

Abstract
Tyramine β-monooxygenase (TβM), the insect homologue of dopamine β-monooxygenase, is a neuroregulatory enzyme that catalyzes the beta hydroxylation of tyramine to yield octopamine. Mutation of the methionine (Met) ligand to CuM of TβM, Met471Cys, yielded a form of TβM that is catalytically active but susceptible to inactivation during turnover [Hess, C. R.; Wu, Z.; Ng, A.; Gray, E. E.; McGuirl, M. M.; Klinman, J. P. (2008) J. Am. Chem. Soc. 130, 11939-11944]. Further, although wild-type (WT) enzyme undergoes coordination of Met471 to CuM in its reduced form, the generation of Met471Cys almost completely eliminates this interaction [Hess, C. R.; Klinman, J. P.; Blackburn, N. J. (2010) J. Biol. Inorg. Chem. 15, 1195-1207]. The aim of this study is to identify the chemical consequence of the poor ability of Cys to coordinate CuM. We show that Met471Cys TβM is ca. 5-fold more susceptible to inactivation than WT enzyme in the presence of the co-substrate/reductant ascorbate and that this process is not facilitated by the substrate tyramine. The resulting 50-fold smaller ratio for turnover to inactivation in the case of Met471Cys prevents full turnover of substrate under all conditions examined. LC-MS/MS analysis of proteolytic digests of inactivated Met471Cys TβM leads to the identification of cysteic acid at position 471. While both Met and Cys side chains are expected to be similarly subject to oxidative damage in proteins, the enhanced reactivity of Met471Cys toward solution oxidants in TβM is attributed to its poor coordination toward Cu(I)M.
Keywords: Copper monooxygenases, inactivation, cysteic acid formation
Tyramine β-monooxygenase (TβM) belongs to a small class of eukaryotic copper-, ascorbate-, and O2-dependent enzymes that includes dopamine β-monooxygenase (DβM) and peptidylglycine α-hydroxylating monooxygenase (PHM) (1-3). TβM is the insect homologue of DβM and plays a neuroregulatory role in insects by catalyzing the β-hydroxylation of tyramine to yield octopamine (eq. 1) (4-6).
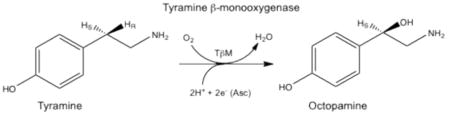 |
(1) |
Studies indicate that TβM expression occurs in octopaminergic neurons (5,6). A significant amount of spectroscopic and kinetic data collected for these enzymes has demonstrated a high conservation of mechanism (7-14). Structural information about TβM is inferred from primary amino acid sequence homology and the X-ray structures solved for PHM (3,15-17). TβM is assumed to use two solvent-exposed, non-coupled mononuclear copper centers, denoted CuM and CuH and separated by ~ 11 Å (Figure 1), to incorporate an oxygen atom into the phenethylamine side chain of tyramine (3,14), eq. 1. O2 activation and substrate hydroxylation occur exclusively at the CuM domain, which is coordinated by two histidines and a Met residue (17,18). CuH, which is ligated by three histidine residues, serves as an electron reservoir to supply the second electron required for insertion of oxygen into the C–H bond by long-range intramolecular electron transfer (7). The combination of structural, kinetic, biochemical, and theoretical evidence supports the formation of a CuM-superoxo intermediate as the active oxygen species responsible for C–H activation at the CuM site (1,17,19-22); by contrast, the precise timing and pathway for electron transfer from CuH to CuM remain under active debate (19,20,22).
Figure 1.
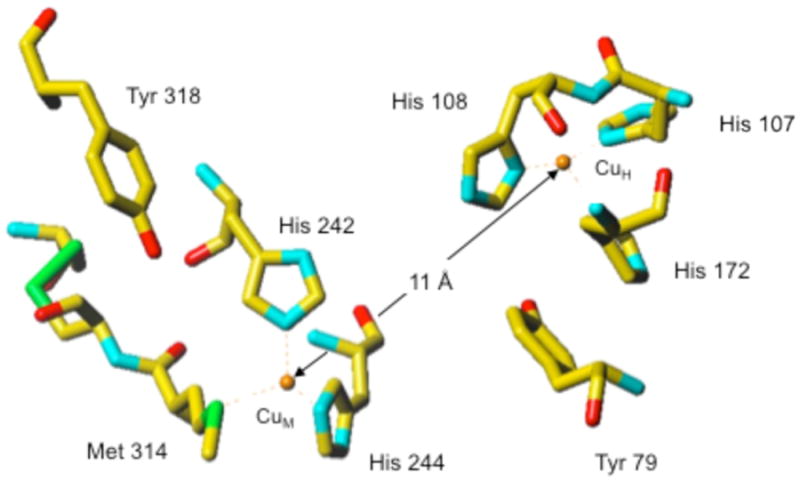
(Recreated from PDB: 1PHM) (15). The ligands at CuH (His107, His108 and His172) and to CuM (His242, His244, Met314) of PHM. Met471 in TβM corresponds to Met314 in PHM.
