses of cisplatin, IR and UV. Cells were incubated with various concentrations of cisplatin for 48 h. Cisplatin inhibited the relative viable cell number in a dose- and time-dependent manner (Figures 1a and 1b). Interestingly, the three NPC cell lines responded differently to cisplatin, with CNE1 being the most sensitive, the relative resistance factor (RRF) was 2.5 and 3.5 times higher in CNE2 and HONE1 cells than in CNE1 cells. In addition, IR and UV were cytotoxic to NPC cell lines in a dose- and time-dependent manner (Figures 1a and 1b). As was the case with cisplatin, CNE1 cells displayed an enhanced IR -and UV -induced cell inhibition among the NPC cell lines tested.
Figure 1.
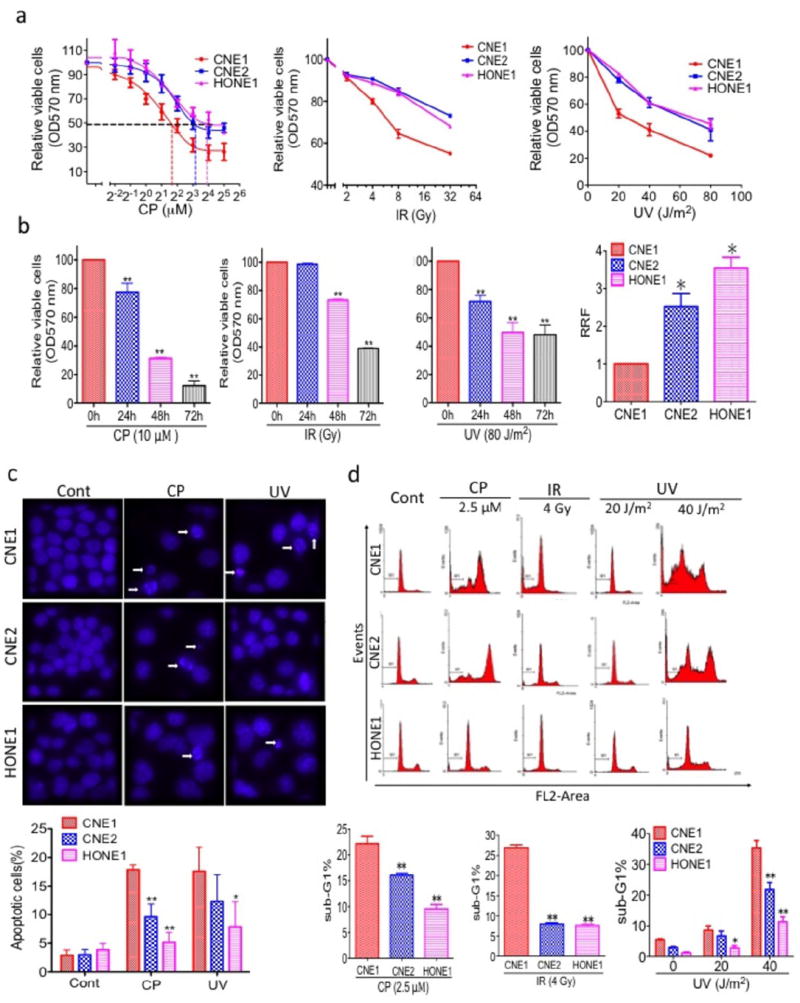
Characterization of NPC cell lines. (a) CNE1, CNE2, and HONE1 cells were treated with various doses of cisplatin (CP, left) or IR (middle) or UV (right) for 48 h. Relative viable cells were measured by the MTT assay, and the relative resistance factors (RRFs) are shown (bottom). (b) Cisplatin, IR and UV inhibit relative viable cells in a time-dependent manner. CNE2 cells were treated with 10 μM CP or 32 Gy IR or 80 J/m2 UV for the indicated times, and relative viable cells was measured by the MTT assay. (c) Apoptosis was measured by Hoechst 33342 staining. (Top) NPC cells were treated with 5 μM CP or 40 J/m2 UV for 72 h, nuclei were stained with Hoechst 33342, and imaging analysis was performed as described in the Materials and Methods. The white arrows indicate apoptotic cells. Original magnification, ×200. (Bottom) Quantification of the cell staining. (d) Measurement of apoptosis by PI staining. (Top) NPC cells were treated with 2.5 μM CP or 4 Gy IR or 20 J/m2 or 40 J/m2 UV for 48 h, followed by PI staining as described in the Materials and Methods. (Bottom) Quantification of PI staining. All data represent three independent experiments, mean ± s.d. *P <0.05, **P <0.01.
To determine whether the cisplatin and UV sensitivity of CNE1 cells is followed by increases in apoptosis, CNE1, CNE2, and HONE1 cells were treated with cisplatin (5 μM) or UV (40 J/m2) and analyzed by Hoechst 33342 staining, which detects condensed nuclei, an indicator of apoptosis. Treatment of NPC cells with cisplatin or UV radiation resulted in a marked increase in the number of apoptotic cells (Figure 1c). However, significantly more CNE1 cells (18% and 17%, respectively) underwent apoptosis after being treated with cisplatin and UV radiation than either CNE2 (10% and 12%, respectively) or HONE1 (5% and 8%, respectively) cells. To confirm these findings with an independent assay, we measured apoptosis by PI staining and flow cytometry. Forty-eight h after cisplatin exposure, 22% of the CNE1 cells had hypodiploid (sub-G1) DNA content, reflecting apoptosis, whereas only 16 % of CNE2 and 9% of HONE1 had hypodiploid DNA (Figure 1d). At the same time, treatment with IR and UV significantly increased the number of CNE1 cells in the sub-G1 phase compared with that of CNE2 and HONE1 cells in the sub-G1 phase (CNE1 vs CNE2 cells, 3.4-fold more at 4 Gy IR, and 1.2- and 1.6-fold more at 20 J/m2 and 40 J/m2 UV radiation, respectively; and CNE1 vs HONE1 cells, 3.5-fold more at 4 Gy IR and 3.1-fold more at both 20 J/m2 and 40 J/m2 UV radiation.
Contribution of Jab1 depletion to cisplatin, IR and UV sensitivity of NPC cells
We first examined the Jab1 expression levels in CNE1, CNE2, and HONE1 cells by western blotting. HONE1 and CNE2 cells expressed higher levels of Jab1 than CNE1 cells (Figure 2a). We next examined whether suppression of Jab1 expression could increase the sensitivity of NPC cells to cisplatin, IR and UV by knocking down Jab1 in HONE1 and CNE2 cells. Western blotting showed that Jab1 expression was successfully knocked down in the Jab1 siRNA–treated HONE1 (Figure 2b) and CNE2 cells (Figure 2c). HONE1 cells with reduced Jab1 expression showed an increase in the efficacy of cisplatin, IR and UV radiation compared with control cells transfected with a scrambled siRNA. The relative viable cells was reduced by 9% and 26% following cisplatin treatment at 8 and 16 μM; by 10% and 12% following IR at 8 and 16 Gy; and by 12% and 25% following UV radiation at 20 and 80 J/m2, respectively (Figure 2b). The Jab1 siRNA–treated CNE2 cells showed increased sensitivity to cisplatin, IR and UV, with approximately 11% and 15% decreases in the relative viable cells when treated with 4 μM and 8 μM cisplatin, and 13% and 18% decreases upon 8 and 16 Gy IR, compared with the scrambled siRNA treated controls, and 9% and 41% decreases in the relative viable cells upon 20 and 80 J/m2 UV radiation, respectively (Figure 2c).
