

Abstract
Fluorescent proteins (FPs) have revolutionized molecular and cellular biology; yet, discrimination over cellular autofluorescence, spectral deconvolution, or detection at low concentrations remain challenging problems in many biological applications. By optically depopulating a photoinduced dark state with orange secondary laser co-excitation, the higher-energy green AcGFP fluorescence is dynamically increased. Modulating this secondary laser then modulates the higher-energy, collected fluorescence; enabling its selective detection by removing heterogeneous background from other FPs. Order-of-magnitude reduction in obscuring fluorophore background emission has been achieved in both fixed and live cells. This longwavelength modulation expands the dimensionality to discriminate FP emitters based on dark state lifetimes and enables signal of interest to be recovered by removing heterogeneous background emitter signals. Thus, AcGFP is not only useful for extracting weak signals from systems plagued by high background, but it is a springboard for further FP optimization and utilization for improving sensitivity and selectivity in biological fluorescence imaging.
Keywords: Fluorescence modulation, cellular imaging, photoisomerization, spectroscopy, signal recovery, GFP
Over the past two decades, the green fluorescent protein (GFP) and its derivatives have emerged as universal, genetically encodable markers for biological imaging.1,2 Proteins fused to GFP typically retain their function and subcellular localization; thus, GFP is widely used for studying intracellular protein dynamics, visualizing protein-protein interactions, and developing biosensors responsive to a broad range of analytes.3-5 The chromophore of GFP, formed through autocatalytic cyclization of a peptide segment, can be readily tuned over a broad range of emission wavelengths by simply altering the protein’s amino acid composition.1 The amino acid residues surrounding the GFP chromophore are especially important in defining the manifold of photoaccessible states, for example, by differentially stabilizing photointerconvertable cis/trans isomers or by altering the chromophore protonation state.6,7 Tuning these relative ground and excited state stabilities as well as the barriers separating them yields a rich array of optically controllable behaviors that continue to fuel development of innovative imaging schemes.8-13 Although photoswitching has been useful in resolving ever-finer features and elucidating some kinetic processes, signals of interest are too-often obscured by the spatially heterogeneous background characteristic of biological imaging. Applicable to both low copy number proteins in cellular imaging and selective recovery of multiple overlapping fluorophore signals, sensitivity improvements that directly distinguish weak signals within high background are crucial to improved cellular imaging. Although reversible short-wavelength photoswitching of Dronpa and of synthetic fluorophores has realized sensitivity gains, they are somewhat tempered as higher energy or high-intensity photorecovery can generate additional fluorescence background14 and photodamage.15 Therefore, these schemes require either dual-color nanoparticles14 or cross-correlation with the emission of an exogenously introduced synthetic dye16,17 to distinguish modulated signals from differently modulated background.18
To address the sensitivity challenges in biological imaging, we recently developed SAFIRe (Synchronously Amplified Fluorescence Image Recovery), a detection scheme that recovers fluorescence by rapidly depopulating non-emissive fluorophore dark states through secondary excitation at wavelengths longer than those of the collected fluorescence.19 Complementary to photoswitch-based optical lock-in detection13,14 and frequency domain photoswitching,14 co-illumination at lower energy reduces phototoxicity and fluorophore decomposition without generating additional background fluorescence or modulating background signals. Further, as transient dark states are optically depopulated to recover emission, the molecules can respond at the modulation frequencies applied to the long-wavelength secondary laser, thereby directly encoding the modulation waveform only on the fluorescence signal of interest, but not on the background. Thus, selective recovery from unmodulated background is achieved upon Fourier transformation or lock-in detection at the modulation frequency and is completely decoupled from the non-modulated background, thereby eliminating the need for cross-correlation with exogenous or control fluorescent signals. As the SAFIRe modulation depth depends on the relative rates in and out of transient dark states, each fluorophore exhibits a characteristic frequency-dependent modulation profile.
Because many important biological processes involve the interaction of proteins expressed at low concentrations, the development of imaging techniques with high detection sensitivity to fluorophores in the presence of heterogeneous background is of critical importance. At present, we have already identified a range of synthetic inorganic and organic fluorophores that offer high modulation depths at frequencies up to 100 kHz.19-21 Herein, we demonstrate that the manifold of photo-accessible states of the fluorescent protein (FP) chromophore, which is responsible for its blinking and switching behavior,22 can be effectively utilized for SAFIRe-based fluorescence modulation to yield greatly improved imaging contrast in fixed and live cells.
