

Abstract
Comprehensive sampling is crucial to DNA barcoding, but it is rarely performed because materials are usually unavailable. In practice, only a few rather than all species of a genus are required to be identified. Thus identification of a given species using a limited sample is of great importance in current application of DNA barcodes. Here, we selected 70 individuals representing 48 species from each major lineage of Solanum, one of the most species-rich genera of seed plants, to explore whether DNA barcodes can provide reliable specific-species discrimination in the context of incomplete sampling. Chloroplast genes ndhF and trnS-trnG and the nuclear gene waxy, the commonly used markers in Solanum phylogeny, were selected as the supplementary barcodes. The tree-building and modified barcode gap methods were employed to assess species resolution. The results showed that four Solanum species of quarantine concern could be successfully identified through the two-step barcoding sampling strategy. In addition, discrepancies between nuclear and cpDNA barcodes in some samples demonstrated the ability to discriminate hybrid species, and highlights the necessity of using barcode regions with different modes of inheritance. We conclude that efficient phylogenetic markers are good candidates as the supplementary barcodes in a given taxonomic group. Critically, we hypothesized that a specific-species could be identified from a phylogenetic framework using incomplete sampling–through this, DNA barcoding will greatly benefit the current fields of its application.
Introduction
DNA barcoding is a species diagnostic technique using standardized DNA regions across all possible forms of life [1], [2]. This method is promising for taxonomy-related studies owing to its rapid and accurate use with micro materials, for which traditional identification is not feasible. DNA barcoding, using the mitochondrial coxI gene (COI), is now well established in animals (e.g.[3]–[5]). However, there is no such single locus to barcode land plants due to the low mutation rate of plant plastid genomes [6]–[8]. In addition, complex evolutionary histories, such as hybridization and polyploidy, are common in plants, and make species boundaries difficult to define [9]–[12]. Thus, multiple genetic loci might be necessarily included in plant barcodes to provide adequate information [13]–[15]. The selection of plant barcode loci involved complex trade-offs between universality and discrimination. The ideal barcodes would require a certain level of variation for discriminatory power. However, they should also be somewhat conservative for universality and ease of alignment. This double standard is a great challenge for the choice and use of a perfect barcode. As a result, a tiered method has been proposed: a first tier composed of a conservative (coding) region shared by all land plants provides resolution at a higher rank (e.g. family or genus) and an additional more variable (coding or noncoding) region provides resolution at species level [16]. Recently, a two-locus combination of matK+rbcL from the chloroplast genome and the nuclear ITS region was successively recommended as the core barcode for land plants [17]–[19]. However, the supplementary barcodes, the choice of which depends on the group itself, are still inconclusive. Therefore, screening and testing supplementary barcodes in certain groups may be an important goal for future DNA barcoding.
One applied field that urgently needs the barcode technique is biomonitoring, in which foreign species are required to be accurately and rapidly distinguished from their close domestic relatives [20], [21]. This process is difficult because the materials used for identification usually lack adequate information and/or sometimes only a fraction of organism is available, thus DNA barcoding will greatly benefit this work. However, to date, applying DNA barcodes to plant biosecurity is challenging. In addition to the problem of the loci chosen in plant barcoding as mentioned above, taxon sampling is another difficulty hindering its rapid use. Quarantine weeds are often exotic species within a large genus distributed worldwide. As a result, constructing a barcode library that includes quarantine weeds and all their relatives, with multiple individuals per species, is extremely difficult. In view of these problems and challenges, one important issue is how to carry out biomonitoring through DNA barcoding without comprehensive sampling.
To date, a large number of taxa have been phylogenetically studied, using efficient markers in sophisticated testing. Can these markers be used as the supplementary barcode regions for a given taxonomic group? If this is possible, it will greatly minimize the amount of work for the next supplementary barcode screening study. More importantly, adding the sequence information of species of interest, such as plants of economic importance, to the existing phylogenetic data matrixes would greatly reduce the sampling work. To some extent this would overcome the sampling difficulty that has always troubled the barcode researcher, especially when studying a large group with worldwide distribution or endangered species with rare materials. This issue, although very important to DNA barcoding, has not been critically investigated.
