

Abstract
An efficient synthetic route to biologically relevant (−)-5-fluorocarbodine 6 was developed. Direct coupling of N6 protected 5-fluorouracil 15 with cyclopentenyl intermediate 13, followed by formation of a macrocycle between the base and the carbocyclic sugar moiety, via ring closing metathesis, allowed for a facial selective hydrogenation of the sugar double bond to give, exclusively, the desired 4’-β stereoisomer.
Carbocyclic nucleosides, in which the furanose ring oxygen atom has been replaced by a carbon atom, are, in general, more chemically and enzymatically stable and some of them have demonstrated interesting biological properties.1 Thus, naturally occurring (−)-aristeromycin (1)2 and (−)-neplanocin A (2)3 manifest cytotoxic and antiviral properties while the drugs abacavir (3)4 and entecavir (4)5 are Food and Drug Administration (FDA) approved for the treatment of human immunodeficiency virus (HIV) and hepatitis B virus (HBV), respectively (Figure 1.).
Figure 1.

Structures of Biologically Relevant Carbocyclic Nucleosides
The carbocyclic cytosine analogs (−)-carbodine (5) and (−)-5-fluorocarbodine (6) was reported to possess significant antitumor and antiviral activities. For instance, compound (6) showed selective submicromolar IC50’s against a human T cell lymphoblast cell line and human B cell lymphoma cell line but also potent inhibition of HCV RNA replication (ΔCt of 6 at 10 µM in a Huh-7 cell based subgenomic replicon assay).6 In addition, compounds (5) and (6) demonstrated selective activity against Venezuela Equine Encephalitis Virus (VEEV)7 and various strains of H5N1 influenza.8
The most recently reported syntheses of (−)-carbodine (5) and (−)-5-fluorocarbodine (6) used a linear approach (Scheme 1) from known cyclopentenyl intermediate including stepwise construction of the heterocyclic base. The key intermediate 7 was prepared from D-ribose in 13 steps by a sequence that used protecting/deprotection strategies and each step required purification by column chromatography on silica gel. After building the pyrimidine base, intermediate 8 was either converted, after a series of protection/deprotection, to its cytosine counterpart then fluorinated (ref 8), or directly fluorinated and then converted to its cytosine analog (ref 6). Overall, both approaches are lengthy (18 and 19 steps) and resulted in the formation of (6) in an overall yield of 26 and 10%.8
Scheme 1.

Linear Approaches for the Synthesis of 5-Fluorocarbodine (6)
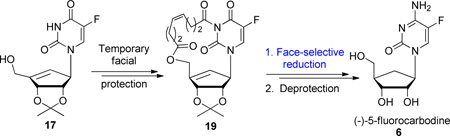
Preparation of (−)-carbodine (5) has also been achieved by either enzymatic hydrolysis9 or selective hydrogenation of cyclopentenyl cytosine.10 However, both of these methods resulted in poor overall yields and low selectivities. In the later approach, the authors demonstrated that the use of diimide as a source of hydrogen was not completely diastereoselective and led to a mixture of 5, along with isocarbodine (10) (Scheme 2). Furthermore, separation of the two isomers appeared problematic and purification of enantiomerically pure 5 could only be achieved using semi-preparative HPLC. With this last result in mind, we hypothesized that the introduction of a temporary sugar-base bridging group on compound A (Scheme 2; X = OH) would act as an umbrella, covering the β face of the cyclopentene and allow for selective α face hydrogenation. Furthermore, we envisioned a sequence to form a cyclic eight member linker using a ring-closing metathesis (RCM)11 reaction that would connect the 5’-O of the sugar and 3-N of the uridine base as depicted in Scheme 2 compound B. Facial selectivity of the subsequent hydrogenation could also be rationalized by the calculated12 increased ring strain of the macrocycle for beta face hydrogenation that would ultimately lead to isocarbodine (10) formation.
Scheme 2.

Facially Selective Hydrogenation
In order to prepare 5-fluorocarbodine (6), the key cyclopentenol derivative 13 was synthesized in 9 steps from d-ribose, using a slightly modified known methodology (Scheme 3).13 Thus, chiral intermediate 11 was selectively protected by treatment with TBDPSCl in presence of DMAP to give compound 12. Diene 12 was then cyclized via a ring-closing metathesis reaction in the presence of catalytic amount of 2nd generation Grubbs’ catalyst D, affording desired cyclopentenol 13. Meanwhile, N3-benzoyl-5-fluorouracil 15 was prepared in 2 steps from 5-fluorouracil 14 (Scheme 4) by benzoylation followed by selective deprotection using K2CO3 in dioxane.14
Scheme 3.

Preparation of Protected Sugar 13
Scheme 4.