The Met residue at the CuM domain appears to be a critical component for catalysis by these enzymes, but the exact function of this residue is not known. The ability of the CuM-Met ligand to move into the coordination sphere upon reduction of the copper domains is supported by spectroscopic and crystallographic analyses (2,7,9-11,15,23). Extended X-ray absorption fine structure (EXAFS) for the Cu(II) state of PHM, DβM and TβM indicates an average of 2.5 nitrogen (His) and 1.5 oxygen or nitrogen ligands per copper at 1.97 Å (2,7,10,11,23) and a weak sulfur ligand detected at 2.71 Å, in agreement with the PHM X-ray crystal structure (15). Enzyme reduction leads to a shorter Met to copper bond distance: 2.27 Å in PHM, 2.25 Å in DβM, and 2.24 Å in TβM (2,10,11,23). Importantly, despite the active site solvent exposure of these enzymes and the requirement for cycling between Cu(I) and Cu(II) during each catalytic cycle, the consumption of O2 and the generation of hydroxylated product are tightly coupled. Neither the use of a very slow substrate with DβM (19) or site-specific mutation in PHM (His172Ala) (20), both of which diminished kcat by nearly three orders of magnitude, has any impact on the 1:1 ratio for O2 uptake and product formation. The aggregate data implicate a dynamic role for the Met ligand to CuM that regulates the equilibrium for generating a copper-superoxo species and prevents the diffusional loss of activated oxygen species during turnover (1,19,22).
Site-directed mutagenesis has been used extensively to understand the significance of the Met ligand to CuM. Studies with PHM in which Met314 was substituted with Ile, His, Asp, and Cys, reported that the hydroxylation of glycine-extended peptides was undetectable in spent media (2,24), whereas a low activity has been reported for Met314His (25). The X-ray structure of the Met314Ile PHM variant was found to lead to a significant disorder at the CuH domain, implicating communication between the ligands at distal metal sites (26). Recently, the analogous Met ligand in TβM was mutated to a Cys, His, and Asp, and only the Met471Cys retained measureable activity. Electron paramagnetic resonance (EPR) characterization of the Cu(II) forms of WT and Met471 variants have shown identical properties. Interestingly, Met471Cys TβM was unable to fully convert substrate to product under any conditions examined. This result led to the proposal of a secondary inactivation pathway during turnover (27). XAS/EXAFS experiments were carried out on the series of Met471 variants, to examine how changes in structure relate to the observed altered reactivity, and these showed no binding of His and Asp to Cu(I)M and only a very weak/small interaction between Cys and the metal site (23). Herein, we demonstrate that the replacement of Met471 by Cys generates an enzyme form that undergoes directed oxidation at position 471. This is attributed to the poor coordination between Cys471 and Cu(I)M, and not to a substrate-dependent inactivation as previously suggested (27).
Experimental Procedures
Materials
Drosophila Schneider S2 cells, Drosophila Expression System, and blasticidin S-HCl were purchased from Invitrogen (Carlsbad, CA); Insect Xpress protein free medium was acquired from Lonza (Basel, Switzerland). Reagents were purchased from Fisher Scientific (Pittsburgh, PA) or Sigma-Aldrich (St. Louis, MO). Catalase was obtained from Roche. Acetonitrile (Fisher Optima grade, 99.9%) and formic acid (Pierce, 1 mL ampules, 99+ %) were purchased from Fisher Scientific and water was purified to a resistivity of 18.2 MΩ·cm (at 25 °C) using a Milli-Q Gradient ultrapure water purification system (Millipore, Billerica, MA). His-tagged Met471Cys TβM was expressed and purified as previously described (14,27). Pure fractions (single banded as determined by SDS-PAGE) were pooled, and the concentration was determined by UV absorbance at 280 nm (ε = 99,210 M-1 cm-1 or A280mg/ml = 1.423).
Product Analysis by HPLC
HPLC methodology used was similar to that first described for product analysis during reactions catalyzed by Met471Cys TβM (27). Separation of tyramine and octopamine from other assay components was achieved using an Alltech Adsorbosphere reversed phase 10 μm C18 column (Grace Discovery Sciences, Deerfield, IL) (4.6 × 250 mm) interfaced to a Beckman-Coulter system Gold HPLC equipped with an autosampler. A mobile phase of 5 mM acetic acid (pH 5.8), 600 μM heptane sulfonic acid, and 15% methanol at 1.1 ml/min was used to achieve separation of substrate and product. Octopamine and tyramine were monitored at 224 and 274 nm, respectively, and under these conditions octopamine eluted at a retention time of ~ 10.5 min and tyramine at ~ 35 min. Assay mixtures (VT = 1 − 2 mL) contained tyramine (250 μM – 10 mM), 50 mM ascorbate, 50 mM KPi, 0.1 M KCl, 100 μg/mL catalase, and enzyme at pH 6. These solutions were mixed for periods of 2 min to 8 h. CuSO4 was maintained in all assay mixtures at a ratio of at least 4:1 Cu:TβM. The reactions were initiated by the addition of enzyme to the assay mixtures. At specific timepoints, 115 μL aliquots were removed from the assay solution and quenched with 1.5 μL of 70 % HClO4. The aliquots were centrifuged at 14,000 rpm (22 min) to remove precipitated protein prior to analysis by high performance liquid chromatography (HPLC). Samples that could not be immediately analyzed by HPLC were frozen in liquid nitrogen and stored at -80 °C.
Standard Curves
Octopamine standard curves were generated using solutions containing varying amounts of octopamine (50 μM – 2 mM). The amount of product formed during end-point and inactivation-based assays was determined by integration of the octopamine peak and comparison to the standards. Generation of a tyramine standard curve and quantification of the loss of substrate were not carried out, due to the broadness of the tyramine peak at 274 nm under the reaction conditions studied. However, the disappearance of tyramine was monitored as a secondary qualitative approach to confirm substrate turnover catalyzed by Met471Cys TβM. The validity of the octopamine standard curve was confirmed with a sample of known octopamine concentration on the same day that reaction samples were analyzed by HPLC.
