

Abstract
Intestinal lymphoid tissues have to simultaneously ensure protection against pathogens and tolerance towards commensals. Despite such vital functions, their development in the colon is poorly understood. Here, we show that the two distinct lymphoid tissues of the colon–colonic patches and colonic SILTs–can easily be distinguished based on anatomical location, developmental timeframe and cellular organization. Furthermore, whereas colonic patch development depended on CXCL13-mediated LTi cell clustering followed by LTα-mediated consolidation, early LTi clustering at SILT anlagen did not require CXCL13, CCR6 or CXCR3. Subsequent dendritic cell recruitment to and gp38+VCAM-1+ lymphoid stromal cell differentiation within SILTs required LTα; B cell recruitment and follicular dendritic cell differentiation depended on MyD88-mediated signalling, but not the microflora. In conclusion, our data demonstrate that different mechanisms, mediated mainly by programmed stimuli, induce the formation of distinct colonic lymphoid tissues, therefore suggesting that these tissues may have different functions.
INTRODUCTION
Secondary lymphoid organs, such as lymph nodes and Peyer’s patches, develop in the sterile environment of the uterus during embryonic life, whereas solitary intestinal lymphoid tissues (SILT) develop early after birth under the continuous exposure to commensals as well as potential pathogens1. In general, lymphoid tissue development depends on the interaction between hematopoietic-derived lymphoid tissue inducer (LTi) cells and stromal lymphoid tissue organizer (LTo) cells and the engagement of the lymphotoxin (LT) signalling pathway1–3. LTi cells express membrane bound LTα1β2, which engages LTβR on stromal organizers, inducing them to synthesize chemokines, adhesion molecules, growth factors and survival signals that further attract and retain LTi cells1–3. Lack of LTβR triggering, as observed in LTβR-, LTα- or LTβ-deficient mice, results in the absence of most lymphoid tissues4–8. Exceptions to this general model are found in the formation of mucosal-associated lymphoid tissue (MALT), which can develop independently of either nuclear retinoic acid-receptor related orphan receptor (RORγ)-dependent LTi cells, helix-loop-helix protein inhibitor of DNA binding 2 (Id2)-dependent LTi cells and/or LT signalling (reviewed in 3). Importantly, in most lymphoid tissues, the initial clustering of LTi cells occurs independently of LT signalling9–13. Indeed, we have recently shown that such clustering, at peripheral lymph node anlagen, is dependent on the retinoic acid-mediated release of the homeostatic chemokine CXCL1314.
Immune cell function and lymphoid tissue development in the intestine, where opposing needs for host defence and nutrient uptake collide, must be tightly regulated. The lymphoid tissues in the small intestine that are available for these functions include Peyer’s patches and SILTs. Peyer’s patches develop during embryogenesis1. Hematopoietic cells start colonizing the developing gut around day E12.515, 16. Among these cells, receptor tyrosine kinase RET-expressing CD45+IL7Rα−CD4−CD3−CD11c+ lymphoid tissue initiator (LTin) cells, which have a key role in Peyer’s patch development, encounter RET ligands and start clustering at discrete locations in the small intestine, leading to stromal cell activation and LTi cell recruitment16.
In contrast to Peyer’s patches, SILT development in the small intestine is strictly post-natal. SILTs, which consist of dynamic lymphoid clusters ranging from small aggregates of lineage-negative cells known as cryptopatches to large clusters rich in B cells known as isolated lymphoid follicles, develop in the intestine’s lamina propria within the first 2 weeks after birth17, 18. The development of these structures depends on LTi cells and LT signalling, as they fail to form both in RORγ-deficient mice19, which lack LTi cells11, as well as in LTα−/−and LTβR−/− mice20, 21. Importantly, SILT development within the small intestine is also dependent on the CXCL13-CXCR5 axis, as young (4 weeks-old) CXCR5-deficient mice completely lack SILTs22. However, such dependency is not absolute as adult (8–10 weeks-old) CXCR5−/− mice develop aberrant SILTs22. Enhanced stimulation by the enteric microbiota, which is likely to increase with age in CXCR5−/− mice due to defective immune responses22, may compensate for the lack of CXCR5 signalling and contribute to lymphoid tissue formation in these mice. In this regard, it should be noted that the homeostatic transition from immature to mature SILTs (cryptopatch to mILF), which is characterized by the enlargement of these structures and the development of organized B cell follicles, is mediated by the microflora present in the intestinal lumen23, 24. Indeed, the recruitment of B cells, which initiates this transition, was shown to be dependent on the CCR6 ligands CCL20 and β-defensin3, which are induced upon the recognition of commensal gram-negative bacteria by the innate receptor NOD-1; and the subsequent organization of the infiltrating B cells into B cell follicles to be further dependent on Toll-like receptors (TLR) and MyD8823.
Lymphoid tissue development in the colon has not been comprehensively addressed so far. As colon and small intestine have considerable differences, discrepancies in lymphoid tissue development may exist. Of particular interest, the microflora, which was shown to influence the formation of lymphoid tissues in the small intestine, is of a different composition in the colon25. Even within the colon itself, the microbiota composition was shown to differ between the proximal and distal parts and to be responsible for regional differences in the expression levels of innate immunity related molecules26. Here we have analyzed colonic lymphoid tissue development during ontogeny. We show that colonic patches and colonic SILTs are distinct tissues, based on their differential 1) anatomical location, 2) cellular organization, 3) developmental time frame and 4) developmental molecular requirements. In addition, we report considerable differences in SILT development in the colon as compared to the small intestine. Altogether, the data presented here highlight the multitude of pathways that are involved in lymphoid tissue development.
RESULTS
Two distinct lymphoid tissues, with distinct developmental kinetics, are present in the colon
In this study, we aimed to characterize the development of lymphoid tissue within the murine colon. Analysis of adult colons confirmed the presence of two distinct lymphoid tissues in the steady-state, which were easily distinguished based on their relative position within the colon and the presence of organized T cell areas. Colonic patches composed of large B cell follicles (on average, two follicles per colonic patch) and separate T cell areas were present within the submucosa of the colon between the two external (longitudinal and circular) muscular layers and the muscularis mucosae, which were delineated with α-smooth muscle actin (αSMA) staining (Fig. 1a and 1b, arrowhead). Their number averaged 3.4 ± 0.9 per colon (mean ± standard deviation; n=14, range 2–5). In addition, numerous small B cell clusters, containing only a few T cells, could be detected in the lamina propria, i.e. from the muscularis mucosae towards the lumen of the colon (Fig. 1a and 1b, arrow). Importantly, these latter structures, which localized predominantly in the distal colon (fig. S1a), strongly resembled the SILTs of the small intestine. Indeed, similar to the reported stages of SILT maturation in the small intestine22, 24, colonic SILTs could be found as a mere collection of CD4+ LTi cells or containing additionally, different degrees of B cell accumulation and organization (Figs. S1b and S1c).
