

Background: Gentamicin causes nephrotoxicity and ototoxicity using megalin as a key cellular uptake site, but no structural data are available.
Results: Gentamicin binds to megalin with low affinity and exploits the common ligand binding motif.
Conclusion: This first structure of human megalin in complex with gentamicin suggests electrostatics to be the main binding determinant.
Significance: Structure-based design of gentamicin antagonists may now be possible.
Keywords: Apolipoproteins, Kidney, Lipoprotein-like Receptor (LRP), Low Density Lipoprotein (LDL), Molecular Docking, Nephrology, NMR, Bacterial Infections, Gentamicin, Megalin
Abstract
Gentamicin is an aminoglycoside widely used in treatments of, in particular, enterococcal, mycobacterial, and severe Gram-negative bacterial infections. Large doses of gentamicin cause nephrotoxicity and ototoxicity, entering the cell via the receptor megalin. Until now, no structural information has been available to describe the interaction with gentamicin in atomic detail, and neither have any three-dimensional structures of domains from the human megalin receptor been solved. To address this gap in our knowledge, we have solved the NMR structure of the 10th complement type repeat of human megalin and investigated its interaction with gentamicin. Using NMR titration data in HADDOCK, we have generated a three-dimensional model describing the complex between megalin and gentamicin. Gentamicin binds to megalin with low affinity and exploits the common ligand binding motif previously described (Jensen, G. A., Andersen, O. M., Bonvin, A. M., Bjerrum-Bohr, I., Etzerodt, M., Thogersen, H. C., O'Shea, C., Poulsen, F. M., and Kragelund, B. B. (2006) J. Mol. Biol. 362, 700–716) utilizing the indole side chain of Trp-1126 and the negatively charged residues Asp-1129, Asp-1131, and Asp-1133. Binding to megalin is highly similar to gentamicin binding to calreticulin. We discuss the impact of this novel insight for the future structure-based design of gentamicin antagonists.
Introduction
Severe infections with Gram-negative bacteria are normally treated with aminoglycosides of various chemical modifications (1). These antibacterial chemicals are affordable and are low resistance drugs with a high potency (2). One of these, gentamicin, is widely used in treatments of enterococcal, mycobacterial, and severe Gram-negative bacterial infections and acts on the bacterial ribosome, interrupting bacterial protein synthesis (3, 4). Application of gentamicin in high doses has, however, resulted in accumulation in the cochlea of the inner ear and the renal proximal tubular cells, and has been associated with both ototoxicity and nephrotoxicity (2, 5) causing the severe side effects of kidney damage and hearing loss (2). Although therapies with aminoglycoside derivatives using once-a-day dosage have limited this effect (6, 7), the clinical use of gentamicin is still a major problem today.
The human megalin receptor (also known as gp330 and LRP2) is a scavenger receptor, belonging to the low-density lipoprotein (LDL) receptor family (8). It is expressed widely throughout the body, including the cochlea of the inner ear (9) and the kidneys (10). In the kidney nephrons, megalin is localized on the apical surface of polarized epithelial cell membranes facing the proximal tubule (11). Its function in the inner ear is, however, unknown. Megalin is reported to bind at least 30 different ligands, including vitamin- and hormone-binding proteins, lipoproteins, and several other carrier proteins, including albumin and hemoglobin (12). Megalin undergoes endocytosis initiated by interactions via the adaptor protein disabled-2 (Dab2), through the third intracellular NPXY motif (13).
Structural work on the megalin receptor is very limited. From sequence analyses, the huge extracellular part consisting of 4400 residues has different domains, the most abundant being the complement type repeat (CR)2 domains. Each of these consists of ∼40 residues and contains a calcium binding site and three disulfide bonds (14). A total of four CR clusters appear in the extracellular part of megalin, containing 7, 8, 10, and 11 CR domains (14). The linker region between each CR domain varies in length and residue composition, suggested to be of importance for the ligand adaptation (15). The four CR clusters are separated by β-propeller domains containing YWTD sequences flanked with EGF-like repeat domains. To date, only a single structure of a megalin CR domain has been solved: the 12th CR domain from rat megalin (CR12r) (16). However, several structures of CR domains from both the LDL and the LRP receptors are available (17–20). Their overall fold is similar, with conservation of the calcium-coordinating residues and six cysteines responsible for the three disulfide bridges between cysteines I–III, II–V, and IV–VI.
Because of the sparse structural information available, little is known regarding the broad ligand binding profile of this family of receptors. Most CR domains bind to receptor-associated protein (RAP) during synthesis in the ER (21, 22). Structural studies of this complex have been reported (18, 23), and only structures of a few complexes other than RAP-CR are known. These include a fusion construct between apolipoprotein E (apoE) and CR17 from human LRP (24), the extracellular domain of the human LDL receptor bound to its β-propeller domain (19), and the very LDL receptor module V3 from humans in complex with rhinovirus HVR2 (25). A common ligand binding motif was suggested from an analysis of these structures (18) harboring a Trp and Asp/Glu pair from the receptor and a Lys and hydrophobic residue from the ligand. The positively charged ligands bind around the calcium binding site, which imposes a large negative surface potential on the CR domains driving the interaction.
It has been firmly established that gentamicin accumulates in the epithelial cells of the proximal renal tubule or the cochlea of the inner ear via megalin (26, 27). Once inside the cell, it accumulates in the lysosomes and the ER, where it binds to calreticulin, leading to elevated levels of misfolded protein in the cell (28, 29). After further accumulation, gentamicin is released into the cytosol, leading to oxidative stress and apoptosis (30), as well as to a reduction in the SGLT-1-dependent glucose transport (31). In vivo experiments using megalin knock-out mice have shown that megalin deficiency offers protection from gentamicin accumulation, indicating that megalin is the major contributor to gentamicin cell accumulation (32). Interestingly, however, megalin deficiency in mice also led to progressive hearing loss and affected the uptake of the steroid hormone β-estrogen (33).
A number of different approaches have been taken to lower the toxic effect of gentamicin. Some have targeted intracellular effects, e.g. by adding antioxidants such as allium or mitoquinone (34, 35), whereas others have attacked the receptor directly using natural ligands such as apoE3 (36) or positively charged peptides derived from, for example, cytochrome c (37, 38). These peptides have, however, not proven potent, and consequently, more efficient and structure-based design of receptor antagonists could prove resourceful. To aid this design, we have solved the solution structure of the 10th CR domain from the human megalin receptor alone and modeled its complex with gentamicin based on NMR chemical shift analyses. Gentamicin binds in the ligand binding site with submillimolar affinity utilizing the common binding motif. We discuss how this rather weak binding affinity may be linked to kidney failure and hearing loss during antibacterial treatment.
EXPERIMENTAL PROCEDURES
Vector Design and Pichia pastoris Electroporation
Stable isotope-labeled CR10 from human megalin (CR10h) was expressed in P. pastoris (Invitrogen) with the cDNA encoding for megalin (1103–1148) (GenBankTM U33837) optimized for P. pastoris expression, inserted into pPICZαC (GeneArt) using the following primers (restriction sites underlined): 5′-CGCCTCGAGAAGAGA-3′ and 5′-TAGTCTAGATTAATGATGATGATGATGATG-3′ (TAG Copenhagen). The correct sequence of the CR10h-pPICZαC was confirmed by DNA sequencing (Eurofins MWG Operon).
CR10h-pPICZαC was linearized using SacI and inserted using electroporation into strain X-33 pretreated with lithium acetate and DTT (39). To isolate multicopy recombinant colonies, a total of 12 colonies were picked from YPDS plates containing 2000 μg/ml ZeocinTM, and their vector was confirmed by colony PCR. The expression levels were tested by small-scale expression before isolating the highest expressing colony.