Synthesis of 5-Fluorocarbodine 6
Coupling of chiral intermediate 13 with 3-N-Bz-5-F-uracil 15 under Mitsunobu conditions provided the desired N-alkylated derivative as a mixture with its O-alkylated isomer and DIAD byproducts (Scheme 4). The partially column purified mixture was directly deprotected by treatmentwith 7 N NH3 in MeOH, followed by NH4F in MeOH, to afford pure carbocyclic 5-fluorouridine derivative 17 along with pure O-alkylated compound 16 (ratio 4:1) in 93% yield over 3 steps. At this stage, introduction of pentenoyl moieties on both 5’-OH and 3-NH positions of compound 17 had to be optimized. While use of pyridine either alone or with DMAP or Et3N gave the desired compound in low yields (the monoalkylated compound being predominant), the use of Et3N in dichloromethane lead to the nearly quantitative formation of dialkylated compound 18. With intermediate 18 in hand, a ring closing metathesis reaction to form the desired macrocycle 19 was investigated. Evaluation of solvents, catalyst loading and reaction time was undertaken. The best results were obtained when compound 18 was treated with 2.0 mol% of 2nd generation Grubbs’ catalyst D (Figure 2) in toluene at 80 °C for 1 h.
As predicted, hydrogenation of unsaturated derivative 19 in the presence of Pd/C in EtOAc lead to complete stereoselective formation of cyclopentane derivative 15 in 85% yield. The absolute stereochemistry of the final product was determined by 1H-NMR and 1D-NOE experiments on compound 21, which was obtained after removal of the linker using NH3 in MeOH. The NOE experiment showed an enhancement of the 4’ proton when the 1’ proton was irradiated indicating that they reside on the same face of the sugar moiety. Finally, carbocyclic 5-F-uridine 21 was converted to its cytosine counterpart following a previously reported method.15 The primary alcohol in compound 21 was first protected using TBSCl. Compound 22 was then being reacted with 2,4,6-triisopropylbenzenesulfonyl chloride followed by NH4OH to give intermediate 23. Final deprotection was achieved using HCl in MeOH providing the target 5-fluoro-carbodine 6 in 98% isolated yield.
CONCLUSION
In summary, an efficient synthetic route to 5-fluoro-carbodine 6 was developed utilizing a key α-face-selective hydrogenation driven by the introduction of a temporary macrocyclic tether. This approach represents a noteworthy improvement to the existing syntheses of biologically relevant compound 6 and allowed us to prepare 6 in 23% overall yield (compared with 2 to 10% yields from previous approaches) with no major purification problems nor use of special synthetic techniques. We are currently evaluating our umbrella approach’s compatibility with other reactions to modify the olefin functionality and further explore the scope of this innovative methodology.
EXPERIMENTAL SECTION
General Considerations
Nuclear magnetic resonance (NMR) spectra (1H, 13C and 19F) were recorded on a 400 MHz FT-NMR spectrometer at ambient temperature, with tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ) are reported in parts per million (ppm), and signals are quoted as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad), dd (doublet of doublets), dt (doublet of triplets) or ddd (doublet of doublets of doublets). High-resolution mass spectra (HRMS) were recorded on a linear ion trap LTQ-FT mass spectrometer with electrospray ionization (ESI). Thin-layer chromatography (TLC) was performed on 0.25 mm silica gel. Purifications were carried out by silica gel column chromatography (60 Å, 63–200 µm, or 40–75 µm).
(R)-1-((4R,5S)-5-(3-(tert-Butyldiphenylsilyloxy)prop-1-en-2-yl)-2,2-dimethyl-1,3-dioxolan-4-yl)prop-2-en-1-ol (12)
To a solution of compound 11 (4.0 g, 20.5 mmol) in 100 mL of anhydrous CH2Cl2 was added DMAP (0.2 g, 1.4 mmol), TBDPSCl (6.3 g, 24.6 mmol) and Et3N (3.1 g, 30.8 mmol) at 0 °C under N2 atmosphere. The reaction mixture was stirred for 12 h at room temperature and quenched with 5.0 mL of MeOH. The solution was adsorbed on silica gel and purified by silica gel column chromatography (hexane:EtOAc = 20:1 to 2:1 v/v) to give compound 12 as a white foam (8.7 g, 19.1 mmol) in 93% yield. HRMS-ESI+ calcd. for C27H37O4Si (M+H+) 453.2462, found 453.2456; 1H NMR (400 MHz, CDCl3) δ 7.71-7.67 (m, 4H), 7.46-7.37 (m, 6H), 6.03-5.95 (m, 1H), 5.47 (dd, J = 13.2, 1.2 Hz, 2H), 5.32 (dt, J = 17.2, 1.2 Hz, 1H), 5.21 (dt, J = 10.4, 1.6 Hz, 1H), 4.65 (d, J = 5.6 Hz, 1H), 4.24 (s, 2H), 4.04-3.99 (m, 1H), 3.94 (dd, J = 8.8, 6.0 Hz, 1H), 2.63 (d, J = 3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H), 1.09 (s, 9H); 13C NMR (100 MHz, CDCl3) δ; 143.2, 138.0, 135.9, 135.8, 132.9, 132.8, 130.2, 130.1, 128.1, 128.0, 116.2, 113.7, 108.2, 80.9, 77.6, 70.3, 66.2, 27.5, 27.0, 25.4, 19.4.