Even prior to the first reports of blinking and switching of GFP emission,22,23 researchers tried to manipulate the characteristic ground and excited state fluorophore potential energy surfaces through targeted mutations and succeeded in altering the balance between short- and long-wavelength excitation/emission profiles.24,25 The palette of currently available fluorescent protein variants is very large,1,5,26 and many mutants offer a diverse set of long-lived27 and spectrally distinct chromophore states that are potentially amenable to SAFIRe modulation. Although producing long-lived states that facilitate photoswitchability and kindling behaviors, photoinduced cis-trans isomerization of the chromophore, by itself, is unlikely to yield sufficient spectral shifts to selectively recover the emissive state by long- or short-wavelength secondary excitation.28-30 The largest GFP spectral shifts typically result from mutating surrounding amino acid residues that influence the balance between the anionic and neutral form of the chromophore.6 Thus, high-energy reversible photoswitches that combine long-lived (millisecond) states31 and large blue-shifts almost certainly involve an anionic to neutral change in protonation state along with cis-trans photoisomerization. Long-wavelength photorecovery from long-lived dark states, however, more directly suggests photoisomerization without anionneutral conversion. In such a case, the kinetically trapped trans photoisomer would eventually relax to the cis ground state, but photorecovery of the original emissive cis-form could also occur with low-energy excitation, regardless of the chromophore’s protonation state (e.g. Scheme 1).1,5,26
Scheme 1.
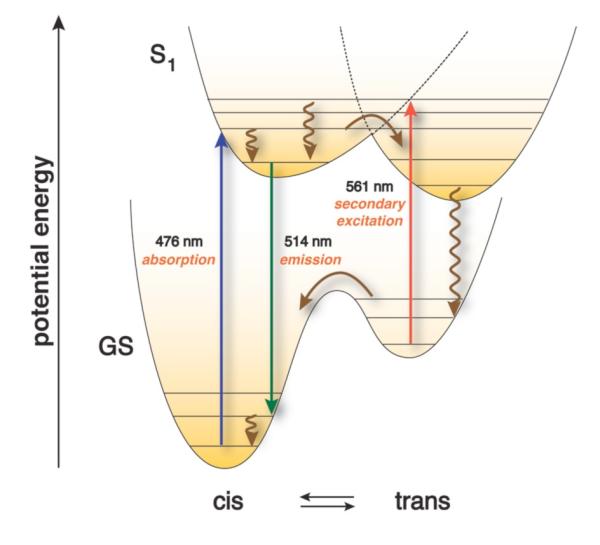
Schematic potential energy diagram rationalizing kinetically trapped trans vs. ground state cis isomers and long-wavelength fluorescence recovery.
Used extensively in short-wavelength photoswitch-based imaging applications,16 Dronpa reversibly photoisomerizes, as confirmed by crystallographic data, to give rise to emissive and dark states of the protein.32-34 As transient absorptions can occur throughout the visible due to drastic changes in chromophore environment, we neglected the well-known ~400 nm co-excitation in favor of illumination at wavelengths longer than those of the collected ~500-nm fluorescence to revert the chromophore back to the emissive cis state. Using spectral lines available, co-illumination with 476 nm for fluorescence excitation and 561 nm for long-wavelength photoreversion did not, however, increase observed fluorescence (data not shown).
Analogous to Dronpa, the highly emissive AcGFP is one mutation away from being an initially dark kindling protein.35,36 Thus, AcGFP was chosen as a promising candidate for long-wavelength activation of the anionic cis/trans chromophore dynamics. Excitation of AcGFP in PBS buffer at 476 nm yields bright green emission (λmax = 514 nm, 2 kW/cm2), with intensity further increasing by ~8% upon steady-state secondary co-illumination at either 561 nm or 594 nm (20 kW/cm2). Secondary illumination alone yielded no emission (at higher or lower energy), nor did co-illumination with the secondary laser alter the photobleaching quantum yield compared to primary illumination alone. Chopping the secondary laser dynamically increased and decreased primary laser-excited AcGFP emission and encoded the long-wavelength secondary laser modulation waveform directly on the higher-energy fluorescence. Demodulated AcGFP signal was recovered from the Fourier amplitude at the modulation frequency (Fig. 1 Inset). In contrast, modulation experiments on enhanced GFP (EGFP, Clontech),37 a mutant with almost identical absorption and emission spectra, fluorescence quantum yield (~0.60), and extinction coefficient (~50,000 M−1cm−1) compared with AcGFP,4,36 showed no fluorescence enhancement or modulation upon secondary laser co-illumination (Fig. S1), underscoring the importance of the chromophore environment to achieve optical modulation.
Figure 1.