Protein Expression
A total of 250 ml of buffered (pH 6.0) minimal growth medium containing 1% (w/v) [13C6]glucose and 0.5% (w/v) [15N](NH4)2SO4 were used to grow P. pastoris to an A600 ∼4 while shaking at 200 rpm, 30 °C. The cells were harvested and dissolved to an A600 ∼1 in 1 liter of buffered induction medium containing 0.5% (w/v) [15N] (NH4)2SO4 and 0.5% (v/v) [13C]methanol. The culture was grown for 3 days, and 0.5% (v/v) [13C]methanol was added once every 24 h. The cell medium was isolated by centrifugation at 3000 × g for 5 min at 5 °C.
Protein Purification and Refolding
The medium was adjusted to pH 8.0, loaded onto a 5-ml HisTrap FF column (GE Healthcare), and eluted in 100 mm imidazole, 10 mm Tris-HCl, 150 mm NaCl, 1 mm CaCl2, pH 7.5. To obtain correctly folded protein, megalin CR10h was subjected to refolding by two protocols. In the first, protein (∼0.1 mg/ml) was reduced in 8 m urea, 50 mm Tris-HCl, 1 mm DTT, 1 mm EDTA, pH 7.8, and dialyzed toward 2 × 5 liters of 50 mm Tris-HCl, 20 mm CaCl2, 1/0.1 mm reduced/oxidized glutathione, pH 8.2, using a molecular weight cut-off filter of 3500 Da at 4 °C, followed by 5 liters of the same buffer without the redox pair present. In the second protocol, adapted from Ref. 40, the yield was significantly higher. Briefly, CR10h (∼0.2 mg/ml) was reduced in 6 m urea, 20 mm Tris-HCl, 50 mm NaCl, 10 mm CaCl2, 14 mm β-mercaptoethanol, pH 8.0, and after 1 h, was further diluted to ∼0.1 mg/ml in 50 mm Tris-HCl, 50 mm NaCl, 10 mm CaCl2, 14 mm β-mercaptoethanol, 8 mm 2-hydroxyethyl disulfide, pH 8.5 (refolding buffer) and refolded by dialysis toward 5 liters of refolding buffer at room temperature with N2 gas bubbling through the first 24 h followed by 24 h at 4 °C without. This was followed by exhaustive dialysis at 4 °C toward 20 mm Tris-HCl, 50 mm NaCl, 5 mm CaCl2, pH 7.8, to remove urea and the redox pair. The refolded protein was captured using a HisTrap FF column as described above. The eluted protein was deglycosylated overnight at room temperature using 0.5 units/μl N-glycosidase F (New England Biolabs) under native conditions.
Refolded, concentrated CR10h was acidified to pH ∼3 by 10% (v/v) TFA and loaded onto a μRPC C2/C18 column (GE Healthcare), eluted in a linear gradient from 0 to 70% (v/v) acetonitrile in 0.1% (v/v) TFA, identified by RAP binding, buffer-exchanged, and concentrated for NMR spectroscopy. Purity and identity were verified by SDS-PAGE, MALDI-TOF mass spectrometry, and N-terminal sequencing (Alphalyse).
NMR Experiments
[13C,15N]CR10h (0.6 mm) in 100 mm NaCl, 50 mm CaCl2, 50 mm Tris-HCl, 10% (v/v) or 100% (v/v) D2O, 0.1 mm 2,2-dimethyl-2-silapentanesulfonic acid, 0.1% (v/v) NaN3, pH 6.5, was analyzed using NMR spectroscopy. An 800-MHz Varian INOVA spectrometer equipped with a 5-mm triple resonance cryoprobe with a Z-field gradient, a 750-MHz Varian INOVA spectrometer equipped with a room temperature probe with a Z-field gradient, or an 800-MHz Bruker Avance equipped with a 5-mm triple resonance TCI cryoprobe were used. RAP binding was tested by adding 1:1 molar ratio of unlabeled RAP domain 1–2 purified as described (18).
The following experiments were recorded at 25 °C using Varian/Agilent BioPack sequences to assign backbone and side chain resonances: 1H,15N HSQC, 1H,13C CT-HSQC, HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH, C(CO)NH, H(CCO)NH, TOCSY 15N HSQC, and HCCH TOCSY spectra. NOE distance information was obtained from assigning and scoring peaks in aliphatic and aromatic NOESY 13C HSQC (mixing times 150 ms) and NOESY 15N HSQC (mixing times 150 ms) spectra. The HCCH TOCSY and the two NOESY 13C HSQC spectra were recorded on the sample buffer exchanged to 100% (v/v) D2O. All recorded free induction decays were processed using nmrPipe (41). Proton chemical shifts were referenced to internal 2,2-dimethyl-2-silapentanesulfonic acid at 0.00 ppm and heteronuclei indirectly using their gyromagnetic ratios.
NMR Assignments and Structure Calculation
Assignments of the CR10h backbone nuclei were performed by manually picking and linking resonances using CcpNMR analysis (42). The NOE assignments and structure calculation were performed using Aria2/CNS (43). Backbone dihedral angles were determined using DANGLE (44). Through eight iterations, a total of 200 structures were calculated in each cycle, selecting the 20 lowest energy models for the next iteration. Initially, structure calculations were performed based exclusively on the NOEs. Subsequently, three disulfide bonds, additional hydrogen bonds, and six calcium coordinating restraints were added. The calcium-oxygen distances were set to 2.3 ± 0.2 Å, based on the mean and S.D. of 389 high resolution (<2.0 Å) crystal structures in the MESPEUS database (45). Finally, the 20 lowest energy structures were refined in explicit water and selected to represent the final solution structure. To define the electrostatics and size of the calcium ion during the water refinement, the force field values for the Ca2+ radius were adjusted to 2.440 Å, and the Lennard-Jones potential well depth (ϵ) was set to 0.41 (46).
Gentamicin Titration
Two samples were sequentially mixed to record chemical shift perturbations in the presence of increasing gentamicin concentration. The gentamicin sulfate obtained from Sigma contains a mixture of three different types: C1 (25–50%), C1a (10–35%), and C2 (25–55%). The first sample contained 100 μm [15N,13C]CR10h dissolved in 100 mm NaCl, 50 mm CaCl2, 50 mm Tris-HCl, 10% (v/v) D2O, 0.1 mm 2,2-dimethyl-2-silapentanesulfonic acid, 0.1% (v/v) NaN3, pH 6.5. The second sample was similar and contained in addition 10 mm gentamicin. 1H,15N HSQC experiments were recorded on each sample, before transferring the gentamicin sample to the one without in five steps. A total of seven HSQC spectra with varying gentamicin concentrations were acquired.
Changes in backbone chemical shifts were used to map the overall binding site; the sizes of the chemical shift changes were calculated using Equation 1
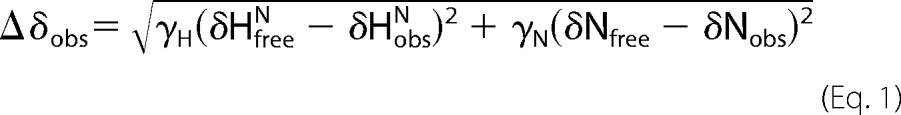 |
where δHfreeN and δHobsN are the unbound and observed proton chemical shift, and δfreeN and δobsN are the unbound and observed nitrogen chemical shift weighted by their respective (numerical) gyromagnetic ratios, γH and γN. The binding constant, Kd, was determined using Equation 2,
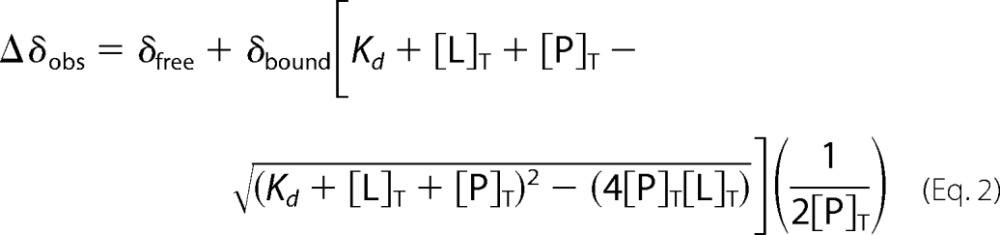 |
where Δδobs is the sum of unbound (δfree) and fully bound (δbound) times a factor of total protein concentration ([P]T) and total ligand concentration ([L]T), as well as the dissociation constant Kd. The observed chemical shift changes were fitted to Equation 2 using MATLAB.