Solanum, with ca. 1400 species distributed worldwide, is the largest genus in the family Solanaceae and within the top ten of the most species-rich genera in seed plants [22], [23]. This taxonomic group contains not only many members of economic importance, such as eggplant and tomato, but also a large number of noxious weeds, among which four species S. carolinense, S. elaeagnifolium, S. rostratum, and S. torvum are of great concern as quarantine pests in China and other countries. These four invasive species are a serious threat to the ecological environment and livestock production owing to their strong adaptability and poisonous substances contained. However, they are difficult to remove artificially as the plants are covered with sharp prickles. For these reasons, they have been listed as the most dangerous weeds and are rigorously monitored by quarantine authorities. These species are, however, not easily distinguished from their relatives because of the shortage of reliable characters. This is especially so for quarantine and inspection staff, who mainly work with their seeds.
In this paper, DNA barcoding of S. carolinense, S. elaeagnifolium, S. rostratum, and S. torvum was studied in the context of biosecurity. Here, we selected two chloroplast gene regions (ndhF, and trnS-trnG) and one nuclear gene region (waxy), which have been widely used in Solanum phylogeny, as the supplementary barcode regions. Therefore, most sequences of the DNA regions used in this study are available in GenBank, and only a few species of interest were needed to add to the study. The general aims of the study were to (1) test the feasibility of using efficient phylogenetic markers as the supplementary barcode and (2) develop and test the hypothesis that barcoding of a single (or a few) species of interest could be realized through incomplete sampling. Thus, our results may provide new insights in the current fields of application of DNA barcoding on how to efficiently use existing phylogenetic information to identify specific species.
Materials and Methods
Sampling Strategy
Previous molecular phylogenetic studies showed that Solanum species can be divided into 13 groups [22], [23]. However, the species of interest are included exclusively in the Leptostemonum group, whose members include most of the spiny Solanum species. [24]. According to these results, we used 70 individuals of 48 Solanum species, including 31 samples from the present study and 39 from Genbank. These samples represent nine of the 13 groups of Solanum, with special emphasis on the Leptostemonum group–of which 35 species were sampled, covering all ten clades of the group [24](Table S1). Most sequences obtained from GenBank were extracted from published articles [22]–[27], and we added a few new individuals of species of interest, especially of the four quarantine species (S. carolinense, S. elaeagnifolium, S. rostratum, and S. torvum) and their possible closest relatives documented in existing phylogenetic studies. The materials used were mainly seeds from seed companies outside of China, weeds intercepted by CIQ (China Inspection and Quarantine) authorities, escaping species around import enterprises, and herbaria species exchanged from abroad. Thus, although most of the original sources of materials were uncertain, their diverse obtained sources guaranteed genetic divergence among the species. A potential outgroup of Jaltomata procumbens was determined from a previous phylogenetic study [23].
DNA Extraction, Amplification and Sequencing
Total genomic DNA was extracted using a modified CTAB protocol [28] or plant DNA Extraction Kit (Tiangen Biotech, Beijing, China). The PCR primer and its reaction conditions for ndhF region were followed from Olmstead and Sweere [29] and Bohs and Olmstead [25]; those for trnS-trnG were according to Hamilton [30] and Levin et al. [26]. The Waxy region was originally amplified and sequenced using primers 181F and 1171R [31], and Solanum specific primers were designed based these sequence (WAXYS: 5′-ACT GCT ATA AAC GTG GGG TTG ATC G-3′; WAXYA2∶5′-TGG AAC CAA CAT AAA ATC AGC-3′). The PCR programs were 94°C for 4 min, followed by 36 cycles of 94°C for 30 s, 53°C for 30 s, and 72°C for 1.5 min, with a single cycle of 72°C for 10 min. PCR products were purified using a Tiangen (Beijing, China) PCR purification kit, and then sequenced bi-directionally on a 3730XL DNA analyzer (Applied Biosystems, Foster City, CA, USA). For some of the waxy regions with poor sequence quality, PCR products were cloned with the pGEM-T EASY Vector System II (Promega), with 6–8 clones per individual selected and bi-directionally sequenced with the primers T7 and SP6.