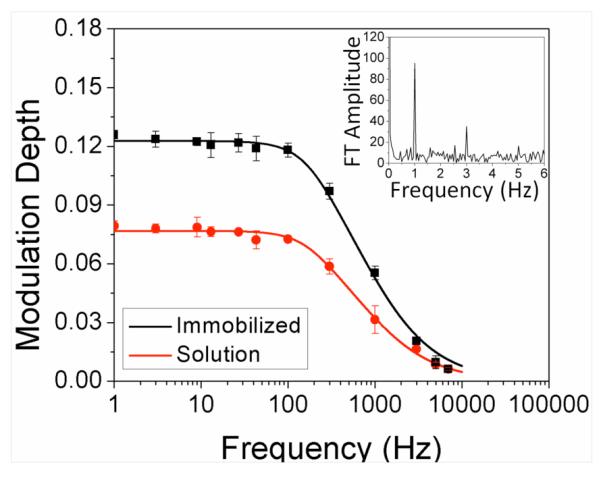
Long-wavelength modulation of AcGFP. Modulation frequency dependence of enhancement for immobilized (black) and diffusing (red) AcGFP molecules. The frequencydependent modulation depth was computed by Fourier transforming each time trace, normalizing by the number of data points, and dividing each by its DC component. Inset: Fourier transform of modulated fluorescence of AcGFP at 1 Hz. Square wave modulation also produces odd harmonics.
Accelerating dark state decay, secondary laser excitation alters relative emissive and dark state populations to establish a new steady-state fluorescence intensity in a time that directly depends on the new rates in and out of the dark state manifold. Analogous to frequency domain lifetimes,38 the buildup and decay of steady state population takes a finite time and results in decreased modulation depths at frequencies higher than the inverse time to establish these populations. The low fractional dark state population achieved with primary illumination alone (~0.1) both limits modulation depth to this same ~10% and requires secondary:primary laser intensity ratios of ~10 for optimal modulation,39 even for equal forward and reverse action cross sections (products of absorption cross section and quantum yields to other manifold of states, Fig. S2). The modulation frequency-dependent fluorescence enhancement (Fig. 1) is well-fit by equation 138
| (1) |
relating modulation depth, m, and modulation frequency, νmod. We define the characteristic frequency as that at which the modulation depth drops to 50% of its original value. Both solution-based and polyacrylamide-immobilized22 AcGFP behaved similarly, with characteristic frequencies of ~760 Hz and ~830 Hz, respectively (Fig. 1). Immobilized proteins show ~twofold higher enhancements relative to those diffusing in solution. These characteristic modulation frequencies directly reflect the dark state lifetime responsible for enhancement, while modulation depth differences result from a combination of residence time in the excitation volume and average excitation rate, thereby leading to increased modulation depth for immobilized species.40
To confirm these characteristic timescales, we measured polyacrylamide gel-immobilized single AcGFP intensity correlations using time-correlated single-photon counting. Autocorrelations of individual molecule fluorescence trajectories revealed average characteristic decays of ~250 μs and ~1.5 ms (Fig. 2). The corresponding on- and off-times41 extracted from these fits are 350 (± 220) μs and 280 (± 15) μs for the faster decay and 24 (± 9) ms and 1.6 (± 0.6) ms for the slower decay, respectively. Secondary laser co-illumination at 561 nm, however, eliminated contrast only at the longer timescale, again indicating that the ~1.5 ms-lived dark state is solely responsible for the observed fluorescence enhancements. Importantly, these millisecond on- and off-times accurately predict our observed enhancement of 8%. As the millisecond lifetime is too long for excited state proton transfer or most triplet state residences, the long-wavelength-modulatable fluorescence in AcGFP most likely involves a photoinduced cis-trans isomerization accompanied by drastic changes of the fluorophore environment. The ability to optically depopulate this long-lived dark state, however, indicates that the trans photoisomer has very different interactions with surrounding residues that confer long wavelength absorption – modifying both ground and excited state energy levels.
Figure 2.
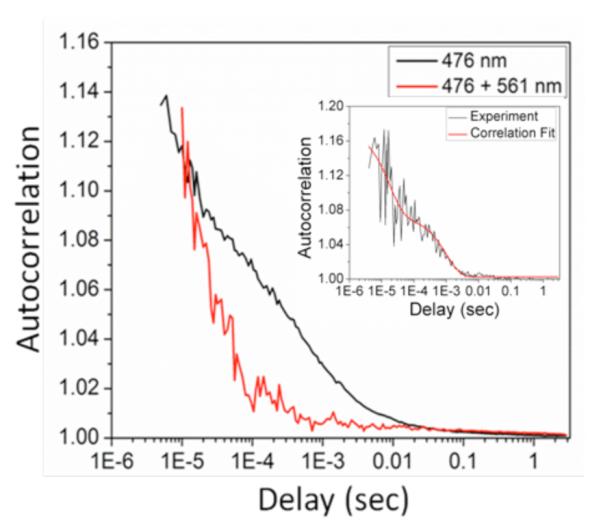
Average single molecule autocorrelations of AcGFP immobilized in polyacrylamide gels with (red) and without (black) secondary illumination at 561 nm. Primary only correlations are averaged over 98 molecules with primary intensity of 2 kW/cm2. Dual laser correlations were averaged over 30 different molecules with the same primary intensity and secondary intensity of 24.3 kW/cm2. Inset: correlation and fit of a typical immobilized single molecule autocorrelation showing fast and slow decays. Only the slow decay (~1.5 ms) is eliminated with secondary illumination. All emitters photobleached in a single step.