Enzyme Kinetic Assays Using HPLC
To determine the apparent Km(Tyr) for Met471Cys TβM, endpoint assays were performed as a function of tyramine concentration. Assay solutions contained a fixed concentration of tyramine (100 μM – 8 mM), 50 mM ascorbate, 100 mM KPi, 0.1 M KCl, 100 μg/mL catalase, 15 μM CuSO4, and 3.0 μM enzyme, pH 6, 35 °C. Enzyme was always added last to initiate the reaction. The rate (kobs) at each tyramine concentration was determined from the slope of the line as a result of plotting the amount of octopamine formed at early timepoints. A background absorbance, representing an 0.3 % contamination of the tyramine stock by octopamine, was substrated from each timepoint.The observed rates were first normalized against the published rate for Met471Cys (14) and the resulting kobs plotted as a function of tyramine concentration. Initial velocities of octopamine formation were fitted to the Michaelis-Menten equation, using the program Kaleidagraph.
Enzyme Activity/Inactivation Assays
For each assay, ~ 2.0 μM Met471Cys TβM was mixed with 50 mM ascorbate, 100 mM KPi pH 6.0, 100 mM KCl, 100 μg/mL catalase, and 10 μM CuSO4 for 0–4 h at room temperature (ca. 23 °C). After incubating Met471Cys TβM with the reaction components for a precise period of time (0–4 h), 1 mM tyramine was added and reactions allowed to proceed for up to 4 h. Aliquots (115 μL) were removed at specific timepoints (2 min – 4 h) during the course of the reaction, added to a 1.5 mL eppendorf tube containing 1.5 μl of 70 % HClO4, and centrifuged at 14,000 rpm (22 min) to remove precipitated protein prior to injection onto the HPLC column. Samples not analyzed on the same day as the assays were stored at -80 °C until analysis by HPLC. These experiments were carried out to quantify the amount of octopamine generated as a function of reaction time, subsequent to enzyme inactivation in the absence of substrate. The rate constant for TβM inactivation was determined by plotting the natural log of the observed rate (ln kobs) versus the time of incubation prior to addition of substrate. Control reactions using 2.0 uM WT enzyme were carried out exactly as described for Met471Cys. A WT enzyme preparation of reduced specific activity allowed the use of identical protein concentrations in the comparison of WT to Met471Cys rates of inactivation.
Control Proteolytic Digest Reactions
Proteolysis of Met471Cys TβM in 100 mM Tris, pH 8.5 was achieved by first denaturing ~ 15 μM enzyme in 8 M urea. All cysteines participating in disulfide bonds were reduced with 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) for ~ 20 min at room temperature, and the free cysteines capped with 10 mM iodoacetic acid (IAA) for ~ 45 min at room temperature. The samples were diluted fourfold with 100 mM Tris pH 8.5 to bring the final urea concentration to ~ 2 M, and 1 mM CaCl2 was added to the solution. 1 μL of L-1-tosylamido-2-phenylethyl chloromethyl ketone (TPCK) trypsin (Sigma-Aldrich) (0.5 μg/μL) was added, and the digestion reaction incubated in the dark at 37 °C overnight. 1 μL of neat formic acid was added to each digest reaction before analysis by mass spectrometry.
Proteolytic Digest of Met471Cys TβM Reactions
For each assay, ~ 15 μM Met471Cys TβM was added to a solution containing ascorbate (1 or 50 mM), 50 mM KPi pH 6.0, 100 mM KCl, 20-100 μg/mL catalase, 45 μM CuSO4, and 0.5 – 10 mM tyramine for 0–2 h at room temperature. At specific timepoints, 8 M urea was added to the assay mixture, which was immediately concentrated using a small volume (0.5 mL) concentrator (YM30, Millipore). This process of denaturing and washing was repeated three times to remove any small molecular reaction components (i.e., KCl, CuSO4, ascorbate, tyramine, octopamine), after which Met471Cys TβM was reconstituted in 100 mM Tris, pH 8.5 and concentrated to ~ 20 μL. Inactivated Met471Cys TβM was prepared for proteolysis using the same procedure as described above for the control reactions.
Liquid Chromatography-Mass Spectrometry
TPCK-trypsin-digested samples of Met471Cys TβM were analyzed with a Waters nanoAcquity ultraperformance liquid chromatograph (UPLC) connected in-line with a quadrupole time-of-flight (Q-Tof) mass spectrometer that was equipped with a nanoelectrospray ionization (nanoESI) source (Q-Tof Premier, Waters, Milford, MA).
Liquid Chromatography
The UPLC was equipped with C18 trapping (20 mm × 180 μm, 5 μm, Waters Symmetry) and analytical (100 mm × 100 μm, 1.7 μm, Waters BEH130) columns with a 10 μL sample loop. Solvent A was 0.1 % formic acid/99.9 % water and solvent B was 0.1 % formic acid/99.9% acetonitrile (v/v). Following sample injection, trapping was performed for 8 min with 100 % A at a flow rate of 3 μL/min. The elution program consisted of a linear gradient from 25 % to 35 % B over 30 min, a linear gradient to 55 % B over 10 min, a linear gradient to 95 % B over 0.33 min, isocratic conditions at 95 % B for 6.67 min, a linear gradient to 1 % B over 0.33 min, and isocratic conditions at 1 % B for 7.67 min, at a flow rate of 500 nL/min. A seal wash period of 5 min was used between sample injections. The injection needle was washed with 500 μL each of solvents A and B to avoid cross-contamination. The analytical column and sample compartment were maintained at 35 °C and 8 °C, respectively.