Data Analysis
Sequence alignments were initially performed with ClustalX [32], and adjusted manually using BioEdit version 7.0.5 [33]. Sequence variation and Kimura 2-parameter (K2P) distance matrix were computed with MEGA 4.0 [34]. Barcoding gaps were evaluated by comparing the inter- and intra-specific genetic divergences [35]. To further show the genetic divergence of each individual, an alternative method was proposed and tested: we compared their K2P distances with each other, analyzed the matrix using a principal components analysis (PCA) module in MVSP (Multi-Variate Statistical Package, http://www.kovcomp.co.uk/mvsp/index.html) and constructed a scatter plot to show the result [36]. Species discrimination was evaluated through tree-based analysis. The Neighbor Joining (NJ) tree recommended as the standard barcoding method [1] was adopted and performed with MEGA 4.0 based on the K2P model [34], and branch support was evaluated with 500 bootstrap replicates. Phylogenetic analyses based on maximum parsimony (MP) were performed using the program PAUP* version 4.0b10 [37]. Bootstrap analyses based on 500 replicates with ten random additions per replicate were used to estimate the confidence of the clades. Unambiguous indels were treated as phylogenetic characters according to the simple indel coding method [38] and performed by GapCoder [39].
Results
Character Analysis of Barcode Sequences
Of the 31 individuals used in this study, PCR amplification was successful for all three loci. The PCR production of the waxy locus yielded a single band, in which 6–8 clone sequences were identical in the cloned samples, confirming the single copy of waxy in Solanum as previously reported [40]. We obtained additional trnS-trnG, ndhF and waxy sequences of 40 species from GenBank– a total of 213 sequences were used in the present study. The lengths of the aligned DNA fragments of trnS-trnG, ndhF and waxy were 681, 1745 and 1527 bp, respectively. Among the three regions, waxy provided the greatest number of variable sites (642) and the highest percentage of both variable characters (42.04%) and parsimony informative characters (21.68%). In addition, this region also showed the greatest mean inter–specific distance (0.0453) for the DNA barcode (Table 1). To get higher discriminatory power, we combined all DNA regions together. The combined matrix ranged from 3760 bp (S. thelopodium) to 3845 bp (S. macrocarpon7) in length, and the aligned length was 3953 bp, with 1080 (27.32%) variable characters and 552 (13.96%) informative characters (Table 1). To infer the putative hybrids among the examined species, the cpDNA and nuclear DNA were separately phylogenetically analyzed–when gaps were coded this produced a total of 2480 and 1621 bp alignment, respectively.
Table 1. Sequence characteristics of the three DNA regions and their combinations in the studied Solanum species.