HADDOCK Modeling
The web server version (47) of the docking program HADDOCK (high ambiguity-driven protein-protein docking) (48) was used to model the complexes between CR10h and gentamicin C1, C1a, and C2. The restraints used for the interaction were generated from the chemical shift perturbations. For CR10h, the solution structure ensemble between the terminal cysteine residues was used (residues 1109–1143). No significant chemical shift changes were observed for residues outside this region. Active and passive residues were analyzed using the program SAMPLEX (49). Active ambiguous interaction restraints (AIRs) included residues Gln-1120–Ile-1122, Lys-1124–Val-1127, Asp-1129–Asn-1132, and Asp-1139. Passive residues were Asn-1116 and Cys-1128. In addition, six calcium-coordinating unambiguous restraints were added to maintain the metal ion. These were defined on the basis of on the measured distances in the CR10h ensemble structure, using the above mentioned parameters.
The three different gentamicin isomers were energy-minimized in explicit water, and structure ensembles containing the 10 lowest energy structures were used in HADDOCK. Charges were calculated using the PRODRG server (50). In separate docking calculations, a total of 2000 structures were sampled during the rigid body docking. During the final iteration, the docking models were refined in explicit water, using the same parameters for defining the Ca2+-ion as described above. The 200 lowest energy structures were clustered on the basis of the pairwise ligand interface r.m.s.d. matrix, using a 2.5 Å cut-off with a minimum of four members per cluster. The clusters were ranked on the basis of the average HADDOCK score of the top four members. The HADDOCK score is a linear combination of restraint (EAIR), van der Waals (Evdw), and electrostatics (Eelec) energies on the basis of the optimized potentials for liquid simulations (OPLS) force field (51) and an empirical desolvation term (Edesol) (52) (HADDOCKscore = 0.1 EAIR + 1.0 Evdw + 0.2 Eelec + 1.0 Edesol).
SPR Measurement of Gentamicin to Purified Full-length Rabbit Megalin
Surface plasmon resonance (SPR) analysis was carried out on a Biacore 3000 instrument (Biacore, Uppsala, Sweden) using a CM5 sensor chip activated with a 1:1 mixture of 0.2 m N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide and 0.05 m N-hydroxysuccinimide in water. Megalin purified from rabbit kidneys (53) was diluted in 10 mm sodium acetate, pH 4.0, and immobilized in separate flow cells by injecting 320 μl of 5 μg/ml protein at 15 μl/min, to obtain a protein density of 21 fmol/mm2. Remaining binding sites were blocked with 1 m ethanolamine, pH 8.5 (70 μl, 5 μl/min). Samples were diluted in 10 mm HEPES, 150 mm (NH4)2SO4, 1.5 mm CaCl2, 1 mm EGTA, 0.005% Tween 20, pH 7.4 (assay buffer), and binding studies were carried out by injecting aliquots of 40 μl of gentamicin, at 16 concentrations between 0.25 and 10 mm, with a flow rate of 5 μl/min. After association bound ligand was dissociated by shifting to assay buffer. The binding signal is indicated as the difference in response units between the immobilized protein flow cell and the corresponding control flow cell (activated and blocked but without protein).
RESULTS
The 10th CR domain from human megalin (CR10h) from the second ligand binding cluster was chosen for the present study for several reasons. Firstly, it harbored the critical residues of the common binding motif (18), and secondly, it has been shown to be part of the ligand binding site for apoE-βVLDL, lactoferrin, bovine pancreatic trypsin inhibitor, lipoprotein lipase, and RAP (54). Finally, when comparing CR10h with CR12r, these have distinctly different residue compositions, particularly with regard to their overall charge, which has been suggested to be of importance in ligand binding.
We chose to express human CR10h using Pichia pastoris, which was expected to provide correctly folded protein. However, the 1H,15N HSQC spectrum of purified CR10h indicated incorrectly formed disulfide bridges, and CR10h was subsequently refolded in vitro and validated by the ability to specifically bind RAP (data not shown).
The CR10h Solution Structure
The solution structure of CR10h was determined on the basis of 408 NOE distance restraints (Table 1). A large dispersion of peaks in the proton dimension of the 1H,15N HSQC spectrum indicated a folded and calcium-loaded CR10h. Initial structure calculations were based solely on NOEs, revealing the expected CR domain disulfide bonding pattern. Subsequently, disulfide bond-, hydrogen bond-, and six calcium-coordinating restraints (20, 55, 56) were included (Table 1).
TABLE 1.
Statistics for the NMR solution structure of megalin CR10h residues 1103–1148
| NOE distance restraints | |
| Total number | 408 |
| Short range | 169 |
| Medium range | 157 |
| Long range | 82 |
| NOE violations | |
| >0.1 Å | 21 |
| >0.3 Å | 0 |
| Torsion angle restraints (DANGLE) | |
| ϕ | 21 |
| ψ | 17 |
| Maximal violation >1° | 10 |
| Maximal violation >3° | 1 |
| Ramachandran plot statistics | |
| Most favorable regions | 72.1% |
| Additional allowed regions | 26.3% |
| Generously allowed regions | 1.1% |
| Disallowed regions | 0.5% |
| r.m.s.d. values | |
| Backbone atoms (all residues) | 2.9 ± 1.2 Å |
| Heavy atoms (all residues) | 3.3 ± 1.1 Å |
| Cys I–VI backbone atoms (Cys-1109–Cys-1143) | 0.7 ± 0.2Å |
| Cys I–VI heavy atoms (Cys-1109–Cys-1143) | 1.1 ± 0.2 Å |
| WHAT-CHECK Z-scores (all residues) | |
| Second generation packing quality | 3.4 ± 1.8 |
| Ramachandran plot appearance | −4.2 ± 0.8 |
| χ-1/χ-2 rotamer normality | −4.9 ± 0.5 |
| Backbone conformation | −2.2 ± 0.8 |
| ResProx (70) | |
| All residues | 2.05 ± 0.19 |
| Cys I–VI (residues Cys-1109–Cys-1143) | 1.68 ± 0.30 |
The structure of CR10h has the same overall fold as homologues domains. The fold resembles a Greek omega letter (Ω) held in place by three disulfide bonds (Fig. 1). From the N terminus, a small β-hairpin (residue Gln-1113–Ser-1123) is followed by a 310 helix turn. This is succeeded by a flexible loop, coordinating the calcium ion in an octahedral arrangement. Both the N termini and the C termini are unstructured. The final bundle of 20 solution structures aligned with a backbone r.m.s.d. of 0.7 ± 0.2 Å for residues between cysteine I–VI.
FIGURE 1.

NMR solution structure ensemble of CR10h. A, stereo view (cross-eyed) of the 20 lowest energy structures aligned between Cys I–VI. The backbone r.m.s.d. is 0.7 ± 0.2 Å. Secondary structures are highlighted with β-strands in orange, 310 helices in red, and the disulfide bonds in purple. The Ca2+-ion is shown in gold. B, schematic representation of CR10h topology. The secondary structure elements are shown and are colored as in A. The octahedral coordination of the calcium ion is illustrated. C, multiple structural alignment of CR10h and all available PDB structures of unbound CR domains (extracted from PDB 1F8Z, 2LGP, 1AJJ, 2I1P, 1F5Y, 1CR8, 1D2J, 1LDL, 1N7D, 1LDR, 1FYJ, and 2KNX). CR10h is shown in solid red, with all other CR domains shown as thin transparent ribbons.
The sequence and structure of CR10h were compared with rat megalin, CR12r (Fig. 2) (16). Both belong to the second ligand binding region (residues 1111–1210) (54). Their alignment within cysteines I–VI gave an r.m.s.d. of 1.3 Å (SALIGN: cutoff 3.5 Å, quality score 35 (57)) for all backbone atoms. Their overall fold is similar with the calcium binding site and the three disulfide bonds conserved. A major difference is seen in the common ligand binding motif where the Trp is replaced by a Tyr, flanked by two Arg residues (Fig. 2A).