Mass Spectrometry
The column exit was connected to a Universal NanoFlow Sprayer nanoESI emitter mounted in the nanoflow ion source of the Q-Tof Premier. The nanoESI source parameters were as follows: nanoESI capillary voltage 2.3 kV, nebulizing gas (nitrogen) pressure 0.15 mbar, sample cone voltage 30 V, extraction cone and ion guide voltages both 4 V, and source block temperature 80 °C. No cone gas was used. The collision cell contained argon gas at a pressure of 8 × 10-3 mbar. The Tof analyzer was operated in “V” mode. Under these conditions, a mass resolving power (28) of 1.0 × 104 (measured at m/z = 771) was routinely achieved, which was sufficient to resolve the isotopic distributions of the singly and multiply charged peptide ions measured in this study. External mass calibration was performed immediately prior to analysis using solutions of sodium formate. Full scan mass spectra were acquired in the positive ion mode over the range m/z = 500 to 2000, in continuum data format, using a 1.45 s scan integration and a 0.05 s interscan delay.
Tandem Mass Spectrometry (MS/MS)
For MS/MS performed in the data-dependent mode, up to five precursor ions occurring within the range m/z = 400 to 1800 and exceeding an intensity threshold of 30 counts/sec (cps) was selected from each survey scan for MS/MS. Real-time deisotoping and charge state recognition were used to select 2+, 3+, 4+, 5+, and 6+ charge state precursor ions for MS/MS. Collision energies for collisionally activated dissociation (CAD) were automatically selected based on the mass and charge state of a given precursor ion. MS/MS spectra were acquired over the range m/z = 50 to 2000 using a 0.95 s scan integration and a 0.05 s interscan delay. Ions were fragmented to achieve a minimum total ion current (TIC) of 18,000 cps in the cumulative MS/MS spectrum for a maximum of 3 s. To avoid the occurrence of redundant MS/MS measurements, real-time dynamic exclusion was used to preclude re-selection of previously analyzed precursor ions over an exclusion width of ± 0.50 m/z unit for a period of 180 s. An include list was specified to preferentially select the (M+3H)3+ ions of the peptide, DDKTAALGGFSISDECCVNYIHYYPATK, which corresponds to residues 456-483 of Met471Cys TβM, in two forms: (1) the form in which both Cys residues are present as CMC (m/z = 1066.47, monoisotopic); and (2) the form in which one Cys residue was present as CMC and the other was cysteic acid (m/z = 1063.13, monoisotopic). The inclusion width was ± 0.20 m/z unit.
Mass Spectrometry Data Analysis
The LC-MS/MS data were processed using ProteinLynx Global Server software (version 2.3, Waters), which performed background subtraction (threshold 35% and fifth order polynomial), smoothing (Savitzky-Golay, 10 times, over three channels), and centroiding (top 80 % of each peak, and minimum peak width at half height = four channels) of the mass spectra and MS/MS spectra. The processed data were searched against the amino acid sequence of Met471Cys TβM using the following criteria: non-specific cleavages, precursor ion mass tolerance 50 ppm, fragment ion mass tolerance 0.1 Da, and the following variable post-translational modifications: Met sulfoxide, CMC, cysteic acid and histidine oxidation. The MS/MS spectra were manually inspected to verify the presence of the fragment ions, which uniquely identify the peptide sequences. The fragment ions were labeled using the nomenclature of Roepstoff and Fohlman (29). Monoisotopic molecular masses are used to identify ions of interest unless stated otherwise.
Results and Discussion
A series of Met471 TβM mutants was recently generated to probe the role of the Met residue bound to CuM. Met471Cys TβM, the only active variant, undergoes a secondary inactivation process during turnover under the conditions examined (27). The fact that the observed octopamine/enzyme ratio is constant (~ 75) at all concentrations of Met471Cys TβM examined led to the proposed partitioning of an intermediate form of enzyme (ES') to generate product (E + P) or inactivated enzyme (E′) (Scheme 1) (27). This analysis allowed for the estimation of a rate of inactivation (kinact) as ~7 × 10-4 s-1. While these earlier experiments demonstrated the greater stability of a thioether linkage at position 471, the source and chemical identity of the inactivation process were not pursued.
Scheme 1.

A. Branching mechanism initially proposed in which partitioning occurs after substrate binding (27). B. Branching mechanism in which the majority of enzyme inactivation occurs with free enzyme.
In this work, the premise of a substrate-mediated inactivation process has been further examined. We first determined the Km(Tyr) for Met471Cys TβM by measuring the initial rate of octopamine formation via HPLC end-point assays as a function of tyramine concentration (Figure S1). The initial rate determined from early time points was plotted against each tyramine concentration (Figure 2). Surprisingly, the observed Km of 4.6 (2.0) mM is ~20-fold greater than the Km for WT enzyme (with the true kcat for Met471Cys, 0.45 (0.09) s-1, now seen to be reduced only ca. 10-fold in relation to WT). The elevated Km means that the assays in which the inactivation was initially observed (27) were carried out using a starting concentration (500 μM tyramine) that is only 11 % of Km.
Figure 2.

Rates of octopamine formation catalyzed by 3.0 μM Met471Cys TβM at varying concentrations of tyramine in reactions containing 50 mM KPi, pH 6, 35 °C, 100 mM KCl, 100 μg/mL catalase, 50 mM ascorbate and 15 μM CuSO4.
Assays were then designed to determine whether solution oxidants alone inactivate Met471Cys TβM. The potential susceptibility of any form of TβM to inactivation by solution oxidants is due to the ability of reduced copper to bind and activate O2. The susceptibility of the WT forms of DβM and PHM to inactivation by ascorbate has been noted in the earlier literature (30-32). This family of enzymes functions in neurosecretory vesicles, which are enriched with copper, ascorbate, and O2. Similarly, the in vitro assays require a source of copper (CuSO4), excess reductant (ascorbate), and O2 (~ 222 μM at 35 °C). Met471Cys was incubated with all assay components except tyramine for 0–4 h, followed by the addition of 1 mM tyramine to assay for the percentage of remaining enzyme activity. Met471Cys TβM is essentially inactive after incubating this form of enzyme for 2 h in the assay solution before the addition of 1 mM tyramine (Figure S2, Table 1).