Motivated by the ability to optically modulate the AcGFP fluorescence by longer wavelength illumination, we investigated selective recovery of AcGFP fluorescence in the presence of a non-modulatable background, distinguishing its emission from that of autofluorescence and from the similarly emitting EGFP. For this purpose, NIH-3T3 cells were co-transfected with two plasmids, the first encoding AcGFP fused to a mitochondrial targeting peptide (mito-AcGFP), and the second to express EGFP as fluorescent background. Imaging was performed in a stage-scanning confocal geometry utilizing 476 nm primary excitation and spatially overlapped, intensity-modulated secondary excitation at 561 nm. Fluorescence was collected between 500 and 530 nm. To generate cell images, the average fluorescence intensity and lock-in amplitude (intensity of the modulated fluorescence component) were recorded at each stage position. No emission was observed from secondary laser illumination alone, and background in blank or EGFP-only cells was non-modulatable.
Lock-in-based fluorescence demodulation at each stage position within the AcGFP-expressing cells improves the visibility of fluorescence associated with mitochondria, primarily by suppressing the heterogeneous, unmodulated background. Control mito-AcGFP-only cells exhibit largely mito-AcGFP fluorescence, with other subcellular locations showing only dim residual background, resulting in modest (<10%) contrast improvements upon backgroundremoving demodulation (Fig. S3). When co-expressed with untargeted EGFP, cells show much higher overall emission that obscures much of the mitochondria-associated AcGFP fluorescence (Fig. 3). As only AcGFP fluorescence is modulated by the secondary laser, selective recovery of the AcGFP signal over EGFP was readily achieved through demodulation, revealing the AcGFP-labeled mitochondria (Fig. 3) with a maximum signal-to-background ratio indistinguishable from that of the AcGFP-only control. The most promising gain in signal visibility was observed in areas with low AcGFP presence and hence weak emission signals. In formaldehyde-fixed co-transfected cells, mitochondria were barely visible over background in the raw fluorescence images (Fig. 3A, left), exhibiting an average 1.4:1 ratio. However, in the demodulated image the ratio increased to an average of 18.8:1, yielding a ~13-fold improved signal visibility (Fig. 3A, right). Similarly, in live co-transfected cells, the raw fluorescence ratio of weakly emissive AcGFP signal was 1.2:1 (Fig. 3B, left). Upon demodulation, the average ratio increased to 12.1:1, again yielding a ~10-fold improvement upon demodulation (Fig. 3B, right).
Figure 3.
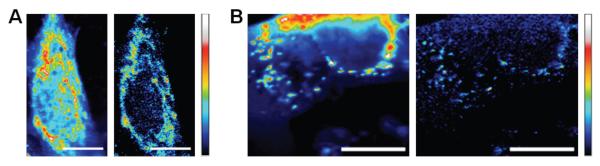
Selective fluorescence recovery of mitochondria-targeted AcGFP in the presence of high EGFP background fluorescence in NIH 3T3 mouse fibroblasts. The primary laser intensity was held at 5.9 kW/cm2 and the secondary intensity (64 kW/cm2) was modulated at 300 Hz. (A) Fixed cells. (Left) Raw fluorescence image of AcGFP-labeled mitochondria and EGFP. Min to max color bar is 0 to 7.5 × 105 photons counts. (Right) Ratio image of lock-in signal to raw fluorescence. (B) Live cells. (Left) Raw fluorescence image of AcGFP-labeled mitochondria and EGFP. Min to max color bar is 0 to 9.6 × 105 photons counts. (Right) Ratio image of lock-in signal to total fluorescence image. The color map is linear from min to max in each image, with no threshold applied. Scale bar: 10 μm.
Selective recovery of mitochondrial signal is also seen when both fluorescent proteins, AcGFP and EGFP are targeted to specific locations. By targeting EGFP to the nucleus, the EGFP emission is much higher than that of AcGFP targeted to the mitochondria (Fig. 4). In formaldehyde-fixed co-transfected cells (Fig. 4A), mitochondria exhibited noticeably weaker fluorescence than did the nuclear EGFP in the raw fluorescence images, yielding an average 0.27:1 ratio. However, in the demodulated image the ratio increased to an average of 6.6:1, yielding a ~24-fold improved signal recovery. Similarly, in live co-transfected cells (Fig. 4B), the raw fluorescence ratio of weakly emissive AcGFP signal was 0.25:1. Upon demodulation, the average ratio increased to 5:1, again yielding a ~20-fold improvement upon demodulation. Thus, based on the different dark state residences and selective long-wavelength optical recovery of fluorescence, AcGFP signals are readily distinguished from both autofluorescent background and from the otherwise indistinguishable EGFP emission in fixed and live cells. While point-by-point scanning was employed here, the demodulation technique could be readily combined with other techniques such as total internal reflection microscopy or spinning disk confocal microscopy to further expand the scope and speed of live cell imaging.
