

Abstract
Significance: Aerobic organisms must exist between the dueling biological metabolic processes for energy and respiration and the obligatory generation of reactive oxygen species (ROS) whose deleterious consequences can reduce survival. Wide fluctuations in harmful ROS generation are circumvented by endogenous countermeasures (i.e., enzymatic and nonenzymatic antioxidants systems) whose capacity decline with aging and are enhanced by disease states. Recent Advances: Substantial efforts on the cellular and molecular underpinnings of oxidative stress has been complemented recently by the discovery that reductive stress similarly predisposes to inheritable cardiomyopathy, firmly establishing that the biological extremes of the redox spectrum play essential roles in disease pathogenesis. Critical Issues: Because antioxidants by nutritional or pharmacological supplement to prevent or mitigate disease states have been largely disappointing, we hypothesize that lack of efficacy of antioxidants might be related to adverse outcomes in responders at the reductive end of the redox spectrum. As emerging concepts, such as reductive, as opposed, oxidative stress are further explored, there is an urgent and critical gap for biochemical phenotyping to guide the targeted clinical applications of therapeutic interventions. Future Directions: New approaches are vitally needed for characterizing redox states with the long-term goal to noninvasively assess distinct clinical states (e.g., presymptomatic, end-stage) with the diagnostic accuracy to guide personalized medicine. Antioxid. Redox Signal. 18, 1114–1127.
General Principles on Oxido-Reductive Stress and Antioxidative Pathways
Molecular di-oxygen (O2) has been exploited through evolution by aerobic organisms and is converted to water via four-electron reduction, through sequential steps catalyzed by membrane-bound complexes within the mitochondria. Terminal electron transfer takes place in the mitochondrial electron transport chain, which generates high levels of ATP by oxidative phosphorylation. This is particularly essential within the mammalian heart, which could not produce enough energy to maintain essential cellular functions under anaerobic conditions (45). However, a small percentage (1%–2%) of the molecular oxygen consumed is incompletely reduced during this process (43, 86), resulting in the generation of highly reactive, partially reduced products of oxygen within the cell. These oxygen-derived pro-oxidants, or reactive oxygen species (ROS) are highly damaging to the cell if allowed to accumulate in large amounts, a process that increases with aging (Fig. 1) (8). Thus, the paradox of aerobic life has arisen: aerobic organisms cannot survive without oxygen, despite it being inherently dangerous to their existence (30).
FIG. 1.
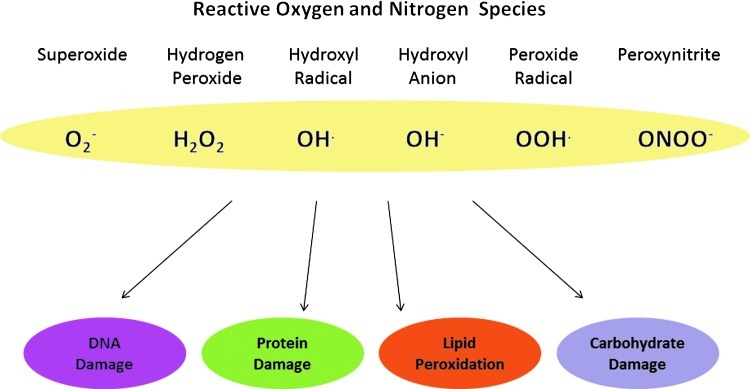
Types of reactive oxygen species.
In addition to the electron transport chain, there are many other potential sources of ROS to which the cell is exposed, both endogenous and exogenous. Thus, ROS may also be produced as by-products of other metabolic processes, including those catalyzed by xanthine oxidase, monamine oxidase, cytochrome P450-based enzymes, and uncoupled nitric oxide (NO) synthase (2, 18, 34). A wide variety of xenobiotics and toxic chemicals similarly produce ROS as by-products of their metabolism in vivo (37, 92, 128). Exposure of cells to ionizing radiation also results in the production of a range of ROS from the ionization of intracellular water, and constitutes a major exogenous source of ROS (110).
Lipids, proteins, carbohydrates, DNA, and RNA are all inherently susceptible to damage by oxidation. In vivo, a significant amount of autoxidation is caused by exposure to molecular oxygen itself, although many of these processes are, in fact, likely to be metal-catalyzed (30). ROS present an even greater danger to the integrity of these macromolecules, and as a consequence, molecular defense mechanisms have evolved to prevent the harmful accumulation of ROS within the cell. These systems are many and varied, reflecting the array of sources and species of ROS, which could potentially damage the cell. These mechanisms may act indirectly, for instance, by controlling the activity of endogenous ROS producing enzymes, such as xanthine oxidase. Alternatively, they may act by enhancing the efficiency of removal and/or repair systems, which act upon the sites of oxidative damage of macromolecules (72, 73).
Antioxidants can be broadly divided into two groups; those that are comprised of enzymes (Table 1), and those that consist of nonenzymatic low-molecular weight (MW) compounds (Table 2). Thus, the superoxide ion can be converted to water by the combined enzymatic activities of the superoxide dismutases (SODs), which catalyze the formation of H2O2 from O2− (83), and the further catalysis by catalase of H2O2 to H2O (75) or by members of the peroxidase family, including glutathione (GSH) peroxidase (GPx) (23). The SODs comprise a family of proteins, which differ in their structure, cellular localization, cofactor requirements, and reaction rate constants. Thus, mammals employ distinct cytoplasmic, extracellular, and mitochondrial forms of SODs, which utilize different transition metals at their active sites (Cu and Zn, or Mn for the mitochondrial SOD) (41). Catalases and peroxidases similarly have very distinct cellular locations and different modes of action and affinities for their substrate of H2O2. In most cells, including cardiomyocytes, catalase is exclusively localized within peroxisomes (52), while GPx is located in the cytoplasm and mitochondria (82). Catalase has a very low affinity for its substrate and so will remove H2O2 only when present at high concentrations. By contrast, GPx uses the reducing power of GSH and will remove H2O2 even when present in low concentrations [reviewed in Ref. (74)].
Table 1.
Endogenous Cellular Antioxidant Systems
| Enzyme systems | |
|---|---|
| Direct | Reaction |
| Superoxide dismutase | O2+O2→H2O2+O2 |
| Catalase | H2O2+H2O2→2H2O2+O2 |
| Peroxidase | 2 GSH+H2O2→GSSG+2H2O |
| Trx | Trx(SH)2+R-S-S-R→Trx-S2+R-(SH)2 |
| Glutaredoxin | 2GSH=R-S-S-R→2R-SH=GSSG |
| Supporting enzymes | Reaction |
| TrxR | Trx-S2+NADPH+H+→Trx–(SH)2+NADP+ |
| Glutathione S-transferase | GSSG+NADPH+H+→2GSH+NADP+ |
| Glucose-6-phosphate dehydrogenase | Glucose-6-phosphate+NADP+→ 6-hophogluconolactone+NADPH+H+ |
| Xanthine oxidase | Xanthine+H2O2→Hypoxantine+H2O+O2 |
| Uric Acid+H2O2→Xanthine+H2O+O2 |
Trx, thioredoxin; TrxR, thioredoxin reductase.
Table 2.
Small Molecule Antioxidants
| |
Solubility |
|
|---|---|---|
| Water | Lipid | |
| Scavengers | ||
| Ascorbic acid | * | |
| Bilirubin | * | |
| Carnosine | * | |
| Carotenoinds | * | |
| Glucose | * | |
| Glutathione | * | |
| Histidine di-peptides | * | |
| Pyruvate | * | |
| Sulfydryl groups | * | |
| Tocopherols | * | |
| Ubiquinol | * | |
| Uric Acid | * | |
| Cheating agents | ||
| Albumin | * | |
| Ceruloplasim | * | |
| Haptoglobin | * | |
| Hemopexin | * | |
| Transferrin | * | |
| Uric Acid | * | |
Common small molecule antioxidants with asterisks denoting whether compounds are water or lipid soluble.
GPx is a member of an important family of enzymatic antioxidants, collectively termed the thioredoxin (Trx) superfamily, which includes Trxs and thioredoxin reductase (TrxR), glutaredoxins, and peroxiredoxins (3). Trx and glutaredoxin are the primary reductants of intracellular disulfide bonds, but Trx and TrxR can additionally directly reduce H2O2, as can the peroxiredoxins [reviewed in Ref. (64)]. As is the case for SOD, distinct isoforms of the different enzymes exist, which demonstrate specificity of intracellular localization. Thus, the cell has developed the capacity to remove ROS that are produced in many different cellular locations at differing concentrations. Further, the expression levels of many of these enzymatic activities are known to be inducible, often in response to specific pro-oxidant insults, suggesting that the regulation of ROS levels within the cell is of vital importance (94).
The low MW (i.e., nonenzymatic) group of antioxidants includes many compounds that can scavenge the oxidizing radical by donating electron(s) directly, such as GSH, histidine dipeptides, uric acid vitamin C (ascorbic acid), and vitamin E (α-tocopherol) [reviewed in Refs. (30, 74)]. These small compounds possess a spectrum of activities against a wide variety of ROS, and their concentrations can be regulated within the cell. Some, such as vitamin E are lipid soluble, while others are aqueous, further demonstrating the adaptability that the cell possesses to regulate tightly the intracellular ROS.