Figure 1. Two distinct lymphoid tissues, with distinct developmental kinetics, are present in the colon.

a,bStitched pictures of adult colons stained for B220 (green) and CD3 (red) (a) and B220 (green) and αSMA (red) (b) depicting a large colonic patch (left immune cell cluster, arrowhead) and a SILT (right immune cell cluster, arrow). Scale bar 250μm. c,d Immunofluorescence characterization of the lymphoid tissues found in wild-type colons at days 0 (c left panels), 7 (c middle panels) and 14 (c right panels; d) post-partum. In c, colonic patches were stained for (I) CD4 (green), αSMA (red) and MAdCAM-1 (blue), (II) CD4 (green), CD3 (red), and RORγ (blue) (insert is a higher magnification of LTi cells expressing RORγ) and (III) CD4 (green), B220 (red), and Lyve-1 (blue). Scale bar 100μm. In d, SILTs were stained for (I) CD4 (green), αSMA (red), and MAdCAM-1 (blue) and (II) CD4 (green), CD11c (red), and B220 (blue). Scale bar 50μm. At least 3 colons per time point were entirely sectioned and the detected lymphoid tissues further analyzed.
Highlighting the different origin of colonic patches and colonic SILTs, we found their development to occur during different timeframes. Development of colonic patches started in embryonic life (Fig. S1d), but was completed only after birth. At day 0 (day of birth), colonic patch anlagen could be observed as clusters of CD3−CD4+RORγ+ LTi cells in the submucosa of the colon (Fig. 1c). With age, these early clusters were progressively infiltrated by B and T lymphocytes, which were initially randomly distributed but by day 14 were organized into separate B and T cell areas (Fig. 1c). Of note, colonic patch development led to the disruption of the muscularis mucosae (Fig. 1c), thereby allowing contact of the colonic patch tissue with the colonic epithelium. In marked contrast, SILT development was entirely post-natal. By days 0 and 7, no clusters of LTi cells could be found in the lamina propria of the colon. Only by day 14, the full spectrum of SILTs, i.e. immature clusters of LTi cells as well as mature clusters with their single well defined B cell follicle and stages in-between, could be detected (Fig. 1d).
The cellular organization within colonic patches and colonic SILTs is distinct
The cellular organization of colonic patches and colonic SILTs also diverged significantly (Fig. 2). Definitive colonic patches had separate B and T cell areas, which contained CD35+ follicular dendritic cells and CD11c+ dendritic cells, respectively (Fig. 2a I–III, V). CD3−CD4+RORγ+ LTi cells persisted within the “mature” colonic patch (Fig. 2a II and data not shown). Importantly, colonic patches also contained CD31+ vessels (Fig. 2a II); a fraction of which expressed the adhesion molecule MAdCAM-1 and showed the typical appearance of high endothelial venules (HEVs) (Fig. 2a IV and data not shown). In contrast, mature SILTs had a single central B cell follicle, which contained CD35+ follicular dendritic cells, and only sparsely distributed T cells that did not form T cell areas (Fig. 2b I–III, V). Indeed, colonic SILTs contained very few CD3+ T cells (Fig. 2b II). The majority of the CD4+ cells present within SILTs were CD3−RORγ+IL7Rα+ (phenotype which classifies them as LTi cells) and colocalized with CD11c+ dendritic cells in a “ring-like” pattern enclosing the SILT (Fig. 2b II, V). Importantly, CD31+ vessels within the SILT usually did not express MAdCAM-1 (Fig. 2b IV) and lacked the appearance of HEVs (data not shown), suggesting that lymphocytes do not leave the circulation directly towards the SILT but rather towards the lamina propria of the colon, which contains flat MAdCAM-1+ vessels (27 and data not shown). Both colonic patches as well as SILTs contained gp38+VCAM-1+ stromal cells (Figs. 2a VI and 2b VI), which were absent in non-lymphoid tissue-associated regions of the colon (data not shown).
Figure 2. Colonic Patches and colonic SILTs are differently organized.

Histological characterization of colonic patches (a) and SILTs (b) present in the colon of 14 days-old wild-type mice. Serial sections of the colon were stained for: (I) CD4 (green), αSMA (red), and B220 (blue); (II) CD4 (green), CD31 (red), and CD3 (blue); (III) CD4 (green), CD35 (red), and B220 (blue); (IV) CD4 (green), MadCAM-1 (red), and Lyve-1 (blue); (V) CD4 (green), IL7Rα (red), and CD11c (blue); and (VI) CD4 (green), VCAM-1 (red), and gp38 (blue). Scale bars (c) 100μm and (d) 50μm. At least 4 colons were entirely sectioned and the detected lymphoid tissues further analyzed.
Early LTi cell attraction to colonic patch and SILT anlagen is lymphotoxin-independent
Having established clear differences between colonic patches and SILTs, we subsequently addressed whether the molecular requirements for their development also differed. Definitive formation of secondary lymphoid tissues depends on the engagement of the LTα1β2-LTβR signalling pathway1–3. To test whether this pathway was involved in the development of colonic lymphoid tissues, we analyzed the colons of LTα−/− mice in the first two weeks of age. At day 0, clusters of CD3−CD4+RORγ+ LTi cells could be observed in the submucosa of the colon (Fig. 3a). Importantly, these colonic patch anlagen were smaller than the ones observed in wild-type mice at the same age (Fig. 1c) and seemed to disintegrate with age, so that, by day 14, colonic patches could no longer be found. This was likely due to impaired differentiation of gp38+VCAM-1+ stromal cells (Fig. 3a III), which may have limited the accumulation of LTi cells at the tissue anlagen. In support of such hypothesis, it is noteworthy that, in some LTα−/− mice, LTi cells accumulated at the distal end of the colon (Fig. S2a).
Figure 3. Lymphotoxin and CXCL13 are both required for colonic patch development, but only lymphotoxin, and not CXCL13, is required for colonic SILT formation.