FIGURE 2.

Structural comparison of CR10h and CR12r. A, sequence alignment of CR10h (Thr-1103–Thr-1148) and CR12r (Gly-1182–Gly-1229). Coloring is black for hydrophobic residues, green for polar and uncharged residues, red for negatively charged residues, and blue for positively charged residues. Asterisks illustrate calcium-coordinating residues. Boxed residues are identical in the two CR domains. B, electrostatic potential of the front and back of CR10h as calculated using APBS (68). The blue, red, and white surfaces illustrate positive, negative, and neutral charge, respectively. The N terminus and the calcium ion are labeled. C, electrostatic potential of the front and back of CR12r colored as in B.
The charged residues result in very different electrostatic surfaces of the two CR domains. CR10h contains eight negatively charged residues and three positively charged residues, His-1119, Lys-1124, and Lys-1141, whereas CR12r contains four positively charged residues, Lys-1194, Arg-1205, Arg-1207, and Arg-1215, and six negatively charged residues. The resulting electrostatic potential plot shows that the surface of CR10h is mainly negatively charged with positive patches, whereas CR12r has a clear-cut positive half and negative half of charged surface (Fig. 2, B and C). This difference could indicate either that CR12r is capable of binding different ligands than CR10h or that the difference in surface charge is necessary for accommodating larger protein ligands spanning multiple CR domains. We note that CR12r has two arginine residues close to the common binding motif, whereas CR10h instead has an asparagine and a valine.
Binding of Gentamicin to Full-length Rabbit Megalin
Megalin and gentamicin interaction is critical for the known nephrotoxic and ototoxic side effects that can occur during drug treatment (5). The commercially available gentamicin contains a mixture of three gentamicins (Fig. 3), and because this product is used in treatments, we applied this mixture to megalin. We initially used SPR to determine an overall, average dissociation constant (Kdavg) for binding of gentamicin, in this case to full-length rabbit megalin. This was purified from rabbit kidneys, solubilized in Triton, and coupled to the chip. We observed saturable binding with a Kdavg of 4.2 ± 0.2 mm (Fig. 4). The binding is of low affinity, indicating that high doses of gentamicin are needed for effective binding, although species variations may be possible.
FIGURE 3.

Chemical structure of gentamicin C1, C1a, and C2. The R-groups for C1 are R1 = H, R = R2 = CH3. The R-groups for C1a are R = R1 = R2 = H. The R-groups for C2 are R = CH3, R1 = R2 = H. The pKa values for the titratable groups are shown in red (59).
FIGURE 4.

SPR binding curve for gentamicin binding to full-length rabbit megalin. Measurements of response units (RU) at 16 different gentamicin sulfate concentrations in the range between 0.25 and 10 mm are shown. The data are fitted to a single binding site with a Kdavg of 4.2 ± 0.2 mm, with fit shown in solid line.
Binding of Gentamicin to CR10h
We followed the gentamicin interaction with CR10h using NMR mapping of the effect on chemical shift and signal intensities. By analyzing the changes in amide backbone and side chain chemical shifts with increasing concentration of gentamicin, the residues in the binding site as well as the binding affinity were determined. Several chemical shift changes were observed in CR10h in the presence of gentamicin (Fig. 5A). The residues Cys-1121, Ser-1123, Lys-1124, Asn-1125, and Trp-1126 (backbone and indole Nϵ residues), Val-1127, Asp-1129, Thr-1130, and Cys-1143 were all perturbed above average, with the indole side chain of Trp-1126 exhibiting the largest chemical shift change (Fig. 5B).
FIGURE 5.

1H,15N HSQC titration and chemical shift perturbation of CR10h with gentamicin. A, overlay of seven 1H,15N HSQC spectra with varying gentamicin concentrations. The spectra of 0 μm, 280 μm, 500 μm, 1 mm, 2 mm, 5 mm, and 10 mm gentamicin are shown in pink, red, yellow, green, cyan, blue, and purple, respectively. Residues perturbed above average are boxed and labeled. The peaks of Glu-1120 and Trp-1126 are shown in the same box. B, the per residue calculated chemical shift perturbation when comparing 0 and 10 mm gentamicin are plotted for CR10h. The perturbation for the Trp-1126 indole Nϵ is shown in red. The horizontal black and red bars illustrate the average (AVG) and the average plus 1 S.D. (AVG+1σ).
The dissociation constant Kd for gentamicin binding to CR10h was determined from nonlinear fits to the chemical shift perturbations observed in the 1H,15N HSQC for all residues perturbed above average plus 1 S.D., except for Asp-1139 (Table 2). The average Kd value was 550 ± 60 μm, indicating a 1:1 binding complex with moderately low affinity. When comparing this value with the measured SPR Kdavg of 4.2 ± 0.2 mm, the difference we observed most likely stems from overall averaging over a number of sites in the full-length receptor and the two-dimensional nature of the SPR method. Species variations may also have an effect.
TABLE 2.
Kd values from perturbed residues in gentamicin CR10h binding
| Residue | Kd |
|---|---|
| μm | |
| Cys-1121 HN-N | 580 ± 60 |
| Lys-1124 HN-N | 540 ± 40 |
| Trp-1126 Nϵ-Hϵ | 530 ± 50 |
| Val-1127 HN-N | 520 ± 40 |
| Thr-1130 HN-N | 600 ± 80 |
| Asp-1131 HN-N | 550 ± 120 |
| Average | 550 ± 60 |
As the titration was performed using a mix of three isoforms of gentamicin, we were interested to infer from the 1H,15N HSQC spectra whether or not it would be possible to 1) determine whether one of the gentamicin isoforms was able to bind tighter than the others and 2) whether the different R-groups of the purpurosamine ring would give rise to different chemical shift populations and hence orient gentamicin on CR10h. The majority of the chemical shift changes are in the fast exchange regime on the NMR time scale, as observed by single weighted-average peaks containing information on the ligand-bound and ligand-free ratio. Thus, neither of these options could be exploited. Similarly, due to an elevated off-rate, it was not possible to obtain conventional intermolecular NOEs (58). Lastly, saturation transfer difference spectroscopy was applied, but the size of CR10h (6 kDa) was too small to eliminate resonances from the protein by a T1ρ filter. Hence, all attempts to obtain experimental data on the interaction, besides the chemical shift changes, were unsuccessful.
HADDOCK Model of the CR10h Gentamicin Complex
The chemical shift perturbations from the titration experiment were used to model the complex between CR10h and gentamicin using HADDOCK (47). We individually modeled gentamicin C1, C1a, and C2 into the binding site and compared the outputs. Active and passive residues in the binding complex were determined from the titration data analyzed by SAMPLEX (49). In all three dockings, the same active and passive residues were chosen (see “Experimental Procedures”).
The clustering based on an r.m.s.d. cutoff of 2.5 Å of the 200 refined structures from each of the three different gentamicin structure docking calculations all resulted in one large and several smaller clusters (Table 3). For the three top clusters, the buried surface area ranged between 495 and 539 Å2. The total solvent-accessible surface area of gentamicin is 643 Å2, with 322 Å2 ± 9 Å2 (∼50%) buried in the interface with CR10h. The lower ranking clusters displayed similar orientations of gentamicin in the binding site as the top cluster, although with fewer hydrogen bonds and salt bridges, and hence lower interaction energies. The orientation of gentamicin was not reversed in any of the top-ranking clusters, suggesting that the overall complementarity of the binding site directs the binding of gentamicin.
TABLE 3.