Figure 2.
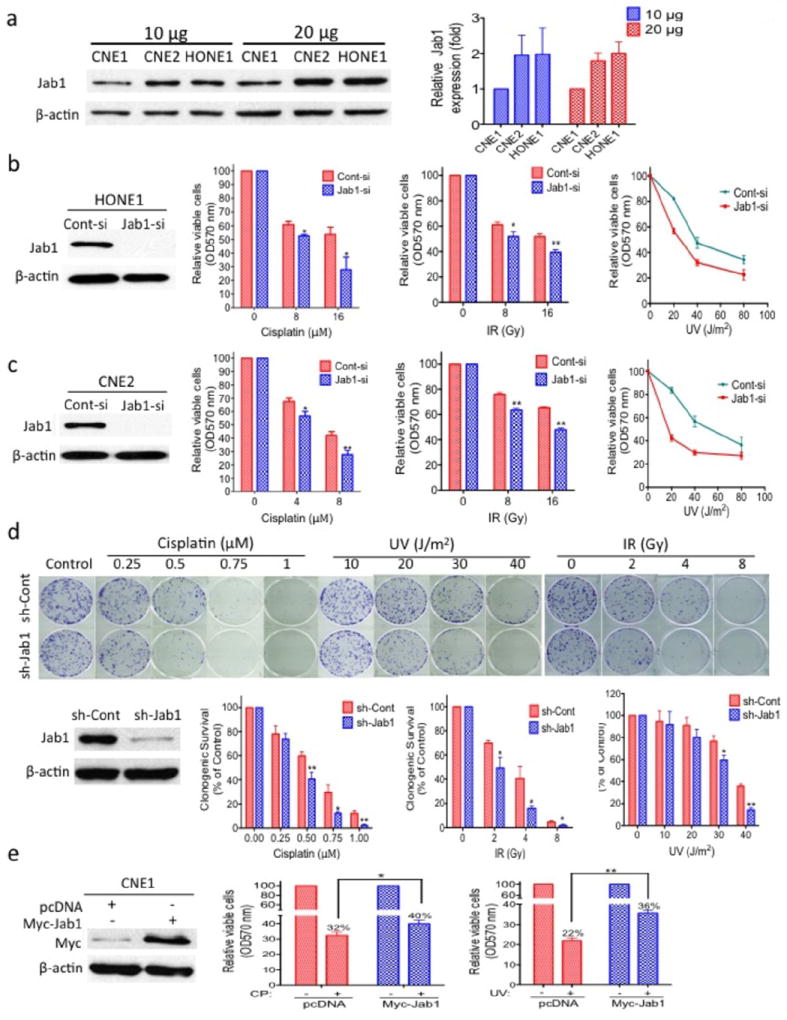
Effect of Jab1 depletion on the sensitivity of NPC cells to cisplatin, IR and UV irradiation. (a) Jab1 expression in NPC cells. CNE1, CNE2, and HONE1 cells in the logarithmic growth phase were collected and lysed, followed by western blotting for Jab1. β-actin was used as a loading control. (Right) Quantification of Jab1 expression. (b, c, d, e) Effect of knockdown or ectopic overexpression of Jab1 on the sensitivity of NPC cells to cisplatin, IR and UV. HONE1 (b) and CNE2 (c) cells were transiently transfected with a Jab1 siRNA (Jab1-si) or scrambled control siRNA (Cont-si), CNE2 (d) cells were stably transfected with Jab1 shRNA (sh-Jab1) or control shRNA (sh-Cont), CNE1 (e) cells were transfected with ectopic Jab1 (Myc-Jab1) or a control vector (pcDNA). Cells were then analyzed for their Jab1 expression level (left) and response to cisplatin (CP), IR irradiation (middle) UV (right) as described in the Materials and Methods. Data represent three independent experiments, mean±s.d. * P <0.05, **P <0.01.
We also established a stable Jab1 shRNA–transfected CNE2 cell line to test the effects of Jab1 depletion on the cells’ response to cisplatin, IR and UV using a colony formation assay. We observed similar results to those described above; CNE2 cells transfected with Jab1 shRNA had fewer colonies when exposed to cisplatin or IR or UV (Figure 2d). Thus, downregulation of Jab1 likely contributes to the increased sensitivity of the NPC cells to cisplatin, IR and UV.
To confirm the above conclusion, we next conducted the reverse experiment, we increased Jab1 expression in CNE1 cells and determined whether it would enhanced the resistance of CNE1 cells to cisplatin and UV radiation treatments. Thus, CNE1 cells were transfected with Myc-Jab1 cDNA, and relative viable cells was measured by the MTT assay (Figure 2e). The CNE1 cells overexpressing Jab1 displayed decreased cisplatin-induced cell inhibition, with a 1.25 times higher relative viable cells than control cells transfected with a vector (Figure 2e). Similar results were seen when the cells were treated with UV irradiation, with a 1.6 times higher relative viable cells than control cells. Taken together, we conclude that Jab1 depletion increases the sensitivity of NPC cells to cisplatin and UV radiation.
Loss of Jab1 results in increased apoptosis induced by cisplatin, IR and UV
As discussed above, CNE1 cells displayed increased cisplatin-, IR- and UV-induced apoptosis compared with CNE2 or HONE1 cells (Figures 1c and 1d). To determine whether Jab1 downregulation mediates this process, we first examined whether knocking down Jab1 in HONE1 cells increased cisplatin-, IR- and UV-induced apoptosis. HONE1 cells transfected with Jab1 siRNA had significantly more apoptosis after cisplatin treatment (1.2- to 1.4-fold more apoptosis, as measured by PI staining; Figure 3a) and IR (1.2- to 1.3-fold more apoptosis) than control siRNA–transfected cells (Figure 3b). Similar results were seen when cells were treated with UV radiation (1.7- to 2.5-fold more apoptosis) (Figure 3c). Because cleavage of PARP and caspase-3 activation are hallmarks of the initiation of apoptosis (17, 18), we further examined the influence of Jab1 on cisplatin-, IR- and UV-induced apoptosis in NPC cells. HONE1 cells transfected with Jab1 siRNA displayed increased cisplatin-, IR- and UV-induced PARP and caspase-3 cleavage compared with control cells when exposed to cisplatin, IR and UV (Figures 3a, 3b and 3c). By contrast, overexpression of Jab1 in CNE1 cells blocked cisplatin- and UV radiation-induced apoptosis, as measured by Hoechst 33342 staining and analysis of PARP and caspase-3 cleavage. Myc-Jab1-treated CNE1 cells had 35% and 25% fewer apoptotic cells with condensed nuclei and less PARP and caspase-3 cleavage than the control pcDNA-treated CNE1 cells after cisplatin or UV radiation treatment (Figure 3d).
Figure 3.