a Immunofluorescence characterization of colonic patch anlagen present in LTα−/− mice at day 0 post-partum, stained for (I) CD4 (green), αSMA (red), and MAdCAM-1 (blue), (II) CD4 (green), CD3 (red), and RORγ (blue) (insert is a higher magnification of LTi cells expressing RORγ), and (III) CD4 (green), VCAM-1 (red), and gp38 (blue). b,c Immunoflourescence characterization of lamina propria lymphoid aggregates present in the colon of LTα−/− (b) and CXCL13−/− (c) mice at day 14 post-partum, stained for (I) CD4 (green), αSMA (red), and MAdCAM-1 (blue), (II) CD4 (green), CD11c (red), and B220 (blue), and (III) CD4 (green), VCAM-1 (red), and gp38 (blue). Scale bars 50μm. At least 3 colons per group were entirely sectioned and the detected lymphoid tissues further analyzed.
Regarding SILTs, at day 14, only small clusters of CD3−CD4+RORγ+ LTi cells were observed in the lamina propria of the colon of LTα−/− mice (Fig. 3b and data not shown). Importantly, these clusters were neither associated with gp38+VCAM-1+ stromal cells nor did they recruit CD11c+ dendritic cells or B220+ B cells, and therefore could not be considered bona fide SILTs (Fig. 3b II, III). Collectively these data show that the LT pathway is required for the retention of LTi cells and with that for the definitive formation of colonic patches and SILTs. However, in both cases, i.e. in both colonic patch as well as SILT organogenesis, it is not required for the initiation phase of lymphoid organ development, namely the early attraction and clustering of LTi cells.
CXCL13 is necessary for the clustering of LTi cells at colonic patch anlagen, but dispensable for their clustering at SILT locations
Given that early LTi cell clustering at lymph node anlagen also occurs independently of LT signalling9–14, in a manner recently reported to be dependent on retinoic acid-induced CXCL13 expression14, we reasoned that lymphoid tissue development within the colon could also be dependent on CXCL13. Confirming this hypothesis, we found no colonic patch anlagen, typified by clusters of LTi cells, in CXCL13−/− mice at day 0. Indeed, although CD45+ hematopoietic cells were clearly observed in the colon (Fig. S2b), indicating that colon infiltration by immune cells is not defective in CXCL13−/− mice, immune cell aggregates in the submucosa could not be found at any time point. In marked contrast, however, immune cell aggregates could easily be detected in the lamina propria by days 7 and 14 (data not shown and Fig. 3c). These aggregates were composed of CD3−CD4+RORγ+ LTi cells as well as CD11c+ dendritic cells and B220+ B cells and were associated with gp38+VCAM-1+ stromal cells (Fig. 3c). Importantly, the immune cells within these SILTs failed to organize into separate domains (Figs. 3c and S1c); and such unorganized SILTs developed much earlier and in higher numbers than the organized SILTs of wild-type mice (data not shown and Fig. S1c). Collectively, these data demonstrate that early colonic patch versus colonic SILT development occurs via different molecular pathways; while CXCL13 is absolutely required for colonic patch development, it is dispensable for LTi cell attraction and clustering and subsequent SILT development in the lamina propria of the colon.
Chemokine receptors on colonic LTi cells and chemokine expression in the colon
CXCL13-independent LTi cell attraction and clustering in the lamina propria, suggested that other chemokine(s) could function as the initial inducer of colonic SILT development. To identify this potential chemokine, we assessed chemokine receptor expression on colon-derived LTi cells. Single cell suspensions were obtained from E18.5 wild-type colons and analyzed by flow cytometry. As shown in Fig. 4a, colonic LTi cells, which we identified as CD45intCD4+CD3−CD11c−IL7Rα+ cells, expressed the homeostatic chemokine receptors CXCR4, CXCR5 and CCR7. Importantly, they did not express the chemokine receptor CCR9, which is used by activated B and T cells to infiltrate mucosal tissues such as the gut28, 29, but they did express CCR6 which is used by B cells for the same purpose30.
Figure 4. Chemokine receptor expression on colonic LTi cells and colonic chemokine expression.

a Chemokine receptor expression on CD45intCD4+IL7Rα+CD3−CD11c− cells LTi cells was assessed by flow cytometry. Grey histograms represent FMO controls in which the primary antibody was omitted, while blue histograms represent the actual staining. At least 8 colons were used per analysis and a representative FACS experiment is shown. The experiment was performed 3 times. b Chemokine mRNA expression in whole colons of wild-type, LTα−/− and CXCL13−/− mice of different ages was assessed by real-time PCR. 4–7 colons were analyzed per group. The expression of each transcript in each sample was normalized to the expression of the transcripts for the housekeeping genes Cyclophilin and Ubiquitin. The mean expression levels of each transcript for sample wild-type day 0 were arbitrarily set at 1. Data were compared with one way non-parametric ANOVA and Dunn’s multiple comparison tests. Mean ± SEM is shown. * p<0.05, ** p<0.01 between different age groups within the same mouse genotype; # p<0.05 between different mouse genotypes within the same age group.
To gain further insight into which chemokine(s) might be involved in SILT genesis, we assessed chemokine expression by PCR. Chemokine mRNA levels were measured in whole colon samples obtained at days 0, 7 and 14 from wild-type, LTα−/− and CXCL13−/− mice. As seen in Fig. 4b, we observed no differences in CXCL12, CCL19 and CCL21 mRNA expression levels between the different genotypes. In contrast, although we had already established it not to be involved in early immune cell clustering in colonic SILTs, we found CXCL13 mRNA expression to be reduced in LTα−/− mice as compared to wild-type mice (CXCL13 mRNA was not detected in CXCL13−/− mice). Of interest, we found CCL20 transcript expression to be reduced in LTα−/− mice as compared to wild-type and CXCL13−/−mice at a time point in which SILTs are fully developed, i.e. day 14 post-partum.
CCR6 is neither required for colonic patch nor colonic SILT development
As B cell recruitment into SILTs in the small intestine was shown to be dependent on a functional CCR6-CCL20 axis23, 30 and LTα−/− mice, which failed to develop colonic lymphoid tissues, had reduced colonic CCL20 expression (Fig. 4b), we hypothesized that this axis would also play a role in lymphoid tissue development in the colon. To test this hypothesis, we analyzed the colon of CCR6−/− mice for the presence of lymphoid tissues. In contrast to the small intestine23, 30, lymphoid tissue formation in the colon of these animals progressed normally when analyzed at day 14 (Fig. 5). CCR6-deficient colonic patches had B cell domains containing CD35+ follicular dendritic cells (Figs. 5a I and III); T cell domains containing CD11c+ dendritic cells (Figs. 5a II and V); CD3−CD4+ LTi cells which persisted at the border of B and T cell domains (Fig. 5a II); and MAdCAM-1+ HEVs dispersed within a network of gp38+VCAM-1+ stromal cells (Fig. 5a II IV and VI).