Table 1.
Octopamine Formed After Met471Cys TβM Inactivation.
| Incubation Time (h)a | [Octopamine] (μM)b 1 h | [Octopamine] (μM)b 4 h | [Octopamine] (μM)b 6 h |
|---|---|---|---|
| 0 | 163.4 | 350.3 | 355.2 |
| 2 | 8.07 | 17.5 | 20.1 |
| 4 | 4.02 | 8.28 | 11.6 |
The number of hours all reaction components were allowed to react before the addition of 1 mM tyramine.
For each assay, 2.0 μM Met471Cys TβM was incubated with buffer (100 mM KPi, pH 6.0), KCl (100 mM), ascorbate (50 mM), catalase (100 μg/mL), CuSO4 (10 μM). Following the addition of 1 mM tyramine, aliquots (115 μL) were removed at 5, 10, 15, 30, 60, 120, 240 and 360 min intervals, quenched with 1.5 μL of 70 % HClO4, and assayed for octopamine formation. Data are shown after 1, 4, and 6 h.
Met471Cys TβM was subsequently exposed to the assay mixture in the absence of tyramine for shorter incubation times (0–45 min) to quantify more precisely the rate of inactivation for Met471Cys TβM-catalyzed reactions and to compare the rate of inactivation for comparison to WT TβM. The slope of the line resulting from the plot of the natural log of kobs, determined by HPLC end-point assays (Figure 3), as a function of incubation time provides an estimate for the rate of inactivation, kinact ~ 3.6 × 10-4 s-1, in relation to the rate for WT of 0.79 × 10-4 s-1 (Figure 3). Replacement of Met471 with a Cys, thus, increases the susceptibility of the enzyme ca. 5-fold to inactivation. This contrasts with the similar second order rate constants for reaction of free Cys and Met toward the oxidant hydrogen peroxide at pH 8 (33,34). While it is difficult to assess the impact of the CuM on the protonation state of Cys in Met471Cys, the current incubation conditions of pH 6 are expected to suppress thiolate formation relative to pH 8 and, hence, possibly even reduce the intrinsic chemical reactivity for Cys in relation to Met.
Figure 3.

Plot of ln(kobs) versus incubation time. For these assays Met471Cys or WT TβM was incubated at 23 °C with 100 mM KPi, pH 6.0, 100 mM KCl, 50 mM ascorbate, and 100 μg/mL catalase (no tyramine) for 0, 40, 80, 120, 160, 200, 240 min before addition of 1 mM tyramine. After the incubation time was complete, end-point assays were executed at 35 °C and product versus time curves generated in order to determine a kobs as a function of incubation time.
In understanding the impact of mutation on the properties of Met471Cys, the key variables are the ratio of turnover (10 % of WT) to inactivation (500 % of WT), leading to a 50 fold greater viability in the naturally occurring Met-containing enzyme. We conclude that Met at position 471 plays a dual role in TβM function: that of controlling the level of the reactive Cu(II)(O2·-) during catalysis via sulfur ligation (cf. 1,19,22), and that of protecting the enzyme against oxidative inactivation. The data in Table 1 indicate further that the role of substrate in enzyme inactivation is distinct from that originally proposed (Scheme 1A). Independent of the length of incubation of Met471Cys TβM in the absence of tyramine (0-4 h), subsequent addition of substrate leads to a ca. doubling of product formation after another 4-h incubation. Of particular note is the fact that a 4-h incubation without substrate virtually eliminates all enzyme activity assayed after 1 h with substrate, whereas incubation of enzyme with substrate for 6 h leads to a substantial level of octopamine production (Table 1). These data indicate a much slower inactivation in the presence of substrate leading to a mechanism in which the majority of inactivation processes occur with free enzyme, Scheme 1B.
In an effort to identify the residues in Met471Cys modified by solution oxidants, limited proteolysis using TPCK-trypsin followed by LC-MS/MS analysis was performed. Numerous tryptic peptides of Met471Cys TβM were analyzed, including several containing active-site residues. In particular, the peptide containing Met471Cys TβM (residues 456-483) was observed in two forms: (1) having both Cys residues, at positions 471 and 472, in the form of CMC (m/z 1066.5, 3+ charge state) and (2) having one CMC and one cysteic acid (m/z 1063.1, 3+ charge state). These peptide ions were well resolved in the precursor ion mass spectrum (Figure 4) and were each individually selected for MS/MS. The fragment ions observed in the MS/MS spectrum for the former precursor ion (m/z 1066.5) identify CMC residues at positions 471 and 472 (Figure 5). Such modification results from reduction and capping of cysteines by TCEP and IAA, respectively, prior to proteolysis. On the other hand, the MS/MS spectrum for the later precursor ion (m/z 1063.1) identifies a CMC solely at position 472, as indicated by the mass difference between the y12/y11 pair fragment ions (161 Da) and the y13/y12 pair fragment ions (151 Da). This difference of 10 Da is predicted for cysteic acid at position 471 (Figure 6).
Figure 4.

Precursor ion mass spectrum of the [M + 3]3+ ion (m/z 1063.4) of the Cys471-cysteic acid modified peptide residues (456–483) (1) and the [M + 3]3+ ion (m/z 1067.7) of the carboxymethyl cysteine (CMC)-modified peptide (456 – 483) (2).
Figure 5.