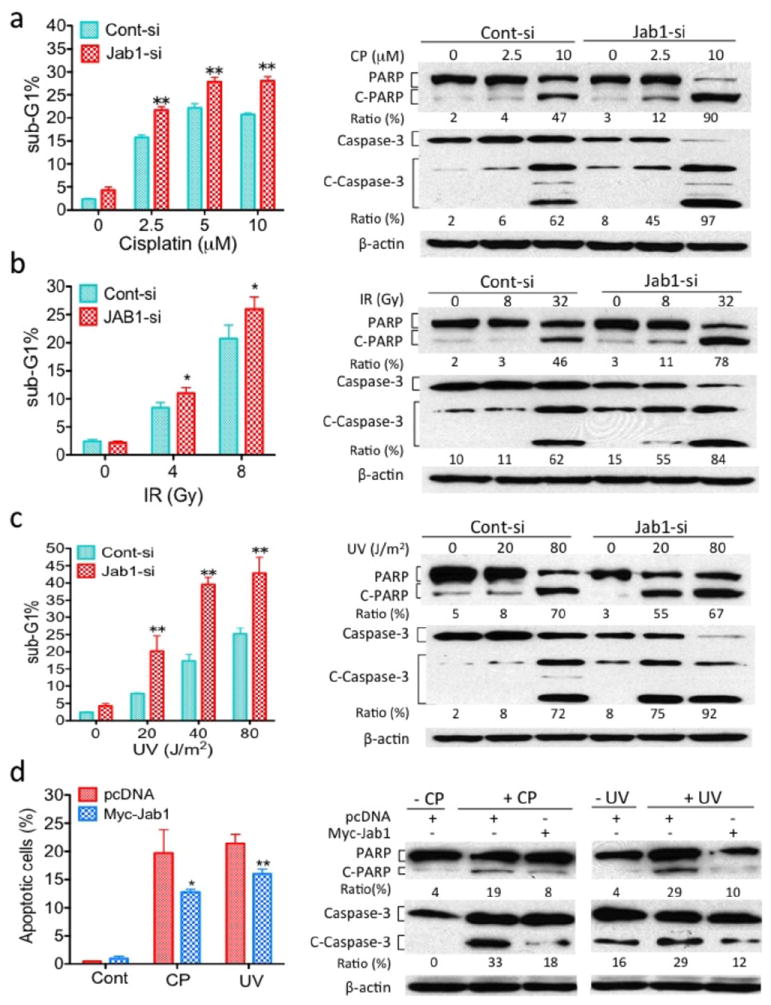
Effect of Jab1 on DNA damage-induced apoptosis. HONE1 cells were transiently transfected with either Jab1 siRNA (Jab1-si) or scrambled control siRNA (Cont-si) and treated with cisplatin (CP) (a) or IR (b) or UV (c) at the indicated doses. Apoptosis was analyzed by PI staining (left) or detection of cleaved PARP and caspase-3 by western blotting (right). (c) CNE1 cells were transfected with ectopic Myc-Jab1 plasmid and treated with cisplatin (CP) or UV for 48 h. Apoptosis was analyzed by Hoechst 33342 staining for condensed nuclei (left) and detection of cleaved PARP and caspase-3 by western blotting (right). Data represent three independent experiments, mean±s.d. *P <0.05, **P <0.01.
The increase in cisplatin-, IR- and UV radiation-induced apoptosis upon Jab1 depletion is not cell line specific
To rule out the possibility that the increase in cisplatin-, IR and UV radiation–induced apoptosis upon Jab1 depletion was specific to the HONE1 cell line, we also determined the effects of knocking down Jab1 expression in CNE1 and CNE2 cells. Loss of Jab1 in both cell lines (Figures 2c and 6a) resulted in greater apoptosis, with 1.4- and 4.4-fold (CNE1 and CNE2, respectively) more apoptosis when cells were treated with cisplatin (Figure 4a).
Figure 6.
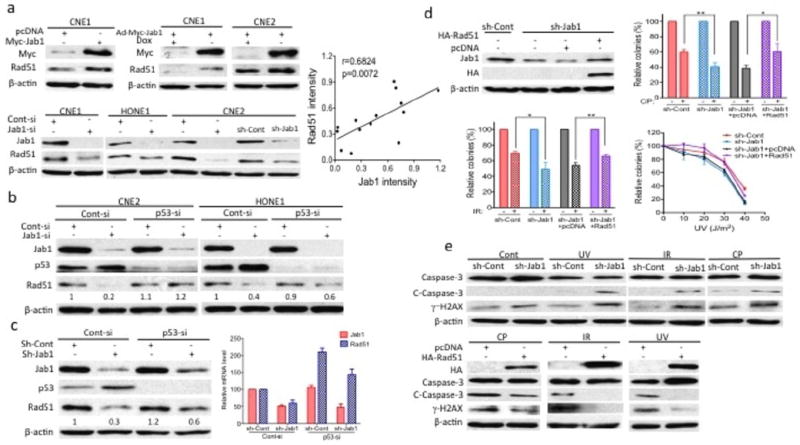
Jab1 associates with Rad51 and contributes to cisplatin (CP), IR and UV resistance. (a) NPC cells were transfected with Myc-Jab1 plasmid DNA or siRNA or transduced with Jab1 adenovirus in the presence or absence of doxycycline (Dox) for 48 h. Cell lysates were prepared 48 h after transfection or from cells stably expressing control (sh-Cont) or Jab1 shRNA (sh-Jab1) and western blotting was performed with the Jab1 and Rad51 antibodies. (Right) Correlation plot of Jab1 and Rad51 blot intensities obtained from control and treated cells. A fair linear correlation was obtained. (b) NPC cells were transfected with siRNA for 48 h, cells lysates were prepared and the levels of Jab1, p53 and Rad51 were determined by western blotting. (c) sh-Cont or sh-Jab1 stable CNE2 cells were transfected with siRNA for 48 h, then total protein and RNA was extracted and expression protein or RNA levels of the Jab1 and p53 and Rad51 were quantified by western blotting (left) or real time qRT-PCR (right). (d) CNE2 cells stably expressing sh-Jab1 were transfected with pcDNA or HA-Rad51 plasmid DNA. (Left) Western blot analyses demonstrated the effective knockdown and ectopic expression of Jab1. (Right and bottom) Colonies were stained with crystal violet 10 days after CP, IR and UV exposure. (e) (Top) CNE2 cells stably expressing sh-Cont or sh-Jab1 were exposed to UV or IR or CP. (Bottom) Cells stably expressing sh-Jab1 were transfected with pcDNA or HA-Rad51 plasmid DNA and then exposed to CP, IR and UV. Cleaved caspase-3 and γ̃H2AX were examined 48 h after exposure. All data represent three independent experiments, mean±s.d. *P <0.05, **P <0.01.
Figure 4.
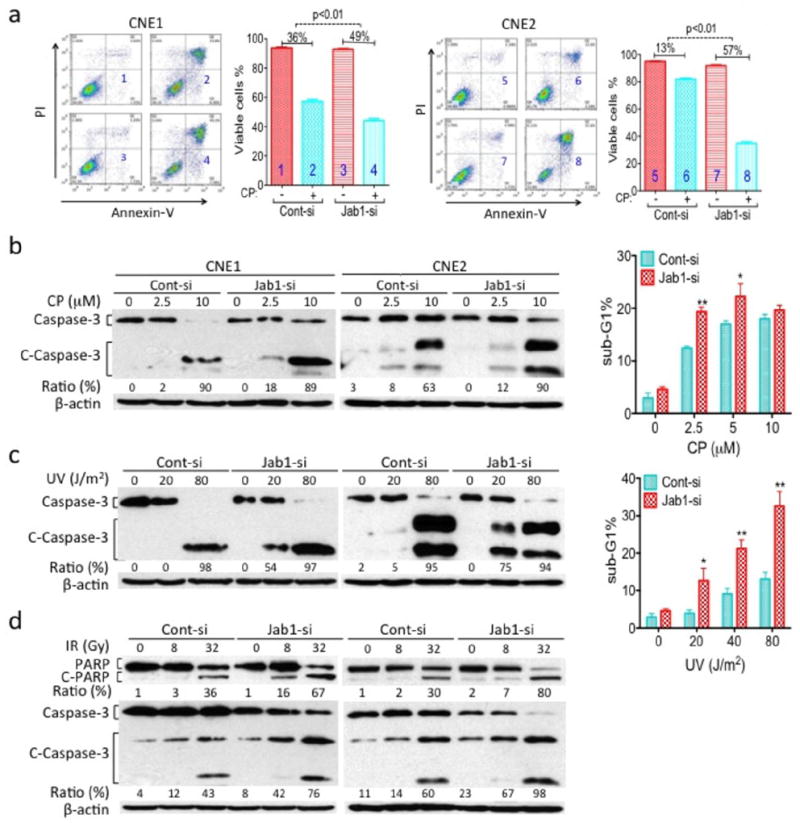
Role of Jab1 in the response of NPC cells to cisplatin (CP), IR and UV. CNE1 and CNE2 cells were transiently transfected with Jab1 siRNA (Jab1-si) or scrambled control siRNA (Cont-si) and treated with CP, IR or UV for 48 h. Apoptosis was analyzed by Annexin-V and PI staining (a), detection of cleaved PARP (C-PARP) and caspase-3 (C-Caspase 3) (b, c and d). The percentage of CNE2 cells in the sub-G1 phase are shown (b and c, right). Data represent three independent experiments, mean±s.d. *P <0.05, **P <0.01.