| Statistic | ndhF | trnS-trnG | Waxy | ndhF+trnSG | ndhF+trnSG+Waxy |
| Length range (bp) | 1741–1744 | 556–610 | 1450–1503 | 2297–2353 | 3760–3845 |
| Aligned length (bp) | 1745 | 681 | 1527 | 2426 | 3953 |
| No. of variable characters (%) | 251 (14.38%) | 187 (27.46%) | 642 (42.04%) | 438 (18.05%) | 1080 (27.32%) |
| No. of parsimony informative characters (%) | 127 (7.28%) | 94(13.80%) | 331 (21.68%) | 221 (9.11%) | 552 (13.96%) |
| Sequence divergence (Pi) | 0.0138 | 0.0284 | 0.0453 | 0.0173 | 0.0277 |
Monophyletic Test Based on Phylogenetic Trees
The 71 individuals were divided into nine clades in the NJ tree, and each clade corresponding to a taxonomic group recognized by previous authors (Figure 1). In the Leptostemonum clade, species were subdivided into ten strongly supported clades, concordant with the complete ten taxonomic clades recognized by Levin et al. [24]. The four quarantine species S. carolinense, S. elaeagnifolium, S. rostratum and S. torvum were nested within the carolinense, elaeagnifolium, Crinitum and torvum clades, respectively. In these clades, the four target quarantine species were successfully identified because their individuals were clustered together into a 100% supported monophyletic group, which separated them from their closest relatives. All Solanum species with multiple individuals were recovered as monophyletic except those of S. luteum, S. macrocarpon and S. virginanum–especially the later two, which fell into two topologically disjunct but individually well supported clades (Figure 1). In order to explain the reasons for these results, the MP analyses were performed based on the cpDNA and nuclear DNA datasets respectively. In the MP tree of nuclear DNA (waxy), the individuals of S. macrocarpon divided into two distinct clades, one (S. macrocarpon) nested within the old world clade, while the other (S. macrocarpon7) fell into the Crinitum clade. However, these two individuals were clustered together in the cpDNA tree, in the same phylogenetic place as that of individual S. macrocarpon in the nuclear tree. These incongruent results indicated that the individual S. macrocarpon7 may be a hybrid species of S. macrocarpon (♀) × S. rostratum (♂). In contrast, the two individuals of S. virginanum were also phylogenetically conflicting but each was consistent in the place of cpDNA and nuclear DNA trees of their own (Figure S1). These results show that at least one of the two individuals was mistakenly identified. In sum, our results showed that the supplementary barcode of trnS-trnG+ndhF (cpDNA)+waxy (nuclear DNA) regions in Solanum had sufficient discriminatory power to not only identify a given species but also their hybrids.
Figure 1. Neighbor joining tree based on the three combined DNA regions (ndhF, trnS-trnG and waxy).

Bootstrap values (>80%) are shown above the branches. Numbers followed taxon names are individual numbers (see Table S1).
Barcode Gap Test
The barcoding gap enables the assignment of unknown individuals to their species with a negligible error rate. The utility of barcoding is based on the hypothesis that genetic variation between species is much larger than those within species, thus generating a barcoding gap. To date, the barcoding gap has been widely used in well-sampled groups to evaluate all-species discriminatory power of a barcode region. However, few studies have considered the application to specific-species resolution given insufficient sample data. In the present study, we identified four given species using a barcoding gap based on the assumption that a specific species can be identified if it was separated from its closest relative, even though the sampling was insufficient. By comparing the K2P distances, we tested whether the barcoding gap of the three combined DNA regions existed among the four quarantine species and their closest relatives. The results showed that three of the quarantine species had barcoding gaps, the exception being S. elaeagnifolium, with an intra- and inter-specific genetic variation overlap (Figure 2). We examined the source data of S. elaeagnifolium and its two closest relatives (S. hindsianum and S. tridynamum) and found that inter-specific genetic variation between S. hindsianum and S. tridynamum was close to the intra-specific genetic variation within S. elaeagnifolium, and thus led to the overlap (Table S2). Moreover, the PCA scatter plot, based on K2P genetic distances, showed that individuals within S. elaeagnifolium were more closely related to one another than any were to S. hindsianum or S. tridynamum (Figure 3). This result demonstrated a genetic gap between S. elaeagnifolium and its closest relatives.
Figure 2. Barcoding gaps between the four quarantine species and their closest relatives.
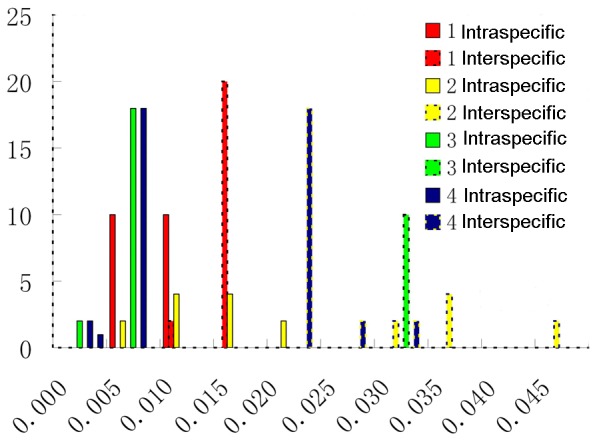
The X-axis relates to the K2P distances of the three combined DNA regions (ndhF, trnS-trnG and waxy) between the four quarantine species and their closest relatives. 1. S. elaeagnifolium; 2. S. carolinense; 3. S. torvum; 4. S. rostratum. The Y-axis corresponds to the number of occurrences.