HADDOCK docking scores
| Gentamicin C1 (Cluster size/HADDOCK score) | Gentamicin C1a (Cluster size/HADDOCK score) | Gentamicin C2 (Cluster size/HADDOCK score) | |
|---|---|---|---|
| Cluster 1 | 103/−105.9 ± 1.5 | 113/−82.8 ± 2.8 | 72/−94.9 ± 0.7 |
| Cluster 2 | 6/−95.8 ± 6.2 | 23/−76.6 ± 3.3 | 36/−81.4 ± 1.8 |
| Cluster 3 | 32/−94.1 ± 0.5 | 32/−74.6 ± 2.9 | 5/−76.4 ± 0.9 |
| Cluster 4 | 10/−90.4 ± 8.0 | 8/−64.2 ± 1.3 | 4/−67.8 ± 3.9 |
| Cluster 5 | 26/−85.5 ± 1.6 | 22/−65.9 ± 2.3 | |
| Cluster 6 | 5/−74.3 ± 2.2 | 5/−65.5 ± 1.4 | |
| Cluster 7 | 5/−72.2 ± 9.7 | 4/−62.7 ± 5.8 | |
| Cluster 8 | 5/−57.4 ± 7.6 |
Gentamicin contains 4–5 positively charged groups at pH 6.5 (59), and the electrostatic surface of CR10h is highly negatively charged. Docking was performed with five positive charges on each gentamicin, and the resulting electrostatic energy potentials of the Cluster 1 structures were −525 ± 5 kcal mol−1 for C1, −455 ± 43 kcal mol−1 for C1a, and −500 ± 12 kcal mol−1 for C2. The electrostatic intermolecular energies are 50–100 times larger than the van der Waals and desolvation energy contributions, indicating that the electrostatic energy is indeed dominating the interaction, as predicted. Examining the common binding motif of CR modules (18), it is evident that gentamicin exploits this motif. In the crystal structure of CR34-RAPd3 (Protein Data Bank (PDB) 2FCW), the positively charged side chains of Lys-256 and Lys-270 create salt bridges to three aspartate side chains in the calcium binding cage (23). Here, in a similar manner, the charged NH3 groups of gentamicin form salt bridges to the side chains of Asp-1129, Asp-1131, and Asp-1133 in CR10h (Fig. 6E).
FIGURE 6.

HADDOCK model of gentamicin binding to CR10h. A–C, LigPlot (60) displaying the interactions between CR10h and gentamicin C1, C1a, and C2. Green striped lines indicate salt bridges with distances in Å. Spiked curves indicate hydrophobic interactions. D, electrostatic surface of CR10h displayed as in Fig. 2B, with the best Cluster 1 model of the three different gentamicin dockings. The calcium ion, Trp-1126, Asp-1129, Asp-1131, and Asp-1133 are labeled. E, zoom on the interaction between the DXDXD motif in CR10h and the charged NH3 groups of gentamicin.
By examining the HADDOCK complexes of the three individual gentamicin molecules and CR10h, one finds both differences and similarities (Fig. 6, A–C). In all models, Asp-1129, Asp-1131, and Asp-1133 participate in interactions with gentamicin C1, C1a, and C2. They are part of a DXDXD motif, found in other CR modules where the central aspartate has been suggested to increase the affinity ∼20 fold toward simple ligands (40). Similarly, in all models, the side chain of Trp-1126 is seen in π-cation interaction with the 2-deoxystreptamine ring from gentamicin C2 and the purpurosamine ring in both gentamicin C1 and gentamicin C1a. The differences in the three HADDOCK clusters are distinctly related to the NH3 groups from gentamicin C1, C1a, and C2, which form salt bridges with Asp-1129, Asp-1131, and Asp-1133. In the HADDOCK model of gentamicin C1, both NH3 groups of the 2-deoxystreptamine ring are involved; in C2, it is the NH3 group closest to the garosamine ring, and in C1a, it is the NH3 group from the purpurosamine ring. Whether this affects individual affinities is not presently known.
As the interaction between CR10h and gentamicin is governed mostly by electrostatics, the structures of the bound and unbound states of CR10h were similar, with r.m.s.d. values ranging between 0.7 and 1.0 Å. The side chain of Trp-1126 has the same orientation in the bound and unbound state.
CR Modules Are Preformed to Bind Ligands
We finally compared CR10h with bound and unbound structures of CR modules from LRP, LDL, and VLDL. The multiple structure alignment of CR10h to unbound CR modules had r.m.s.d. values between 1.1 (PDB 1F8Z) and 2.3 Å (PDB 1N7D). Apparently, the structures aligning with low r.m.s.d. to CR10h align with a low r.m.s.d. to each other, and vice versa for the structures with the highest observed r.m.s.d. values. The largest measured r.m.s.d. of 2.3 Å between CR10h and the R2 CR module from PDB 1N7D is in the N-terminal region of the LDL receptor crystal structure with missing electron density, explaining the poor r.m.s.d. The three additional CR modules from this structure (R3, R6, and R7) all align within 1.8 Å to CR10h. The most similar structure to CR10h is the sixth CR module from the LDL receptor (PDB 1F8Z). This module has, similarly to CR10h, 35 residues between cysteines I–VI, resulting in two very homologous structures. We did not find any correlation between alignment and overall surface charges (data not shown).
The comparison between CR10h and structures of bound CR domains (Table 4) reveals similar r.m.s.d. values as to the unbound structures (data not shown). CR10h aligns with an r.m.s.d. between 0.9 (PDB 2FCW) and 2.5 Å (PDB 2FYL), indicating that the binding-competent state of CR modules is the ground state and that side chain and backbone orientations do not change significantly during binding. For instance, the structures of free and bound CR56 to RAPd3 (PDB 2FYJ/2FYL) align with an r.m.s.d. of 1.9/2.3 Å in the free-state and with 1.8/2.5 Å in the bound state. Hence, from these comparisons, it appears that CR modules in general are preformed to bind their ligands.
TABLE 4.
Multiple structural alignment of bound CR modules
| CR10h | 2FCW (LB3) | 2FCW (LB4) | 1N7D (R4) | 1V9U | 1N7D (R5) | 2KNY | 2FYL (CR5) | 2FYL (CR6) | |
|---|---|---|---|---|---|---|---|---|---|
| CR10h | 0 | 0.907 | 0.939 | 1.503 | 1.657 | 1.740 | 1.744 | 1.835 | 2.483 |
| 2FCW (LB3) | 0 | 0.814 | 1.437 | 1.218 | 1.528 | 1.669 | 1.694 | 2.433 | |
| 2FCW (LB4) | 0 | 1.411 | 1.460 | 1.574 | 1.714 | 1.628 | 2.349 | ||
| 1N7D (R4) | 0 | 1.845 | 1.910 | 1.912 | 2.152 | 2.704 | |||
| 1V9U | 0 | 1.884 | 2.042 | 1.949 | 2.693 | ||||
| 1N7D (R5) | 0 | 1.980 | 2.134 | 2.826 | |||||
| 2KNY | 0 | 1.957 | 2.639 | ||||||
| 2FYL (CR5) | 0 | 2.476 | |||||||
| 2FYL (CR6) | 0 |
DISCUSSION
The megalin receptor presents itself as a fascinating receptor system as it is expressed widely throughout the body and binds a plethora of different types of ligands, yet it still maintains specificity within its ligand binding domains. Here, we present the first structure of the human megalin receptor, a CR module from the second ligand binding region, CR10h. We examined the similarities and differences between this domain and other structures of CR modules, in both their bound and their unbound states, and studied how it interacts with the aminoglycoside gentamicin.
Gentamicin nephrotoxicity and ototoxicity arise from unwanted cellular uptake of the drug, mainly via megalin. Once gentamicin enters the cell, it finds its way to the ER and binds to the lectin site of calreticulin, which leads to misfolding of newly synthesized proteins and subsequent oxidative stress in the cell, ultimately inducing apoptosis (29). Here, we examined the interaction between gentamicin and megalin and found that both the full-length megalin and a single CR domain have low affinity (μm–mm) for gentamicin. So how can gentamicin still accumulate in the kidney as well as in the inner ear?