Figure 4.
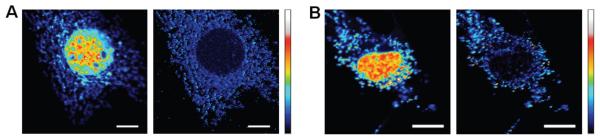
Selective fluorescence recovery of mitochondria-targeted AcGFP from nuclear targeted EGFP fluorescence in NIH 3T3 mouse fibroblasts. The primary laser intensity was held at 5.9 kW/cm2 and the secondary intensity (64 kW/cm2) was modulated at 300 Hz. (A) Fixed cells. (Left) Raw fluorescence image of AcGFP-labeled mitochondria and EGFP. Min to max color bar is 0 to 4.2 × 105 photons counts. (Right) Ratio image of lock-in signal to raw fluorescence. (B) Live cells. (Left) Raw fluorescence image of AcGFP-labeled mitochondria and EGFP. Min to max color bar is 0 to 5.9 × 105 photons counts. (Right) Ratio image of lockin signal to total fluorescence image. The color map is linear from min to max in each image, with no threshold applied. Scale bar: 10 μm.
Expanding SAFIRe to fluorescent proteins offers both greatly improved sensitivity by removing the spatially heterogeneous background typically encountered in biological imaging and the opportunity to further distinguish signals from multiple proteins with strongly overlapping spectra. Because secondary illumination is lower in energy than the collected fluorescence, no additional background is introduced, and heterogeneous cellular fluorescence and obscuring fluorophore signals are readily removed to yield order-of-magnitude increased signal visibility in fixed and live cells. The already-demonstrated 10- to 20-fold sensitivity improvement provides motivation for even more dramatic gains as proteins are bred for modulation depth and characteristic frequency, thereby greatly expanding signal recovery applications in biological and medical imaging. Additionally, long-wavelength modulation expands the dimensionality to discriminate FP emitters based on dark state lifetimes. Once fully mechanistically understood and optimized, this would allow for similarly emitting modulatable proteins to be selectively modulated by varying the modulation frequency, thereby inherently offering an additional dimension for multispectral imaging.
EXPERIMENTAL METHODS
Protein Preparation
DH5αPRO cells containing the gene encoding AcGFP fused with His6-tag (a gift from Prof. A. Bommarius, Georgia Institute of Technology) were grown at 37°C in the presence of chloramphenicol (20 mg/L final concentration) to an optical density at 600 nm of 0.5–0.6. Protein expression was induced with anhydrotetracycline (ATC) (1mg/L final concentration). The cells were further grown for 15 hours at 37°C before harvesting. The cell pellet was resuspended in lysis buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 10 mM imidazole) and lysed by sonication. AcGFP was purified on a Co2+-based IMAC column (Clontech). Fractions containing pure protein were confirmed by Laemmli-SDS-PAGE and stored at −80°C. Protein concentrations were quantified using Bradford protein assays with BSA as a standard (Pierce). The final protein concentration was 3.6 μg/μL. Samples were diluted in phosphate-buffered saline (PBS) pH 7.4 (Invitrogen).
Cell Culture
NIH 3T3 mouse fibroblast cells were cultured at 37°C (5% CO2) in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% bovine calf serum, 4 mM L-glutamine, 200 units/mL penicillin, and 200 μg/mL streptomycin. Plasmids pAcGFP-mito (Clontech), pEGFP-C2 (Clontech), and H2B-GFP (Addgene) were transiently transfected with Turbofect (Fermentas) into NIH 3T3 cells grown on glass cover slips. After a recovery period of 24 h, the cells were fixed with 3.7% formaldehyde and mounted on slides with ProLong Gold antifade reagent (Invitrogen). For live cell experiments, NIH 3T3 cells were cultured in glassbottom dishes and the growth medium was replaced with phenol red-free DMEM containing 25 mM HEPES buffer and 5% fetal calf serum (Invitrogen) prior to imaging.
Fluorescence Imaging
Microscopy was performed on an inverted microscope (Olympus IX71) using a 60x water-immersion objective (Olympus 1.2 NA). All solution data were taken by focusing 30μm into solution. The signal was collected in a confocal arrangement with a 100-μm multimode fiber serving as the pinhole and directing the emission to a photon counting avalanche photodiode (APD, Perkin-Elmer). Intensity trajectories were recorded using a Becker-Hickl (SPC 630) or TimeHarp 100 (Picoquant) photon counting module. Continuous wave primary excitation was used near the excitation maximum of AcGFP (476 nm) using a linetunable Ar+ laser (Coherent). A 561 nm solid-state laser (Coherent, Compass) was used as secondary excitation. Appropriate band pass filters centered near the emission wavelength of AcGFP were used to efficiently block both primary and secondary laser excitation. For dual-laser excitation experiments, lasers were overlapped using a dichroic mirror prior to entering the microscope. Modulation of the secondary laser was performed with an electro-optical modulator (EOM, ConOptics, Model 350-210) with square wave input.