Figure 3. Scatter plot of K2P genetic distances of S. elaeagnifolium and its closest relatives.

S. hindsianum and S. tridynamum. Thus, the results indicated that the heterogeneous nature of intra- and inter-specific genetic variations across all taxa blurred the barcode gap boundary and thus made all species identified through barcode gaps more difficult (Figure S2). In contrast, the barcoding gap between a given species and its closest relatives was always clear cut and could be used unambiguously to identify specific species (Figure 2 and 3).
Discussion
Unlike coxI, which is well established in animal DNA barcoding, there is no such single region with adequate efficiency to barcode all plant species. Although the two-loci combination of rbcL+matK and ITS have been proposed as the core barcode for land plants, no one marker or their combinations have discriminatory power of >80% [17]. This low resolving power of the core-barcode markers or their combinations limits their use to identifying ‘species group’ (e.g. family and genus) rather than species. As a result, using these markers to identify geographically diverse taxa in a given site where many samples are not necessarily closely related, and/or a large-scale taxonomic group with distantly related species, is workable [41]–[44], whereas, DNA barcoding of closely related species and/or taxonomically complex groups frequently fail [45]. Therefore, it is necessary to develop supplementary barcodes for particularly narrowly circumscribed taxonomic groups. In the present study, we selected the ndhF, trnS-trnG and waxy regions – the phylogenetic markers commonly used in Solanum studies – as the supplementary barcode to test discriminatory power. Theoretically, powerful phylogenetic markers do not always equate to an efficient DNA barcode, because variable characters of parsimony informative utility are not always the unique changes used as ‘species markers’ [13]. However, in our three DNA regions, the percentage of the variable characters increased with the parsimony informative characters increasing. Accordingly, the sequence divergence also increased with the variable characters. In the NJ tree, all individuals of a single species (except those of S. luteum, S. macrocarpon and S. virginanum, discussed below) clustered together in a monophyletic group. Furthermore, all of the three regions were 100% successful in PCR amplification. These results suggest that the phylogenetic markers are so efficient that they would be good candidates for DNA barcodes.
To date, plant barcodes have been based exclusively on the chloroplast genome. However, hybridization and polyploidy are common in plants [11], [46]–[48], which seriously affects the use of any uniparental inheritance locus for species boundary delimitation. Thus, a combination of DNA markers with different modes of inheritance is necessary in plant barcodes. Although the ITS region has been exclusively proposed as a nuclear marker, knotty problems, such as incomplete concerted evolution, fungal contamination and low recovery in some groups reduced its utility [49], [50]. Therefore, it is necessary to add additional nuclear regions as complementary markers in barcoding of a given taxonomic group. In the present study, we used both the chloroplast genes trnS-trnG and ndhF and the nuclear gene waxy to explore the species boundary, and the contradiction between different gene trees implied that the individual S. macrocarpon7 was a hybrid and one sample of S. virginanum had been misidentified. These results have universal significance, because both of the phenomena are common in plant systematics [13]. Thus, our results confirmed the importance of using multiple markers from different genomes for plant barcodes.