The gentamicin binding site is located around the calcium binding site of the CR module, in full agreement with the common binding motif for the LRP receptor. Although the affinity in the μm–mm range is moderate to low, a single intravenous gentamicin dosage in treatment is as high as 5 mg/kg/day in adults, resulting in serum levels varying from 4 to 18 μg/ml (8–38 μm) or even higher (61). When gentamicin in the blood reaches the glomerulus, it passes through the Bowman's capsule via ultrafiltration due to its size and positive charges. Megalin then captures gentamicin in the proximal tubule. As all CR modules contain several negatively charged surface-exposed residues, the governing type of interaction between CR modules and its ligands is electrostatic. This was clearly revealed during HADDOCK docking of CR10h and gentamicin where the electrostatic energy term was more than 50-fold larger than the sum of the other terms, showing specific salt bridge formation to aspartic acid side chains of megalin. The higher levels of gentamicin in the tubular cells when compared with plasma (62), in combination with the low affinity for megalin, may explain the toxic effect observed during drug treatment. The release of gentamicin intracellularly is most likely due to a concentration gradient between the renal filtrate and the brush border cells of the kidney or in the cochlea in the inner ear.
We found indications that there are important common features in the CR modules that increase the affinity for gentamicin. First of all, gentamicin exploits the common binding motif found in all CR modules. However, the DXDXD motif, commonly found in many but far from all CR domains, and which has been shown to have ∼20-fold higher affinity for positively charged ligands (40), is only present in 12 out of 36 modules. This indicates that gentamicin potentially has 11 other binding sites similar to CR10h. In this respect, and taking the overall surface potential into account, we suggest that gentamicin is probably not a ligand for CR12r. As the Kd value for gentamicin binding to the intact receptor is lower than gentamicin binding to CR10h, we cannot exclude the possibility that other CR modules of megalin may have even higher affinity for gentamicin than CR10h. For instance, CR13h has a more negatively charged surface than CR10h and may bind gentamicin with higher affinity.
We also considered the possibility that the EGF-like and β-propeller domains could bind gentamicin as these domains contain negatively charged surface residues. However, the β-propeller domains probably do not bind gentamicin as their binding loops mostly contain polar and positively charged residues (63–65). We are also not aware of any reported interaction between gentamicin and the EGF-like or the β-propeller domains.
Gentamicin Binding to Calreticulin When Compared with CR10h
Once gentamicin is transported into the cell via endocytosis, it accumulates in the endosomes, from where it can be shuttled to the Golgi complex and the ER (66). In the ER, gentamicin binds the folding chaperone calreticulin, as determined by bovine kidney microsomes binding to a gentamicin-Sepharose column (29). SPR studies suggest a binding affinity in the μm range between calreticulin and gentamicin (29). Hence, the gentamicin affinity gradient from megalin to calreticulin suggests that once gentamicin is transported into the cell, other, higher affinity interaction partners are available.
A molecular model of gentamicin in complex with calreticulin was built by rigid body using DS 2.0 LigandFit, based on the 1.95 Å crystal structure of the carbohydrate complex of the lectin domain of calreticulin (PDB 3O0W) (67) stripped of its ligand (28). The suggested gentamicin binding site on calreticulin was confirmed by mutation studies of calreticulin and by measuring chaperone activity. It was proposed that gentamicin acts as a competitive inhibitor of sugar binding to calreticulin. When comparing our HADDOCK model of the gentamicin CR10h complex with that of gentamicin and calreticulin, we observe a high degree of similarity. Both have salt bridges involving three aspartate residues (Asp-125, Asp-135, and Asp-317 in calreticulin) and π-stacking with a tryptophan indole ring (Trp-319 in calreticulin) (28). Furthermore, in calreticulin, a tyrosine hydroxyl (Tyr-109) hydrogen-bonds with the NH3 group in the purpurosamine ring, potentially providing an explanation for the higher affinity when compared with megalin.
Receptor Antagonist Design
Attempts to inhibit gentamicin uptake by designed receptor antagonists have included positively charged peptides derived from natural megalin ligands such as lysozyme, demonstrating competition between gentamicin and lysozyme for megalin in proximal tubular cells (69). Gentamicin administration increases the concentration of vitamin D-binding protein and calcium, both of which are natural ligands of megalin, in the urine (62). This indicates that gentamicin administration also inhibits uptake of natural megalin ligands. The high similarity in structural architecture of CR modules furthermore poses the risk that administration of high affinity ligands also blocks natural ligand binding. This in turn may lead to proteinuria from lack of protein rescue from the kidney and irreversible loss of hair cells in the inner ear, leading to hearing loss. From the current structure of CR10h, it may now be possible to target ligands that specifically block binding to this module by taking advantage of both the common denominators of the motifs (Asp and Trp) as well as nonconserved residues lining the binding sites. From our analyses, we suggest that residues Lys-1124, Val-1127, and Asp-1131 may be interesting positions to include in an extended antagonist design as this combination of residues is only seen in CR10h.
Conclusions
In this work, we have solved the solution structure of a CR domain from the ligand binding region of human megalin. By comparing its structure with other CR domains, we have shown that a high degree of structural conservation exists. The overall charge, however, varies in the different modules, although they all contain negatively charged residues to accommodate Ca2+ binding. By SPR and NMR titrations, we demonstrated that full-length megalin and the single CR10h module bind the aminoglycoside gentamicin with low affinity in the μm–mm range. We propose a molecular model of the interaction between the three isomers of gentamicin and CR10h in which all are found to exploit the common ligand binding motif of CR domains (18). Residues Lys-1124, Val-1127, and Asp-1131 were identified as specific interacting residues from CR10h, which could be exploited in targeted design of a receptor antagonist to protect against gentamicin accumulation.
Supplementary Material
Acknowledgments
We thank Line Hyltoft Kristensen, Simon Erlendsson, Kaare Teilum, and Johan G. Olsen for fruitful discussions and Kresten Lindorff-Larsen for help with the water refinements. Signe Sjørup and Michael Nielsen are thanked for their excellent technical assistance. The Worldwide e-infrastructure for NMR and structural biology (WeNMR) project (European FP7 e-Infrastructure grant, Contract 261572), supported by the national grid Initiatives of Belgium, Italy, Germany, the Netherlands (via the Dutch BiG Grid project), Portugal, the United Kingdom, South Africa, Taiwan, and the Latin America grid infrastructure via the Gisela project, is acknowledged for the use of web portals, computing, and storage facilities. We also thank the Danish Instrument Center at the Carlsberg Research Center for spectrometer time.
This work was supported by a grant from the Oticon Foundation.

The 4 lowest energy structures from the HADDOCK model of CR10h and gentamicin C1, C1a, and C2 are available as supplemental information.
The atomic coordinates and structure factors (code 2M0P) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Chemical shift assignments for CR10h at pH 6.5 have been deposited in the Biological Magnetic Resonance Bank (http://www.bmrb.wisc.edu/) under accession number 18816.