(3aS,4S,6aR)-6-((tert-Butyldiphenylsilyloxy)methyl)-2,2-dimethyl-4,6a-dihydro-3aH-cyclopenta[d][1,3]dioxol-4-ol (13)
To a solution of compound 12 (8.2 g, 18.1 mmol) in 500 mL of anhydrous CH2Cl2 was added a solution of 2nd Grubb’s catalyst (0.1 mol%, 0.02 g, 0.02 mmol) in 100 mL of anhydrous CH2Cl2 at room temperature under argon atmosphere. After stirring for 24 h, the reaction mixture was treated with 5.0 mL of DMSO, additionally stirred for 1 h at room temperature and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:EtOAc = 10:1 to 1:1 v/v) to give compound 13 as a white foam (7.40 g, 17.43 mmol) in 96% yield. HRMS-ESI+ calcd. for C25H33O4Si (M+H+) 425.2149, found 425.2143; 1H NMR (400 MHz, CDCl3) δ 7.69-7.65 (m, 4H), 7.44-7.34 (m, 6H), 5.84 (d, J = 1.6 Hz, 1H), 4.86 (d, J = 5.6 Hz, 1H), 4.74 (t, J = 5.2 Hz, 1H), 4.56 (m, 1H), 4.38 (dd, J = 15.2, 20.0 Hz, 2H), 2.70 (d, J = 10.0 Hz, 1H), 1.35 (s, 3H), 1.33 (s, 3H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 145.5, 135.7, 133.5, 129.9, 129.5, 127.9, 112.7, 83.0, 78.2, 73.5, 61.0, 27.8, 27.0, 26.9, 19.5.
N3-Benzoyl-5-fluorouracil (15)
To a solution of 5-fluorouracil 14 (3.6 g, 27.3mmol) in 50 mL of anhydrous CH3CN was added BzCl (9.3 g, 68.2 mmol), pyridine (10.8 g, 136.5mmol) at 0 °C under N2 atmosphere. The solution was stirred for 24 h at room temperature and concentrated under reduced pressure. The residue was dissolved in 200 mL of CH2Cl2 and then washed with cold water (50 mL × 3) and concentrated under reduced pressure. The residue was re-dissolved in 50 mL of 1,4-dioxane and treated with aqueous K2CO3 (20 mL, 0.5 M in water solution) at 0 °C. After stirring for 30 min at room temperature, the pH of the solution was lowered to ca. 5.0 by careful addition of glacial acetic acid. The solution was concentrated in vacuo and the residue was treated with a saturated solution of NaHCO3 (100 mL). After stirring for 1 h, the white solid was filtered and washed with cold water (20 mL × 5) then dried under vacuum for 48 h. The crude product was purified by silica gel column chromatography (hexane:EtOAc = 1:1 to EtOAc) to give compound 15 as a white amorphous solid (5.6 g, 23.7 mmol) in 87% yield. MS-ESI+m/z 235 (M+H+); 1H NMR (400 MHz, DMSO-d6) δ 11.54 (br, 1H), 8.06 (d, J = 6.4 Hz, 1H), 8.01 (d, J = 8.4 Hz, 2H), 7.77 (t, J = 7.6 Hz, 1H), 7.58 (dd, J = 8.4, 7.6 Hz, 2H).