Figure 5. Colonic Patch and colonic SILT development is independent of CCR6.

Histological characterization of the colonic patches (a) and SILTs (b) present in the colon of 14 days-old CCR6−/− mice. Colon’s serial sections were stained for: (I) CD4 (green), αSMA (red), and B220 (blue); (II) CD4 (green), CD31 (red), and CD3 (blue); (III) CD4 (green), CD35 (red), and B220 (blue); (IV) CD4 (green), MadCAM-1 (red), and Lyve-1 (blue); (V) CD4 (green), IL7Rα (red), and CD11c (blue); and (VI) CD4 (green), VCAM-1 (red), and gp38 (blue). Scale bars (c) 100μm and (d) 50μm. At least 3 colons were entirely sectioned and the detected lymphoid tissues further analyzed.
CCR6-deficient SILTs had well defined B cell follicles containing CD35+ follicular dendritic cell networks (Figs. 5b I and III). The few CD3+ T cells present within SILTs were sparsely distributed among their parenchyma of gp38+VCAM-1+ stromal cells (Figs. 5b II and VI). CD11c+ dendritic cells accumulated, in a ring-like pattern, at the periphery (Fig. 5b V). Similarly, vessels expressing MAdCAM-1 were normally found at the periphery of SILTs rather than within them (Fig. 5b IV). Collectively, these data showed that, in contrast to the small intestine, the CCL20-CCR6 axis is neither required for colonic SILT nor colonic patch development.
CXCR3-deficient SILTs develop in different locations
LTi cell responsiveness to CXCR3 ligands is contextual, as they do not migrate towards CXCL10 between embryonic days E12.5 and E14.514, but readily do so soon after birth (Fig. S3a). This particularity suggested that CXCR3 ligands could be involved in the post-natal development of lymphoid tissues. Supporting this hypothesis, CXCL10 mRNA levels increased significantly with age in wild-type mice, but failed to do so in LTα−/− and CXCL13−/−mice (Fig. S3b). Therefore, to determine whether CXCR3 had a role in the development of colonic lymphoid tissues, we analyzed CXCR3-deficient colons. Both colonic patches and colonic SILTs developed in these mice (Fig.6). Colonic patches had separate B and T cell areas, containing CD35+ follicular dendritic cells and CD11c+ dendritic cells, respectively; MAdCAM-1+ HEVs and gp38+VCAM-1+ stromal cells were also present (Fig. 6a). CXCR3-deficient SILTs also seemed to develop normally, with mature SILTs having well defined B cell follicles that contained CD35+ follicular dendritic cells (Fig. 6b). Furthermore, they contained sparsely distributed CD3+ T cells; CD11c+ dendritic cells that colocalized with CD3−CD4+ LTi cells; and gp38+VCAM-1+ stromal cells (Fig. 6b). Significantly, a large fraction of CXCR3-deficient SILTs also included MAdCAM-1+ vessels within their parenchyma (Figs. 6b IV and S3c). This feature contrasted with the organization of SILTs in wild-type mice, in which MAdCAM-1+ vessels were usually not present within the SILT itself but rather at its periphery (Figs. 2b and S3c). Importantly, the number and maturation status of SILTs did not differ between wild-type and CXCR3−/− mice (Fig. S1c) and MAdCAM-1+ vessels were found equally represented among the parenchyma of the different SILT maturation stages in CXCR3−/− mice (Fig. S3c). These data suggested that, while LTi cell clustering in the lamina propira of the colon may be altered in the absence of CXCR3-guided migration, colonic SILT development upon LTi clustering proceded undisturbed. Altogether, although CXCR3 seemed dispensable for colonic patch development, during colonic SILT development CXCR3 appeared to be involved in defining the exact location of SILT formation within the lamina propria.
Figure 6. CXCR3-guided LTi migration influences SILT location within the colon’s lamina propria.

Histological characterization of the colonic patches (a) and SILTs (b) present in the colon of 14 days-old CXCR3−/− mice. Serial sections of the colon were stained for: (I) CD4 (green), αSMA (red), and B220 (blue); (II) CD4 (green), CD31 (red), and CD3 (blue); (III) CD4 (green), CD35 (red), and B220 (blue); (IV) CD4 (green), MadCAM-1 (red), and Lyve-1 (blue); (V) CD4 (green), IL7Rα (red), and CD11c (blue); and (VI) CD4 (green), VCAM-1 (red), and gp38 (blue). Scale bars (c) 100μm and (d) 50μm. At least 3 colons were entirely sectioned and the detected lymphoid tissues further analyzed.
SILT maturation is dependent on non-microbial induced MyD88 signalling
Since lymphoid tissue development in the colon was largely post-natal and thus, occurred simultaneously with the establishment of the gastro-intestinal microbiota, we hypothesized that its development could be influenced by the recognition of the colonizing bacteria. Therefore, to test the role of bacterial colonization in colonic lymphoid tissue development, we analyzed the lymphoid tissues present within the colons of germ-free mice. We found that, in these mice, colonic patches developed normally (data not shown). In contrast, SILT development seemed to be moderately influenced by the presence of commensal bacteria, as a larger proportion of the SILTs was found to be immature in germ-free mice as compared to animals maintained in SPF conditions (fig. 7a). Importantly, the total number of colonic SILTs did not differ between germ-free and SPF mice (fig. 7a), suggesting that LTi cell clustering in the lamina propria of the colon progresses independently of the intestinal micorbiota. In these particular studies, we used C3H mice, which we found to have many more colonic lymphoid tissues as compared to C57BL/B6 mice (14.75 ± 1.5 vs. 3.6 ± 1.1 colonic patches in C3H (n=4) and B6 (n=7) animals, respectively; and figs. 7a vs. 7b). Therefore, to rule out the possibility that our data were being affected by the genetic background of the mice, we decided to verify them by analyzing C57BL/B6 mice in which bacterial colonization of the intestine had been prevented by treatment with a cocktail of antibiotics since embryonic day E14 until sacrifice at 2 weeks post-partum. This protocol clearly reduced the amount of bacteria present in the colon (fig. S4). However, to our surprise it influenced neither the number nor the maturation status of colonic SILTs (fig. 7b). As bacterial-derived products are recognized by pattern-recognition receptors, such as Toll-like receptors (TLR), which signal mainly via the adaptor molecule MyD88, we decided to further confirm our data by analyzing the development of SILTs in the colon of MyD88−/− mice. In these mice, colonic patches developed normally (fig. S5a and data not shown). SILT development, however, seemed arrested at an immature stage (Figs. 7b and S5b). MyD88-deficient SILTs were smaller and had fewer B cells as compared to wild-type SILTs (Figs. 7b and S5b); and, likely as a consequence of defective B cell recruitment, had reduced CD35+ follicular dendritic cell networks (fig. S5b). Altogether, our data suggest that in contrast to the small intestine, bacterial recognition in the colon does not play a major role in SILT maturation. Furthermore, they imply that MyD88-dependent B cell recruitment and SILT maturation are not a consequence of bacterial recognition via TLRs.