For photobleaching experiments, AcGFP was immobilized in polyacrylamide and illuminated at either 476 nm or co-illuminated with 476 nm and 561 nm. Fluorescence intensity vs. time was recorded for multiple spots; each was fit to a biexponential decay.
For cellular imaging, a piezo stage-scanning (Mad City Labs) confocal arrangement was used with 200-nm or 300-nm step sizes. Fluorescence was collected through a 50-μm optical fiber that was approximately equally split onto two separate APDs. The output of one APD was directed to a photon counting module (National Instruments) for recording raw fluorescence. The other APD output was directed to a digital lock-in amplifier (Stanford Research Systems). The lock-in amplifier was synchronized to the function generator controlling the EOM. The secondary laser was modulated at 300 Hz and the lock-in time constant was 30 ms. The amplitude of the lock-in signal and raw fluorescence were simultaneously recorded at each stage position in LabView. The dwell time at each pixel was 50 ms. For live cell imaging, the cells were placed on a heated stage (Bioptechs Stable “Z” Specimen Warmer) so that cells in the imaging area were held at 37°C. Images had constant background subtracted from each pixel and then intensity values were set linearly from the minimum to the maximum recorded values. A false-color LUT “Royal” (ImageJ) was applied. For ratio images, individually normalized demodulated image and fluorescence images were used with the resulting ratio image being presented with the LUT “Royal”.
Data Analysis
Modulated sample time traces were binned at a rate at least 2.2 times faster than the highest modulation frequency used. Fourier transformation of each time trace reveals the modulation frequency amplitude. The modulation frequency amplitude is divided by the DC amplitude and multiplied by two to determine the enhancement (accounting for the equal amplitude peaks at positive and negative modulation frequency). Reported average fluorescence ratios were calculated from the average fluorescence intensity of mitochondria signals and dividing by the average fluorescence intensity within the nucleus. The same pixel locations were then used to calculate demodulated ratios. Ratio images (Figs. 3 & 4) are simply the normalized raw fluorescence image divided by the normalized demodulated image.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge financial support from NIH-R21EB009976 (RMD) and from NIH-R01GM086195 (CJF and RMD). The authors also appreciate the gift of the AcGFP plasmid from A. Bommarius and R. Vegh, as well as Dronpa preparation by Irina Issaeva.
Footnotes
Supporting Information. EGFP controls, AcGFP-mitochondria cells, secondary laser dependence of modulation depth, and demodulated images for cells. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Shaner NC, Lin MZ, Mckeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. Improving the Photostability of Bright Monomeric Orange and Red Fluorescent Proteins. Nat. Meth. 2008;5:545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Remington SJ. Fluorescent Proteins: Maturation, Photochemistry and Photophysics. Curr. Opin. Struct. Biol. 2006;16:714–721. doi: 10.1016/j.sbi.2006.10.001. [DOI] [PubMed] [Google Scholar]
- (3).Patterson GH, Lippincott-Schwartz J. A Photoactivatable GFP for Selective Photolabeling of Proteins and Cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- (4).Zimmer M. Green Fluorescent Protein (GFP): Applications, Structure, and Related Photophysical Behavior. Chem. Rev. 2002;102:759–781. doi: 10.1021/cr010142r. [DOI] [PubMed] [Google Scholar]
- (5).Newman RH, Fosbrink MD, Zhang J. Genetically Encodable Fluorescent Biosensors for Tracking Signaling Dynamics in Living Cells. Chem. Rev. 2011;111:3614–3666. doi: 10.1021/cr100002u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Conyard J, Kondo M, Heisler IA, Jones G, Baldridge A, Tolbert LM, Solntsev KM, Meech SR. Chemically Modulating the Photophysics of the GFP Chromophore. J. Phys. Chem. B. 2011;115:1571–1576. doi: 10.1021/jp111593x. [DOI] [PubMed] [Google Scholar]