5-Fluoro-2-(((3aS,4R,6aR)-6-(hydroxymethyl)-2,2-dimethyl-4,6a-dihydro-3aH-cyclopenta[d][1,3]dioxol-4-yl)oxy)pyrimidin-4(3H)-one (16) and 5-Fluoro-1-((3aS,4R,6aR)-6-(hydroxymethyl)-2,2-dimethyl-4,6a-dihydro-3aH-cyclopenta[d][1,3]dioxol-4-yl)pyrimidine-2,4(1H,3H)-dione (17)
To a solution of Ph3P (11.1 g, 42.4 mmol) in 150 mL of CH3CN was added DIAD (8.6 g, 42.4 mmol) at 0 °C under N2 atmosphere. After 30 min, to the reaction mixture was added a solution of compound 13 (7.2 g, 17.0 mmol) in 150 mL of CH3CN and N-Bz-5-fluorouracil 15 (6.0 g, 25.4 mmol) at −78 °C. The reaction mixture was stirred for 24 h at room temperature and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:EtOAc = 4:1 to 1:1 v/v) to give a non separable mixture of N-/O-alkylated products along with DIAD byproducts. This mixture was dissolved in 100 mL of MeOH and treated with 100 mL of 7 N NH3 in MeOH at 0 °C. The solution was stirred for 12 h at room temperature and concentrated under reduced pressure. The residue was then directly treated with a solution of ammonium fluoride (1.6 g, 42.4 mmol) in 200 mL of MeOH at room temperature and the solution was stirred at 50 °C for 6 h. After removal of the volatiles under vacuum, the residue was purified by silica gel column chromatography (hexane:EtOAc = 1:5 to EtOAc v/v) to give 16 as an off white gum (0.94 g, 3.2 mmol) and 17 as an off white gum (3.8 g, 12.6 mmol) in 93% yield over 3 steps. Compound 16: MS-ESI+m/z 299 (M+H+); HRMS-ESI+calcd. for C13H16FN2O5 (M+H+) 299.1039, found 299.1035; 1H NMR (400 MHz, CD3OD) δ 7.77 (d, J = 3.2 Hz, 1H), 5.86 (d, J = 1.6 Hz, 1H), 5.72 (s, 1H), 5.19 (d, J = 5.6 Hz, 1H), 4.73 (d, J = 5.6 Hz, 1H), 4.24 (dd, J = 16.0, 10.8 Hz, 2H), 1.38 (s, 3H), 1.34 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 154.7, 149.3, 146.8, 136.0, 123.7, 113.7, 87.3, 85.0, 84.4, 59.00, 27.8, 26.2; Compound 17: MS-ESI+m/z 299 (M+H+); HRMS-ESI+calcd for C13H16FN2O5 (M+H+) 299.1038, found 299.1036; 1H NMR (400 MHz, CD3OD) δ 7.54 (d, J = 6.4 Hz, 1H), 5.60 (m, 1H), 5.36 (s, 1H), 5.24 (d, J = 6.0 Hz, 1H), 4.62 (d, J = 5.6 Hz, 1H), 4.29 (s, 2H), 1.40 (s, 3H), 1.33 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 155.0, 151.5, 143.1, 140.8, 127.8, 122.6, 113.5, 85.6, 84.8, 68.9, 60.0, 27.7, 26.1; 19F NMR (376 MHz, CD3OD) δ −169.69 (d, J = 6.0 Hz).
((3aR,6R,6aS)-6-(3-But-3-enoyl-5-fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,2-dimethyl-6,6a-dihydro-3aH-cyclopenta[d][1,3]dioxol-4-yl)methyl but-3-enoate (18)
To a solution of compound 17 (1.4 g, 4.7mmol) in 40 mL of anhydrous CH2Cl2 was added Et3N (2.4 g, 23.5 mmol) and 4-pentenoyl chloride (1.7 g, 14.1 mmol) at 0 °C under argon atmosphere. After stirring for 4 h at room temperature, the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:EtOAc = 4:1 to 1:1 v/v) to give a compound 18 as a white foam (2.1 g, 4.5 mmol) in 97% yield. MS-ESI+m/z 463 (M+H+); HRMS-ESI+calcd for C23H27FN2O7Na (M+Na+) 485.1705, found 485.1696; 1H NMR (400 MHz, CDCl3) δ 7.09 (d, J = 5.6 Hz, 1H), 5.90-5.78 (m, 2H), 5.57 (s, 1H), 5.39 (s, 1H), 5.21 (d, J = 5.6 Hz, 1H), 5.15-5.01 (m, 4H), 4.82 (q, J = 15.2 Hz, 2H), 4.59 (d, J = 5.6 Hz, 1H), 2.97 (t, J = 7.2 Hz, 2H), 2.51 (m, 4H), 2.43 (m, 2H), 1.43 (s, 3H), 1.35 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 173.9, 172.8, 158.7, 149.3, 148.0, 136.6, 135.6, 124.9, 122.9, 116.5, 116.1, 113.4, 94.6, 84.1, 83.5, 68.1, 60.9, 39.8, 33.4, 28.9, 27.5, 27.4, 25.9; 19F NMR (376 MHz, CDCl3) δ −165.50 (d, J = 4.5 Hz).