Figure 7. SILT maturation is microflora-independent, but MyD88-dependent.
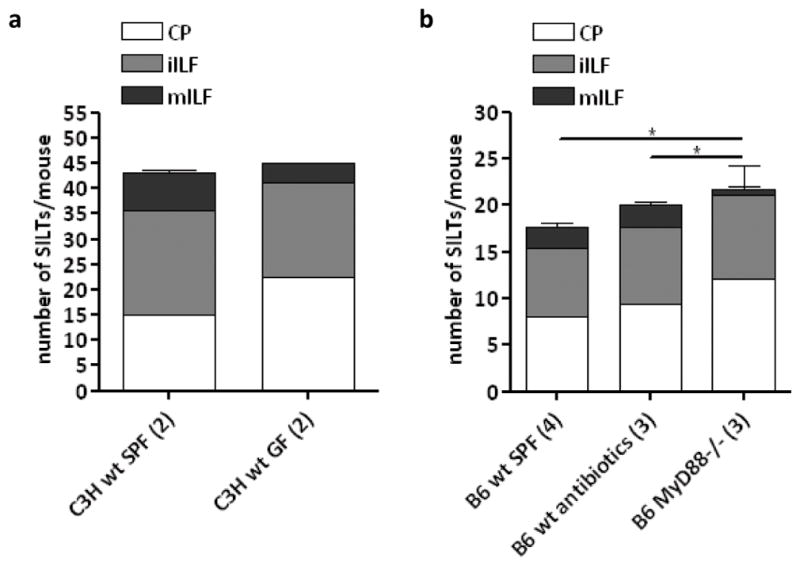
Total number and maturation status of SILTs in C3H mice maintained under specific-pathogen free or germ-free conditions (a) and wild-type C57BL/B6 mice maintained under SPF conditions treated or not with antibiotics since embryonic day 14 until analysis at day 14 post-partum and MyD88−/− mice (b). The number of animals analyzed is depicted between parentheses in front of the respective group. Data were compared with one way non-parametric ANOVA and Dunn’s multiple comparison tests. * p<0.05 between the numbers of mILF in the different mice.
DISCUSSION
In this study, we analyzed the development of lymphoid tissues within the murine colon. We showed that the steady-state colon contains two distinct lymphoid tissues: colonic patches, that are embryonically programmed, and SILTs, that develop postnatally and depend on the adaptor molecule MyD88, but not microbial colonization, for their full maturation.
Colonic patches were located in the submucosa, between the external longitudinal and circular muscular layers and the muscularis mucosae. The muscularis mucosae was disrupted at sites of colonic patches allowing for contact of the colonic patch tissue with the colonic epithelial layer, which should permit the sampling of colon’s luminal contents by the antigen-presenting cells present within the colonic patch. In contrast, SILTs were positioned in the lamina propria of the colon, i.e. from the muscularis mucosae towards the lumen, directly contacting the intestinal epithelium.
Colonic patches were composed of B and T cells, segregated into clearly distinct compartments, and LTi cells that persisted at the border of these compartments. B cell follicles contained CD35+ follicular dendritic cells. T cell areas had CD11c+ conventional dendritic cells and MAdCAM-1+ HEVs. Non-hematopoietic gp38+VCAM-1+ stromal cells were present throughout the colonic patch parenchyma. In contrast to the uniform and highly structured appearance of colonic patches, colonic SILTs appeared in different developmental/maturation stages. In the same animal, they could range from simple collections of CD4+IL7Rα+ LTi and CD4−IL7Rα+ pre-LTi cells surrounded by CD11c+ dendritic cells up to clusters containing a single well organized B cell follicle and CD35+ follicular dendritic cells. gp38+VCAM-1+ stromal cells were present throughout SILTs. In contrast to colonic patches, MAdCAM-1+ vessels were usually not seen within SILTs, suggesting that immune cell migration into these tissues does not occur directly from the circulation but rather through the lamina propria.
The programmed development of colonic patches started during embryonic life, as LTi cells and/or their precursors associated with MAdCAM-1+ stromal organizer cells in the colonic submucosa. Similarly as to the formation of other secondary lymphoid organs in mice1–3, such programmed development ceased only after birth, when infiltration of the colonic patch anlagen by B and T cells occurred. Importantly, B and T cells were initially randomly distributed within the colonic patch but subsequently segregated into separate compartments. This contrasts with Peyer’s patch development in the small intestine in which, upon recruitment, B and T lymphocytes promptly segregate into their respective domains31 and therefore suggests that Peyer’s patch and colonic patch development is differentially regulated.
Developmental regulation of colonic patches mirrored the molecular cues used during lymph node development (reviewed in 1–3). LTi cells clustered in the submucosa of the colon in a CXCL13-dependent, LTα-independent manner. Whether early expression of CXCL13 at the colonic patch anlagen is induced by retinoic acid, as shown for lymph node development14, warrants further investigation. In the colonic patch anlagen, LTi cells likely interacted with and supported the differentiation of gp38+VCAM-1+ stromal cells via the engagement of the LT signalling pathway. Importantly, sustained colonic expression of the chemokines CXCL13, CCL20 and CXCL10 was controlled by the LT pathway (our data and 32). However, and in contrast with CXCL13, these latter two chemokines were not essential for colonic patch development.
SILT development, in contrast to colonic patch development, was entirely post-natal. By day 14, the complete developmental/maturation spectrum of colonic SILTs was present and included both small as well as large clusters of immune cells. Despite common features, such as dependence on the LT pathway23, 33, 34, the process of SILT formation differed substantially from that of colonic patches. CXCL13 was not essential for colonic SILT development as numerous SILTs were found in CXCL13−/− mice. However, CXCL13-deficient SILTs were aberrantly formed as B-cell follicles never developed, despite B cell recruitment. This situation contrasts with the development of SILTs in the small intestine in which absence of CXCL13 prevents B cell recruitment into the few aberrant SILTs that develop35. Additionally, colonic SILTs also formed earlier and more abundantly in CXCL13−/− mice than in wild-type mice, which could reflect an increased number of circulating LTi cells as a result of absence of peripheral lymphoid tissues3, 36.