Central to the mechanisms involved in redox signaling are thiol/disulfide couples, such as GSH/GSSG, Trxs, and cysteine/cystine (Cys/CySS), which must be maintained at distinct redox potentials to regulate the redox states of the reactive thiols (or so-called sulfur switches) of specific regulatory proteins (24). Among the major thiol–disulfide redox buffers within the cell, the GSH/GSSG system is considered the most important, since the concentration of GSH within the cytosol (1–11 mM) is far higher than most other redox-active compounds (44, 107). For decades, however, the intracellular levels of GSH are known to regulate redox-dependent cellular differentiation, proliferation, and apoptosis and are, therefore, of central importance to the function of the cell (4, 6, 39, 40). Until recently, the central dogma has been that low MW nonenzymatic thiols exclusively serve to counterbalance sources of ROS and to prevent (patho)physiological processes. However, there are increasing examples of elevated reducing equivalents (e.g., NADPH/NADP, GSH/GSSG ratios) resulting in cellular dysfunction and cardiac diseases, a condition termed “reductive, as opposed to oxidative, stress.” We will address, in greater depth, the initiating factors and pathogenic mechanisms of this emerging concept in cardiac biology in a later section of the review.
The concept of the redox state of a cell, as a measure of the intracellular balance between pro-oxidants and antioxidants, is embedded in the term “oxidative stress,” which represents a tilt in cellular redox imbalance in favor of the pro-oxidants, leading to oxidative damage (5, 10, 111). In contrast, reductive stress is defined as an excessive amount of reducing equivalents, in the forms of NAD(P)H and/or GSH, in the presence of intact oxido-reductive systems. The global cellular redox state as a measure of the balance between oxidative and reductive processes within a cell is further misleading, since it is now clear that the central thiol/disulphide couples are maintained at distinct redox potentials in different organelles (62, 107). These redox couples within the different cellular compartments are maintained at distinct, nonequilibrium steady states, and are central to the specialized redox-dependent functions of the individual organelles (69).
In the heart, the importance of the cell's antioxidant systems has become increasingly recognized with the emergence of the strong correlations between the progression to cardiac pathologies, and cellular damage caused by pro-oxidant species, such as ROS (33, 68). In addition, many in vitro and animal studies have demonstrated increased ROS in the cardiovascular system in response to various stressors and in the failing heart (14, 20, 56, 104, 115). As a result, antioxidants have garnered considered attention as potential therapeutic defenses against heart (and other) diseases (81). However, many large-scale interventional trials with antioxidant free radical scavengers have failed to demonstrate any health benefits, and in some cases have shown deleterious effects (113). In contrast to the highly sensitive and specific biomarkers for ischemic myocardial injury (e.g., creatine kinase MB isoform, troponins), similar diagnostic tools, among the plethora of redox markers, are inadequate to guide therapeutic interventions in diverse pathological oxido-reductive states. Indeed, this review undertakes such conceptual hurdles and explores how dynamic shifts of individual redox couples within specific intracellular compartments alter signaling and function, in relation to both normal physiology and pathophysiology, pose important challenges and opportunities for research into redox biology of the cardiovascular system.
Redox Metabolism and Small MW Heat Shock Proteins
Redox metabolism, in theory, refers to the integrated networks of catabolic and anabolic biochemical pathways for oxidative phosphorylation and energy production, which critically depend on the flow of free electrons and ROS/reactive nitrogen species. Dynamic shifts in redox metabolism associated redox couples (e.g., NAD/NADH+, GSH/GSSG) add the system's versatility at the level of redox-sensitive biomolecules, which ultimately are governed by the redox signaling events within membrane-bound compartments. Current understanding suggests that these subcellular compartments maintain mostly reductive and/or oxidative environments in a tissue-specific manner. Although oxidative damage from excessive production of ROS, in particular, at the level of the mitochondria (8, 50, 95), is counterbalanced through redox couples [NAD(P)H/NAD(P)+, GSH/GSSG], much less attention has focused on the mechanisms for buffering ROS by vital proteostasis networks engaged in protein synthesis and repair (69, 107). Molecular chaperones, exemplified by the multigene family of heat shock proteins (HSPs), are effective cytoprotective agents against the proteotoxic effects of oxidative stress. In the cardiovascular field, several recent reviews have highlighted the general properties of HSPs in ischemic cardioprotection and their unique roles in redox homeostasis and cardiovascular function (26, 106, 116). In particular, the subfamily of small MW HSPs (sHSPs) has emerged to play key roles in redox metabolism/state, the field of redox biology, and for providing new understanding about disease pathogenesis.
sHSPs, averaging between 15 and 30 kDa in MW, comprise a heterogeneous group of ATP-independent molecular chaperones. There are 11 mammalian sHSPs recognized by the HUGO Gene Nomenclature Database (65), which share a central α-crystallin domain and whose diverse biological roles have been extensively studied in development, differentiation and, more recently, in maintaining redox balance. These are shown in Table 3, listed in the order they were discovered.
Table 3.
Small Molecular Weight Heat Shock Proteins in Health and Disease
| Protein | Mass (kDa) | Localization | Cardiac diseases | Other pathological conditions |
|---|---|---|---|---|
| HSPB1 (HSP27/HSP25) | 22.3 | Ubiquitous | Dox-induced Heart Failure, I/R (119) | Neuropathy, cancer (7, 91) |
| HSPB2 (MKBP) | 20.2 | Heart and muscle | Myopathy, I/R (15, 47) | Alzheimer's disease and hereditary cerebral hemorrhage with amyloidosis, Dutch type (127) |
| HSPB3 (HSPL27) | 17.0 | Heart and muscle | A point mutation correlates with the development of axonal motor neuropathy (12) | |
| HSPB4 (CRYAA) | 19.9 | Eye lens | Cataract (27) | |
| HSPB5 (CRYAB) | 20.2 | Ubiquitous | I/R, point mutations lead to cardiomyopathy and age-related HF (42) | Neuropathy, desmin-related myopathy, breast cancer, cataract (54, 114) |
| HSPB6 (HSP20/p20) | 17.1 | Ubiquitous | I/R, cardiac hypertrophy, myocardial infarction, and heart failure (38) | Platelet aggregation, atherosclerosis, insulin resistance, Alzheimer's disease (109) |
| HSPB7 (cvHSP) | 18.6 | Heart and muscle | Polymorphisms associated with heart failure, idiopathic DCM (112) | |
| HSPB8 (HSP22, H11 kinase) | 21.6 | Ubiquitous | Ischemia, cardiac hypertrophy (1) | Neuropathy, cancer (1, 59) |
| HSPB9 (CT51) | 17.5 | Testis | Cancer (31, 67) | |
| HSPB10 (ODF1) | 28.4 | Testis | Infertility with decapitated sperm heads (130) | |
| HSPB11 (HSP16.2) | 16.3 | Placenta | Cancer (97) |
I/R, ischemia/reperfusion.
The storied history of HSPs, best known as molecular chaperones for facilitating protein folding and the prevention of protein aggregation, has many unanticipated twists and serendipitous links for the field of redox biology. Arrigo and coworkers were the first to identify an inverse relationship between GSH levels and the sHSP25/27 (HspB1) (85). In fibroblast cells, overexpression of HSPB1 elevated levels of GSH and increased the expression and enzymatic activity of glucose 6 phosphate dehydrogenase (G6PD), the rate-limiting enzyme of the pentose phosphate pathway. Although a more direct causal relationship between HSPB1 expression and G6PD activity was not formally tested at the molecular level, these studies have provided the foundations for exploring potential roles sHSPs play in modulating ROS in both in vitro and in vivo models. These associative studies were modestly compelling for evidence that HSPB1 regulated iron metabolism and potential promoted the enzymatic functions of G6PD, the rate-limiting enzyme in the pentose phosphate pathway. However, disease-modeling of the inheritable cardiomyopathy caused by the human αB-crystallin (CryAB) (R120G) in the mice was a milestone not only for the discovery of R120G-induced increased GSH levels, but that this form of reduced stress (i.e., increased GSH/GSSG ratios) was pathogenically linked to human disease. As expected, R120G overexpression serves as a stress signal to induce increased HSPB1 expression, oxidative stress, and levels of antioxidative metabolic enzymes. Because G6PD deficiency rescued the cardiomyopathic phenotypes (i.e., hypertrophy, heart failure, and mortality), these studies for the first time established that G6DP was both necessary and sufficient for mediating pathologic reductive stress. These counterintuitive studies on the relationship between sHSPs and redox state were elegantly supported by transgenic (TG) HSPB1/Hsp27 overexpression per se caused increased GSH levels and cardiomyopathy. Taken together, there is compelling evidence for a causal relationship between sHSPs and reduced state for generating a redox imbalance in the cardiac tissue, tilting the balance toward the reductive spectrum.
In recent years, biomarkers have garnered considerable attention, but oftentimes lack the sensitivity and specificity for specific disease states. In an integrated systems approach using gene expression profiling, we identified a biosignature, whose features can be validated to predict the onset, rate of progression, and clinical outcome of R120GCryAB cardiomyopathy (100).
Our study confirmed our previous findings that reductive stress is a causal mechanism for hR120G CryAB cardiomyopathy and demonstrated that alteration in GSH pathway gene expression is an early biosignature with utility for presymptomatic detection. Increased levels of sHSPs are well-documented after physiological and, more commonly, pathophysiological stimuli. However, most, but not all HSPs are intracellular molecules whose multifunctional roles are widely thought to be the first line of defense against proteotoxic damage, increased proteolysis of irreversibly damaged protein, prevent protein aggregation, activation of autophagy, and maintenance of survival pathways. In many instances, the physiological expression of HSPs might impart specialized functions associated in a tissue-specific manner without necessarily affecting redox metabolic pathways. Likewise, we think that dysregulation of antioxidative pathways associated with HSPs are highly context and tissue-dependent. Altogether, tissue sampling by an invasive procedure will be necessary to capture the critical biochemical and cellular information of reductive stress. In the likely event there are therapeutic interventions, we think that the benefits will outweigh the risks in the era of genomic and personalized medicine. However, the answers to the following questions can pave a way to a more targeted therapeutic intervention of the protein aggregation-mediated diseases (i) to what extent are these proteins effective in dealing with ROS generation/abrogation, (ii) what are the redox metabolic pathways used directly or indirectly by sHSPs, and (iii) can sHSPs serve as pharmacological targets in rescuing the oxido-reductive stress? Recent studies primarily using gain-of-function maneuvers for either wild-type or mutant sHSPs in genetically engineered murine models are beginning to provide answers.
