

Abstract
Prolyl 3-hydroxylase1 (P3H1) is a collagen modifying enzyme which hydroxylates certain prolines in the Xaa position of conventional GlyXaaYaa triple helical sequence. Recent investigations have revealed that mutations in the LEPRE1 (gene encoding for P3H1) cause severe osteogenesis imperfecta (OI) in humans. Similarly LEPRE1 knockout mice display an OI-like phenotype. Significant hearing loss is a common problem for people with osteogenesis imperfecta. Here we report that hearing of the P3H1 null mice is substantially affected. Auditory brainstem responses (ABRs) of the P3H1 null mice show an average increase of 20–30 dB in auditory thresholds. Three dimensional reconstructions of the mutant middle ear bones by Micro-scale X-ray computed tomography (Micro-CT) demonstrate abnormal morphology of the incudostapedial and incudomalleal joints. We establish the LEPRE1 knockout mouse as a valuable model system to investigate the mechanism of hearing loss in recessive OI.
Keywords: Osteogenesis Imperfecta, Prolyl 3-Hydroxylase 1, Mouse, Hearing loss
1. Introduction
Osteogenesis imperfecta is a generalized connective tissue and skeletal disorder characterized by bone fragility. OI phenotypes range from very severe to mild forms. Originally Sillence proposed a classification of Osteogenesis Imperfecta in four types (Sillence and Rimoin, 1978; Sillence et al., 1979). These types are caused by mutations in COL1A1 and COL1A2 genes. Recently more genes such as CRTAP, LEPRE1, PPIB, SERPINH1, SERPINF1, FKBP10, PLOD2, SP7 responsible for autosomal recessive forms of the disease were identified (Shaheen et al., 2011) (Alanay et al., 2010; Christiansen et al.; Homan et al.; Kelley et al., 2011; Morello et al., 2006; Schwarze et al.; van Dijk et al., 2009; Vranka et al., 2010). Three of these genes, namely LEPRE1, CRTAP, and PPIB code for proteins that form the molecular complex required for type I collagen 3-hydroxylation. It has been demonstrated that mutations in P3H1 cause severe osteogenesis imperfecta in humans (Marini et al., 2010) and a similar but more mild phenotype in mice (Vranka et al., 2010). The P3H1•CRTAP•CypB complex is also a potent molecular chaperone for procollagen biosynthesis (Ishikawa et al., 2009). Recently one more newly identified collagen chaperone FKBP65 (Ishikawa et al., 2008) was found to cause a severe form of osteogenesis imperfecta (Alanay et al., 2010).
Approximately half of the OI patients experience hearing loss at different points of their lifetime (Paterson et al., 2001). Hearing problems may start at an early age and be progressive. Cases of purely conductive, mixed and purely sensorineural types of hearing loss are documented (Pillion and Shapiro, 2008). Early onset is generally conductive. Sensorineural or mixed types are usually characterized by later onsets (Swinnen et al., 2012b). Problems with hearing are usually attributed to the mild forms of osteogenesis imperfecta. All currently known cases of hearing loss were observed in patients with autosomal dominant types of OI. However, recessive forms of OI are usually severe, often prenatal or neonatal lethal which precludes audiological evaluation. An appropriate mouse model system to study the pathogenic mechanisms underlying auditory dysfunction in OI is vitally needed. Recently a number of OI mouse models have become available. However until now there was only one attempt to study hearing sensitivity in OI mice. The Mov-13 mouse has served as an animal model of type I OI by virtue of exhibiting variable transcriptional block of the COL1A1 gene. ABR measurements of Mov-13 mice demonstrated profound hearing loss ranging from 20 to 60 dB depending on age and frequency (Altschuler et al., 1991; Stankovic et al., 2007). Remodeling of the otic capsule similar to that in otosclerosis have been demonstrated in Mov-13 mice but the exact molecular mechanisms leading to the pathological changes remain unclear. While the Mov-13 mouse was used as a model system for dominant forms of OI until now no mouse model was proposed to study hearing loss in recessive OI. In a very recent study (Swinnen et al., 2012a) the degree of the hearing loss was linked to the bone mineral density (BMD) in OI patients. Recessive forms of OI are characterized by very low BMDs. Hearing loss is expected for patients with P3H1 mutations and therefore an animal model to study its pathogenesis is crucially important.