The other striking difference between SILT development in the small intestine and colon is the independence on the CCR6-CCL20 axis for colonic SILT formation. Even though colonic LTi cells expressed CCR6 and CCL20 expression was reduced in mice unable to properly cluster immune cells in the colonic lamina propria, colonic SILTs developed normally in CCR6−/− mice.
Another chemokine receptor functionally expressed on LTi cells is CXCR3 (our data and 37). Even though in the absence of CXCR3, SILT development and organization was mostly normal, MAdCAM-1+ vessels were regularly found in CXCR3-deficient SILTs, therefore suggesting that SILT localization might be altered in the absence of CXCR3-guided LTi cell migration. Alternatively, as chemokine receptors were found to be expressed on endothelial cells and their deficiency found to cause vascular malformations38, 39, it may be that abnormal development of the colonic vasculature leads to the presence of MAdCAM-1+ vessels within the parenchyma of colonic SILTs.
Colonic SILT development is likely directed by numerous chemokines with overlapping roles and this may explain the inability to completely abrogate these structures in mice deficient for a single chemokine(receptor). Redundancy in the chemokine system may even explain some of the discrepancies in SILT development between small intestine and colon described here. In this regard, it is important to recognize that SILT development concurred with the onset of microbial colonization of the intestine and exposure to bacterial components induces multiple chemokines. Therefore, similarly to the small intestine in which SILTs are smaller in germ-free mice23, 24, it was likely that the recognition of the colonizing bacteria would also promote the development of lymphoid tissues in the colon. In contrast, however, we found the overall process of colonic SILT development to be independent of the colonic microflora. Notwithstanding, we found SILT maturation, i.e. B cell recruitment, follicular organization and follicular dendritic cell network development, to require MyD88 signalling. As bacteria were not required for SILT development, this suggests that either IL1R signalling or the release of endogenous TLR ligands may induce the maturation of SILTs. The release of endogenous TLR ligands would reconcile the published observations showing a role for both TLR2/4 and MyD88 in colonic SILT maturation23.
In conclusion, we showed that the murine colon harbours two developmentally distinct lymphoid tissues. Colonic patches started their development during embryogenesis, whereas colonic SILTs developed only after birth in a unique way, dissimilar to the formation of colonic patches, lymph nodes and small intestinal SILTs. Furthermore, we showed a hitherto unrecognized role for CXCR3 in the localization of SILTs within the lamina propria of the colon and a role for MyD88 in SILT maturation, which was independent of the colonic microflora. Altogether, the data presented here highlights the multitude of different molecular pathways involved in the genesis of lymphoid tissues and provides a useful framework for future assessment of intestinal lymphoid tissue development during inflammatory conditions.
MATERIALS AND METHODS
Mice
C57BL/B6, LTα−/−, CXCL13−/−, CCR6−/−, CXCR3−/− and MyD88−/− mice were kept under specific-pathogen free (SPF) conditions; C3H mice were kept under SPF or germ-free conditions. For breeding and prenatal analysis purposes, breeding pairs were housed together overnight and the day of vaginal plug detection recorded as embryonic day E0.5. For post-natal analysis, the day of pup delivery was considered day 0. All experiments were approved by the Vrije University Scientific and Animal Ethics Committees.
Antibiotic treatment
To prevent bacterial colonization of the intestine, wild-type mice were treated with a “cocktail” of antibiotics containing streptomycin (5g/l), colistin (1g/l), ampicilin (1g/l) and sucrose (2.5% wt/vol) (all from Sigma-Aldrich) in the drinking water starting at embryonic day E14 and continuing until analysis at day 14 post-partum. Antibiotic treatment was renewed every week.
Immunofluorescence histology
Colons were dissected at the appropriate age, embedded in optimum cutting temperature compound (OCT; Sakura Finetek) and frozen in liquid nitrogen. Frozen blocks were cut into 7 μm thick serial sections. Detection of organized gut-associated lymphoid tissue was performed on every 10th section (20th section for adult colons) stained for CD4 and MAdCAM-1 in combination with either B220 or α-smooth muscle actin (αSMA); and SILT enumeration on every 50th section (350μm apart) stained for CD4, αSMA and B220. Cryptopatches (CP) were defined as clusters of CD4+ cells present in the lamina propria of the colon; immature isolated lymphoid follicles (iILF) as CD4+cell clusters containing unorganized B cells; and mature isolated lymphoid follicles (mILF) as CD4+cell clusters containing organized B cell follicles. The procedure to enumerate colonic SILTs will establish only a rough estimation of their number, allowing the comparison between the different mouse genotypes. Importantly, it may give a biased representation of their maturation status as small SILTs will extend over 50 sections and may thus occasionally be missed. For staining, slides were first fixed in acetone, hydrated with EBSS (Gibco) and blocked with the blocking reagent of the TSA signal amplification kit (Invitrogen Life Technologies) diluted in EBSS. Endogenous biotin/avidin activity was blocked with a biotin/avidin-blocking kit (Vector Lab). Slides were them incubated with the appropriate antibodies for periods of 45 minutes at room temperature, washed extensively with EBSS and finally mounted in polyvinyl alcohol containing DAPI (Sigma-Aldrich). For RORγ staining, slides were incubated 90 minutes at room temperature with the primary antibody in the blocking reagent containing 0.1% saponin (Sigma). Pictures were taken on a DM6000 Leica immunofluorescent microscope (Leica Microsystems), using the stitch function for overview imaging and analyzed with Leica AF6000 imaging software (Leica Microsystems).
Cell isolation and Flow cytometry
Colons were dissected from E18.5 C57BL/B6 wt embryos. Single cell suspensions were prepared by cutting the colons into small pieces which were subsequently digested at 37°C for 30 minutes with 0.05 mg/ml Liberase Blendzyme 2 (Roche) and 25 U/ml DNAse I (Roche) in Ca2+/Mg2+-free HBSS/1.5% HEPES (Gibco), while continuously stirring. After digestion, cells were extensively washed in PBS/2% FCS (Sigma-Aldrich) and clumps removed by filtration over a 100 μm nylon mesh. Stainings were performed on ice for 30 minutes in PBS/2% FCS. 7AAD (Invitrogen) was used to discriminate between live and dead cells. Cells were analyzed on a Cyan ADP flow cytometer (DakoCytomation) with FlowJo software (TreeStar). Fluorescence minus one controls were used to assess the expression threshold of each individual marker.