A Genetic Model of Human HSPB1 Overexpression Mimics Reductive Stress in Mice
In the cardiovascular system, HSPB1 remains the best characterized sHSP, and has been shown to play an important role in regulating intracellular redox homeostasis. Thus, moderate levels of HSPB1 expression protect the myocardium against doxorubicin-induced cardiac dysfunction and attenuate lipopolysaccharide-induced myocardial injury (35, 79). Although HSPB1 influences the intracellular redox homeostasis by upholding reduced GSH and decreasing intracellular iron levels (11), direct physiological evidence for such proreducing properties remain speculative. To test the hypothesis that HSPB1 overexpression is necessary and sufficient to generate a proreducing state, Zhang and coworkers generated TG mice harboring cardiac-specific overexpression of HSPB1 in TG mice (135). Among the TG lines, mild (∼2-fold) to moderate (∼7-fold) overexpression (OE) of HSPB1 was found to have no effect on cardiac growth, whereas high (∼15-fold) HSPB1 OE caused spontaneous hypertrophy, which progressed into overt heart failure by 43 weeks (135). TG mice with high HSPB1 OE exhibited significantly increased GSH and GSH/GSSG ratio, which correlated reciprocally with a decrease in protein carbonylation and with elevated GPx, consistent with maintaining GSH in the reduced state and a general shift in the redox imbalance toward the reductive spectrum. Of interest, HSPB1 OE was not accompanied by elevations of either the activity or expression of G6PD.
In comparison with the established roles of HSPs as cardioprotective agents, the molecular mechanisms for aforementioned functions in redox homeostasis are less well-understood. Among members of the multigene HSP family, the mechanisms for their cardioprotective properties are likely multifactorial—(i.e., prevention of protein aggregation, protection against ROS-induced DNA, lipid and protein damage, etc.). In the specific of HSPB1 (i.e., Hsp25/25) overexpression, we speculate that potential protein–protein interactions between chaperone-like functions of HSPB1 and G6PD likely contribute to alterations of the GSH redox potential, both in vitro and in vivo. Indeed, such questions are being actively investigated worldwide. Notwithstanding, these results establish that HSPB1 OE could promote pathological shifts in redox homeostasis in a dose-dependent manner, thus validating earlier studies and implicating unspecified pathways involving the redox sensitive enzymes, transcription factors, and signaling molecules in disease pathogenesis.
Mutant HSPB5 (R120G) Causes Protein Aggregation Cardiomyopathy and a Gain of Toxic Function-Induced Reductive Stress
HSPB5 (CryAB) is a bona fide HSP and a well-characterized member of the crystallin family, characterized initially as lens proteins for their refractive and structural properties (16, 36). With the discovery by Dubin and colleagues (36) that murine HSPB5 was not restricted to the ocular lens, the full spectrum of HSPB5 expression was established in tissues with high rates of oxidative metabolism, such as the heart, type I, and type IIa skeletal muscle fibers, brain, and oxidative regions of the kidney. HSPB5 alone constitutes up to 3% of the soluble cardiac homogenate, suggesting a prominent role of this protein in maintaining cardiac function. In striated tissues, the chaperone-like functions of HSPB5 are recruited for the prevention of protein misfolding and aggregation of the intermediate filament protein, desmin, thereby providing protein quality control in metabolically active tissues (56, 57, 84). In contrast to the HSPB1 OE TG mouse model, TG mice with HSPB5 OE are significantly protected against ischemia/reperfusion (I/R) injury compared with the non-TG littermates (102). However, knockout mice harboring double deletions of both HSPB2/HSPB5 are viable, indicating that HSPB5 per se is dispensable for survival and raising the intriguing possibility of genetic redundancy among sHSPs in vivo (15, 63).
While HSPB5 expression is dispensable for cardiac metabolism, an inheritable missense mutation of HSPB5 (abbreviated as R120G) identified by Vicart et al. (121) causes a multisystem disorder, termed desmin-related myopathy (DRM). Biochemical studies, characterized by chaperone activity, protein stability, and aggregation-prone insolubilization, indicate R120G has loss-of-function consequences (125). To determine the molecular mechanisms of R120G expression, TG mice harboring cardiac-specific overexpression were shown to recapitulate the human-specific phenotype of DRM characterized histologically by formation of large aggresomes and progressive heart failure (99, 126). The human R120G model characterized by Rajasekaran et al. exhibited cardiac hypertrophy, increased GSH levels, and the GSH/GSSG ratio or reductive stress, which was attributed to the increased G6PD expression and activity (99). Further, the intercross of R120G mice with mice harboring a mutant G6PD allele that lowers its activity by ∼20% rescued the hypertrophic phenotype with concomitant decrease in the mutant aggregate formation. Because G6PD deficiency abrogated the pro-reductive GSH levels of R120G expression, these findings provide genetic evidence for a causal role of this metabolic pathway in R120G cardiomyopathy (Fig. 2). Along with the molecular link between R120G- and G6PD-mediated GSH production, HSPB1 was upregulated at the transcriptional and translational levels.
FIG. 2.

Redox couples in relationship to thiol-dependent small HSPs.
Mitochondria, by far the most vital organelle for energy production and redox adaptation (56, 120), are organized spatially at the A and I bands of the sarcomere, thereby providing proximity of ATP production to the proximal contractile apparatus (28, 84). Independently, Maloyan and coworkers demonstrated severely compromised mitochondrial function and disrupted mitochondrial organization of mouse R120G compared with HSPB5 WT littermates, suggesting that the mutant R120G expression might disrupt ROS production during progressive heart failure (80). In fact, we have found that human R120G induces significant levels of ROS at 3 months of age (>∼3-fold vs. controls) assessed by both electron paramagnetic resonance probes, but that ROS were elevated to comparable between TG and non-TG littermates at 6 months (101). Because gene expression profiling revealed that distinct categories of antioxidants: catalase, G6PD, GPx1, GSH reductase (GR), and NAD(P)H quinone oxidoreductase 1 (NQO1) were increased significantly in the TG mice (100), we hypothesized that elevated ROS levels during compensatory hypertrophy (i.e., 3 months) could trigger upregulation of key antioxidants, in a nuclear erythroid 2-related factor 2 (Nrf2) antioxidant response element (ARE)-dependent transactivation manner (Fig. 3) (80). The role of Nrf2 in upregulating the antioxidant enzymes, especially in the setting of excess reducing equivalents or reductive stress, will be discussed further in the following section (90, 101).
FIG. 3.

Role of Nrf2 in R120G metabolism (101). Reprinted with permission.
ROS, Reductive Stress, and Nrf2 Pathways
Nrf2 was initially identified from a screen of a cDNA expression library for DNA binding proteins that bound to the beta-globin cluster of genes (87). Nrf2 is a basic leucine zipper protein that belongs to the Cap n’ Collar family of transcription factors (87). Nrf2 governs an inducible cytoprotective system designed to allow cells to adapt to stress induced by oxidants and electrophiles. Adaptation to these stresses ensures acute control over the cellular redox balance; therefore, Nrf2 functions as an intracellular redox sensor that allows sensitive adjustments to be made to regulate redox homeostasis. This ensures that a steady-state pool of Nrf2 is readily available to promote constitutive gene expression and to respond to stress stimuli by coordinating the expression of an arsenal of antioxidant and cytoprotective genes in response to oxidants, xenobiotics, and electrophiles. Nrf2 is subject to activation by ROS, which act to regulate its localization, phosphorylation, and protein degradation (13). Levels of Nrf2 expression correlate with genes encoding the pentose phosphate pathway responsible for NADPH production, supporting its transcriptional control by redox-dependent mechanisms (17). We have preliminarily determined that oxidative stress precedes the development of reductive stress, but direct evidence either for this exclusive relationship or for the reciprocal relationship remains speculative.
In unstressed cells under balanced redox conditions, Nrf2 is normally negatively regulated by the Kelch-like ECH-associated protein 1 (Keap1), a redox-sensitive, cysteine-rich adaptor protein (60). Homodimeric Keap1 binds Nrf2 via its Kelch-like domain, sequestering it in the cytoplasm, and thereby inhibiting its activation and nuclear translocation (66). Additionally, Keap1 regulates the turnover and, therefore, stability of the Nrf2 protein by acting as a bridge between the Nrf2 and a Cul3-dependent E3 ubiquitin ligase complex (Cullin3-Rbx1) (29, 71). This complex ubiquitinates Nrf2, and subsequently targets it for proteasome-mediated degradation ensuring a low steady-state level in unstressed cells [reviewed in Refs. (13, 93)]. Keap1-independent pathways, which regulate Nrf2 through microRNA-mediated translational inhibition (131) and phosphorylation of Nrf2 by stress-activated kinases, have also been described (117).