- (7).Stoner-Ma D, Jaye AA, Ronayne KL, Nappa J, Meech SR, Tonge PJ. An Alternate Proton Acceptor for Excited-State Proton Transfer in Green Fluorescent Protein: Rewiring GFP. J. Am. Chem. Soc. 2008;130:1227–1235. doi: 10.1021/ja0754507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- (9).Rust MJ, Bates M, Zhuang XW. Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM) Nat. Meth. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hofmann M, Eggeling C, Jakobs S, Hell SW. Breaking the Diffraction Barrier in Fluorescence Microscopy at Low Light Intensities by Using Reversibly Photoswitchable Proteins. Proc. Natl. Acad. Sci. U.S.A. 2005;102:17565–17569. doi: 10.1073/pnas.0506010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dertinger T, Colyer R, Iyer G, Weiss S, Enderlein J. Fast, Background-Free, 3D Super-resolution Optical Fluctuation Imaging (SOFI) Proc. Natl. Acad. Sci. U.S.A. 2009;106:22287–22292. doi: 10.1073/pnas.0907866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dertinger T, Heilemann M, Vogel R, Sauer M, Weiss S. Superresolution Optical Fluctuation Imaging with Organic Dyes. Angew. Chem., Int. Ed. 2010;49:9441–9443. doi: 10.1002/anie.201004138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dedecker P, Mo GCH, Dertinger T, Zhang J. Widely Accessible Method for Superresolution Fluorescence Imaging of Living Systems. Proc. Natl. Acad. Sci. U.S.A. 2012;109:10909–10914. doi: 10.1073/pnas.1204917109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tian Z, Wu W, Wan W, Li ADQ. Single-Chromophore-Based Photoswitchable Nanoparticles Enable Dual-Alternating-Color Fluorescence for Unambiguous Live Cell Imaging. J. Am. Chem. Soc. 2009;131:4245–4252. doi: 10.1021/ja805150g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ringemann C, Schoenle A, Giske A, Von Middendorff C, Hell SW, Eggeling C. Enhancing Fluorescence Brightness: Effect of Reverse Intersystem Crossing Studied by Fluorescence Fluctuation Spectroscopy. ChemPhysChem. 2008;9:612–624. doi: 10.1002/cphc.200700596. [DOI] [PubMed] [Google Scholar]
- (16).Marriott G, Mao S, Sakata T, Ran J, Jackson D. Optical Lock-In Detection Imaging Microscopy for Contrast-Enhanced Imaging in Living Cells. Proc. Natl. Acad. Sci. U.S.A. 2008;105:17789–17794. doi: 10.1073/pnas.0808882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mao S, Benninger R, Yan Y, Petchprayoon C, Jackson D, Easley C, Piston D, Marriott G. Optical Lock-In Detection of FRET Using Synthetic and Genetically Encoded Optical Switches. Biophys. J. 2008;94:4515–4524. doi: 10.1529/biophysj.107.124859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Li ADQ, Zhan C, Hu D, Wan W, Yao J. Photoswitchable Nanoprobes Offer Unlimited Brightness in Frequency-Domain Imaging. J. Am. Chem. Soc. 2011;133:7628–7631. doi: 10.1021/ja1108479. [DOI] [PubMed] [Google Scholar]
- (19).Richards CI, Hsiang J-C, Dickson RM. Synchronously Amplified Fluorescence Image Recovery (SAFIRe) J. Phys. Chem. B. 2010;114:660–665. doi: 10.1021/jp909167j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Richards CI, Hsiang J-C, Khalil AM, Hull NP, Dickson RM. FRET-Enabled Optical Modulation for High Sensitivity Fluorescence Imaging. J. Am. Chem. Soc. 2010;132:6318–6323. doi: 10.1021/ja100175r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Richards CI, Hsiang J-C, Senapati D, Patel S, Yu J, Vosch T, Dickson RM. Optically Modulated Fluorophores for Selective Fluorescence Signal Recovery. J. Am. Chem. Soc. 2009;131:4619–4621. doi: 10.1021/ja809785s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dickson RM, Cubitt AB, Tsien RY, Moerner WE. On/Off Blinking and Switching Behaviour of Single Molecules of Green Fluorescent Protein. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- (23).Yokoe H, Meyer T. Spatial Dynamics of GFP-Tagged Proteins Investigated by Local Fluorescence Enhancement. Nat. Biotechnol. 1996;14:1252–1256. doi: 10.1038/nbt1096-1252. [DOI] [PubMed] [Google Scholar]