Compound 19
To a solution of compound 18 (1.0 g, 2.16 mmol) in 500 mL of anhydrous toluene was added 2nd Generation Grubbs’ catalyst D (2.0 mol%, 0.037 g, 0.043 mmol) at room temperature under an argon atmosphere. After stirring for 1 h at 80 °C, the reaction mixture was treated with 2.0 mL of DMSO at room temperature and additionally stirred for 1 h. The solution was concentrated under reduced pressure and then purified by silica gel column chromatography (hexane:EtOAc = 2:1 v/v) to give compound 19 as a white foam (0.85 g, 2.0 mmol) in 90% yield. MS-ESI+m/z 435 (M+H+); HRMS-ESI+calcd for C21H24FN2O7 (M+H+) 435.1562, found 435.1560; 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 5.2 Hz, 1H), 5.62 (s, 1H), 5.56-5.40 (m, 3H), 5.25 (d, J = 11.6 Hz, 1H), 4.90 (d, J = 5.6 Hz, 1H), 4.42 (s, 1H), 4.25 (d, J = 11.2 Hz, 1H), 3.07 (ddd, J = 18.0, 8.0, 2.4 Hz, 1H), 2.86 (ddd, J = 18.0, 10.4, 2.4 Hz, 1H), 2.60 (m, 1H), 2.50-2.30 (m, 3H), 2.28-2.15 (m, 2H), 1.40 (s, 3H), 1.36 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 174.5, 172.7, 155.9, 146.9, 141.1, 138.7, 129.8, 129.1, 127.7, 125.5, 112.3, 84.5, 82.7, 74.9, 58.6, 39.4, 32.8, 27.4, 25.8; 19F NMR (376 MHz, CDCl3) δ −165.63 (d, J = 6.0 Hz), −166.20 (d, J = 6.0 Hz).
Compound 20
To a solution of compound 19 (2.5 g, 5.8 mmol) in 100 mL of EtOAc was added Pd/C catalyst (10 wt %, 0.25 g). The solution was stirred for 12 h under H2 atmosphere (1 atm) and then treated with Celite. After stirring for 30 min, the suspension was carefully filtered, the filtrate concentrated under vacuum, and then purified by silica gel column chromatography (hexane:EtOAc = 1:1 v/v) to give compound 20 as a white foam (2.1 g, 4.9 mmol) in 85% yield. MS-ESI+m/z 439 (M+H+); HRMS-ESI−calcd for C21H27FN2O7Cl (M+Cl−) 473.1496, found 473.1499; 1H NMR (400 MHz, CDCl3) δ 7.28 (d, J = 5.6 Hz, 1H), 5.00 (dd, J = 6.4, 2.4 Hz, 1H), 4.88 (t, J = 5.6 Hz, 1H), 4.70 (d, J = 12.0, 1.2 Hz, 1H), 3.99 (dd, J = 12.0, 1.2 Hz, 1H), 3.91 (m, 1H) 2.95 (ddd, J = 16.0, 8.4, 3.2 Hz, 1H), 2.73 (ddd, J = 16.0, 9.6, 3.2 Hz, 1H), 2.55 (ddd, J = 16.0, 8.4, 5.2 Hz, 1H), 2.33-2.17 (m, 4H), 1.90 (m, 1H), 1.79 (s, 1H), 1.66-1.40 (m, 6H), 1.39-1.25 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 174.4, 174.1, 147.6, 141.1, 138.7, 130.1, 129.8, 113.0, 94.6, 83.4, 80.3, 69.7, 61.0, 45.7, 40.1, 33.6, 31.2, 28.1, 27.1, 25.6, 24.4; 19F NMR (376 MHz, CDCl3) δ −166.59 (d, J = 4.5 Hz),
5-Fluoro-1-((3aS,4R,6R,6aR)-6-(hydroxymethyl)-2,2-dimethyltetrahydro-3aH-cyclopenta[d][1,3]dioxol-4-yl)pyrimidine-2,4(1H,3H)-dione (21)
A solution of compound 20 (2.0 g, 4.58 mmol) in 50 mL of MeOH was treated with 20 mL of 7 N NH3 in MeOH at 0 °C in a sealed tube. After stirring for 12 h at 80 °C, the solution was concentrated under reduced pressure and the residue was purified on a silica gel pad (CH2Cl2:MeOH = 30:1 to 10:1 v/v) to give compound 21 as a beige gum (1.3 g, 4.3 mmol) in 94% yield. MS-ESI+m/z 301 (M+H+); HRMS-ESI+calcd for C13H18FN2O5 (M+H+) 301.1194, found 301.1193; 1H NMR (400 MHz, CDCl3) δ 10.01 (br, 1H), 7.48 (d, J = 5.6 Hz, 1H), 4.77 (dd, J = 7.2, 5.6 Hz, 1H), 4.61 (dd, J = 7.2, 4.8 Hz, 1H), 4.57 (m, 1H), 3.83 (dd, J = 10.4, 4.8 Hz, 1H), 3.74 (dd, J = 10.8, 4.8 Hz, 1H), 2.80 (br, 1H) 2.37-2.27 (m, 2H), 2.12-2.03 (m, 1H), 1.54 (s, 3H), 1.31 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 157.5, 157.2, 149.8, 141.9, 139.6, 127.9, 127.6, 113.2, 83.0, 81.4, 64.2, 63.3, 45.6, 32.0, 27.7, 25.4; 19F NMR (376 MHz, CDCl3) δ −169.67 (d, J = 6.0 Hz).