LC-MS/MS analysis of the [M + 3]3+ ion (m/z 1067.7) of the CMC-modified peptide (456–483) from Met471Cys TβM. Met471Cys TβM reactions for proteolytic digest and mass spectral analysis were carried out under the following conditions: Met471Cys TβM (15 μM), 50 mM KPi, pH 6 0.1 M KCl, 50 mM ascorbate, 45 μM CuSO4, and 100 μg/mL catalase. The reactions were quenched with 8M urea, reduced with 5 mM TCEP, and capped with 10 mM IAA. Proteolysis was initiated with TPCK trypsin (0.5 μg/mL) and allowed to react overnight at 37 °C. Neat formic acid (1 μL) was added to each reaction before analysis.
Figure 6.

LC-MS/MS analysis of the [M + 3]3+ ion (m/z 1063.4) of the Cys471-cysteic acid modified peptide (456–483) from Met471Cys TβM. Conditions were as described in Legend to Figure 5. These data are for the 60-min timepoint.
It was possible to monitor the fraction of Cys471 in an oxidized, cysteic acid form as a function of time by monitoring the relative mass spectral intensities of the two precursor ions at m/z 1066.5 and 1063.1. As summarized in Table 2, the fraction of cysteic acid was 0.28 (± 0.09), 0.66 (± 0.11), and 0.71 (± 0.07) at incubation times of 0 min, 30 min, and 60 min, respectively, for assay mixtures containing copper and 50 mM ascorbate. The fact that only 71 % of the Cys471 was oxidized at the time of 100 % inactivation (Figure 3) is attributed to low level, non-specific oxidations throughout the rest of the protein (35). Such non-specific oxidations within tryptic peptides containing amino acid residues inherently susceptible to oxidation (i.e., Met, Cys, His), but not located at the active site of the enzyme, varied from < 1 % to 20 % and remained relatively constant within experimental error under all conditions examined. Control experiments in which copper was excluded from the assay solution resulted in nearly identical extents of oxidation compared to those obtained using enzyme without ascorbate and copper. In addition, the extent of oxidation of Cys471 obtained with an assay solution containing a low ascorbate concentration (1 mM) in the presence of copper was almost indistinguishable from control experiments. These measurements support an inactivation process that is primarily the result of time-dependent oxidation of Cys471 by products formed from copper and high ascorbate. As will be discussed in a forthcoming study of mutants of TβM at CuH (Osborne et al., unpublished), WT-enzyme does not appear susceptible to a targeted oxidation at Met471.
Table 2.
Fraction of Cysteic Acid for M471C TβM Proteolytic Fragment 459-483.
| 0 mina | 30 min | 60 min | |
|---|---|---|---|
| 50 mM Ascb | 0.28 ± 0.09 | 0.66 ± 0.11 | 0.71 ± 0.07 |
| 1 mM Asc | 0.28 ± 0.09 | 0.28 ± 0.05 | 0.40 ± 0.07 |
| 50 mM Asc, 500 μM Tyrc | 0.28 ± 0.09 | 0.56 ± 0.05 | 0.72 ± 0.03 |
For the ‘0 min’ timepoint Met471Cys TβM (15 μM) was denatured in urea (8 M) and Tris buffer, pH 8.5 (100 mM), reduced with TCEP (5 mM), and capped with IAA (10 mM). Proteolysis was subsequently initiated by TPCK-trypsin (0.5 μg/mL) and allowed to react overnight at 37 °C. Neat formic acid (1 μL) was added to each reaction before mass spectral analysis. The fraction of cysteic acid detected at 0 min is attributed to the inherent instability at this position during protein purification and work up for the analyses.
Met471Cys TβM (15 μM) was incubated for 30 and 60 min with the following: KPi, pH 6.0 (0.1 M), KCl (0.1 M), ascorbate (50 mM), catalase (20 μg/mL), and CuSO4 (30 μM). These reactions were subjected to the same proteolysis conditions as described above.
These are the conditions of the earlier experiment (27).
It can be seen that Cys at position 471 results in an unstable hydroxylase that is susceptible to extensive inactivation concomitant with cysteic acid formation (Figure 3, Table 2). Previously, XAS/EXAFS experiments indicated that replacement of Met471 with a His or Asp eliminates binding to CuM in both the oxidized and reduced states of enzyme, whereas the XAS/EXAFS spectra for Met471Cys are consistent with a very minor binding component of Cys471 to Cu(I)M (23). The catalytic activity of Met471Cys TβM is ca. 10 % of the WT enzyme (Figure 2), and the altered activity can be rationalized by a significantly reduced percentage of enzyme in which Cys471 binds to the Cu(I)M (23). The impaired binding of Cys471 to Cu(I)M, and its resulting increased exposure to oxidants also offer an explanation for the impact of Cys replacement on enzyme inactivation. Multiple pathways for Cys oxidation are possible (cf. 35, 36), although a dominant involvement of solution-derived H2O2 is unlikely due to the presence of catalase in all incubations and assays. A major unanswered structure/function question regards the highly specific manner in which Met at CuM both stabilizes and activates all members of the TβM/DβM/PHM family of enzymes.
Conclusions
The small class of mononuclear dicopper monooxygenases that includes TβM has been proposed to function via a catalytic mechanism in which the strength of binding of Met471 to CuM limits the extent of superoxide accumulation at this domain prior to substrate activation (23). The present study, which provides a kinetic and chemical rationalization for the previously reported time-dependent activation of Met471Cys (27), highlights an additional role for Met471 in stabilizing enzyme in the presence of oxidants. We propose that the poor ability of Cys at position 471 to coordinate to CuM in either its oxidized or reduced state (23) leaves this residue vulnerable to oxidative processes, resulting in a ratio of kcat to kinact that is reduced ≥ 500-fold for Met471Cys relative to the WT enzyme.