In this study we demonstrate for the first time that auditory thresholds are elevated in P3H1 null mice. Our micro-CT data show abnormal morphology of the middle ear bone joints in the mutant animals. We conclude that the P3H1 knockout mouse can serve as a useful model to investigate the molecular mechanisms underlying hearing loss in OI.
2. Results
2.1. Auditory brain stem responses of the P3H1 null versus wild type mice
We tested the hearing of the P3H1 null mice in comparison to the age-matched wild types by measuring auditory brainstem responses for both groups. The results of ABR measurements are graphically represented in Figure 1 and summarized in Table 1. The data are represented as mean values of hearing thresholds at 4, 8, 16 and 32 kHz. Sixteen ears in each group were tested. Tone burst stimuli at the four frequencies were presented to cover a large portion of the range of mouse hearing. Measurements were done at P28. This time point falls within a window of normal hearing for the C57/BL6 mouse strain. Prior to 3 weeks of age, their hearing has not fully matured. After several months of age, however, these mice begin to display age-related hearing loss (Ison et al., 2007). ABR thresholds for the P3H1 null mice were found to be increased for every measured frequency (see figure 1). As shown in table 1 the differences in mean values between nulls and wild types vary from 14.7 dB to 26.7 dB depending on the frequency. Two independent ears were found to be unresponsive at 100dB SPL at a frequency of 32 kHz. Thus, the auditory thresholds of the P3H1 null mice are significantly statistically elevated in comparison to age-matched, wild type controls.
Figure 1. ABR thresholds in adult (P28) wild type (C57Bl6) and P3H1 null mice.

Average ABR thresholds in decibels referenced to sound pressure level (dB SPL) are plotted against frequency for each group. Frequencies tested were 4, 8, 16, and 32 kHz. Standard errors of the mean ranges are plotted for each data point. A statistically significant threshold shift at each of the frequencies is seen between the mutant (P3H1 null) mouse group and the controls (P < 0.001).
Table 1.
Average auditory brain stem responses (ABRs) measured for P3H1 null and wild type mice. Averaging is done over 16 ears for each genotype.
| Frequency | 4 kHz | 8kHz | 16kHz | 32kHz |
|---|---|---|---|---|
| Wild types (dB SPL) | 66.6± .3 | 63.0 ± 3.6 | 41.8 ± 6.7 | 43.9 ± 3.2 |
| P3H1 nulls (dB SPL) | 87.3 ± 9.0 | 77.7 ± 8.3 | 59.3 ± 17.5 | 70.6 ± 23.6a |
| Mean (null) – Mean (wt) (dB SPL) | 20.7 | 14.7 | 17.4 | 26.7 |
| Statistical values (two sample t-test) |
df = 26 t = 3.5 p = 0.0008 |
df = 26 t = 3.6 p = 0.0006 |
df = 26 t = 3.5 p = 0.0009 |
df = 22 t = 3.5 p = 0.0008 |
| Mean (null) – Mean (wt) used for t-test (dB SPL) | > 12 | > 6 | > 0 | > 4 |
two ears were found unresponsive at 32kHz up to 100dB and were not included in the statistics.
2.2. Micro-CT data demonstrate thinning of the middle ear bone joints in P3H1 knockout mice
To evaluate the middle ear bone morphology we applied Micro-scale X-ray computed tomography (micro-CT). Mice used for the ABR study were sacrificed and their heads were subsequently scanned. Overall 8 P3H1 null ears and 4 wild type control ears were analyzed at P28. In addition 4 mutant and 2 control ears were scanned at P15. At P28 seven out of eight null ears showed different degrees of dysmorphogenesis in the middle ear bone joints as described below. Figure 2 shows representative scans of the mutant mouse middle ear bulb compared with an age-matched, wild type control. There appear to be no grossly detectable abnormalities in the overall bone morphology of the control middle ears. By contrast, the mutant temporal bones appeared thinner compared to the wild type. When measured from the 3-dimensional view as indicated on fig. 2A by arrows, the thickness of P3H1 null bone is 0.07 mm and that of the wild type is 0.11 mm. The bones of the P3H1 knockouts appeared more porous (see circled area on fig. 2A). The overall size of the mutant ear bulb is smaller and its shape is more round compared to the control. The measurements were taken using 2-dimensional cross sections (fig. 2B). These findings are consistent with the OI phenotype of the P3H1 null mouse: we have previously documented the reduced bone mineral densities and craniofacial phenotype of these mice (Vranka et al., 2010).