1-((3aS,4R,6R,6aR)-6-((tert-Butyldiphenylsilyloxy)methyl)-2,2-dimethyltetrahydro-3aH-cyclopenta[d][1,3]dioxol-4-yl)-5-fluoropyrimidine-2,4(1H,3H)-dione (22)
To a solution of compound 21 (1.3 g, 4.5 mmol) in 30 mL of anhydrous CH2Cl2 was added imidazole (0.74 g, 10.9mmol) and TBDPSCl (1.7 g, 5.4 mmol) at 0 °C under N2 atmosphere. The reaction mixture was stirred for 12 h at room temperature and treated with 5.0 mL of MeOH. After stirring for 1 h at room temperature, the solution was concentrated under reduced pressure and purified by silica gel column chromatography (hexane:EtOAc = 2:1 to 1:4 v/v) to give compound 22 as an off white foam (2.3 g, 4.3mmol) in 98% yield. MS-ESI+m/z 539 (M+H+);HRMS-ESI+calcd for C29H36FN2O5Si (M+H+) 539.2372, found 539.2368; 1H NMR (400 MHz, CDCl3) δ 9.78 (br, 1H), 7.66-7.62 (m, 4H), 7.40-7.36 (m, 6H), 7.23 (d, J = 5.6 Hz, 1H), 4.71 (m, 1H), 4.63 (dd, J = 6.8, 6.0 Hz, 1H), 4.51 (dd, J = 7.2, 6.8 Hz, 1H), 3.79 (d, J = 5.2 Hz, 2H), 2.33 (m, 1H), 2.25 (m, 1H), 2.00 (q, J = 12.0 Hz, 1H), 1.53 (s, 3H), 1.29 (s, 3H), 1.09 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 157.2, 157.0, 149.7, 142.1, 139.7, 135.83, 135.78, 133.5, 133.4, 130.1, 128.0, 126.6, 126.3, 113.9, 82.8, 80.7, 64.3, 63.8, 45.2, 32.4, 27.8, 27.2, 25.5, 19.6; 19F NMR (376 MHz, CDCl3) δ −165.85 (d, J = 4.5 Hz).
4-Amino-1-((3aS,4R,6R,6aR)-6-((tert-butyldiphenylsilyloxy)methyl)-2,2-dimethyltetrahydro-3aH-cyclopenta[d][1,3]dioxol-4-yl)-5-fluoropyrimidin-2(1H)-one (23)
To a solution of compound 22 (2.2 g, 4.1 mmol) and DMAP (2.0 g, 16.4 mmol) in 50 mL of anhydrous CH3CN was added 2,4,6-trisiopropyl benzenesulfonyl chloride (3.1 g, 10.3mmol) and Et3N (1.7 g, 16.4 mmol) at room temperature under N2 atmosphere. The reaction mixture was stirred for 48 h at room temperature and treated with 25 mL of 30% NH4OH solution. After stirring for 4 h at room temperature, the solution was poured into 150 mL of a cold water-CHCl3 solution (1:2 v/v) and then extracted with CHCl3 (30 mL × 3). The combined organic layers were washed with a saturated NH4Cl aqueous solution (100 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2 to CH2Cl2:MeOH = 10:1 v/v) to give compound 23 as a gum (2.0 g, 3.7mmol) in 89% yield. MS-ESI+m/z 538 (M+H+); HRMS-ESI+calcd for C29H37FN3O4Si (M+H+) 538.2532, found 538.2525; 1H NMR (400 MHz, CDCl3) δ 8.83, (br, 1H), 7.66-7.63 (m, 4H), 7.45-7.36 (m, 6H), 7.24 (d, J = 5.6 Hz, 1H), 5.60 (br, 1H), 4.80 (dd, J = 7.2, 5.2 Hz, 1H), 4.52-4.44 (m, 2H), 3.82 (dd, J = 10.0, 4.4 Hz, 1H), 3.74 (dd, J = 10.0, 6.0 Hz, 1H), 2.33-2.25 (m, 2H), 2.16 (m, 1H), 1.50 (s, 3H), 1.26 (s, 3H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 158.1, 154.6, 137.8, 135.8, 135.7, 135.4, 133.7, 133.6, 129.9, 128.4, 128.1, 127.9, 113.3, 83.0, 81.1, 66.1, 64.8, 46.2, 32.8, 27.8, 27.1, 25.5, 19.5; 19F NMR (376 MHz, CDCl3) δ −169.48 (d, J = 7.5 Hz).