Supplementary Material
Acknowledgments
We would like to thank Ann Fischer of UC Berkeley for assistance and advice on cell culturing.
Abbreviations
- TβM
tyramine β-monooxygenase
- DβM
dopamine β-monooxygenase
- PHM
peptidylglycine α-hydroxylating monooxygenase
- Met
methionine
- Cys
cysteine
- Ile
isoleucine
- His
histidine
- Asp
aspartate
- Tyr
tyrosine
- XAS
X-ray absorption spectroscopy
- EXAFS
extended X-ray absorption fine structure
- WT
wild-type
- HPLC
high performance liquid chromatography
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
- IAA
iodoacetic acid
- TPCK
L-1-tosylamido-2-phenylethyl chloromethyl ketone
- UPLC
ultraperformance liquid chromatograph
- CMC
carboxymethyl cysteine
- CAD
collisionally activated dissociation
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- Q-Tof
quadrupole time-of-flight
- TIC
total ion current
- cps
counts per second
Footnotes
Supported by grants from the NIH (GM025765 to J.P.K. and 1F32 GM082095-O1A1 to R.L.O.
Supporting Information: This includes graphical analysis of product vs. time curves for the reaction of Met471Cys TβM at varying concentrations of tyramine, comparative time courses for the stability of Met471Cys TβM (± tyramine), and raw data for the early time dependence of Met471Cys inactivation (– tyramine). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Klinman JP. The copper-enzyme family of dopamine β-monooxygenase and peptidylglycine α-hydroxylating monooxygenase: resolving the chemical pathway for substrate hydroxylation. J Biol Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 2.Eipper BA, Quon ASW, Mains RE, Boswell JS, Blackburn NJ. The catalytic core of peptidylglycine α -hydroxylating monooxygenase: Investigation by site-directed mutagenesis, Cu x-ray absorption spectroscopy, and electron paramagnetic resonance. Biochemistry. 1995;34:2857–2865. doi: 10.1021/bi00009a016. [DOI] [PubMed] [Google Scholar]
- 3.Gray EE, Small SN, McGuirl MA. Expression and characterization of recombinant tyramine β-monooxygenase from Drosophila: A monomeric copper-containing hydroxylase. Protein Expression Purif. 2006;47:162–170. doi: 10.1016/j.pep.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 4.Roeder T. Tyramine and octopamine: Ruling behavior and metabolism. Annu Rev Entomol. 2005;50:447–477. doi: 10.1146/annurev.ento.50.071803.130404. [DOI] [PubMed] [Google Scholar]
- 5.Lehman HK, Schulz DJ, Barron AB, Wraight L, Hardison C, Whitney S, Takeuchi H, Paul RK, Robinson GE. Division of labor in the honey bee (Apis mellifera): The role of tyramine β-hydroxylase. J Exp Biol. 2006;209:2774–2784. doi: 10.1242/jeb.02296. [DOI] [PubMed] [Google Scholar]
- 6.Monastirioti M. Distinct octopamine cell population residing in the CNS abdominal ganglion controls ovulation in Drosophila melanogaster. Dev Biol. 2003;264:38–49. doi: 10.1016/j.ydbio.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 7.Blackburn NJ, Pettingill TM, Seagraves KS, Shigeta RT. Characterization of a carbon monoxide complex of reduced dopamine β-hydroxylase. Evidence for inequivalence of the Cu(I) centers. J Biol Chem. 1990;265:15383–15386. [PubMed] [Google Scholar]
- 8.Klinman JP. Mechanisms whereby mononuclear copper proteins functionalize organic substrates. Chem Rev. 1996;96:2541–2561. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- 9.Boswell JS, Reedy BJ, Kulathila R, Merkler D, Blackburn NJ. Structural investigations on the coordination environment of the active-site copper centers of recombinant bifunctional peptidylglycine α-amidating enzyme. Biochemistry. 1996;35:12241–12250. doi: 10.1021/bi960742y. [DOI] [PubMed] [Google Scholar]
- 10.Reedy BJ, Blackburn NJ. Preparation and characterization of half-apo dopamine-β-hydroxylase by selective removal of CuA. Identification of a sulfur ligand at the dioxygen binding site by EXAFS and FTIR spectroscopy. J Am Chem Soc. 1994;116:1924–1931. [Google Scholar]
- 11.Scott RA, Sullivan RJ, DeWolf WE, Dolle RE, Kruse LI. The copper sites of dopamine β-hydroxylase: An x-ray absorption spectroscopic study. Biochemistry. 1988;27:5411–5417. doi: 10.1021/bi00415a005. [DOI] [PubMed] [Google Scholar]
- 12.Miller SM, Klinman JP. Magnitude of intrinsic isotope effects in the dopamine β-monooxygenase reaction. Biochemistry. 1983;22:3091–3096. doi: 10.1021/bi00282a011. [DOI] [PubMed] [Google Scholar]
- 13.Francisco WA, Merkler DJ, Blackburn NJ, Klinman JP. Kinetic mechanism and intrinsic isotope effects for the peptidylglycine α-amidating enzyme reaction. Biochemistry. 1998;37:8244–8252. doi: 10.1021/bi973004y. [DOI] [PubMed] [Google Scholar]
- 14.Hess CR, McGuirl MM, Klinman JP. Mechanism of the insect enzyme, tyramine β-monooxygenase, reveals differences from the mammalian enzyme, dopamine β-monooxygenase. J Biol Chem. 2008;283:3042–3049. doi: 10.1074/jbc.M705911200. [DOI] [PubMed] [Google Scholar]