Figure 2. Micro-CT scans of P3H1 null and wild type control ears.
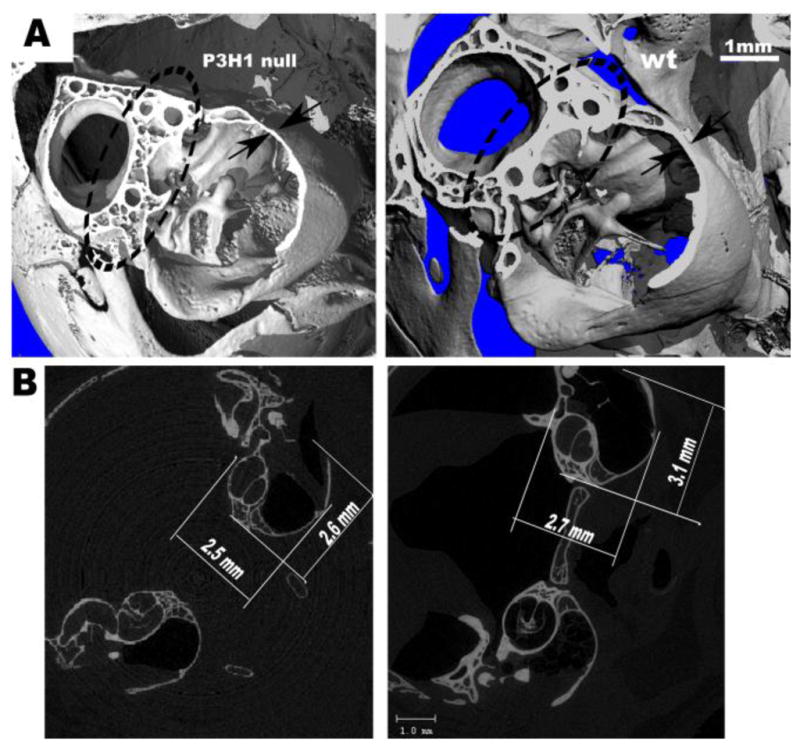
Panel A represents a 3-D reconstructed cut section of the P3H1 null and age-matched wild type control temporal bones. Arrows indicate the measurements of the temporal bone thickness (0.07 mm for mutant, and 0.11 mm for wild type). Note more porous bone in the circled area for the null.
Panel B shows corresponding 2-dimentional crossections. Temporal bone of the P3H1 tend to be less elongated compare to the control (measurements are indicated on the figure).
Incudostapedial joint dislocation and stapes fixation are the most common anomalies among ossicular malformations (Suzuki et al., 2008). We took a closer look at the middle ear bones in the P3H1 null mice. Figure 3 represents isolated ossicular chains of the P3H1 nulls compared to the age-matched controls. Panel A shows ear bones of the one month old (P28) mice (used for ABR measurements), panel B demonstrates two null ears and one P3H1 heterozygous control ear at two weeks of age (P15) and panel C represents one year old ossicular chains. At every investigated developmental stage P3H1 nulls show abnormalities in either incudo-malleal or incudo-stapedial joints. The incudo-stapedial joint of the knockout mouse in figure 3A displays abnormal morphology. The joint area between the long process of the incus (red) and the head of the stapes (yellow) is greatly reduced compared to the control (fig. 3A, circled). This joint appears to be dramatically thinned in the P3H1 null mouse. The connection between the incus and stapes in the mutant is insubstantial, appearing very slender. Similarly in Fig 3B (at P15) one of the P3H1 null incudo-stapedial joints appear much thinner in comparison with the control. The other null ear on Fig. 3B has rather normal incudo-stapedial joint but very abnormal incudo-malleal joint (left, arrowhead). Dramatic dysmorphology in the mutant incudo-malleal joint is also observed in a one year old mouse (Fig. 3C, arrowhead). In both cases the articular facets of the incus (red) and malleus (green) appear separated. In contrast, in the control mice (Fig. 3B and 3C), the incudo-malleal joints show close association of the articular facet of the incus and the malleus. In an attempt to investigate the pre-hearing formation of the middle ear bones in the mutant mice we scanned P9 ears. Figure 4 shows 3D reconstructions for the P3H1 null and heterozygous littermate control. As evident from the reconstructions the degree of bone calcification differs substantially at this stage of development. In fact, the stapes is undetectable in the mutant while it is already clearly present in the control.