Taxa sampling, directly related to both the inter- and intra-specific divergence, is critically important in DNA barcoding. Although there is agreement that barcoding performs poorly in incomplete samples [35], [51] how many specimens are needed to construct a reliable reference for species identification is still inconclusive. Some authors suggested sampling 5–10 individuals per species (http://www.boldsystems.org/index.php/Login/page), but this is rarely done [52]. Identification of all species of a taxonomic group using complete sampling has been intensively investigated, but is it necessary to barcode a single or a few species using all congeneric species? If not, what is the minimum number and which is necessary needed? This is rarely assessed. In the present study, we explored a two-step sampling strategy to identify four quarantine species from a species-rich genus comprising ca. 1400 species. The first step was clade-sampling– sampling representive species from each primary evolutionary clade of the genus according to the previous phylogeny. Theoretically, genetic distances within clades are much smaller than those between clades, thus generating a genetic gap between clades. As a result, each clade of the group can be identified. We call this step ‘clade barcoding’. If an unknown species is nested within a given clade, we can then conduct the second step– adding species and individuals in this clade–until the unknown species is nested exclusively within a species’ monophyletic group. Thus, specific species can be identified. In using this sampling method, we densely sampled individuals of the target species and closest relatives, but only selectively sampled representatives of distinct relatives. As a result, a large number of unrelated species were removed from the analysis. This taxa sampling strategy, in combination with the utility of the previous phylogenetic markers, from which many sequence are available in Genbank, further reduce the sampling number in DNA barcoding.
Supporting Information
Comparison of cpDNA (left) and nuclear DNA tree (right) using maximum parsimony (MP) method. Bootstrap values (>75%) are shown above the branches. Numbers followed taxon names are individual numbers.
(TIF)
Distribution of inter- and intra-specific K2P distance of combined DNA regions in all studied species.
(TIF)
List of species used in this study.
(DOC)
Comparisons of sequence divergence (Kimura 2-parameter distance) among individuals of S. elaeagnifolium and its closest relatives S. hindsianum and S. tridynamum .
(DOC)
Acknowledgments
We thank Xiuling Shao and Xianxing Chen for providing materials; Yufen Xiong for laboratory assistance.
Funding Statement
This work was supported by a grant from the General Administration of Quality Supervision, Inspection and Quarantine, P. R. China (grant number 2010IK268) to XHF and by a grant from the Ministry of Science and Technology, P. R. China (grant number 2012BAK11B03). Independent innovation youth fund of Shandong University (Weihai) (1070501312002; 1070511300001) and collaborative innovation system construction fund of Shandong University (Weihai) (2012ZRXT004). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hebert PDN, Cywinska A, Ball SL, deWaard JR (2003) Biological identifications through DNA barcodes. Proceedings of the Royal Society of London, Series B 270: 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Savolainen V, Cowan RS, Vogler AP, Roderick GK, Lane R (2005) Towards writing the encyclopaedia of life: an introduction to DNA barcoding. Philosophical Transactions of the Royal Society B 360: 1805–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hebert PDN, Stoeckle MY, Zemlak TS, Francis CM (2004) Identification of birds through DNA barcodes. PLoS Biology 2: e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ward RD, Zemlak TS, Innes BH, Last PR, Hebert PDN (2005) DNA barcoding Australia's fish species. Philosophical Transactions of the Royal Society B 360: 1847–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hajibabaei M, Janzen DH, Burns JM, Hallwachs W, Hebert PDN (2006) DNA barcodes distinguish species of tropical Lepidoptera. Proceedings of the National Academy of Sciences 103: 968–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wolfe KH, Li WH, Sharp PM (1987) Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proceedings of the National Academy of Sciences 84: 9054–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wolfe KH, Sharp PM, Li WH (1989) Rates of synonymous substitution in plant nuclear genes. Journal of Molecular Evolution 29: 208–211. [Google Scholar]
- 8. Fazekas AJ, Burgess KS, Kesanakurti PR, Graham SW, Newmaster SG, et al. (2008) Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS One 3: e2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wendel JF, Doyle JJ (1998) Phylogenetic incongruence: window into genome history and molecular evolution. In: Soltis DE, Soltis PS, Doyle JJ, editors. Molecular Systematics of Plants II: DNA Sequencing. Dordrecht: Kluwer Academic. 265–296.