Activation of Nrf2, upon dissociation and release from Keap1, results in its translocation to the nucleus, where it binds to cis-acting AREs or electrophile response elements and induces the transcription of an array of antioxidant and cytoprotective genes, such as GSH-S-transferase (GST) (103), heme oxygenase-1 (58), Trx (70), NQO1 (32), and glutamate-cysteine ligase (GCL) (89). Both Keap1 and Nrf2 are ROS-sensitive proteins. Key cysteine residues (Cys-151/273/288) within the Keap1 protein act as stress sensors that, upon oxidation, result in a conformational change in Keap1 and promote the dissociation and activation of Nrf2 (122, 129). Nrf2, in turn, is itself regulated by oxidation of a specific cysteine residue (Cys-183) within its transactivation domain (TAD). Oxidation of this cysteine masks a nuclear export signal in the TAD of Nrf2 and promotes its nuclear localization (78). Additionally, phosphorylation of Nrf2 (Ser-40) by protein kinase C (PKC) can also promote its dissociation from Keap1 and induce nuclear translocation and Nrf2-dependent transcription upon oxidative stress (55).
Within the heart, oxidative stress inducers are believed to be important mediators of the pathological progression toward cardiac dysfunction and disease. Primarily, Nrf2 responses oppose these detrimental increases in ROS production and result in increased oxidant handling and detoxification abilities of the cell. This allows a higher oxidant load to be tolerated and alleviates the negative effects of increased ROS. The potential importance of Nrf2 as a regulator of redox homeostasis is suggested in a study of failing human hearts where, concomitant with persistent increases in ROS levels, the activity of Nrf2-regulated antioxidant enzymes were shown to decline (105).
Many studies have been performed recently to investigate the function of Nrf2 in animal models of cardiac dysfunction. In unstressed conditions, Nrf2-null mice are phenotypically normal during growth and development (22), and with regard to cardiac structure and function (76). However, Nrf2-null mice are highly susceptible to a variety of oxidative stresses that can directly or indirectly affect cardiac function, including high-glucose-induced oxidative cardiomyocyte damage (51), bleomycin-induced pulmonary fibrosis (25, 124), and hyperoxic lung injury (21). In response to various stress insults, which are known to cause increased ROS formation, endogenous Nrf2 normally becomes activated and ablation of Nrf2 has proven to be detrimental to cardiac function. For instance, in a mouse model of chronic transverse aortic constriction, Nrf2−/− mice developed greater cardiac dysfunction stemming from increased cardiac hypertrophy and interstitial fibrosis (76). Similarly, angiotensin II (Ang II)-induced cardiac hypertrophy (77), and cell death caused by I/R injury (136) were both exacerbated in Nrf2-null mice. In the case of Ang II-induced hypertrophy, a functional interaction between Nrf2 and the cell cycle regulator, p27, in the inhibition of hypertrophy was demonstrated (77). In general, the deleterious effects of Nrf2 ablation upon cardiac function have been ascribed to decreased levels of antioxidant and detoxifying enzymes resulting in excess oxidant-mediated damage after ROS-induced cardiac stress.
The precise sources of ROS believed to contribute to cardiac pathologies remain largely unknown, but are likely to be important determinants of the Nrf2 response. The NADPH oxidase (NOX) family of enzymes generates endogenous ROS and has been shown to be critical mediators of oxidative signaling during cardiac remodeling (9). Moreover, we have shown that mice, which specifically overexpress NOX4 in cardiomyocytes, have been shown to activate directly Nrf2 signaling pathways and to result in the increased expression of Nrf2 target genes, especially those involved in GSH synthesis and recycling, such as GCL and GR (17). The increased ROS produced by NOX4 in this system caused a change in the intracellular GSH redox state to generate a more reduced intracellular environment (i.e., >GSH/GSSG ratio), which was ablated in an Nrf2−/− background (17). This demonstrates that, while an initial stimulus might be oxidative in nature, the response of the cell can subsequently result in an overall more reducing cellular environment. It is presumed in this case that the ROS-mediated activation of Nrf2 may occur directly through the oxidation of regulatory cysteines in Keap1. Alternatively, the ROS may act indirectly through activation of other signaling molecules. One such molecule that might link ROS generated by NOXs with Nrf2 activation is the mitogen-activated protein kinase, ERK-1. Thus, it has been shown that increased ROS from NOX enzymes can activate ERK-1 signaling and activate Nrf2 (96).
Any change in redox potential, elicited by a pro-oxidant, would be expected to return to the steady-state equilibrium after the appropriate antioxidant cellular response has been taken. However, we previously have shown that the sustained activation of Nrf2 can result in reductive stress in which the reductive capacity of the cell, or the concentration of reducing equivalents, exceeds ROS production (101). Therefore, the sustained activation of Nrf2 could drive reductive stress by increasing GSH levels (Fig. 3). Consistent with this hypothesis, the cardiomyocytes of experimental mouse hearts exhibiting increased protein aggregation showed an increased recycling of oxidized glutathione (GSSG) to reduced glutathione (GSH). This was shown to be a result of sequestration of Keap1 within protein aggregations, and the consequent loss of inhibition of Nrf2 (99, 101). Similarly, in Keap1-null mice, Nrf2 constitutively accumulated in the nucleus and stimulated the transcription of cytoprotective genes (123). Keap1-null mice die early postnatally (∼20 days), primarily from foregut malformations. Cardiac dysfunction in these mice was not reported; however, as cardiomyopathy took 6 months to develop in the protein aggregation model mentioned above, the early death of Keap1-null mice may have provided insufficient time for cardiac dysfunction to occur. The generation of conditional Keap1 null mice might be an alternative approach to clarify this important question.
However, as mentioned in the introduction, cell compartments do not maintain the same redox environment relative to each other. Mitochondria and nuclei have a more reducing environment than does, for instance, the endoplasmic reticulum (62). These differences in redox environment are important for the correct functioning of these compartments and are primarily buffered by GSH levels (46) and, hence, are potentially influenced by the Nrf2 activity. Further studies are therefore needed to determine the causes and effects of oxidative/reductive insults and Nrf2-mediated responses within specific compartments of the cell. However, it is clear that the Nrf2 pathway is an important mediator of cellular redox homeostasis that allows the conversion of a pro-oxidant signal into an appropriate antioxidant response. A key to this regulation appears to be maintaining the Nrf2 activity at optimal levels to deal with an oxidative stimulus. Dysregulation of this response, either through insufficient or excess activation of Nrf2, is associated with a variety of cardiac pathologies.
Lastly, we think that foregoing instances of reductive stress do not preclude its likely roles in the pathogenesis of other disease states most notably I/R, diabetes, and decompensated heart failure. For example, the pathophysiological circumstances during ischemic without reperfusion will promote reductive stress when insufficient oxygen availability drives the accumulation of unpaired electrons and highly energy reducing equivalents [i.e., NAD(P)H]. By definition, we postulate that pathological conditions associated with exaggerated high levels of reducing equivalents in the form of GSH and NAD(P)H and redox shifts.
Pharmacologically or Genetically Induced Reductive Stress and Cell Death Pathways
Reductive stress has predominantly been described as an altered ratio of reduced:oxidized GSH (GSH:GSSG), with an increased ratio (>GSH:GSSG) being indicative of reductive stress (23, 118). GSH is synthesized from cysteine, glutamate, and glycine (48, 61). Many of the antioxidant genes regulated by Nrf2 are enzymes involved in the biosynthesis and regeneration of GSH, including GCL, GSH synthase (GS), GR, and G6PD (61, 88). GCL catalyzes the rate-limiting step in GSH synthesis by catalyzing the formation of γ-glutamylcysteine from glutamine and cysteine and an increase in its expression leads to higher GSH levels (61, 108). GSH becomes oxidized to a disulfide GSSG form in the presence of ROS. To recycle GSSG back to the reduced GSH form, GR catalyzes the NADPH-dependent reduction of GSSG to GSH. This process is dependent on the sufficient availability of NADPH as an enzymatic cofactor. G6PD can provide this NADPH by catalyzing the oxidation of glucose-6-phosphate to 6-phosphogluconate, while reducing NADP+ to generate NADPH (98).
We have recently provided new evidence for GSH-induced reductive stress, which is causally linked to mitochondrial oxidation and cytotoxicity (133). In these studies, two complementary approaches were undertaken for examining the effects of a more reducing redox potential generated by either pharmacologic or genetic maneuvers. In H9c2 cells, treatment with 4 mM N-acetyl-L-cysteine (NAC), a precursor of GSH, led to significant (∼3-fold) increases in total levels of GSH, a decrease in the GSH/GSSG ratio due to correspondingly more oxidized GSH, but a 7mV more reducing Ehc (GSSG/2GSH) by the Nernst equation (−197±1.6 mV compared to −190±2.6 mV, p<0.05). Similar to NAC, we demonstrated that the individualized actions of GCL and GS overexpression, which are required for the biosynthesis of reduced GSH in the cytosol, causes a more reducing Ehc (GSSG/2GSH), mitochondrial oxidation, and cytotoxicity. Such pro-oxidative consequences of GSH-mediated reductive stress in mitochondria were corroborated by redox biosensors, mitochondrial reduction–oxidation green fluorescent protein (roGFP), or the oxidation of mitochondrial thioredoxin (Trx2). These findings are significant, since they challenge the conventional wisdom for increasing GSH levels through NAC, which have been shown to protect against oxidative stress-induced cell death (53, 132), by providing evidence for reductive stress, defined by a more reducing GSH redox potential (Ehc), linked to cell death.