- CR
- complement type repeat
- RAP
- receptor-associated protein
- ER
- endoplasmic reticulum
- LRP
- LDL receptor-related protein
- HSQC
- heteronuclear single quantum correlation
- TOCSY
- total correlation spectroscopy
- AIR
- ambiguous interaction restraint
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Chen L. F., Kaye D. (2009) Current use for old antibacterial agents: polymyxins, rifamycins, and aminoglycosides. Infect. Dis. Clin. North Am. 23, 1053–1075 [DOI] [PubMed] [Google Scholar]
- 2. Forge A., Schacht J. (2000) Aminoglycoside antibiotics. Audiol. Neurootol. 5, 3–22 [DOI] [PubMed] [Google Scholar]
- 3. Blanchard S. C., Fourmy D., Eason R. G., Puglisi J. D. (1998) rRNA chemical groups required for aminoglycoside binding. Biochemistry 37, 7716–7724 [DOI] [PubMed] [Google Scholar]
- 4. Wong C. H., Hendrix M., Priestley E. S., Greenberg W. A. (1998) Specificity of aminoglycoside antibiotics for the A-site of the decoding region of ribosomal RNA. Chem. Biol. 5, 397–406 [DOI] [PubMed] [Google Scholar]
- 5. Quiros Y., Vicente-Vicente L., Morales A. I., López-Novoa J. M., López-Hernández F. J. (2011) An integrative overview on the mechanisms underlying the renal tubular cytotoxicity of gentamicin. Toxicol. Sci. 119, 245–256 [DOI] [PubMed] [Google Scholar]
- 6. Rybak M. J., Abate B. J., Kang S. L., Ruffing M. J., Lerner S. A., Drusano G. L. (1999) Prospective evaluation of the effect of an aminoglycoside dosing regimen on rates of observed nephrotoxicity and ototoxicity. Antimicrob. Agents Chemother 43, 1549–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turnidge J. (2003) Pharmacodynamics and dosing of aminoglycosides. Infect. Dis. Clin. North Am. 17, 503–528 [DOI] [PubMed] [Google Scholar]
- 8. Raychowdhury R., Niles J. L., McCluskey R. T., Smith J. A. (1989) Autoimmune target in Heymann nephritis is a glycoprotein with homology to the LDL receptor. Science 244, 1163–1165 [DOI] [PubMed] [Google Scholar]
- 9. Mizuta K., Saito A., Watanabe T., Nagura M., Arakawa M., Shimizu F., Hoshino T. (1999) Ultrastructural localization of megalin in the rat cochlear duct. Hear. Res. 129, 83–91 [DOI] [PubMed] [Google Scholar]
- 10. Kerjaschki D., Farquhar M. G. (1982) The pathogenic antigen of Heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush border. Proc. Natl. Acad. Sci. U.S.A. 79, 5557–5561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chatelet F., Brianti E., Ronco P., Roland J., Verroust P. (1986) Ultrastructural localization by monoclonal antibodies of brush border antigens expressed by glomeruli. I. Renal distribution. Am. J. Pathol. 122, 500–511 [PMC free article] [PubMed] [Google Scholar]
- 12. Christensen E. I., Birn H. (2002) Megalin and cubilin: multifunctional endocytic receptors. Nat. Rev. Mol. Cell Biol. 3, 256–266 [DOI] [PubMed] [Google Scholar]
- 13. Gallagher H., Oleinikov A. V., Fenske C., Newman D. J. (2004) The adaptor disabled-2 binds to the third ψ xNPxY sequence on the cytoplasmic tail of megalin. Biochimie 86, 179–182 [DOI] [PubMed] [Google Scholar]
- 14. Saito A., Pietromonaco S., Loo A. K., Farquhar M. G. (1994) Complete cloning and sequencing of rat gp330/“megalin,” a distinctive member of the low density lipoprotein receptor gene family. Proc. Natl. Acad. Sci. U.S.A. 91, 9725–9729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Russell D. W., Brown M. S., Goldstein J. L. (1989) Different combinations of cysteine-rich repeats mediate binding of low density lipoprotein receptor to two different proteins. J. Biol. Chem. 264, 21682–21688 [PubMed] [Google Scholar]
- 16. Wolf C. A., Dancea F., Shi M., Bade-Noskova V., Rüterjans H., Kerjaschki D., Lücke C. (2007) Solution structure of the twelfth cysteine-rich ligand-binding repeat in rat megalin. J. Biomol. NMR 37, 321–328 [DOI] [PubMed] [Google Scholar]
- 17. Guttman M., Komives E. A. (2011) The structure, dynamics, and binding of the LA45 module pair of the low-density lipoprotein receptor suggest an important role for LA4 in ligand release. Biochemistry 50, 11001–11008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jensen G. A., Andersen O. M., Bonvin A. M., Bjerrum-Bohr I., Etzerodt M., Thøgersen H. C., O'Shea C., Poulsen F. M., Kragelund B. B. (2006) Binding site structure of one LRP-RAP complex: implications for a common ligand-receptor binding motif. J. Mol. Biol. 362, 700–716 [DOI] [PubMed] [Google Scholar]
- 19. Rudenko G., Henry L., Henderson K., Ichtchenko K., Brown M. S., Goldstein J. L., Deisenhofer J. (2002) Structure of the LDL receptor extracellular domain at endosomal pH. Science 298, 2353–2358 [DOI] [PubMed] [Google Scholar]
- 20. North C. L., Blacklow S. C. (2000) Solution structure of the sixth LDL-A module of the LDL receptor. Biochemistry 39, 2564–2571 [DOI] [PubMed] [Google Scholar]
- 21. Bu G., Geuze H. J., Strous G. J., Schwartz A. L. (1995) 39 kDa receptor-associated protein is an ER resident protein and molecular chaperone for LDL receptor-related protein. EMBO J. 14, 2269–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Willnow T. E., Armstrong S. A., Hammer R. E., Herz J. (1995) Functional expression of low density lipoprotein receptor-related protein is controlled by receptor-associated protein in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 4537–4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fisher C., Beglova N., Blacklow S. C. (2006) Structure of an LDLR-RAP complex reveals a general mode for ligand recognition by lipoprotein receptors. Mol. Cell 22, 277–283 [DOI] [PubMed] [Google Scholar]
- 24. Guttman M., Prieto J. H., Handel T. M., Domaille P. J., Komives E. A. (2010) Structure of the minimal interface between ApoE and LRP. J. Mol. Biol. 398, 306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Verdaguer N., Fita I., Reithmayer M., Moser R., Blaas D. (2004) X-ray structure of a minor group human rhinovirus bound to a fragment of its cellular receptor protein. Nat. Struct. Mol. Biol. 11, 429–434 [DOI] [PubMed] [Google Scholar]
- 26. Tauris J., Christensen E. I., Nykjaer A., Jacobsen C., Petersen C. M., Ovesen T. (2009) Cubilin and megalin co-localize in the neonatal inner ear. Audiol. Neurootol. 14, 267–278 [DOI] [PubMed] [Google Scholar]
- 27. Christensen E. I., Gliemann J., Moestrup S. K. (1992) Renal tubule gp330 is a calcium binding receptor for endocytic uptake of protein. J. Histochem. Cytochem. 40, 1481–1490 [DOI] [PubMed] [Google Scholar]
- 28. Hariprasad G., Kumar M., Rani K., Kaur P., Srinivasan A. (2012) Aminoglycoside induced nephrotoxicity: molecular modeling studies of calreticulin-gentamicin complex. J. Mol. Model. 18, 2645–2652 [DOI] [PubMed] [Google Scholar]
- 29. Horibe T., Matsui H., Tanaka M., Nagai H., Yamaguchi Y., Kato K., Kikuchi M. (2004) Gentamicin binds to the lectin site of calreticulin and inhibits its chaperone activity. Biochem. Biophys. Res. Commun. 323, 281–287 [DOI] [PubMed] [Google Scholar]
- 30. Lopez-Novoa J. M., Quiros Y., Vicente L., Morales A. I., Lopez-Hernandez F. J. (2011) New insights into the mechanism of aminoglycoside nephrotoxicity: an integrative point of view. Kidney Int. 79, 33–45 [DOI] [PubMed] [Google Scholar]
- 31. Takamoto K., Kawada M., Usui T., Ishizuka M., Ikeda D. (2003) Aminoglycoside antibiotics reduce glucose reabsorption in kidney through down-regulation of SGLT1. Biochem. Biophys. Res. Commun. 308, 866–871 [DOI] [PubMed] [Google Scholar]
- 32. Schmitz C., Hilpert J., Jacobsen C., Boensch C., Christensen E. I., Luft F. C., Willnow T. E. (2002) Megalin deficiency offers protection from renal aminoglycoside accumulation. J. Biol. Chem. 277, 618–622 [DOI] [PubMed] [Google Scholar]