Antibodies
The following antibodies were used for immunofluorescence histology: α-B220 (clone 6B2), α-CD4 (GK1.5), α-CD31 (ERMP12), α-CD35 (8c12), α-CD45 (MP33), α-CD127 (A7R34) and α-MAdCAM-1 (MECA367) were purified from hybridoma cell culture supernatants with protein G-Sepharose (Pharmacia) and labelled with Alexa-Fluor488, Alexa-Fluor555 and Alexa-Fluor647 (Invitrogen); mouse α-mouse alpha smooth muscle actin (αSMA; clone 1A4; Sigma-Aldrich), armenian hamster α-mouse nuclear retinoic acid-related orphan receptor-γ (ROR-γ; AFKJS-9) and rat α-mouse VCAM-1 (429(MVCAM.A); BD Biosciences) were developed with goat anti-mouse IgG2a Alexa-Fluor488 or Alexa-Fluor555 (both from Invitrogen), goat anti-armenian hamster Cy3 or DyLight649 (both from Jackson Lab), and goat anti-rat Alexa-Fluor555 (Invitrogen), respectively; rat α-mouse CD3 (KT3) and hamster α-mouse gp38/podoplanin (8.1.1) containing unpurified supernatants together with secondary antibodies goat anti-rat Alexa-Fluor555 and goat anti-hamster Alexa-Fluor647 antibodies (Invitrogen) were used to detect CD3 and gp38/podoplanin, respectively; CD3 was also visualized with the directly labelled antibody α-CD3e eFluor660 (17A2) from eBioscience; directly labelled α-Lyve1 eFluor660 (ALY7) was obtained from eBioscience; biotin-labelled α-CD11c (N418; BioLegend) and α-CD45 (30-F11; eBioscience) were visualized with streptavidin conjugated to Alexa-Fluor555 or Alexa-Fluor647 (both from Invitrogen); biotin-labelled α-CCL19, α-CCL20, α-CCL21, α-CXCL10, α-CXCL12 and α-CXCL13 (all polyclonal goat IgGs from R&D systems) were developed with the TSA signal amplification kit (Invitrogen) with HRP-streptavidin and Alexa-Fluor546 tyramide. For flow cytometry, the antibodies used were: α-CD45 (MP33) and α-CD127 (A7R34), purified from hybridoma supernatants and labelled with Alexa-Fluor488 and Alexa-Fluor647, respectively; α-CD3 APC.eFluor780 (17A2), α-CD4 PE.Cy7 (GK1.5), α-CD11c Alexa450 (N418) and α-CXCR4 PE (2B11) purchased from eBioscience; α-CXCR5 PE (2G8; BD Biosciences); biotin labelled α-CXCR3 (Y-16; SantaCruz) and α-CCR7 (4B12; Ebioscience) were visualized with streptavidin-PE (Jackson Lab); unlabelled α-CCR6 (140706; R&D Systems) and α-CCR9 (7E7; Abcam) were visualized with a goat anti-rat PE antibody (Jackson Lab).
RNA isolation, cDNA synthesis and real-time PCR
For gene/transcript expression analysis, colons were dissected at the appropriate age and stored in Trizol (Gibco) at −80°C. RNA was isolated, upon colon homogenization with an Ultra-Turrax T10 (IKA), by precipitation with isopropanol. Contaminating DNA was removed from the RNA preparation by treating the samples with DNAse I (Fermentas Life Sciences). cDNA was synthesized from 1 μg RNA using the RevertAid First Strand cDNA Synthesis Kit (Fermentas Life Sciences) according to the manufacturer’s protocol. Real-time PCR was performed on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems). Primers were designed across exon-intron boundaries using Primer Express software (Applied Biosystems). To correct for primer efficiency, standard curves for each primer set were generated with cDNA from pooled non-activated peripheral lymph nodes or pooled steady-state intestines. The expression level of each transcript was analyzed and normalized for the expression of selected housekeeping genes with geNORM v3.4 software.
Statistics
Statistical analysis was conducted with GraphPad Prism version v4.00 (GraphPad Software Inc.). One way non-parametric ANOVA (Kruskal-Wallis test) and Dunn’s multiple comparison test were used for the analysis of overall variance and the assessment of differences between individual pairs of data, respectively. For analysis of mRNA expression, results are expressed as mean ± SEM, with the mean value for day 0 wild-type samples arbitrarily set as 1, and were considered significant when p<0.05.
Supplementary Material
Acknowledgments
We thank B. Beuger, D. Schut and E. Keuning for animal care; and D. Littman, P. Leenen and S. Nighikawa for antibodies.
This work was supported by grants from Fundação para a Ciência e Tecnologia – Portugal (SFRH/BD/33247/2007 to APB), Dutch Digestive Disease Foundation (to MK), US National Institutes of Health (R01CA069212 to ADL; and AI061511, AI072689 and HL069409 to TDR) and Netherlands Organization for Scientific Research (VIDI grant 917.10.377 to TC; and VICI grant 918.56.612 to REM).
Footnotes
Disclosure: The authors have no competing financial interests.