Because NAC treatment has been shown to induce cell death in vascular smooth muscle cells and to enhance apoptosis during hypoxia in murine embryonic fibroblasts, the rationale that NAC functions solely as an antioxidant should be reappraised, particularly, in relation to the dynamic events underlying oxido-reductive signals and redox-dependent pathways. How the reductive GSH potential Ehc (GSSG/2GSH) triggers mitochondrial oxidation and cytotoxicity is presently unknown, but several plausible hypotheses exist. First, we observed that neither levels of ROS production nor dichlorodihydrofluorescin diacetate were significantly elevated compared with controls, indicating that the reductive GSH potential was associated with lower levels of either superoxide or H2O2. Superoxide generation from the electron transport chain could be secondary either to increased mitochondrial production from GSH biosynthesis or alternatively, we hypothesize the glutathionylation of mitochondrial target(s), perhaps, influenced by either oxidative stress and/or the GSH/GSSG ratio. Ongoing studies are seeking to determine whether NAC-induced oxidation might trigger glutathionylation of Trx as one possible target (19).
Conclusions and Perspectives
The field of redox biology embodies storied histories ranging from longstanding endeavors on the oxidative theory of aging to more recent fundamental advances of the dual specificity roles for ROS in signaling pathways and disease pathogenesis. Spectacular progress for sophisticated measurements of ROS using electron spin resonance microscopy at the subatomic levels has been matched with global surveys of oxidative modifications of the entire proteome. Notwithstanding numerous clinical trials, therapeutic interventions for mitigating oxidative stress have been uniformly disappointing and even deleterious. Without advances in redox diagnostics, the existing strategies to combat ROS imbalances are seriously flawed, since current experimental approaches rely exclusively on global end points, which lack critical sensitivity and specificity at the subcellular levels. By establishing that alterations of GSH and, perhaps, NADPH homeostasis tilt the redox status toward the reductive phase, which is critically linked to survival and cell death fates, our recent studies have stimulated much interest to develop complementary model systems, mimicking experimental reductive stress. This review was primarily designed to explore the conceptual, biochemical, and molecular consequences—at the reductive ends of the redox spectrum—mediated by key effector pathways (Fig. 4). Although earlier the deleterious effects of reductive stress have been explored in both unicellular eukaryotic and mammalian cells (49, 99, 118, 134), there remains a paucity of robust experimental models beyond the examples described herein (99, 135). We embrace the widespread use of redox biosensors, such as roGFPs, Trx1, and Trx2, which are well suited to independently assess GSH depletion, after induction of either H2O2 or superoxide production at subcellular levels. Future studies are needed to identify genetic polymorphisms that either increased susceptibility or resistance to oxido-reductive and clinical manifestations. Along with improved diagnostic tools for assessment of reducing GSH redox potential or reductive stress, there is considerable excitement that the field of redox biology will turn the corner at an accelerated pace into the genomic personalized medicine.
FIG. 4.
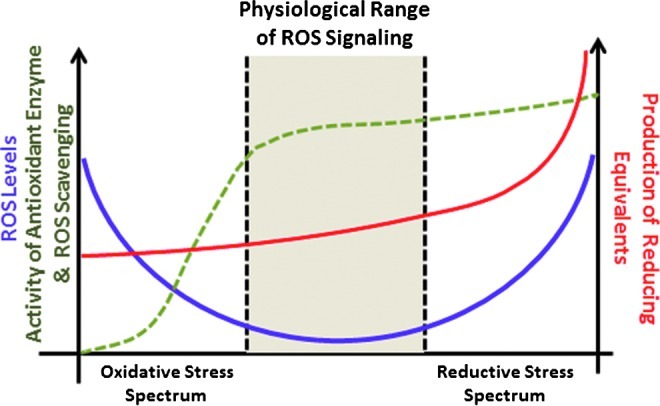
Schematic illustration of the spectrum ranging from oxidative to reductive stress. Adapted from (9a).
Abbreviations Used
- Ang II
angiotensin II
- ARE
antioxidant response element
- CryAB
αB-crystallin
- DRM
desmin-related myopathy
- G6PD
glucose 6-phosphate dehydrogenase
- GCL
glutamate-cysteine ligase
- GPx
glutathione peroxidase
- GR
glutathione reductase
- GS
glutathione synthase
- GSH
glutathione
- GST
glutathione transferase
- HSP
heat shock protein
- I/R
ischemia/reperfusion
- Keap1
Kelch-like ECH-associated protein 1
- MW
molecular weight
- NAC
N-acetyl-L-cysteine
- NO
nitric oxide
- NOX
NADPH oxidase
- NQO1
NAD(P)H quinone oxioreductase 1
- Nrf2
nuclear erythroid 2-related factor 2
- PKC
protein kinase C
- roGFP
reduction–oxidation green fluorescent protein
- ROS
reactive oxygen species
- sHSP
small MW heat shock protein
- SOD
superoxide dismutase
- TAD
transactivation domain
- TG
transgenic
- Trx
thioredoxin
- TrxR
thioredoxin reductase
Acknowledgments
This work was supported by National Heart, Lung and Blood Institute (ARRA Award 2 R01 HL063834-06 to I.J.B.), 2009 NIH Director's Pioneer Award 1DP1OD006438-02, VA Merit Review Award (I.J.B.), NHLBI 5R01HL074370-03 (I.J.B.), American Heart Association Beginning Grant In-Aid Award #0865015F (N.S.R), British Heart Foundation (BFH) programme grant RG/08/011/25922 (A.B.), BHF Programme grant PG/08/110/26228 (AB), BHF project grant PG/11/124/29318 (A.B.), and BHF Centre of Research Excellence grant RE/08/003 (A.B.). We thank Elisabeth Christians for her critical review of the manuscript. Alison Ausman has provided excellent editorial assistance during preparation of this manuscript.
References
- 1.Acunzo J. Katsogiannou M. Rocchi P. Small heat shock proteins HSP27 (HspB1), alphaB-crystallin (HspB5) and HSP22 (HspB8) as regulators of cell death. Int J Biochem Cell Biol. 2012;44:1622–1631. doi: 10.1016/j.biocel.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Addabbo F. Montagnani M. Goligorsky MS. Mitochondria and reactive oxygen species. Hypertension. 2009;53:885–892. doi: 10.1161/HYPERTENSIONAHA.109.130054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahsan MK. Lekli I. Ray D. Yodoi J. Das DK. Redox regulation of cell survival by the thioredoxin superfamily: an implication of redox gene therapy in the heart. Antioxid Redox Signal. 2009;11:2741–2758. doi: 10.1089/ars.2009.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen RG. Balin AK. Oxidative influence on development and differentiation: an overview of a free radical theory of development. Free Radic Biol Med. 1989;6:631–661. doi: 10.1016/0891-5849(89)90071-3. [DOI] [PubMed] [Google Scholar]
- 5.Allen RG. Tresini M. Oxidative stress and gene regulation. Free Radic Biol Med. 2000;28:463–499. doi: 10.1016/s0891-5849(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 6.Allen RG. Venkatraj VS. Oxidants and antioxidants in development and differentiation. J Nutr. 1992;122:631–635. doi: 10.1093/jn/122.suppl_3.631. [DOI] [PubMed] [Google Scholar]
- 7.Almeida-Souza L. Asselbergh B. d'Ydewalle C. Moonens K. Goethals S. de Winter V. Azmi A. Irobi J. Timmermans JP. Gevaert K. Remaut H. Van Den Bosch L. Timmerman V. Janssens S. Small heat-shock protein HSPB1 mutants stabilize microtubules in Charcot-Marie-Tooth neuropathy. J Neurosci. 2011;31:15320–15328. doi: 10.1523/JNEUROSCI.3266-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ames BN. Shigenaga MK. Hagen TM. Mitochondrial decay in aging. Biochim Biophys Acta. 1995;1271:165–170. doi: 10.1016/0925-4439(95)00024-x. [DOI] [PubMed] [Google Scholar]
- 9.Anilkumar N. Sirker A. Shah AM. Redox sensitive signaling pathways in cardiac remodeling, hypertrophy and failure. Front Biosci. 2009;14:3168–3187. doi: 10.2741/3443. [DOI] [PubMed] [Google Scholar]
- 9a.Aon MA. Cortassa S. O'Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. 2010;1797:865–877. doi: 10.1016/j.bbabio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arrigo AP. Small stress proteins: chaperones that act as regulators of intracellular redox state and programmed cell death. Biol Chem. 1998;379:19–26. [PubMed] [Google Scholar]
- 11.Arrigo AP. Virot S. Chaufour S. Firdaus W. Kretz-Remy C. Diaz-Latoud C. Hsp27 consolidates intracellular redox homeostasis by upholding glutathione in its reduced form and by decreasing iron intracellular levels. Antioxid Redox Signal. 2005;7:414–422. doi: 10.1089/ars.2005.7.414. [DOI] [PubMed] [Google Scholar]
- 12.Asthana A. Raman B. Ramakrishna T. Rao CM. Structural aspects and chaperone activity of human HspB3: role of the “C-terminal extension”. Cell Biochem Biophys. 2012;64:61–72. doi: 10.1007/s12013-012-9366-x. [DOI] [PubMed] [Google Scholar]
- 13.Baird L. Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 14.Bendall JK. Cave AC. Heymes C. Gall N. Shah AM. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 15.Benjamin IJ. Guo Y. Srinivasan S. Boudina S. Taylor RP. Rajasekaran NS. Gottlieb R. Wawrousek EF. Abel ED. Bolli R. CRYAB and HSPB2 deficiency alters cardiac metabolism and paradoxically confers protection against myocardial ischemia in aging mice. Am J Physiol Heart Circ Physiol. 2007;293:H3201–H3209. doi: 10.1152/ajpheart.01363.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhat SP. Nagineni CN. alpha B subunit of lens-specific protein alpha-crystallin is present in other ocular and non-ocular tissues. Biochem Biophys Res Commun. 1989;158:319–325. doi: 10.1016/s0006-291x(89)80215-3. [DOI] [PubMed] [Google Scholar]