To confirm this finding, we evaluated the levels of cleaved PARP and caspase-3 by western blotting and apoptosis by PI staining. After treating cells with cisplatin, IR and UV, both CNE1 and CNE2 cells transfected with Jab1 siRNA displayed higher levels of either cleaved PARP or caspase-3 than the control cells (Figure 4b, 4c and 4d). Moreover, we exposed Jab1 siRNA–treated CNE2 cells with different doses of cisplatin and found a increases in apoptotic cells compared with the control cells (1.1 to 1.6 folds more apoptosis; Figure 4b, right). Similar results were found when the cells were treated with UV radiation (2.3- to 3.2-fold more apoptosis; Figure 4c, right). Thus, we conclude that the role of Jab1 depletion in cisplatin, IR and UV radiation was not cell-line specific.
Jab1-deficient cells have increased sensitivity to DNA damage stimuli
Enhanced DNA damage repair is one of the mechanisms of cisplatin or irradiation resistance (19). To identify the effect of Jab1 on DNA damage and repair, reverse-phase protein microarray (RPPA) analysis was performed to examine the activation status of several key signaling molecules related to DNA damage and repair that are induced upon genotoxic stress. Quantitative assessment of total- and phospho-protein levels in Jab1-knockdown cell lines, by RPPA analysis showed much larger changes in the levels of apoptosis markers (cleaved caspase-3, 7, 8 and PARP, Bax, Bak, Bcl-xL) and DNA damage markers (p53, p-Chk2 and NF-kB) and DNA damage repair gene Rad51 than in the control siRNA-treated cells upon exposure to cisplatin and UV radiation (Figure 5a and 5b). The results of the RPPA analysis agree with our western blotting data, confirming that Jab1 depletion promotes apoptosis in NPC cells after cisplatin and UV radiation treatment.
Figure 5.
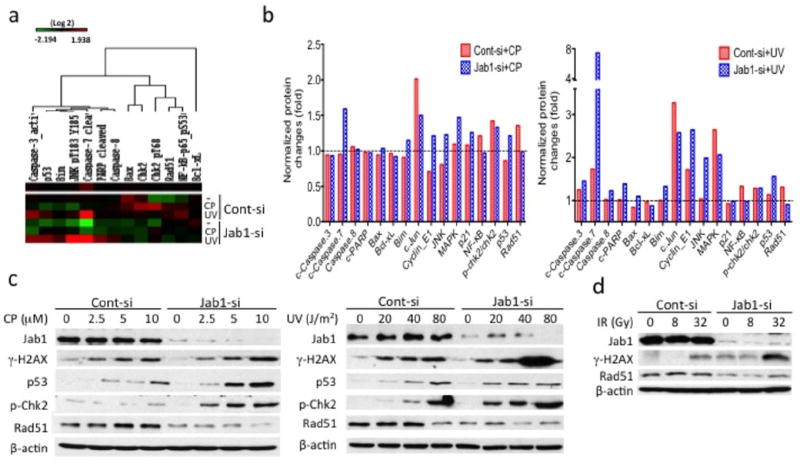
Knockdown of Jab1 impairs DNA repair and decreases expression of the DNA repair gene Rad51. (a) (Left) RPPA analysis of cisplatin (CP)- and UV- treated NPC cells. Cell lysates were harvested after treatment with 5 μM CP or 40 J/m2 UV for 48 h. Samples were organized by treatment, which is indicated on the right side of the heat-map. (Middle and Right) The response of the cells to CP or UV was normalized against the response of untreated cells. (c, d) HONE1 cells were transfected with Jab1 or control siRNA 48 h before CP, UV or IR exposure. Cell lysates were prepared 48 h after CP, UV or IR exposure and western blotting was performed with the indicated antibodies related to DNA damage and repair proteins.
To confirm the results of the RPPA analysis, Jab1 siRNA–treated cells were exposed to cisplatin or UV radiation, and the levels of several crucial DNA damage and repair proteins involved in both cisplatin and UV induction, including phosphor-specific (Ser-139) histone H2AX (γ-H2AX), phospho-Chk2, p53 and Rad51 (20-25), were analyzed. Jab1 siRNA–treated HONE1 cells exhibited higher levels of γ-H2AX, which are early indicators of the presence of DNA double-strand breaks (Figure 5c). γ-H2AX spans megabases of DNA flanking a DNA damage site and allows the recruitment of DNA damage repair proteins. Similarly, the levels of phospho-Chk2, a key molecule in transducing DNA damage signals (26), were increased after cisplatin and UV exposure regardless of whether the cells were treated with Jab1 siRNA (although the phospho-Chk2 levels were higher in Jab1-deficient cells). By contrast, levels of Rad51, a key protein for DNA repair (27), were reduced in Jab1 siRNA-treated NPC cells 48 h after the cells were treated with cisplatin or UV radiation (Figure 5c). Additionally, we also found that γ-H2AX levels were higher, whereas the Rad51 levels were lower in Jab1 siRNA-treated cells than in control siRNA-treated cells upon IR exposure (Figure 5d). These results indicate that Jab1 deficiency enhances DNA damage and decreases DNA repair after exposure to DNA damage stimuli.
Decreased DNA repair in Jab1 knockdown cells correlated with reduced expression of the DNA repair gene Rad51
To further induce the interaction between Jab1 and Rad51, we transfected NPC cells with an ectopic Jab1 plasmid or Jab1 siRNA or transduced them with Jab1 adenovirus in the presence or absence of doxycycline. As expected, 48 h after transfection, Jab1 protein levels were substantially increased in the Jab1 plasmid– and adenovirus–treated cells or decreased in the Jab1 siRNA–treated cells (Figure 6a). The exogenous overexpression of Jab1 was associated with an increase in Rad51 levels, while reduced expression of endogenous Jab1 protein was associated with a decrease in Rad51 levels in the NPC cell lines. Moreover, a correlation plot of the Jab1 and Rad51 blot intensities (using data pooled from control- and Jab1 siRNA– or Myc-Jab1 plasmid–transfected or Jab1 adenovirus–transduced cells) showed that there is a linear correlation between the levels of the two proteins (r= 0.6824; p=0.0072), confirming that the levels of Jab1 are proportional to the Rad51 levels in NPC cells.