Figure 3. Three dimensional micro-CT reconstructions of the mouse middle ear bones at different postnatal stages.

Figure A represents isolated middle ear bones of one month old P3H1 null and wild type. B shows two P3H1 null and oneP3H1 heterozygous ossicular chains from P15 animals. Figure C represents ossicular chains of the 1 year old P3H1 null and wild type mice. Genotypes are indicated. Incudo–stapedial joints are circled and incudo–malleal joints are indicated by arrowheads. Middle ear bones are labeled and color coded: malleus is green, incus is red, and stapes is yellow. Incudo-stapedial joint of the 1 month old P3H1 null (A, left, circled) is dramatically thinner than that of the wild type control. Similar picture is seen in B for one of the mutant ears. The incudo-malleal joints of the P3H1 null mice shown in B (left, arrowhead) and in C display severe abnormalities. The angle between malleus and incus in the affected joint (B) is dramatically different from that of the wild type control (B, bottom view).
Figure 4.
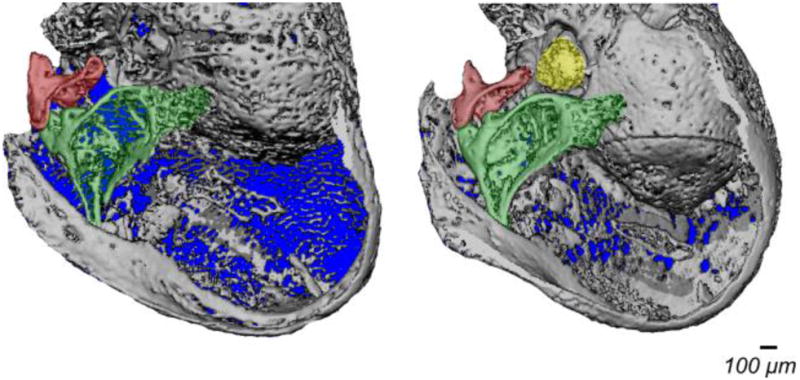
Three dimensional micro-CT reconstructions of the whole ear bulbs from P3H1−/− and P3H1+/− littermates at P9. Middle ear bones are color coded as in Fig. 3 (malleus is green, incus is red, and stapes is yellow). The stapes is not identified in the P3H1 null ear due to the insufficient bone calcification.
Our results show that P3H1 null mice have elevated auditory thresholds and abnormal morphology of the incudo-stapedial and incudo-malleal joints. Our data represent the first demonstration of abnormalities in the middle ear joints in an OI mouse model and establish the P3H1 null mouse as a model system for interrogating the etiology of hearing loss in OI.
3. Discussion
Alterations in the structure of the incudo-stapedial and incudo-mallelal joints demonstrated by our data are predicted to reduce the efficacy of mechanical vibrations from the middle ear to the cochlea and may contribute to the significantly elevated auditory thresholds observed. This is the first demonstration of defects in middle ear joints in OI. The most common cause of hearing loss in humans with OI is mixed conductive and sensorineural. Otosclerosis is one of the major causes of conductive hearing loss. There are similarities between OI associated hearing loss and otosclerosis. Both OI and otosclerosis are characterized by abnormal remodeling of the otic capsule (McKenna et al., 1998; Pedersen et al., 1985). In the most common case the sclerosis invades the stapedio-vestibular joint and complicates or completely blocks the motion of stapes. Compared with otosclerosis, patients with OI develop hearing loss earlier; have more severe middle ear disease; and a higher incidence of sensorineural deafness (Kuurila et al., 2002; Shapiro et al., 1982). The pathogenesis of otosclerosis remains poorly understood but some genetic linkages have been found. The α-1 chain of type I collagen was the first genetic association (McKenna et al., 1998). Reduced levels of COL1A1 mRNA have also been found in some of the otosclerosis patients (McKenna et al., 2002). However no molecular mechanism to explain how a reduced amount of COL1A1 can lead to the abnormal bone formation has been proposed.