- 17.Brewer AC. Murray TV. Arno M. Zhang M. Anilkumar NP. Mann GE. Shah AM. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic Biol Med. 2011;51:205–215. doi: 10.1016/j.freeradbiomed.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown GC. Borutaite V. There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion. 2012;12:1–4. doi: 10.1016/j.mito.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Casagrande S. Bonetto V. Fratelli M. Gianazza E. Eberini I. Massignan T. Salmona M. Chang G. Holmgren A. Ghezzi P. Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proc Natl Acad Sci U S A. 2002;99:9745–9749. doi: 10.1073/pnas.152168599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cesselli D. Jakoniuk I. Barlucchi L. Beltrami AP. Hintze TH. Nadal-Ginard B. Kajstura J. Leri A. Anversa P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001;89:279–286. doi: 10.1161/hh1501.094115. [DOI] [PubMed] [Google Scholar]
- 21.Chan K. Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci U S A. 1999;96:12731–12736. doi: 10.1073/pnas.96.22.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan K. Lu R. Chang JC. Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chance B. Sies H. Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 24.Charles RL. Eaton P. Redox signalling in cardiovascular disease. Proteomics Clin Appl. 2008;2:823–836. doi: 10.1002/prca.200780104. [DOI] [PubMed] [Google Scholar]
- 25.Cho HY. Reddy SP. Yamamoto M. Kleeberger SR. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004;18:1258–1260. doi: 10.1096/fj.03-1127fje. [DOI] [PubMed] [Google Scholar]
- 26.Christians ES. Benjamin IJ. Proteostasis and REDOX state in the heart. Am J Physiol Heart Circ Physiol. 2012;302:H24–H37. doi: 10.1152/ajpheart.00903.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark AR. Lubsen NH. Slingsby C. sHSP in the eye lens: crystallin mutations, cataract and proteostasis. Int J Biochem Cell Biol. 2012;44:1687–1697. doi: 10.1016/j.biocel.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 28.Costa ML. Escaleira R. Cataldo A. Oliveira F. Mermelstein CS. Desmin: molecular interactions and putative functions of the muscle intermediate filament protein. Braz J Med Biol Res. 2004;37:1819–1830. doi: 10.1590/s0100-879x2004001200007. [DOI] [PubMed] [Google Scholar]
- 29.Cullinan SB. Gordan JD. Jin J. Harper JW. Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–8486. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davies KJ. Oxidative stress: the paradox of aerobic life. Biochem Soc Symp. 1995;61:1–31. doi: 10.1042/bss0610001. [DOI] [PubMed] [Google Scholar]
- 31.de Wit NJ. Verschuure P. Kappe G. King SM. de Jong WW. van Muijen GN. Boelens WC. Testis-specific human small heat shock protein HSPB9 is a cancer/testis antigen, and potentially interacts with the dynein subunit TCTEL1. Eur J Cell Biol. 2004;83:337–345. doi: 10.1078/0171-9335-00396. [DOI] [PubMed] [Google Scholar]
- 32.Dhakshinamoorthy S. Jaiswal AK. Small maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:quinone oxidoreductase1 gene. J Biol Chem. 2000;275:40134–40141. doi: 10.1074/jbc.M003531200. [DOI] [PubMed] [Google Scholar]
- 33.Dhalla NS. Golfman L. Takeda S. Takeda N. Nagano M. Evidence for the role of oxidative stress in acute ischemic heart disease: a brief review. Can J Cardiol. 1999;15:587–593. [PubMed] [Google Scholar]
- 34.Di Lisa F. Kaludercic N. Carpi A. Menabo R. Giorgio M. Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66(Shc) and monoamine oxidase. Basic Res Cardiol. 2009;104:131–139. doi: 10.1007/s00395-009-0008-4. [DOI] [PubMed] [Google Scholar]
- 35.Dong GZ. Youn H. Park MT. Oh ET. Park KH. Song CW. Choi EK. Park HJ. Heat shock increases expression of NAD(P)H:quinone oxidoreductase (NQO1), mediator of beta-lapachone cytotoxicity, by increasing NQO1 gene activity and via Hsp70-mediated stabilisation of NQO1 protein. Int J Hyperthermia. 2009;25:477–487. doi: 10.1080/02656730903049836. [DOI] [PubMed] [Google Scholar]
- 36.Dubin RA. Wawrousek EF. Piatigorsky J. Expression of the murine alpha B-crystallin gene is not restricted to the lens. Mol Cell Biol. 1989;9:1083–1091. doi: 10.1128/mcb.9.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elsayed NM. Omaye ST. Klain GJ. Korte DW., Jr. Free radical-mediated lung response to the monofunctional sulfur mustard butyl 2-chloroethyl sulfide after subcutaneous injection. Toxicology. 1992;72:153–165. doi: 10.1016/0300-483x(92)90109-r. [DOI] [PubMed] [Google Scholar]
- 38.Fan GC. Kranias EG. Small heat shock protein 20 (HspB6) in cardiac hypertrophy and failure. J Mol Cell Cardiol. 2011;51:574–577. doi: 10.1016/j.yjmcc.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernandes RS. Cotter TG. Apoptosis or necrosis: intracellular levels of glutathione influence mode of cell death. Biochem Pharmacol. 1994;48:675–681. doi: 10.1016/0006-2952(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 40.Frank L. Groseclose EE. Preparation for birth into an O2-rich environment: the antioxidant enzymes in the developing rabbit lung. Pediatr Res. 1984;18:240–244. doi: 10.1203/00006450-198403000-00004. [DOI] [PubMed] [Google Scholar]
- 41.Fridovich I. Superoxide dismutases. An adaptation to a paramagnetic gas. J Biol Chem. 1989;264:7761–7764. [PubMed] [Google Scholar]
- 42.Garrido C. Paul C. Seigneuric R. Kampinga HH. The small heat shock proteins family: the long forgotten chaperones. Int J Biochem Cell Biol. 2012;44:1588–1592. doi: 10.1016/j.biocel.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 43.Genova ML. Pich MM. Biondi A. Bernacchia A. Falasca A. Bovina C. Formiggini G. Parenti Castelli G. Lenaz G. Mitochondrial production of oxygen radical species and the role of Coenzyme Q as an antioxidant. Exp Biol Med (Maywood) 2003;228:506–513. doi: 10.1177/15353702-0322805-14. [DOI] [PubMed] [Google Scholar]
- 44.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 45.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Go YM. Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780:1273–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Golenhofen N. Redel A. Wawrousek EF. Drenckhahn D. Ischemia-induced increase of stiffness of alphaB-crystallin/HSPB2-deficient myocardium. Pflugers Arch. 2006;451:518–525. doi: 10.1007/s00424-005-1488-1. [DOI] [PubMed] [Google Scholar]
- 48.Ha KN. Chen Y. Cai J. Sternberg P., Jr. Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: implication for protection against oxidative stress. Invest Ophthalmol Vis Sci. 2006;47:2709–2715. doi: 10.1167/iovs.05-1322. [DOI] [PubMed] [Google Scholar]
- 49.Halvey PJ. Watson WH. Hansen JM. Go YM. Samali A. Jones DP. Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem J. 2005;386:215–219. doi: 10.1042/BJ20041829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hansen JM. Go YM. Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol. 2006;46:215–234. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- 51.He X. Kan H. Cai L. Ma Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J Mol Cell Cardiol. 2009;46:47–58. doi: 10.1016/j.yjmcc.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 52.Herzog V. Fahimi HD. Microbodies (peroxisomes) containing catalase in myocardium: morphological and biochemical evidence. Science. 1974;185:271–273. doi: 10.1126/science.185.4147.271. [DOI] [PubMed] [Google Scholar]
- 53.Hockenbery DM. Oltvai ZN. Yin XM. Milliman CL. Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 54.Hu Z. Li T. HspB5/alphaB-crystallin: properties and current progress in neuropathy. Curr Neurovasc Res. 2008;5:143–152. doi: 10.2174/156720208784310222. [DOI] [PubMed] [Google Scholar]
- 55.Huang HC. Nguyen T. Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 56.Ide T. Tsutsui H. Kinugawa S. Utsumi H. Kang D. Hattori N. Uchida K. Arimura K. Egashira K. Takeshita A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–363. doi: 10.1161/01.res.85.4.357. [DOI] [PubMed] [Google Scholar]
- 57.Inagaki N. Hayashi T. Arimura T. Koga Y. Takahashi M. Shibata H. Teraoka K. Chikamori T. Yamashina A. Kimura A. Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem Biophys Res Commun. 2006;342:379–386. doi: 10.1016/j.bbrc.2006.01.154. [DOI] [PubMed] [Google Scholar]
- 58.Inamdar NM. Ahn YI. Alam J. The heme-responsive element of the mouse heme oxygenase-1 gene is an extended AP-1 binding site that resembles the recognition sequences for MAF and NF-E2 transcription factors. Biochem Biophys Res Commun. 1996;221:570–576. doi: 10.1006/bbrc.1996.0637. [DOI] [PubMed] [Google Scholar]