- (24).Chattoraj M, King BA, Bublitz GU, Boxer SG. Ultra-Fast Excited State Dynamics in Green Fluorescent Protein: Multiple States and Proton Transfer. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8362–8367. doi: 10.1073/pnas.93.16.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Brejc K, Sixma TK, Kitts PA, Kain SR, Tsien RY, Ormo M, Remington SJ. Structural Basis for Dual Excitation and Photoisomerization of the Aequorea victoria Green Fluorescent Protein. Proc. Natl. Acad. Sci. U.S.A. 1997;94:2306–2311. doi: 10.1073/pnas.94.6.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Giepmans BNG, Adams SR, Ellisman MH, Tsien RY. The Fluorescent Toolbox for Assessing Protein Location and Function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- (27).Dean KM, Lubbeck JL, Binder JK, Schwall LR, Jimenez R, Palmer AE. Analysis of Red-Fluorescent Proteins Provides Insight into Dark-State Conversion and Photodegradation. Biophys. J. 2011;101:961–969. doi: 10.1016/j.bpj.2011.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dong J, Solntsev KM, Tolbert LM. Solvatochromism of the Green Fluorescence Protein Chromophore and Its Derivatives. J. Am. Chem. Soc. 2006;128:12038–12039. doi: 10.1021/ja063128a. [DOI] [PubMed] [Google Scholar]
- (29).Stavrov SS, Solntsev KM, Tolbert LM, Huppert D. Probing the Decay Coordinate of the Green Fluorescent Protein: Arrest of Cis-Trans Isomerization by the Protein Significantly Narrows the Fluorescence Spectra. J. Am. Chem. Soc. 2006;128:1540–1546. doi: 10.1021/ja0555421. [DOI] [PubMed] [Google Scholar]
- (30).Usman A, Mohammed OF, Nibbering ETJ, Dong J, Solntsev KM, Tolbert LM. Excited-State Structure Determination of the Green Fluorescent Protein Chromophore. J. Am. Chem. Soc. 2005;127:11214–11215. doi: 10.1021/ja0520520. [DOI] [PubMed] [Google Scholar]
- (31).Quercioli V, Bosisio C, Daglio SC, Rocca F, D’Alfonso L, Collini M, Baldini G, Chirico G, Bettati S, Raboni S, Campanini B. Photoinduced Millisecond Switching Kinetics in the GFPMut2 E222Q Mutant. J. Phys. Chem. B. 2010;114:4664–4677. doi: 10.1021/jp910075b. [DOI] [PubMed] [Google Scholar]
- (32).Habuchi S, Ando R, Dedecker P, Verheijen W, Mizuno H, Hofkens J. Reversible Single-Molecule Photoswitching in the GFP-like Fluorescent Protein Dronpa. Proc. Natl. Acad. Sci. U.S.A. 2005;102:9511–9516. doi: 10.1073/pnas.0500489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Habuchi S, Dedecker P, Hotta J.-i., Flors C, Ando R, Mizuno H, Miyawaki A, Hofkens J. Photo-Induced Protonation/Deprotonation in the GFP-like Fluorescent Protein Dronpa: Mechanism Responsible for the Reversible Photoswitching. Photochem. Photobiol. Sci. 2006;5:567–576. doi: 10.1039/b516339k. [DOI] [PubMed] [Google Scholar]
- (34).Andersen M, Stiel AC, Trowitzsch S, Weber G, Eggeling C, Wahl MC, Hell SW, Jakobs S. Structural Basis for Reversible Photoswitching in Dronpa. Proc. Natl. Acad. Sci. U.S.A. 2007;104:13005–13009. doi: 10.1073/pnas.0700629104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Gurskaya NG, Fradkov AF, Pounkova NI, Staroverov DB, Bulina ME, Yanushevich YG, Labas YA, Lukyanov S, Lukyanov KA. A Colourless Green Fluorescent Protein Homologue from the Non-Fluorescent Hydromedusa Aequorea coerulescens and Its Fluorescent Mutants. Biochem. J. 2003;373:403–408. doi: 10.1042/BJ20021966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Pletneva NV, Pletnev VZ, Lukyanov KA, Gurskaya NG, Goryacheva EA, Martynov VI, Wlodawer A, Dauter Z, Pletnev S. Structural Evidence for a Dehydrated Intermediate in Green Fluorescent Protein Chromophore Biosynthesis. J. Biol. Chem. 2010;285:15978–15984. doi: 10.1074/jbc.M109.092320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Haupts U, Maiti S, Schwille P, Webb WW. Dynamics of Fluorescence Fluctuations in Green Fluorescent Protein Observed by Fluorescence Correlation Spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13573–13578. doi: 10.1073/pnas.95.23.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Spencer R, Weber G. Measurements of Subnanosecond Fluorescence Lifetimes with a Cross-Correlation Phase Fluorometer. Ann. N. Y. Acad. Sci. 1969;158:361–376. [Google Scholar]
- (39).Petty JT, Fan C, Story SP, Sengupta B, Sartin M, Hsiang J-C, Perry JW, Dickson RM. Optically Enhanced, Near-IR, Silver Cluster Emission Altered by Single Base Changes in the DNA Template. J. Phys. Chem. B. 2011 doi: 10.1021/jp202024x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Fan C, Hsiang J-C, Dickson RM. Optical Modulation and Selective Recovery of Cy5 Fluorescence. ChemPhysChem. 2012;13:1023–1029. doi: 10.1002/cphc.201100671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vosch T, Antoku Y, Hsiang J-C, Richards CI, Gonzalez JI, Dickson RM. Strongly Emissive Individual DNA-Encapsulated Ag Nanoclusters as Single-Molecule Fluorophores. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12616–12621. doi: 10.1073/pnas.0610677104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.