References
- 1.Randall TD, Carragher DM, Rangel-Moreno J. Development of secondary lymphoid organs. Annu Rev Immunol. 2008;26:627–650. doi: 10.1146/annurev.immunol.26.021607.090257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol. 2003;3:292–303. doi: 10.1038/nri1054. [DOI] [PubMed] [Google Scholar]
- 3.van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–674. doi: 10.1038/nri2832. [DOI] [PubMed] [Google Scholar]
- 4.Alimzhanov MB, et al. Abnormal development of secondary lymphoid tissues in lymphotoxin beta-deficient mice. Proc Natl Acad Sci U S A. 1997;94:9302–9307. doi: 10.1073/pnas.94.17.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De TP, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 6.Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59–70. doi: 10.1016/s1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- 7.Koni PA, et al. Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta-deficient mice. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- 8.Rennert PD, James D, Mackay F, Browning JL, Hochman PS. Lymph node genesis is induced by signaling through the lymphotoxin beta receptor. Immunity. 1998;9:71–79. doi: 10.1016/s1074-7613(00)80589-0. [DOI] [PubMed] [Google Scholar]
- 9.Benezech C, et al. Ontogeny of stromal organizer cells during lymph node development. J Immunol. 2010;184:4521–4530. doi: 10.4049/jimmunol.0903113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coles MC, et al. Role of T and NK cells and IL7/IL7r interactions during neonatal maturation of lymph nodes. Proc Natl Acad Sci U S A. 2006;103:13457–13462. doi: 10.1073/pnas.0604183103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eberl G, et al. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 12.Vondenhoff MF, et al. LTbetaR signaling induces cytokine expression and up-regulates lymphangiogenic factors in lymph node anlagen. J Immunol. 2009;182:5439–5445. doi: 10.4049/jimmunol.0801165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida H, et al. Different cytokines induce surface lymphotoxin-alphabeta on IL-7 receptor-alpha cells that differentially engender lymph nodes and Peyer’s patches. Immunity. 2002;17:823–833. doi: 10.1016/s1074-7613(02)00479-x. [DOI] [PubMed] [Google Scholar]
- 14.van de Pavert SA, et al. Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol. 2009;10:1193–1199. doi: 10.1038/ni.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshida H, et al. IL-7 receptor alpha+ CD3(-) cells in the embryonic intestine induces the organizing center of Peyer’s patches. Int Immunol. 1999;11:643–655. doi: 10.1093/intimm/11.5.643. [DOI] [PubMed] [Google Scholar]
- 16.Veiga-Fernandes H, et al. Tyrosine kinase receptor RET is a key regulator of Peyer’s patch organogenesis. Nature. 2007;446:547–551. doi: 10.1038/nature05597. [DOI] [PubMed] [Google Scholar]
- 17.Hamada H, et al. Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J Immunol. 2002;168:57–64. doi: 10.4049/jimmunol.168.1.57. [DOI] [PubMed] [Google Scholar]
- 18.Kanamori Y, et al. Identification of novel lymphoid tissues in murine intestinal mucosa where clusters of c-kit+ IL-7R+ Thy1+ lympho-hemopoietic progenitors develop. J Exp Med. 1996;184:1449–1459. doi: 10.1084/jem.184.4.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eberl G, Littman DR. Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science. 2004;305:248–251. doi: 10.1126/science.1096472. [DOI] [PubMed] [Google Scholar]
- 20.Lorenz RG, Chaplin DD, McDonald KG, McDonough JS, Newberry RD. Isolated lymphoid follicle formation is inducible and dependent upon lymphotoxin-sufficient B lymphocytes, lymphotoxin beta receptor, and TNF receptor I function. J Immunol. 2003;170:5475–5482. doi: 10.4049/jimmunol.170.11.5475. [DOI] [PubMed] [Google Scholar]
- 21.Taylor RT, Lugering A, Newell KA, Williams IR. Intestinal cryptopatch formation in mice requires lymphotoxin alpha and the lymphotoxin beta receptor. J Immunol. 2004;173:7183–7189. doi: 10.4049/jimmunol.173.12.7183. [DOI] [PubMed] [Google Scholar]
- 22.Velaga S, et al. Chemokine receptor CXCR5 supports solitary intestinal lymphoid tissue formation, B cell homing, and induction of intestinal IgA responses. J Immunol. 2009;182:2610–2619. doi: 10.4049/jimmunol.0801141. [DOI] [PubMed] [Google Scholar]
- 23.Bouskra D, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–510. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- 24.Pabst O, et al. Adaptation of solitary intestinal lymphoid tissue in response to microbiota and chemokine receptor CCR7 signaling. J Immunol. 2006;177:6824–6832. doi: 10.4049/jimmunol.177.10.6824. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Heazlewood SP, Krause DO, Florin TH. Molecular characterization of the microbial species that colonize human ileal and colonic mucosa by using 16S rDNA sequence analysis. J Appl Microbiol. 2003;95:508–520. doi: 10.1046/j.1365-2672.2003.02005.x. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, et al. Regional mucosa-associated microbiota determine physiological expression of TLR2 and TLR4 in murine colon. PLoS One. 2010;5:e13607. doi: 10.1371/journal.pone.0013607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeurissen SH, Duijvestijn AM, Sontag Y, Kraal G. Lymphocyte migration into the lamina propria of the gut is mediated by specialized HEV-like blood vessels. Immunology. 1987;62:273–277. [PMC free article] [PubMed] [Google Scholar]
- 28.Mora JR, et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006;314:1157–1160. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- 29.Svensson M, et al. CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. J Clin Invest. 2002;110:1113–1121. doi: 10.1172/JCI15988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDonald KG, et al. CC chemokine receptor 6 expression by B lymphocytes is essential for the development of isolated lymphoid follicles. Am J Pathol. 2007;170:1229–1240. doi: 10.2353/ajpath.2007.060817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hashi H, et al. Compartmentalization of Peyer’s patch anlagen before lymphocyte entry. J Immunol. 2001;166:3702–3709. doi: 10.4049/jimmunol.166.6.3702. [DOI] [PubMed] [Google Scholar]
- 32.Rumbo M, Sierro F, Debard N, Kraehenbuhl JP, Finke D. Lymphotoxin beta receptor signaling induces the chemokine CCL20 in intestinal epithelium. Gastroenterology. 2004;127:213–223. doi: 10.1053/j.gastro.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 33.Dohi T, et al. Elimination of colonic patches with lymphotoxin beta receptor-Ig prevents Th2 cell-type colitis. J Immunol. 2001;167:2781–2790. doi: 10.4049/jimmunol.167.5.2781. [DOI] [PubMed] [Google Scholar]
- 34.Kweon MN, et al. Prenatal blockage of lymphotoxin beta receptor and TNF receptor p55 signaling cascade resulted in the acceleration of tissue genesis for isolated lymphoid follicles in the large intestine. J Immunol. 2005;174:4365–4372. doi: 10.4049/jimmunol.174.7.4365. [DOI] [PubMed] [Google Scholar]
- 35.McDonald KG, McDonough JS, Dieckgraefe BK, Newberry RD. Dendritic cells produce CXCL13 and participate in the development of murine small intestine lymphoid tissues. Am J Pathol. 2010;176:2367–2377. doi: 10.2353/ajpath.2010.090723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ansel KM, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 37.Lugering A, et al. CCR6 identifies lymphoid tissue inducer cells within cryptopatches. Clin Exp Immunol. 2010;160:440–449. doi: 10.1111/j.1365-2249.2010.04103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cha YR, et al. Chemokine signaling directs trunk lymphatic network formation along the preexisting blood vasculature. Dev Cell. 2012;22:824–836. doi: 10.1016/j.devcel.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerrits H, et al. Early postnatal lethality and cardiovascular defects in CXCR7-deficient mice. Genesis. 2008;46:235–245. doi: 10.1002/dvg.20387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.