- 59.Irobi J. Holmgren A. Winter VD. Asselbergh B. Gettemans J. Adriaensen D. Groote CC. Coster RV. Jonghe PD. Timmerman V. Mutant HSPB8 causes protein aggregates and a reduced mitochondrial membrane potential in dermal fibroblasts from distal hereditary motor neuropathy patients. Neuromusc Disord. 2012;22:699–711. doi: 10.1016/j.nmd.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 60.Itoh K. Wakabayashi N. Katoh Y. Ishii T. Igarashi K. Engel JD. Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 62.Jones DP. Go YM. Redox compartmentalization and cellular stress. Diabetes Obes Metab. 2010;12(Suppl 2):116–125. doi: 10.1111/j.1463-1326.2010.01266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kadono T. Zhang XQ. Srinivasan S. Ishida H. Barry WH. Benjamin IJ. CRYAB and HSPB2 deficiency increases myocyte mitochondrial permeability transition and mitochondrial calcium uptake. J Mol Cell Cardiol. 2006;40:783–789. doi: 10.1016/j.yjmcc.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 64.Kalinina EV. Chernov NN. Saprin AN. Involvement of thio-, peroxi-, and glutaredoxins in cellular redox-dependent processes. Biochemistry (Mosc) 2008;73:1493–1510. doi: 10.1134/s0006297908130099. [DOI] [PubMed] [Google Scholar]
- 65.Kampinga HH. Hageman J. Vos MJ. Kubota H. Tanguay RM. Bruford EA. Cheetham ME. Chen B. Hightower LE. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang MI. Kobayashi A. Wakabayashi N. Kim SG. Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci U S A. 2004;101:2046–2051. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kappe G. Verschuure P. Philipsen RL. Staalduinen AA. Van de Boogaart P. Boelens WC. De Jong WW. Characterization of two novel human small heat shock proteins: protein kinase-related HspB8 and testis-specific HspB9. Biochim Biophys Acta. 2001;1520:1–6. doi: 10.1016/s0167-4781(01)00237-8. [DOI] [PubMed] [Google Scholar]
- 68.Keith M. Geranmayegan A. Sole MJ. Kurian R. Robinson A. Omran AS. Jeejeebhoy KN. Increased oxidative stress in patients with congestive heart failure. J Am Coll Cardiol. 1998;31:1352–1356. doi: 10.1016/s0735-1097(98)00101-6. [DOI] [PubMed] [Google Scholar]
- 69.Kemp M. Go YM. Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic Biol Med. 2008;44:921–937. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim YC. Yamaguchi Y. Kondo N. Masutani H. Yodoi J. Thioredoxin-dependent redox regulation of the antioxidant responsive element (ARE) in electrophile response. Oncogene. 2003;22:1860–1865. doi: 10.1038/sj.onc.1206369. [DOI] [PubMed] [Google Scholar]
- 71.Kobayashi A. Kang MI. Okawa H. Ohtsuji M. Zenke Y. Chiba T. Igarashi K. Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kohen R. Skin antioxidants: their role in aging and in oxidative stress—new approaches for their evaluation. Biomed Pharmacother. 1999;53:181–192. doi: 10.1016/S0753-3322(99)80087-0. [DOI] [PubMed] [Google Scholar]
- 73.Kohen R. Gati I. Skin low molecular weight antioxidants and their role in aging and in oxidative stress. Toxicology. 2000;148:149–157. doi: 10.1016/s0300-483x(00)00206-7. [DOI] [PubMed] [Google Scholar]
- 74.Kohen R. Nyska A. Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol Pathol. 2002;30:620–650. doi: 10.1080/01926230290166724. [DOI] [PubMed] [Google Scholar]
- 75.Lardinois OM. Reactions of bovine liver catalase with superoxide radicals and hydrogen peroxide. Free Radic Res. 1995;22:251–274. doi: 10.3109/10715769509147544. [DOI] [PubMed] [Google Scholar]
- 76.Li J. Ichikawa T. Villacorta L. Janicki JS. Brower GL. Yamamoto M. Cui T. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arterioscler Thromb Vasc Biol. 2009;29:1843–1850. doi: 10.1161/ATVBAHA.109.189480. [DOI] [PubMed] [Google Scholar]
- 77.Li J. Zhang C. Xing Y. Janicki JS. Yamamoto M. Wang XL. Tang DQ. Cui T. Up-regulation of p27(kip1) contributes to Nrf2-mediated protection against angiotensin II-induced cardiac hypertrophy. Cardiovasc Res. 2011;90:315–324. doi: 10.1093/cvr/cvr010. [DOI] [PubMed] [Google Scholar]
- 78.Li W. Yu SW. Kong AN. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J Biol Chem. 2006;281:27251–27263. doi: 10.1074/jbc.M602746200. [DOI] [PubMed] [Google Scholar]
- 79.Liu L. Zhang X. Qian B. Min X. Gao X. Li C. Cheng Y. Huang J. Over-expression of heat shock protein 27 attenuates doxorubicin-induced cardiac dysfunction in mice. Eur J Heart Fail. 2007;9:762–769. doi: 10.1016/j.ejheart.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 80.Maloyan A. Sanbe A. Osinska H. Westfall M. Robinson D. Imahashi K. Murphy E. Robbins J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha-B-crystallin desmin-related cardiomyopathy. Circulation. 2005;112:3451–3461. doi: 10.1161/CIRCULATIONAHA.105.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maxwell SR. Anti-oxidant therapy: does it have a role in the treatment of human disease? Expert Opin Investig Drugs. 1997;6:211–236. doi: 10.1517/13543784.6.3.211. [DOI] [PubMed] [Google Scholar]
- 82.Mbemba F. Houbion A. Raes M. Remacle J. Subcellular localization and modification with ageing of glutathione, glutathione peroxidase and glutathione reductase activities in human fibroblasts. Biochim Biophys Acta. 1985;838:211–220. doi: 10.1016/0304-4165(85)90081-9. [DOI] [PubMed] [Google Scholar]
- 83.McCord JM. Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 84.McLendon PM. Robbins J. Desmin-related cardiomyopathy: an unfolding story. Am J Physiol Heart Circ Physiol. 2011;301:H1220–H1228. doi: 10.1152/ajpheart.00601.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mehlen P. Kretz-Remy C. Preville X. Arrigo AP. Human hsp27, Drosophila hsp27 and human alphaB-crystallin expression-mediated increase in glutathione is essential for the protective activity of these proteins against TNFalpha-induced cell death. EMBO J. 1996;15:2695–2706. [PMC free article] [PubMed] [Google Scholar]
- 86.Miwa S. Brand MD. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem Soc Trans. 2003;31:1300–1301. doi: 10.1042/bst0311300. [DOI] [PubMed] [Google Scholar]
- 87.Moi P. Chan K. Asunis I. Cao A. Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moinova HR. Mulcahy RT. An electrophile responsive element (EpRE) regulates beta-naphthoflavone induction of the human gamma-glutamylcysteine synthetase regulatory subunit gene. Constitutive expression is mediated by an adjacent AP-1 site. J Biol Chem. 1998;273:14683–14689. doi: 10.1074/jbc.273.24.14683. [DOI] [PubMed] [Google Scholar]
- 89.Moinova HR. Mulcahy RT. Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem Biophys Res Commun. 1999;261:661–668. doi: 10.1006/bbrc.1999.1109. [DOI] [PubMed] [Google Scholar]
- 90.Muthusamy VR. Kannan S. Sadhaasivam K. Gounder SS. Davidson CJ. Boeheme C. Hoidal JR. Wang L. Rajasekaran NS. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic Biol Med. 2012;52:366–376. doi: 10.1016/j.freeradbiomed.2011.10.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Norton JA. Weinberger PM. Waller JL. Merkley MA. Jackson LL. Dynan WS. Significance of HSPB1 expression in head and neck squamous cell carcinoma: a meta-analysis of published literatures. The Laryngoscope. 2010;120(Suppl 4):S172. doi: 10.1002/lary.21636. [DOI] [PubMed] [Google Scholar]
- 92.Obata T. Yamanaka Y. Kinemuchi H. Oreland L. Release of dopamine by perfusion with 1-methyl-4-phenylpyridinium ion (MPP(+)) into the striatum is associated with hydroxyl free radical generation. Brain Res. 2001;906:170–175. doi: 10.1016/s0006-8993(01)02238-7. [DOI] [PubMed] [Google Scholar]
- 93.Osburn WO. Kensler TW. Nrf2 signaling: an adaptive response pathway for protection against environmental toxic insults. Mutat Res. 2008;659:31–39. doi: 10.1016/j.mrrev.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Owuor ED. Kong AN. Antioxidants and oxidants regulated signal transduction pathways. Biochem Pharmacol. 2002;64:765–770. doi: 10.1016/s0006-2952(02)01137-1. [DOI] [PubMed] [Google Scholar]
- 95.Page MM. Robb EL. Salway KD. Stuart JA. Mitochondrial redox metabolism: aging, longevity and dietary effects. Mech Ageing Dev. 2010;131:242–252. doi: 10.1016/j.mad.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 96.Papaiahgari S. Kleeberger SR. Cho HY. Kalvakolanu DV. Reddy SP. NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells. J Biol Chem. 2004;279:42302–42312. doi: 10.1074/jbc.M408275200. [DOI] [PubMed] [Google Scholar]
- 97.Pozsgai E. Gomori E. Szigeti A. Boronkai A. Gallyas F., Jr. Sumegi B. Bellyei S. Correlation between the progressive cytoplasmic expression of a novel small heat shock protein (Hsp16.2) and malignancy in brain tumors. BMC Cancer. 2007;7:233. doi: 10.1186/1471-2407-7-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Preville X. Salvemini F. Giraud S. Chaufour S. Paul C. Stepien G. Ursini MV. Arrigo AP. Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp Cell Res. 1999;247:61–78. doi: 10.1006/excr.1998.4347. [DOI] [PubMed] [Google Scholar]