4-Amino-1-((1R,2S,3R,4R)-2,3-dihydroxy-4-(hydroxymethyl)cyclopentyl)-5-fluoropyrimidin-2(1H)-one (6)
A solution of compound 23 (2.5 g, 4.7mmol) in 100 mL of MeOH was treated with 50 mL of HCl in MeOH at 0 °C. After stirring for 3 h at room temperature, the acidic solution was concentrated under reduced pressure. The residue was dissolved in water (50 mL) and washed with EtOAc (20 mL × 5). The aqueous layer was concentrated under reduced pressure. The residue was dissolved in EtOH (10 mL) and the solution was concentrated under reduced pressure. After this process was repeated 5 times, the final residue was dried under vacuum for 72 h at room temperature to give pure compound 6 as a white amorphous solid (1.4 g, 4.6 mmol) as a HCl salt in 98% yield. MS-ESI+m/z 260 (M+H+); HRMS-ESI+calcd for C10H15FN3O4 (M+H+) 260.1041, found 260.1039; 1H NMR (400 MHz, CD3OD) δ 8.35 (d, J = 7.2 Hz, 1H), 4.70 (q, J = 9.2 Hz, 1H), 4.12 (dd, J = 5.6, 9.2 Hz, 1H), 3.85 (dd, J = 5.6, 3.2 Hz, 1H), 3.53 (d, J = 5.2 Hz, 2H), 2.19 (m, 1H), 2.05 (m, 1H), 1.46 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 154.65, 154.42, 148.52, 139.97, 137.78, 133.74, 133.46, 75.44, 73.49, 64.83, 64.53, 46.50, 28.67; 1H NMR (400 MHz, DMSO-d6) δ 9.75 (br, 1H), 9.14 (br, 1H), 8.53 (d, J = 7.2 Hz, 1H), 5.32 (br, 3H), 4.65 (q, J = 9.2 Hz, 1H), 4.01 (dd, J = 9.2, 5.6 Hz, 1H), 3.72 (dd, J = 4.8, 2.4 Hz, 1H), 3.42 (dd, J = 10.4, 6.4 Hz, 1H), 3.35 (dd, J = 10.4, 5.6 Hz, 1H), 2.04 (m, 1H), 1.93 (m, 1H), 1.28 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 153.1, 152.9, 147.8, 136.2, 133.9, 131.8, 131.5, 73.4, 71.2, 62.9, 61.8, 44.81, 27.8; 19F NMR (376 MHz, CD3OD) δ −170.99 (d, J = 7.52 Hz).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by NIH grant 5P30-AI-50409 (CFAR) and by the Department of Veterans Affairs. Dr. Schinazi is the founder and a major shareholder of RFS Pharma, LLC. Emory received no funding from RFS Pharma, LLC to perform this work and vice versa.
Footnotes
Supporting Information
Copies of NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.a) Marquez VE, Lim M-I. Med. Res. Rev. 1986;6:1–40. doi: 10.1002/med.2610060102. [DOI] [PubMed] [Google Scholar]; b) Borthwick AD, Biggadike K. Tetrahedron. 1992;48:571–623. [Google Scholar]; c) Agrofoglio L, Suhas E, Farese A, Condom R, Challand R, Earl RA, Guedj R. Tetrahedron. 1994;50:10611–10670. [Google Scholar]; d) Jenkins GN, Turner NJ. Chem. Soc. Rev. 1995;24:169–176. [Google Scholar]; e) Trost BM, Van Vranken DL. Chem. Rev. 1996;96:395–422. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; f) Ferrero M, Gotor V. Chem. Rev. 2000;100:4319–4347. doi: 10.1021/cr000446y. [DOI] [PubMed] [Google Scholar]; g) Schneller SW. Curr. Top. Med. Chem. 2002;2:1087–1092. doi: 10.2174/1568026023393228. [DOI] [PubMed] [Google Scholar]; h) Pathak T. Chem. Rev. 2002;102:1623–1667. doi: 10.1021/cr0104532. [DOI] [PubMed] [Google Scholar]; i) Rodriguez JB, Comin MJ. Mini Rev. Med. Chem. 2003;3:95–114. doi: 10.2174/1389557033405331. [DOI] [PubMed] [Google Scholar]; j) Agrofoglio LA, Gillaizeau I, Saito Y. Chem. Rev. 2003;103:1875–1916. doi: 10.1021/cr010374q. [DOI] [PubMed] [Google Scholar]; k) Jeong LS, Lee JA. Antiviral Chem. Chemother. 2004;15:235–250. doi: 10.1177/095632020401500502. [DOI] [PubMed] [Google Scholar]; l) Kurteva VB, Afonso CAM. Chem. Rev. 2009;109:6809–6857. doi: 10.1021/cr900169j. [DOI] [PubMed] [Google Scholar]