- 15.Prigge ST, Eipper BA, Mains RE, Amzel LM. Amidation of bioactive peptides: The structure of peptidylglycine α-hydroxylating monooxygenase. Science. 1997;278:1300–1305. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 16.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Substrate-mediated electron transfer in peptidylglycine α-hydroxylating monooxygenase. Nature. 1999;6:976–983. doi: 10.1038/13351. [DOI] [PubMed] [Google Scholar]
- 17.Prigge ST, Eipper BA, Mains RE, Amzel LM. Dioxygen binds end-on to mononuclear copper in a precatalytic complex. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 18.Miller SM, Klinman JP. Secondary isotope effects and structure-reactivity correlations in the dopamine beta-monooxygenase reaction: Evidence for a chemical mechanism. Biochemistry. 1985;24:2114–2127. doi: 10.1021/bi00330a004. [DOI] [PubMed] [Google Scholar]
- 19.Evans JP, Ahn K, Klinman JP. Evidence that dioxygen and substrate activation are tightly coupled in dopamine beta-monooxygenase. Implications for the reactive oxygen species. J Biol Chem. 2003;278:49691–49698. doi: 10.1074/jbc.M300797200. [DOI] [PubMed] [Google Scholar]
- 20.Evans JP, Blackburn NJ, Klinman JP. The catalytic role of the copper ligand H172 of peptidylglycine α-hydroxylating monooxygenase: A kinetic study of the H172A mutant. Biochemistry. 2006;45:15419–15429. doi: 10.1021/bi061734c. [DOI] [PubMed] [Google Scholar]
- 21.Bauman AT, Yukl ET, Alkevich K, McCormack AL, Blackburn NJ. The hydrogen peroxide reactivity of peptidylglycine monooxygenase supports a Cu(II)-superoxo catalytic intermediate. J Biol Chem. 2006;281:4190–4198. doi: 10.1074/jbc.M511199200. [DOI] [PubMed] [Google Scholar]
- 22.Chen P, Solomon EI. Oxygen activation by the noncoupled binuclear copper site in peptidylglycine alpha-hydroxylating monooxygenase. Reaction mechanism and role of the noncoupled nature of the active site. J Am Chem Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 23.Hess CR, Klinman JP, Blackburn NJ. The copper centers of tyramine β-monooxygenase and its catalytic-site methionine variants: an X-ray absorption study. J Biol Inorg Chem. 2010;15:1195–1207. doi: 10.1007/s00775-010-0677-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolhekar AS, Keutmann HT, Mains RE, Quon AS, Eipper BA. Peptidylglycine α-hydroxylating monooxygenase: active site residues, disulfide linkages, and a two-domain model of the catalytic core. Biochemistry. 1997;36:10901–10909. doi: 10.1021/bi9708747. [DOI] [PubMed] [Google Scholar]
- 25.Bauman AT, Broers BA, Kline CD, Blackburn NJ. A copper methionine interaction controls the pH-dependent activation of peptidylglycine monooxygenase. Biochemistry. 2011;50:10819–10828. doi: 10.1021/bi201193j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siebert X, Eipper BA, Mains RE, Prigge ST, Blackburn NJ, Amzel LM. The catalytic copper of peptidylglycine α-hydroxylating monooxygenase also plays a critical structural role. Biophys J. 2005;89:3312–3319. doi: 10.1529/biophysj.105.066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hess CR, Wu Z, Ng A, Gray EE, McGuirl MM, Klinman JP. Hydroxylase activity of Met471Cys tyramine β-monooxygenase. J Am Chem Soc. 2008;130:11939–11944. doi: 10.1021/ja800408h. [DOI] [PubMed] [Google Scholar]
- 28.Marshall AG, Hendrickson CL. High-resolution mass spectrometers. Annu Rev Anal Chem. 2008;1:579–599. doi: 10.1146/annurev.anchem.1.031207.112945. [DOI] [PubMed] [Google Scholar]
- 29.Roepstorff P, Fohlman J. Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 30.Slama P, Jabre F, Tron T, Reglier M. Dopamine β-hydroxylase inactivation generates a proton-bound quinone derivative. FEBS Lett. 2001;491:55–58. doi: 10.1016/s0014-5793(01)02147-0. [DOI] [PubMed] [Google Scholar]
- 31.Skotland T, Lyones T. Evidence from the acceleration of cytochrome-C reduction for the formation of ascorbate free radical by dopamine β-monnoxygenase. Arch Biochem Biophys. 1980;201:81–87. [Google Scholar]
- 32.Miller DA, Sayad KI, Kulathila R, Beaudry GH, Merkler DJ, Bertelsen AH. Characterization of a bifunctional peptidylglycine α-amidating enzyme expressed in chick hamster ovary cells. Arch Biochem Biophys. 1992;298:380–388. doi: 10.1016/0003-9861(92)90425-v. [DOI] [PubMed] [Google Scholar]
- 33.Regino CAS, Richardson DE. Bicarbonate-catalyzed hydrogen peroxide oxidation of cysteine and related thiols. Inorganica Chimica Acta. 2007;360:3971–3977. [Google Scholar]
- 34.Chu JW, Yin J, Wang DIC, Trout BL. Molecular dynamics simulation and oxidation rates of methionine residues of granulocyte colony-stimulating factor at different pH values. Biochemistry. 2004;43:1019–1029. doi: 10.1021/bi0356000. [DOI] [PubMed] [Google Scholar]
- 35.Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 36.Kachur AV, Koch CJ, Biaglow JE. Mechanism of copper-catalyzed autoxidation of cysteine. Free Rad Res. 1999;31:23–34. doi: 10.1080/10715769900300571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.