Because increased levels of p53 were found in Jab1-deficient cells exposed to cisplatin or UV radiation and because p53 has been reported to regulate Rad51 expression (28, 29), we investigated whether the reduction in Rad51 upon knockdown of Jab1 was mediated by negative regulation of p53. As expected, we found that the levels of Rad51 were lower in Jab1 siRNA and shRNA–treated NPC cells than in control siRNA and shRNA–treated cells (Figure 6b and 6c). However, after knocking down p53 expression, there was no or less reduction in the Rad51 levels in the Jab1 siRNA and shRNA–treated NPC cells (Figure 6b and 6c). To further confirm this results, we also extracted the total RNA from siRNA transfected stable cell lines and performed the real time PCR, again, we found that Rad51 RNA level was lower accompanied by decreased level of Jab1 in shRNA-treated CNE2 cells; however, the Rad51 RNA level was increased after p53 siRNA transfected, regardless of Jab1 or control shRNA treatment (Figure 6c, right). These results indicate that reduction of Rad51 levels in Jab1-deficient cells is at least partly p53-dependent.
Furthermore, Jab1 depletion not only decreased the level of Rad51 but also affected its activity, as indicated by loss of Rad51 causing extensive chromosomal breaks, leading to apoptosis (30). A colony formation assay showed that Jab1-deficient cells were less efficient at forming colonies after cisplatin or IR or UV treatment, and this effect was attributable to the reduction of Rad51 (Figure 6d). In addition, transiently transfecting Rad51 plasmid DNA into Jab1-deficient cells altered and reversed their survival curves: the pattern of colony formation in these cells was similar to that of the control shRNA–treated cells, indicating that adding back Rad51 can rescue the DNA damage repair function in Jab1-deficient cells (Figure 6d). Western blotting confirmed the ectopic expression of Rad51 in the siRNA-transfected cells (Figure 6d, left panel). To determine whether rescue of the DNA repair function by the expression of ectopic Rad51 was reflected in decreased DNA damage and thus led to reduced apoptosis, we measured levels of apoptosis and DNA damage repair proteins by western blot. Jab1 shRNA–treated cells had higher levels of cleaved caspase-3 and γ-H2AX after cisplatin, IR and UV exposure (Figure 6e, Top), supporting our hypothesis that Jab1 depletion enhances genotoxic stress-induced apoptosis and DNA damage. Furthermore, ectopic Rad51 expression reduced levels of cleaved caspase-3 and γ-H2AX induced by cisplatin, IR and UV (Figure 6e, bottom). These results suggest that Jab1 depletion reduces the expression of Rad51, leading to a reduction in the ability of cells to repair DNA lesions and an increase in apoptosis, thus sensitizing NPC cells to cisplatin, IR and UV.
Discussion
Here, we have shown that overexpression of Jab1 plays an important role in cisplatin and irradiation resistance in NPC cells by positively regulating the levels of the DNA repair gene Rad51. These findings suggest that Jab1 is a major contributor to the cisplatin, IR and UV resistance of NPC and indicate that Jab1 expression could be used as a biomarker in predicting treatment outcomes of patients with NPC treated with cisplatin- and irradiation-based therapy.
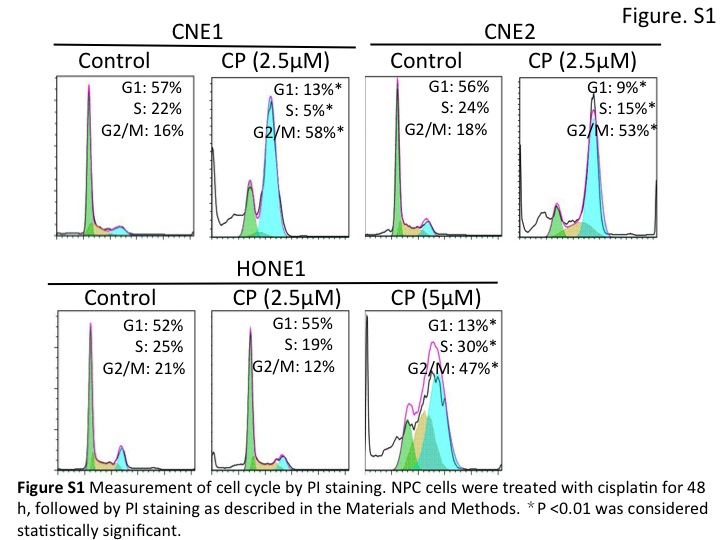
Chemotherapy and radiotherapy are the most common treatments for cancers, but patients frequently do not respond to these treatments (8). Resistance to chemotherapy or radiotherapy may result from failure of the apoptosis pathways that are activated in response to drug treatment or irradiation. Our data showed that NPC cell lines have different responses to cisplatin, IR and UV radiation. CNE2 and HONE1 cells are relatively more resistant to cisplatin, IR and UV than CNE1 cells. It has been widely reported that cisplatin interfere with the cell cycle to act as anti-cancer agents. Cisplatin has been found to arrest the cell cycle at the G2/M phase in several tumor cell types, including NPC (31). Consistent with these reports, we also observed an increase in G2/M cell population following cisplatin treatment in NPC cells (Fig. S1). Interestingly, we found that a lower cisplatin concentration (2.5 μM) did not significantly affect cell cycle progression in HONE1 cells. However, a more substantial cisplatin concentration (5 μM) arrested HONE1 cells at the G2/M phase approximately 2-fold by a mechanism that was not sensitive to cisplatin. Here it is noteworthy that DNA damage is thought to contain an amplification step that converts minor DNA damage into a full cell cycle arrest response (32). The fact that we observed cell cycle arrest only in response to major, but not minor, cisplatin-induced DNA damage also agrees with previous reports that only extensive DNA damage, and not minor damage, induced mitotic checkpoint activation (31, 33). DNA damage stimuli can promote apoptosis in tumor cells, but cells may survive or experience G1 or G2/M cell cycle arrest upon DNA damage because of their inability to undergo apoptosis (34). In our study, we found CNE1 cells had a remarkable sub-G1 content characteristic of apoptotic cells in response to cisplatin, IR or UV. However, both CNE2 and HONE1 cells had less cell apoptosis in response to the same treatment. Thus, in our next experiments we determined whether loss of Jab1 promotes apoptosis of NPC cells in response to DNA damage. Our data indicate that CNE1 cells are more sensitive to cisplatin, a result that is also supported by Zizhen et al. (35). However, the cause of this treatment resistance is not fully understood. Our study revealed that Jab1 depletion contributes to increased UV and IR and cisplatin sensitivity by increasing DNA damage and suppressing DNA repair, which in turn, contributes to an increase in cisplatin-, IR-, and UV-induced apoptosis. This outcome establishes Jab1 as a potential marker for predicting the response of NPC to cisplatin and irradiation, helping to guide future treatment of NPC patients.