- 10. Rieseberg LH, Wood TE, Baack EJ (2006) The nature of plant species. Nature 440: 524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rieseberg LH, Willis JH (2007) Plant speciation. Science 317: 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fazekas AJ, Kesanakurti PR, Burgess KS, Percy DM, Graham SW, et al. (2009) Are plant species inherently harder to discriminate than animal species using DNA barcoding markers? Molecular Ecology Resources 9: 130–139. [DOI] [PubMed] [Google Scholar]
- 13. Chase MW, Salamin N, Wilkinson M, Dunwell JM, Kesanakurthi RP, et al. (2005) Land plants and DNA barcodes: short-term and long-term goals. Philosophical Transactions of the Royal Society B 360: 1889–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chase MW, Fay MF (2009) Barcoding of plants and fungi. Science 325: 682–683. [DOI] [PubMed] [Google Scholar]
- 15. Hollingsworth PM (2011) Refining the DNA barcode for land plants. Proceedings of the National Academy of Sciences 108: 19451–19452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Newmaster SG, Fazekas AJ, Ragupathy S (2006) DNA barcoding in land plants: evaluation of rbcL in a multigene tiered approach. Canadian Journal of Botany 84: 335–341. [Google Scholar]
- 17. Li DZ, Gao LM, Li HT, Wang H, Ge XJ, et al. (2011) Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proceedings of the National Academy of Sciences 108: 19641–19646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hollingsworth PM, Forrest LL, Spouge JL, Hajibabaei M, Ratnasingham S, et al. (2009) A DNA barcode for land plants. Proceedings of the National Academy of Sciences 106: 12794–12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, et al. (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proceedings of the National Academy of Sciences 109: 6241–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Armstrong KF, Ball SL (2005) DNA barcodes for biosecurity: invasive species identification. Philosophical Transactions of the Royal Society B 360: 1813–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Darling JA, Blum MJ (2007) DNA-based methods for monitoring invasive species: a review and prospectus. Biological Invasions 9: 751–765. [Google Scholar]
- 22.Bohs L (2005) Major clades in Solanum based on ndhF sequence data. In: Keating RC, Hollowell VC, Croat TB, editors. A festschrift for William G. D’Arcy: the legacy of a taxonomist. St. Louis: Missouri Botanical Garden Press. 27–49.
- 23. Weese TL, Bohs L (2007) A three-gene phylogeny of the genus Solanum (Solanaceae). Systematic Botany 32: 445–463. [Google Scholar]
- 24. Levin RA, Myers NR, Bohs L (2006) Phylogenetic relationships among the ‘spiny Solanums’ (Solanum subgenus Leptostemonum, Solanaceae). American Journal of Botany 93: 157–169. [Google Scholar]
- 25. Bohs L, Olmstead RG (1997) Phylogenetic relationships in Solanum (Solanaceae) based on ndhF sequences. Systematic Botany 22: 5–17. [Google Scholar]
- 26. Levin RA, Watson K, Bohs L (2005) A four-gene study of evolutionary relationships in Solanum section Acanthophora. American Journal of Botany 92: 603–612. [DOI] [PubMed] [Google Scholar]
- 27. Olmstead RG, Bohs L, Migid HA, Santiago-Valentin E, Garcia VF, et al. (2008) A molecular phylogeny of the Solanaceae. Taxon 57: 1159–1181. [Google Scholar]
- 28. Doyle JJ (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19: 11–15. [Google Scholar]
- 29. Olmstead RG, Sweere JA (1994) Combining data in phylogenetic systematics: an empirical approach using three molecular data sets in the Solanaceae. Systematic Biology 43: 467–481. [Google Scholar]
- 30. Hamilton MB (1999) Four primer pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Molecular Ecology 8: 521–523. [PubMed] [Google Scholar]
- 31. Walsh BM, Hoot SB (2001) Phylogenetic relationships of Capsicum (Solanaceae) using DNA sequences from two noncoding regions: the chloroplast atpB-rbcL spacer region and nuclear waxy introns. International Journal of Plant Sciences 162: 1409–1418. [Google Scholar]
- 32. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research 25: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95–98. [Google Scholar]
- 34. Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Molecular Biology and Evolution 24: 1596–1599. [DOI] [PubMed] [Google Scholar]
- 35. Meyer CP, Paulay G (2005) DNA barcoding: error rates based on comprehensive sampling. PLoS Biology 3: e422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kovach WL (1999) A Multivariate Statistical Package for Windows, ver 3.1. Pentraeth, UK: Kovach Computing Services.