- 2.a) Kusaka T, Yamamoto H, Shibata M, Murori M, Hicki T, Mizuno K. J. Antibiot. 1968;21:255–263. doi: 10.7164/antibiotics.21.255. [DOI] [PubMed] [Google Scholar]; b) Shealy YF, Clayton JD. J. Pharm. Sci. 1973;62:858–859. doi: 10.1002/jps.2600620547. [DOI] [PubMed] [Google Scholar]
- 3.a) Yaginuma S, Tsujino M, Sudate Y, Hayashi M, Otani M. J. Antibiot. 1981;34:359–366. doi: 10.7164/antibiotics.34.359. [DOI] [PubMed] [Google Scholar]; b) De Clercq E. Antimicrob. Agents Chemother. 1985;28:84–89. doi: 10.1128/aac.28.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Kimberlain DW, Coen DM, Biron KK, Cohen JI, Lamb RA, Mckinlay M, Emini EA, Whitley RJ. Antiviral Res. 1995;26:369–401. doi: 10.1016/0166-3542(95)00027-j. [DOI] [PubMed] [Google Scholar]; b) Foster RH, Faulds D. Drugs. 1998;55:729–736. doi: 10.2165/00003495-199855050-00018. [DOI] [PubMed] [Google Scholar]
- 5.a) Bisacchi GS, Chao ST, Bachard C, Daris JP, Innaimo S, Jacobs GA, Kocy O, Lapointe P, Martel A, Merchant Z, Slusarchyk WA, Sundeen JE, Young MG, Colonno R, Zahler R. Bioorg. Med. Chem. Lett. 1997;7:127–132. [Google Scholar]; b) Yamanaka G, Wilson T, Innaimo S, Bisacchi GS, Egli P, Rinehart JK, Zahler R, Colonno RJ. Antimicrob. Agents Chemother. 1999;43:190–193. doi: 10.1128/aac.43.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stuyver L, Watanabe KA. WO 02/32920 A2. Patent. 2002
- 7.Julander JG, Bowen RA, Rao JR, Day C, Shafer K, Smee DF, Morrey JD, Chu CK. Antiviral Res. 2008;80:309–315. doi: 10.1016/j.antiviral.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu CK, Rao J. WO 2008/124157 A1. Patent. 2008
- 9.Ohno M. Nucleosides Nucleotides. 1985;4:21. [Google Scholar]
- 10.Russ PL, Hegedus L, Kelley JA, Barchi JJ, Jr, Marquez VE. Nucleosides Nucleotides. 1992;11:351. [Google Scholar]
- 11.For a review on metathesis strategy in nucleoside chemistry see: Amblard F, Nolan SP, Agrofoglio LA. Tetrahedron. 2005;61:7067.
- 12.The two possible reduction products were energy minimized with the MM2 force field in Chem 3D Ultra version 10. 0.
- 13.Cho JH, Bernard DL, Sidwell RW, Kern ER, Chu CK. J. Med. Chem. 2006;49:1140–1148. doi: 10.1021/jm0509750. [DOI] [PubMed] [Google Scholar]
- 14.a) Cruickshank K, Jiricny J, Reese CB. Tetrahedron Lett. 1984;25:681–684. [Google Scholar]; b) Frieden M, Giraud M, Reese CB, Song Q. J. Chem. Soc. Perkin.Trans. 1. 1998:2827–2832. [Google Scholar]; c) Lucey NM, McCormick JE, McElhinney RS. J. Chem. Soc. Perkin. Trans. 1. 1990:795–802. [Google Scholar]; d) Lee Y-S, Kim BH. Bioorg. Med. Chem. Lett. 2002;12:1395–1397. doi: 10.1016/s0960-894x(02)00182-8. [DOI] [PubMed] [Google Scholar]; e) Shatila RS, Bouhadir KH. Tetrahedron Lett. 2006;47:1767–1770. [Google Scholar]; f) Ludek OR, Meier C. Eur. J. Org. Chem. 2006:941–946. [Google Scholar]
- 15.a) Shuto S, Obara T, Saito Y, Andrei G, Snoeck R, De Clercq E, Matsuda A. J. Med. Chem. 1996;39:2392–2399. doi: 10.1021/jm950853f. [DOI] [PubMed] [Google Scholar]; b) Jin YH, Liu P, Wang J, Baker R, Huggins J, Chu CK. J. Org. Chem. 2006;68:9012–9018. doi: 10.1021/jo034999v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.