- 99.Rajasekaran NS. Connell P. Christians ES. Yan LJ. Taylor RP. Orosz A. Zhang XQ. Stevenson TJ. Peshock RM. Leopold JA. Barry WH. Loscalzo J. Odelberg SJ. Benjamin IJ. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rajasekaran NS. Firpo MA. Milash BA. Weiss RB. Benjamin IJ. Global expression profiling identifies a novel biosignature for protein aggregation R120GCryAB cardiomyopathy in mice. Physiol Genomics. 2008;35:165–172. doi: 10.1152/physiolgenomics.00297.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rajasekaran NS. Varadharaj S. Khanderao GD. Davidson CJ. Kannan S. Firpo MA. Zweier JL. Benjamin IJ. Sustained activation of nuclear erythroid 2-related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice. Antioxid Redox Signal. 2011;14:957–971. doi: 10.1089/ars.2010.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ray PS. Martin JL. Swanson EA. Otani H. Dillmann WH. Das DK. Transgene overexpression of alphaB crystallin confers simultaneous protection against cardiomyocyte apoptosis and necrosis during myocardial ischemia and reperfusion. FASEB J. 2001;15:393–402. doi: 10.1096/fj.00-0199com. [DOI] [PubMed] [Google Scholar]
- 103.Rushmore TH. Pickett CB. Glutathione S-transferases, structure, regulation, and therapeutic implications. J Biol Chem. 1993;268:11475–11478. [PubMed] [Google Scholar]
- 104.Sabri A. Hughie HH. Lucchesi PA. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid Redox Signal. 2003;5:731–740. doi: 10.1089/152308603770380034. [DOI] [PubMed] [Google Scholar]
- 105.Sam F. Kerstetter DL. Pimental DR. Mulukutla S. Tabaee A. Bristow MR. Colucci WS. Sawyer DB. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J Card Fail. 2005;11:473–480. doi: 10.1016/j.cardfail.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 106.Santos CX. Anilkumar N. Zhang M. Brewer AC. Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med. 2011;50:777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schafer FQ. Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 108.Seelig GF. Simondsen RP. Meister A. Reversible dissociation of gamma-glutamylcysteine synthetase into two subunits. J Biol Chem. 1984;259:9345–9347. [PubMed] [Google Scholar]
- 109.Seit-Nebi AS. Gusev NB. Versatility of the small heat shock protein HSPB6 (Hsp20) Cell Stress Chaperones. 2010;15:233–236. doi: 10.1007/s12192-009-0141-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shadyro OI. Yurkova IL. Kisel MA. Radiation-induced peroxidation and fragmentation of lipids in a model membrane. Int J Radiat Biol. 2002;78:211–217. doi: 10.1080/09553000110104065. [DOI] [PubMed] [Google Scholar]
- 111.Sies H. Oxidative stress: from basic research to clinical application. Am J Med. 1991;91:31S–38S. doi: 10.1016/0002-9343(91)90281-2. [DOI] [PubMed] [Google Scholar]
- 112.Stark K. Esslinger UB. Reinhard W. Petrov G. Winkler T. Komajda M. Isnard R. Charron P. Villard E. Cambien F. Tiret L. Aumont MC. Dubourg O. Trochu JN. Fauchier L. Degroote P. Richter A. Maisch B. Wichter T. Zollbrecht C. Grassl M. Schunkert H. Linsel-Nitschke P. Erdmann J. Baumert J. Illig T. Klopp N. Wichmann HE. Meisinger C. Koenig W. Lichtner P. Meitinger T. Schillert A. Konig IR. Hetzer R. Heid IM. Regitz-Zagrosek V. Hengstenberg C. Genetic association study identifies HSPB7 as a risk gene for idiopathic dilated cardiomyopathy. PLoS Genet. 2010;6:e1001167. doi: 10.1371/journal.pgen.1001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Steinhubl SR. Why have antioxidants failed in clinical trials? Am J Cardiol. 2008;101:14D–19D. doi: 10.1016/j.amjcard.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 114.Su CH. Liu LC. Hsieh YH. Wang HC. Tsai CW. Chang WS. Ho CY. Wu CI. Lin CH. Lane HY. Bau DT. Association of Alpha B-Crystallin (CRYAB) genotypes with breast cancer susceptibility in Taiwan. Cancer Genomics Proteomics. 2011;8:251–254. [PubMed] [Google Scholar]
- 115.Suematsu N. Tsutsui H. Wen J. Kang D. Ikeuchi M. Ide T. Hayashidani S. Shiomi T. Kubota T. Hamasaki N. Takeshita A. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation. 2003;107:1418–1423. doi: 10.1161/01.cir.0000055318.09997.1f. [DOI] [PubMed] [Google Scholar]
- 116.Sumandea MP. Steinberg SF. Redox signaling and cardiac sarcomeres. J Biol Chem. 2011;286:9921–7727. doi: 10.1074/jbc.R110.175489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sykiotis GP. Bohmann D. Stress-activated cap‘n'collar transcription factors in aging and human disease. Sci Signal. 2010;3:re3. doi: 10.1126/scisignal.3112re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Trotter EW. Grant CM. Thioredoxins are required for protection against a reductive stress in the yeast Saccharomyces cerevisiae. Mol Microbiol. 2002;46:869–878. doi: 10.1046/j.1365-2958.2002.03216.x. [DOI] [PubMed] [Google Scholar]
- 119.Vedam K. Nishijima Y. Druhan LJ. Khan M. Moldovan NI. Zweier JL. Ilangovan G. Role of heat shock factor-1 activation in the doxorubicin-induced heart failure in mice. Am J Physiol Heart Circ Physiol. 2010;298:H1832–H1841. doi: 10.1152/ajpheart.01047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ventura-Clapier R. Garnier A. Veksler V. Energy metabolism in heart failure. J Physiol. 2004;555:1–13. doi: 10.1113/jphysiol.2003.055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vicart P. Caron A. Guicheney P. Li Z. Prevost MC. Faure A. Chateau D. Chapon F. Tome F. Dupret JM. Paulin D. Fardeau M. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 122.Wakabayashi N. Dinkova-Kostova AT. Holtzclaw WD. Kang MI. Kobayashi A. Yamamoto M. Kensler TW. Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wakabayashi N. Itoh K. Wakabayashi J. Motohashi H. Noda S. Takahashi S. Imakado S. Kotsuji T. Otsuka F. Roop DR. Harada T. Engel JD. Yamamoto M. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 124.Walters DM. Cho HY. Kleeberger SR. Oxidative stress and antioxidants in the pathogenesis of pulmonary fibrosis: a potential role for Nrf2. Antioxid Redox Signal. 2008;10:321–332. doi: 10.1089/ars.2007.1901. [DOI] [PubMed] [Google Scholar]
- 125.Wang X. Klevitsky R. Huang W. Glasford J. Li F. Robbins J. AlphaB-crystallin modulates protein aggregation of abnormal desmin. Circ Res. 2003;93:998–1005. doi: 10.1161/01.RES.0000102401.77712.ED. [DOI] [PubMed] [Google Scholar]
- 126.Wang X. Osinska H. Klevitsky R. Gerdes AM. Nieman M. Lorenz J. Hewett T. Robbins J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 127.Wilhelmus MM. Boelens WC. Kox M. Maat-Schieman ML. Veerhuis R. de Waal RM. Verbeek MM. Small heat shock proteins associated with cerebral amyloid angiopathy of hereditary cerebral hemorrhage with amyloidosis (Dutch type) induce interleukin-6 secretion. Neurobiol Aging. 2009;30:229–240. doi: 10.1016/j.neurobiolaging.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 128.Wormser U. Sintov A. Brodsky B. Nyska A. Topical iodine preparation as therapy against sulfur mustard-induced skin lesions. Toxicol Appl Pharmacol. 2000;169:33–39. doi: 10.1006/taap.2000.9056. [DOI] [PubMed] [Google Scholar]
- 129.Yamamoto T. Suzuki T. Kobayashi A. Wakabayashi J. Maher J. Motohashi H. Yamamoto M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang K. Meinhardt A. Zhang B. Grzmil P. Adham IM. Hoyer-Fender S. The small heat shock protein ODF1/HSPB10 is essential for tight linkage of sperm head to tail and male fertility in mice. Mol Cell Biol. 2012;32:216–225. doi: 10.1128/MCB.06158-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yang M. Yao Y. Eades G. Zhang Y. Zhou Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res Treat. 2011;129:983–991. doi: 10.1007/s10549-011-1604-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zafarullah M. Li WQ. Sylvester J. Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang H. Limphong P. Pieper J. Liu Q. Rodesch CK. Christians E. Benjamin IJ. Glutathione-dependent reductive stress triggers mitochondrial oxidation and cytotoxicity. FASEB J. 2012;26:1442–1451. doi: 10.1096/fj.11-199869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang H. Rajasekaran NS. Orosz A. Xiao X. Rechsteiner M. Benjamin IJ. Selective degradation of aggregate-prone CryAB mutants by HSPB1 is mediated by ubiquitin-proteasome pathways. J Mol Cell Cardiol. 2010;49:918–930. doi: 10.1016/j.yjmcc.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang X. Min X. Li C. Benjamin IJ. Qian B. Ding Z. Gao X. Yao Y. Ma Y. Cheng Y. Liu L. Involvement of reductive stress in the cardiomyopathy in transgenic mice with cardiac-specific overexpression of heat shock protein 27. Hypertension. 2010;55:1412–1417. doi: 10.1161/HYPERTENSIONAHA.109.147066. [DOI] [PubMed] [Google Scholar]
- 136.Zhang Y. Sano M. Shinmura K. Tamaki K. Katsumata Y. Matsuhashi T. Morizane S. Ito H. Hishiki T. Endo J. Zhou H. Yuasa S. Kaneda R. Suematsu M. Fukuda K. 4-hydroxy-2-nonenal protects against cardiac ischemia-reperfusion injury via the Nrf2-dependent pathway. J Mol Cell Cardiol. 2010;49:576–586. doi: 10.1016/j.yjmcc.2010.05.011. [DOI] [PubMed] [Google Scholar]