Recently, increased DNA damage repair has been reported to be one of the mechanisms of cisplatin or irradiation resistance (19). In eukaryotes, the DNA-repairing protein Rad51 plays a central role in DNA damage repair by forming nucleoprotein filaments and mediating strand exchange between DNA duplexes (27). Rad51 is essential for embryonic survival in response to exogenous DNA-damaging agents (36) and for the repair of spontaneously occurring chromosome breaks in proliferating cells (37). Aberrant levels of Rad51 have also been observed in a number of transformed cell types that may induce malignant transformation (38). Furthermore, overexpression of Rad51 can enhance spontaneous recombination frequency and increase resistance to double-strand break-inducing cancer therapies (29, 39). Inhibiting Rad51, therefore, has been explored as a way to sensitize cancer cells to chemotherapy and radiotherapy (40, 41). Our results showed that increased levels of γ-H2AX, a marker of DNA damage, were correlated with reduced expression of Rad51, indicating that a DNA repair defect was the cause of the increase in DNA damage in Jab1-deficient cells. This result could explain the impaired growth and increased apoptosis seen in Jab1-deficient cells exposed to DNA damaging stimuli. Furthermore, Jab1 positively regulates Rad51 in NPC, explaining why Jab1 depletion sensitizes NPC cells to cisplatin, IR and UV. By contrast, ectopic expression of Rad51 increased resistance to cisplatin, IR and UV in Jab1-deficient cells. This depletion of Jab1 resulting in the deregulation of the Rad51 and defects in DNA repair, contribute to the increased sensitivity of the cells to cisplatin, IR and UV. Our results also demonstrated that p53 is essential for the Jab1-mediated regulation of Rad51. The significance of the Jab1-mediated regulation of Rad51 needs to be explored in future mechanistic studies.
In conclusion, we demonstrated that Jab1 contributes to the sensitivity/resistance of NPC cells to cisplatin and irradiation. Jab1 depletion results in more cisplatin-, IR- and UV-induced apoptosis and DNA damage in NPC cells. The current finding that Jab1 positively regulates Rad51 and contributes to the response of NPC cells to cisplatin, IR and UV suggests that assessing the Jab1 level in patients may help predict response to cisplatin and radiation treatments, allowing the design of individualized treatment strategies for patients with NPC.
Materials and methods
Reagents
Cell culture media were purchased from Mediatech Inc (Mannassas, VA, USA) and fetal bovine serum (FBS) was obtained from Gibco (Grand Island, NY, USA). Antibodies to the following proteins were used: Jab1 (Santa Cruz, CA, USA); PARP and p53 (BD Biosciences PharMingen, San Diego, CA, USA); Myc-tag (Roche Applied Biosciences, Indianapolis, IN, USA); caspase-3, γ-H2AX (p-Ser-139), and phosphor-Chk2 (Cell Signaling Technology); Rad51 (Calbiochem, San Diego, CA, USA); and β-actin (Sigma-Aldrich, St. Louis, MO, USA). TaqMan® gene expression assays for the human Jab1/CSN5 (Hs00272789_m1), human Rad51 (Hs00153418_m1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Hs99999905_m1) were from Applied Biosystems/Life Technology (Carlsbad, CA, USA). Lipofectamine Plus reagent and Oligofectamine reagent were purchased from Invitrogen (Carlsbad, CA, USA). Western Lightning Chemiluminescence Plus reagent was purchased from Thermo Scientific Pierce. The Annexin V/propidium iodide (PI) kit was purchased from BD Biosciences. A cell proliferation assay kit was purchased from Roche.
Cell cultures, plasmid and siRNA transfection
The human NPC cell lines CNE1 (well-differentiated, obtained from the Cancer Center, Sun Yat-sen University, China), CNE2 (poorly differentiated, given from the Cancer Center, Sun Yat-sen University), and HONE1 (poorly differentiated, the generous gift of Professor Ronald Glaser The Ohio State University Medical Center (Columbus, OH, USA), were cultured in RPMI-1640 medium containing 10% FBS and penicillin-streptomycin sulfate. The Myc-Jab1-pcDNA3.1 plasmid has been described previously (13). Cells were transfected with the plasmid using the Lipofectamine Plus reagent. The negative control small interfering RNA (siRNA; siControl 3) and siRNA targeting human Jab1/CSN5 (siCOPS5 #18546) were purchased from Ambion, Inc. (Austin, TX, USA). Transient transfections of NPC cells were performed using the Oligofectamine (Invitrogen) protocol with 5 nmol siRNA in RPMI-1640 with 10% FBS and no penicillin-streptomycin.
Adenoviral vectors and gene transduction
A recombinant adenoviral vector expressing a doxycycline-regulated (Tet-Off) form of human Jab1 (Ad-Myc-Jab1) was constructed, as previously described (13, 42). NPC cells were transduced for 48 h with a regulatory virus (adeno-X Tet-Off, Clontech, Palo Alto, CA) and Ad-Myc-Jab1 at a multiplicity of infection of 50 in a growth medium containing 10% FBS in the presence or absence of 1 ug/mL doxycycline, a tetracycline analogue.
Establishment of small hairpin RNA (shRNA) stable cells
To generate stable cell lines with knockdown of Jab1, Jab1 shRNA oligonucleotides were cloned into a retrovirus pSIREN-RetroQ system (Clontech, Moutainview, CA, USA). The packaging cell line 293T was cotransfected with Jab1 shRNA–vector DNA along with the helper vectors pCGP and pVSVG using the Lipofectamine PLUS reagent. The supernatant was collected 48 h after transfection, filtered through a 0.45μm syringe filter, supplemented with 1.2 μg/ml of Polybrene, and used to infect target cells. The medium was replaced by RPMI-1640 with 10% FBS after 12 h. Stable clones were selected following treatment with puromycin at 0.8 μg/mL for 2 weeks. Positive clones were further confirmed by immunoblot analysis and maintained in 0.2 μg/mL puromycin.
Cell proliferation assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to evaluate relative viable cells. Cells were UV irradiated in a FB-UVXL-1000 UV crosslinker (Fisher) or γ-irradiated using a J. L. Shepherd and Associates (CA) Mark I-30 137Cs irradiator at MD Anderson Cancer Center. Briefly, 48 h after transfection, NPC cells were seeded in 96-well plates or 24-well plates in RPMI-1640 medium with 10% FBS, overnight, then cells were exposed to different concentrations of cisplatin or doses of UV or IR as indicated. The MTT labeling reagent was added, and the spectrophotometric absorbance of the samples was read using a microplate (ELISA) reader at 570 nm. The data were analyzed using GraphPad Prism 4 (GraphPad Software, La Jolla, CA, USA) to obtain the IC50. The relative resistance factor (RRF) was calculated by dividing the IC50 of cells with altered Jab1 expression by the IC50 of control cells.
Colony formation assay
CNE2 cells with shRNA-mediated knockdown of Jab1 and control cells (400 cells/well) were plated in six-well plates with RPMI-1640 medium and 10% FBS for growth analysis. The following day, the cells were treated with or without indicated doses of cisplatin or UV or IR for 48 h. After 10 days, the cells were fixed, stained with 0.1% crystal violet, and scored by counting the number of colonies with an inverted microscope, using the standard definition that a colony consists of 50 or more cells.
Hoechst 33342 staining
To detect apoptosis, nuclear staining was performed as described previously (43) using 10 mg/ml Hoechst 33342, and cells were analyzed with a fluorescence microscope (magnification x200 for nuclear analysis and x100 for morphologic analysis). Apoptotic cells were identified by morphologic features and by the presence of condensed and fragmented nuclei. The percentage of apoptotic cells was calculated as the number of apoptotic cells divided by the number of total cells multiplied by 100. Three independent experiments were conducted, and at least 300 cells were counted for each experiment.
Flow-cytometry analysis of the cell cycle and apoptosis
PI staining was performed as described previously (43). Briefly the treated cells were fixed overnight, washed in cold phosphate-buffered saline (PBS), labeled with PI, and analyzed after staining using a FACScan flow cytometer (BD Biosciences).