- 37.Swofford DL (2002) PAUP * : Phylogenetic Analysis Using Parsimony ( * and other methods), Version 4b10. Sunderland, Massachusetts: Sinauer Associates.
- 38. Simmons MP, Ochoterena H (2000) Gaps as characters in sequence-based phylogenetic analyses. Systematic Biology 49: 369–381. [PubMed] [Google Scholar]
- 39. Young ND, Healy J (2003) GapCoder automates the use of indel characters in phylogenetic vanalysis. Bmc Bioinformatics 4: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Van Der Leij FR, Visser RGF, Ponstein AS, Jacobsen E, Feenstra WJ (1991) Sequence of the structural gene for granule-bound starch synthase of potato (Solarium tuberosum L.) and evidence for a single point deletion in the amf allele. Molecular and General Genetics 228: 240–248. [DOI] [PubMed] [Google Scholar]
- 41. Kress WJ, Erickson DL, Jones FA, Swenson NG, Perez R, et al. (2009) Plant DNA barcodes and a community phylogeny of a tropical forest dynamics plot in Panama. Proceedings of the National Academy of Sciences 106: 18621–18626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kress WJ, Wurdack KJ, Zimmer EA, Weigt LA, Janzen DH (2005) Use of DNA barcodes to identify flowering plants. Proceedings of the National Academy of Sciences 102: 8369–8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lahaye R, Van Der Bank M, Bogarin D, Warner J, Pupulin F, et al. (2008) DNA barcoding the floras of biodiversity hotspots. Proceedings of the National Academy of Sciences 105: 2923–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen S, Yao H, Han J, Liu C, Song J, et al. (2010) Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS One 5: e8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Spooner DM (2009) DNA barcoding will frequently fail in complicated groups: An example in wild potatoes. American Journal of Botany 96: 1177–1189. [DOI] [PubMed] [Google Scholar]
- 46. Rieseberg LH, Carney SE (1998) Plant hybridization. New Phytologist 140: 599–624. [DOI] [PubMed] [Google Scholar]
- 47. Wood TE, Takebayashi N, Barker MS, Mayrose I, Greenspoon PB, et al. (2009) The frequency of polyploid speciation in vascular plants. Proceedings of the National Academy of Sciences 106: 13875–13879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Soltis PS, Soltis DE (2009) The role of hybridization in plant speciation. Annual Review of Plant Biology. 60: 561–588. [DOI] [PubMed] [Google Scholar]
- 49. Alvarez I, Wendel JF (2003) Ribosomal ITS sequences and plant phylogenetic inference. Molecular Phylogenetics and Evolution 29: 417–434. [DOI] [PubMed] [Google Scholar]
- 50. Hollingsworth PM, Graham SW, Little DP (2011) Choosing and using a plant DNA barcode. PLoS One 6: e19254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wiemers M, Fiedler K (2007) Does the DNA barcoding gap exist?-case study in blue butterflies (Lepidoptera: Lycaenidae). Frontiers in Zoology 4: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Prendini L (2005) Comment on “Identifying spiders through DNA barcodes”. Canadian Journal of Zoology 83: 498–504. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of cpDNA (left) and nuclear DNA tree (right) using maximum parsimony (MP) method. Bootstrap values (>75%) are shown above the branches. Numbers followed taxon names are individual numbers.
(TIF)
Distribution of inter- and intra-specific K2P distance of combined DNA regions in all studied species.
(TIF)
List of species used in this study.
(DOC)
Comparisons of sequence divergence (Kimura 2-parameter distance) among individuals of S. elaeagnifolium and its closest relatives S. hindsianum and S. tridynamum .
(DOC)