- 33. König O., Rüttiger L., Müller M., Zimmermann U., Erdmann B., Kalbacher H., Gross M., Knipper M. (2008) Estrogen and the inner ear: megalin knockout mice suffer progressive hearing loss. FASEB J. 22, 410–417 [DOI] [PubMed] [Google Scholar]
- 34. Nabavi S. F., Nabavi S. M., Moghaddam A. H., Naqinezhad A., Bigdellou R., Mohammadzadeh S. (2012) Protective effects of Allium paradoxum against gentamicin-induced nephrotoxicity in mice. Food Funct. 3, 28–29 [DOI] [PubMed] [Google Scholar]
- 35. Ojano-Dirain C. P., Antonelli P. J. (2012) Prevention of gentamicin-induced apoptosis with the mitochondria-targeted antioxidant mitoquinone. Laryngoscope 122, 2543–2548 [DOI] [PubMed] [Google Scholar]
- 36. Takamoto K., Kawada M., Ikeda D., Yoshida M. (2005) Apolipoprotein E3 (apoE3) safeguards pig proximal tubular LLC-PK1 cells against reduction in SGLT1 activity induced by gentamicin C. Biochim. Biophys. Acta 1722, 247–253 [DOI] [PubMed] [Google Scholar]
- 37. Watanabe A., Nagai J., Adachi Y., Katsube T., Kitahara Y., Murakami T., Takano M. (2004) Targeted prevention of renal accumulation and toxicity of gentamicin by aminoglycoside binding receptor antagonists. J. Control Release 95, 423–433 [DOI] [PubMed] [Google Scholar]
- 38. Nagai J., Saito M., Adachi Y., Yumoto R., Takano M. (2006) Inhibition of gentamicin binding to rat renal brush-border membrane by megalin ligands and basic peptides. J. Control Release 112, 43–50 [DOI] [PubMed] [Google Scholar]
- 39. Wu S., Letchworth G. J. (2004) High efficiency transformation by electroporation of Pichia pastoris pretreated with lithium acetate and dithiothreitol. BioTechniques 36, 152–154 [DOI] [PubMed] [Google Scholar]
- 40. Gettins P. G., Dolmer K. (2012) A proximal pair of positive charges provides the dominant ligand-binding contribution to complement-like domains from the LRP (low-density lipoprotein receptor-related protein). Biochem. J. 443, 65–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 42. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 43. Rieping W., Habeck M., Bardiaux B., Bernard A., Malliavin T. E., Nilges M. (2007) ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 23, 381–382 [DOI] [PubMed] [Google Scholar]
- 44. Cheung M. S., Maguire M. L., Stevens T. J., Broadhurst R. W. (2010) DANGLE: A Bayesian inferential method for predicting protein backbone dihedral angles and secondary structure. J. Magn. Reson. 202, 223–233 [DOI] [PubMed] [Google Scholar]
- 45. Hsin K., Sheng Y., Harding M. M., Taylor P., Walkinshaw M. D. (2008) MESPEUS: a database of the geometry of metal sites in proteins. J. Appl. Crystallogr. 41, 963–968 [Google Scholar]
- 46. Aaqvist J. (1990) Ion-water interaction potentials derived from free energy perturbation simulations. J. Phys. Chem. 94, 8021–8024 [Google Scholar]
- 47. de Vries S. J., van Dijk M., Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 48. Dominguez C., Boelens R., Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 49. Krzeminski M., Loth K., Boelens R., Bonvin A. M. (2010) SAMPLEX: automatic mapping of perturbed and unperturbed regions of proteins and complexes. BMC Bioinformatics 11, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schüttelkopf A. W., van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 51. Jorgensen W. L., Tirado-Rives J. (1988) The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110, 1657–1666 [DOI] [PubMed] [Google Scholar]
- 52. Fernández-Recio J., Totrov M., Abagyan R. (2004) Identification of protein-protein interaction sites from docking energy landscapes. J. Mol. Biol. 335, 843–865 [DOI] [PubMed] [Google Scholar]
- 53. Moestrup S. K., Nielsen S., Andreasen P., Jørgensen K. E., Nykjaer A., Røigaard H., Gliemann J., Christensen E. I. (1993) Epithelial glycoprotein-330 mediates endocytosis of plasminogen activator-plasminogen activator inhibitor type-1 complexes. J. Biol. Chem. 268, 16564–16570 [PubMed] [Google Scholar]
- 54. Orlando R. A., Exner M., Czekay R. P., Yamazaki H., Saito A., Ullrich R., Kerjaschki D., Farquhar M. G. (1997) Identification of the second cluster of ligand-binding repeats in megalin as a site for receptor-ligand interactions. Proc. Natl. Acad. Sci. U.S.A. 94, 2368–2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Atkins A. R., Brereton I. M., Kroon P. A., Lee H. T., Smith R. (1998) Calcium is essential for the structural integrity of the cysteine-rich, ligand-binding repeat of the low-density lipoprotein receptor. Biochemistry 37, 1662–1670 [DOI] [PubMed] [Google Scholar]
- 56. Fass D., Blacklow S., Kim P. S., Berger J. M. (1997) Molecular basis of familial hypercholesterolaemia from structure of LDL receptor module. Nature 388, 691–693 [DOI] [PubMed] [Google Scholar]
- 57. Braberg H., Webb B. M., Tjioe E., Pieper U., Sali A., Madhusudhan M. S. (2012) SALIGN: a web server for alignment of multiple protein sequences and structures. Bioinformatics 28, 2072–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Breeze A. L. (2000) Isotope-filtered NMR methods for the study of biomolecular structure and interactions. Prog. Nucl. Magn. Reason. Spectrosc. 36, 323–372 [Google Scholar]
- 59. Lesniak W., Mc Laren J., Harris W. R., Pecoraro V. L., Schacht J. (2003) An isocratic separation of underivatized gentamicin components, 1H NMR assignment and protonation pattern. Carbohydr. Res. 338, 2853–2862 [DOI] [PubMed] [Google Scholar]
- 60. Wallace A. C., Laskowski R. A., Thornton J. M. (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 [DOI] [PubMed] [Google Scholar]
- 61. Verpooten G. A., Giuliano R. A., Verbist L., Eestermans G., De Broe M. E. (1989) Once-daily dosing decreases renal accumulation of gentamicin and netilmicin. Clin. Pharmacol. Ther. 45, 22–27 [DOI] [PubMed] [Google Scholar]
- 62. Nagai J., Tanaka H., Nakanishi N., Murakami T., Takano M. (2001) Role of megalin in renal handling of aminoglycosides. Am. J. Physiol. Renal Physiol. 281, F337–344 [DOI] [PubMed] [Google Scholar]
- 63. Cheng Z., Biechele T., Wei Z., Morrone S., Moon R. T., Wang L., Xu W. (2011) Crystal structures of the extracellular domain of LRP6 and its complex with DKK1. Nat. Struct. Mol. Biol. 18, 1204–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ahn V. E., Chu M. L., Choi H. J., Tran D., Abo A., Weis W. I. (2011) Structural basis of Wnt signaling inhibition by Dickkopf binding to LRP5/6. Dev. Cell 21, 862–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bourhis E., Wang W., Tam C., Hwang J., Zhang Y., Spittler D., Huang O. W., Gong Y., Estevez A., Zilberleyb I., Rouge L., Chiu C., Wu Y., Costa M., Hannoush R. N., Franke Y., Cochran A. G. (2011) Wnt antagonists bind through a short peptide to the first β-propeller domain of LRP5/6. Structure 19, 1433–1442 [DOI] [PubMed] [Google Scholar]
- 66. Sandoval R. M., Molitoris B. A. (2004) Gentamicin traffics retrograde through the secretory pathway and is released in the cytosol via the endoplasmic reticulum. Am. J. Physiol. Renal Physiol. 286, F617–F624 [DOI] [PubMed] [Google Scholar]
- 67. Kozlov G., Pocanschi C. L., Rosenauer A., Bastos-Aristizabal S., Gorelik A., Williams D. B., Gehring K. (2010) Structural basis of carbohydrate recognition by calreticulin. J. Biol. Chem. 285, 38612–38620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nagai J., Katsube T., Murakami T., Takano M. (2002) Effect of gentamicin on pharmacokinetics of lysozyme in rats: interaction between megalin substrates in the kidney. J. Pharm. Pharmacol. 54, 1491–1496 [DOI] [PubMed] [Google Scholar]
- 70. Berjanskii M., Zhou J., Liang Y., Lin G., Wishart D. S. (2012) Resolution-by-proxy: a simple measure for assessing and comparing the overall quality of NMR protein structures. J. Biomol. NMR 53, 167–180 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.