For dual Annexin V-fluoroisothiocyanate (FITC) and PI staining, siRNA-treated cells were exposed to 5 μmol/l cisplatin for 48 h and then collected and resuspended in binding buffer containing Annexin V-FITC and PI according to the manufacturer’s recommendations. Annexin V-FITC and PI staining were quantified using a FACScan flow cytometer.
Cell extracts and immunoblotting
The treated cells were collected, washed twice in cold PBS, and lysed as described previously (15). Proteins in the total cell lysates were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and probed with anti-Jab1, anti-Myc, anti-PARP, anti-caspase-3, anti-Rad51, anti-p-Chk2, and anti-γ-H2AX antibodies. β-actin was used as the internal positive control for all immunoblots. Immunoreactive bands were detected using horseradish peroxidase (HRP)-conjugated secondary antibodies with the Western Lightning Chemiluminescence Plus reagent (Thermo Scientific Pierce, Rockford, IL, USA). The protein levels were quantified using ImageJ software (National Institute of Mental Health, Bethesda, MD, USA; http://rsb.info.nih.gov/ij). PARP and caspase-3 activity were measured as the percentage of cleaved PARP or caspase-3 in the samples. This was calculated as the ratio of the intensity of the bands corresponding to the cleaved proteins to the intensity of the bands corresponding to the total proteins as follows: % PARP or caspase-3 = 100% × Tc/Tt, where Tc is the intensity value of the cleaved protein bands, and Tt is the intensity value of total protein bands.
RNA extraction and quantitative PCR
Total RNA was extracted using Trizol regent, and assessed by quantitative PCR using commercially available TaqMan® gene expression assays from Applied Biosystems for the Jab1, Rad51, and GAPDH. The thermal profile for quantitative real-time PCR (Q-PCR) reactions in the Step One instrument (Eppendorf) was 30 min at 48°C, 10 min at 95°C, 40 cycles of 15 s at 95°C, and 1 min at 60°C. All samples were normalized to internal controls and fold changes were calculated through relative quantification (2-ΔΔCt) following the supplier’s protocol.
Reverse-phase protein array analysis
Jab1 siRNA– or control siRNA–treated NPC cells were seeded in six-well plates overnight, treated with 40 J/m2 UV radiation or 5 μM of cisplatin, and harvested after 48 h. The preparation of protein lysates for RPPA analysis has been described previously (44). The RPPA analysis was performed by the Functional Proteomics Core Facility at The University of Texas MD Anderson Cancer Center. Samples were analyzed for the expression of 136 protein markers using RPPA-validated antibodies (Supplementary Table S1). Results were normalized as previously described (44). After correcting for sample loading differences, the protein expression levels were normalized against those of the untreated control cells. Clustering analysis was performed using Cluster 2.1 and visualized using Treeview software (http://rana.lbl.gov/EisenSoftware.html).
Statistical analysis
Statistical analysis of the results was done using Student t test when only two groups were compared, or one-way analysis of variance when more than two groups were compared. Differences between groups were stated to be statistically significant when P < 0.05. All computations were carried out with SPSS 19.0 (SPSS, Chicago, IL, USA)
Supplementary Material
{kind=link}
Acknowledgments
We thank Zahid H. Siddik, Do Youn Oh for helpful discussions and Ronald Glaser for kindly providing HONE1 cells. This work was supported by a fellowship from China Scholarship Council (2010638087) (YP), a grant from National Cancer Institute (RO1-CA90853) (FXC); The University of Texas M.D. Anderson Functional Proteomics Core Facility is supported by an NCI Cancer Center Support Grant (CA16672); National Natural Science Foundation of China (81071837; 30670627); Natural Science Foundation of Guangdong Province, China (9251008901000005; 06021210) and Scientific and Technological Project of Guangdong, China (2008A030201009; 2010B050700016) (HY). We thank Kate J. Newberry and Sunita Patterson for editing the manuscript.
Footnotes
Conflict of interest statement: The authors have no conflicts of interest to declare.
References
- 1.Wei WI, Sham JS. Nasopharyngeal carcinoma. Lancet. 2005 Jun 11-17;365(9476):2041–2054. doi: 10.1016/S0140-6736(05)66698-6. [DOI] [PubMed] [Google Scholar]
- 2.Lo KW, Chung GT, To KF. Deciphering the molecular genetic basis of NPC through molecular, cytogenetic, and epigenetic approaches. Semin Cancer Biol. 2012 Jan 10; doi: 10.1016/j.semcancer.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Spano JP, Busson P, Atlan D, Bourhis J, Pignon JP, Esteban C, et al. Nasopharyngeal carcinomas: an update. Eur J Cancer. 2003 Oct;39(15):2121–2135. doi: 10.1016/s0959-8049(03)00367-8. [DOI] [PubMed] [Google Scholar]
- 4.Lo KW, To KF, Huang DP. Focus on nasopharyngeal carcinoma. Cancer Cell. 2004 May;5(5):423–428. doi: 10.1016/s1535-6108(04)00119-9. [DOI] [PubMed] [Google Scholar]
- 5.Hui EP, Ma BB, Leung SF, King AD, Mo F, Kam MK, et al. Randomized phase II trial of concurrent cisplatin-radiotherapy with or without neoadjuvant docetaxel and cisplatin in advanced nasopharyngeal carcinoma. J Clin Oncol. 2009 Jan 10;27(2):242–249. doi: 10.1200/JCO.2008.18.1545. [DOI] [PubMed] [Google Scholar]
- 6.Le QT, Tate D, Koong A, Gibbs IC, Chang SD, Adler JR, et al. Improved local control with stereotactic radiosurgical boost in patients with nasopharyngeal carcinoma. Int J Radiat Oncol Biol Phys. 2003 Jul 15;56(4):1046–1054. doi: 10.1016/s0360-3016(03)00117-2. [DOI] [PubMed] [Google Scholar]
- 7.Yip KW, Mocanu JD, Au PY, Sleep GT, Huang D, Busson P, et al. Combination bcl-2 antisense and radiation therapy for nasopharyngeal cancer. Clin Cancer Res. 2005 Nov 15;11(22):8131–8144. doi: 10.1158/1078-0432.CCR-05-1266. [DOI] [PubMed] [Google Scholar]
- 8.Liu RY, Dong Z, Liu J, Yin JY, Zhou L, Wu X, et al. Role of eIF3a in regulating cisplatin sensitivity and in translational control of nucleotide excision repair of nasopharyngeal carcinoma. Oncogene. 2011 May 30;30(48):4814–4823. doi: 10.1038/onc.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Claret FX, Hibi M, Dhut S, Toda T, Karin M. A new group of conserved coactivators that increase the specificity of AP-1 transcription factors. Nature. 1996 Oct 3;383(6599):453–457. doi: 10.1038/383453a0. [DOI] [PubMed] [Google Scholar]
- 10.Shackleford TJ, Claret FX. JAB1/CSN5: a new player in cell cycle control and cancer. Cell Div. 2010;5:26. doi: 10.1186/1747-1028-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kouvaraki MA, Rassidakis GZ, Tian L, Kumar R, Kittas C, Claret FX. Jun activation domain-binding protein 1 expression in breast cancer inversely correlates with the cell cycle inhibitor p27(Kip1) Cancer Res. 2003 Jun 1;63(11):2977–2981. [PubMed] [Google Scholar]
- 12.Rassidakis GZ, Claret FX, Lai R, Zhang Q, Sarris AH, McDonnell TJ, et al. Expression of p27(Kip1) and c-Jun activation binding protein 1 are inversely correlated in systemic anaplastic large cell lymphoma. Clin Cancer Res. 2003 Mar;9(3):1121–1128. [PubMed] [Google Scholar]
- 13.Kouvaraki MA, Korapati AL, Rassidakis GZ, Tian L, Zhang Q, Chiao P, et al. Potential role of Jun activation domain-binding protein 1 as a negative regulator of p27kip1 in pancreatic adenocarcinoma. Cancer Res. 2006 Sep 1;66(17):8581–8589. doi: 10.1158/0008-5472.CAN-06-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan Y, Zhang Q, Tian L, Wang X, Fan X, Zhang H, et al. Jab1/CSN5 Negatively Regulates p27 and Plays a Role in the Pathogenesis of Nasopharyngeal Carcinoma. Cancer Res. 2012 Apr 1;72(7):1890–1900. doi: 10.1158/0008-5472.CAN-11-3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian L, Peng G, Parant JM, Leventaki V, Drakos E, Zhang Q, et al. Essential roles of Jab1 in cell survival, spontaneous DNA damage and DNA repair. Oncogene. 2010 Nov 18;29(46):6125–6137. doi: 10.1038/onc.2010.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010 Oct 22;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994 Sep 22;371(6495):346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 18.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995 Jul 6;376(6535):37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 19.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003 Oct 20;22(47):7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 20.Cruet-Hennequart S, Villalan S, Kaczmarczyk A, O’Meara E, Sokol AM, Carty MP. Characterization of the effects of cisplatin and carboplatin on cell cycle progression and DNA damage response activation in DNA polymerase eta-deficient human cells. Cell Cycle. 2009 Sep 15;8(18):3039–3050. [PubMed] [Google Scholar]
- 21.Lee JH, Kang Y, Khare V, Jin ZY, Kang MY, Yoon Y, et al. The p53-inducible gene 3 (PIG3) contributes to early cellular response to DNA damage. Oncogene. 2010 Mar 11;29(10):1431–1450. doi: 10.1038/onc.2009.438. [DOI] [PubMed] [Google Scholar]
- 22.Pabla N, Ma Z, McIlhatton MA, Fishel R, Dong Z. hMSH2 recruits ATR to DNA damage sites for activation during DNA damage-induced apoptosis. J Biol Chem. 2011 Mar 25;286(12):10411–10418. doi: 10.1074/jbc.M110.210989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pedram A, Razandi M, Evinger AJ, Lee E, Levin ER. Estrogen inhibits ATR signaling to cell cycle checkpoints and DNA repair. Mol Biol Cell. 2009 Jul;20(14):3374–3389. doi: 10.1091/mbc.E09-01-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Revet I, Feeney L, Bruguera S, Wilson W, Dong TK, Oh DH, et al. Functional relevance of the histone gammaH2Ax in the response to DNA damaging agents. Proc Natl Acad Sci U S A. 2011 May 24;108(21):8663–8667. doi: 10.1073/pnas.1105866108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dmitrieva NI, Cui K, Kitchaev DA, Zhao K, Burg MB. DNA double-strand breaks induced by high NaCl occur predominantly in gene deserts. Proc Natl Acad Sci U S A. 2011 Dec 20;108(51):20796–20801. doi: 10.1073/pnas.1114677108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buscemi G, Perego P, Carenini N, Nakanishi M, Chessa L, Chen J, et al. Activation of ATM and Chk2 kinases in relation to the amount of DNA strand breaks. Oncogene. 2004 Oct 7;23(46):7691–7700. doi: 10.1038/sj.onc.1207986. [DOI] [PubMed] [Google Scholar]
- 27.Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992 May 1;69(3):457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 28.Arias-Lopez C, Lazaro-Trueba I, Kerr P, Lord CJ, Dexter T, Iravani M, et al. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006 Feb;7(2):219–224. doi: 10.1038/sj.embor.7400587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hannay JA, Liu J, Zhu QS, Bolshakov SV, Li L, Pisters PW, et al. Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: a role for p53/activator protein 2 transcriptional regulation. Mol Cancer Ther. 2007 May;6(5):1650–1660. doi: 10.1158/1535-7163.MCT-06-0636. [DOI] [PubMed] [Google Scholar]
- 30.Qing Y, Yamazoe M, Hirota K, Dejsuphong D, Sakai W, Yamamoto KN, et al. The epistatic relationship between BRCA2 and the other RAD51 mediators in homologous recombination. PLoS Genet. 2011 Jul;7(7):e1002148. doi: 10.1371/journal.pgen.1002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheung HW, Jin DY, Ling MT, Wong YC, Wang Q, Tsao SW, et al. Mitotic arrest deficient 2 expression induces chemosensitization to a DNA-damaging agent, cisplatin, in nasopharyngeal carcinoma cells. Cancer Res. 2005 Feb 15;65(4):1450–1458. doi: 10.1158/0008-5472.CAN-04-0567. [DOI] [PubMed] [Google Scholar]
- 32.Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996 Dec 6;274(5293):1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 33.Mikhailov A, Cole RW, Rieder CL. DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr Biol. 2002 Oct 29;12(21):1797–1806. doi: 10.1016/s0960-9822(02)01226-5. [DOI] [PubMed] [Google Scholar]
- 34.Ma W, Lin Y, Xuan W, Iversen PL, Smith LJ, Benchimol S. Inhibition of p53 expression by peptide-conjugated phosphorodiamidate morpholino oligomers sensitizes human cancer cells to chemotherapeutic drugs. Oncogene. 2012 Feb 23;31(8):1024–1033. doi: 10.1038/onc.2011.300. [DOI] [PubMed] [Google Scholar]
- 35.Feng Z, Xu S, Liu M, Zeng YX, Kang T. Chk1 inhibitor Go6976 enhances the sensitivity of nasopharyngeal carcinoma cells to radiotherapy and chemotherapy in vitro and in vivo. Cancer Lett. 2010 Nov 28;297(2):190–197. doi: 10.1016/j.canlet.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 36.Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, et al. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci U S A. 1996 Jun 25;93(13):6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, et al. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. Embo J. 1998 Jan 15;17(2):598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002 Jan 1;62(1):219–225. [PubMed] [Google Scholar]
- 39.Vispe S, Cazaux C, Lesca C, Defais M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res. 1998 Jun 15;26(12):2859–2864. doi: 10.1093/nar/26.12.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collis SJ, Tighe A, Scott SD, Roberts SA, Hendry JH, Margison GP. Ribozyme minigene-mediated RAD51 down-regulation increases radiosensitivity of human prostate cancer cells. Nucleic Acids Res. 2001 Apr 1;29(7):1534–1538. doi: 10.1093/nar/29.7.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Russell JS, Brady K, Burgan WE, Cerra MA, Oswald KA, Camphausen K, et al. Gleevec-mediated inhibition of Rad51 expression and enhancement of tumor cell radiosensitivity. Cancer Res. 2003 Nov 1;63(21):7377–7383. [PubMed] [Google Scholar]
- 42.Zhang Q, Tian L, Mansouri A, Korapati AL, Johnson TJ, Claret FX. Inducible expression of a degradation-resistant form of p27Kip1 causes growth arrest and apoptosis in breast cancer cells. FEBS Lett. 2005 Jul 18;579(18):3932–3940. doi: 10.1016/j.febslet.2005.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, et al. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003 May 23;278(21):19245–19256. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 44.Davies MA, Stemke-Hale K, Lin E, Tellez C, Deng W, Gopal YN, et al. Integrated Molecular and Clinical Analysis of AKT Activation in Metastatic Melanoma. Clin Cancer Res. 2009 Dec 15;15(24):7538–7546. doi: 10.1158/1078-0432.CCR-09-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.