The abnormal morphology of the ossicular chain joints suggests conductive component to the pathogenesis of hearing loss in P3H1 null mice. Our data does not allow differentiating between the conductive, mixed or sensorineural mechanisms. It is however intriguing that collagen is present in many structures important for the normal hearing. For example type II collagen has been identified in basilar membrane (Tsuprun and Santi, 1999). Collagen II is a prominent substrate for prolyl 3-hyroxylase 1 and 3-hydroxylation might affect its structural and mechanical properties. Therefore the stiffness of the basilar membrane may be altered in the mutant mice which could contribute significantly to the hearing loss observed. The P3H1 mutant represents an important new tool for delineating the pathogenesis of hearing loss in a mouse model of human OI.
The molecular mechanism leading to abnormal morphology of the incudo-stapedial and incudo-mallelal joints in P3H1 mice remains unknown. During mouse development the incudomalleal joint is initially a contiguous structure formed from a single Sox9/ColII expressing condensation which subdivides into two separate ossicles (Amin et al., 2007). Initially uniformly distributed collagen II expressing cells later migrate out of the developing joint. It is not yet clear what specific function 3-hydroxyl group plays in collagens. However it is possible that the lack of the 3-hydroxylation in collagen II may influence signaling during cell migration as well as cause structural defects in cartilage.
Tendons are always involved whenever a joint pathology occurs. We have previously shown that the tail tendon in P3H1 knockout mouse has severe structural defects (Vranka et al., 2010). Type I collagen is overmodified with excessive sugars in P3H1 null mouse which sequentially leads to abnormal fibrillogenesis. These observations lead us to speculate that the P3H1 mutation may affect tendon differentiation or maturation, potentially predisposing the middle ear ossicles to mechanical stress and maladaptive morphological changes. In addition insufficiently calcified ear bones at the time of hearing onset (around P10) (Ehret, 1976) might cause difficulties to withstand mechanical stress in the mutant mice.
Mice start to hear at postnatal day 10. Although the degree of calcification does not allow seeing middle ear joints with mico-CT at this stage our data collected at P15 shows the same joints malformations as it is seen in one month old animals. Therefore the hearing problems in P3H1 null mice seem to be rather congenital than degenerative. This fits well with the finding of the early onset of the conductive hearing loss in the OI patients.
The availability of a mouse model to study the etiology of hearing loss in recessive forms of OI may provide insight into the pathogenesis responsible for hearing loss in dominant forms of OI. Further investigations of the molecular mechanisms involved in the etiology of hearing loss of P3H1 knockout mice may extend our understanding of hearing loss in OI patients. Ultimately, the mouse model will serve as a starting point for defining rational therapies to preserve or even restore auditory function in OI patients.
4. Materials and Methods
4.1. Auditory brain-stem response testing
Mice were anesthetized (0.5 mg/kg, ip, Avertin; Sigma) and placed in a custom head holder located in a sound attenuating chamber (IAC, New York, NY, USA). Prior to the ABR recording, sound pressure at the entrance to the external auditory meatus was calibrated for every subject at each stimulus frequency with a probe tube microphone (Type 4133; Bruel & Kjaer, Denmark) and a spectrum analyzer (SR770, FFT network analyzer; Stanford Research Systems, Sunnyvale, CA, USA). ABR recordings were collected from subdermal electrodes placed at the left mastoid prominence (active electrode), vertex (reference electrode), and rump (ground electrode).
Stimulus generation and response recordings were made and analyzed using the TDT System II signal processing package (Tucker Davis Technology, Gainesville, FL, USA). Tone bursts with a duration of 4 ms (1 ms rise/fall, 2 ms plateau, 50-ms intervals) were presented at a rate of 20/s in a free field with a digital-to-analog converter connected to an electrostatic speaker. The stimulus intensity was controlled via an electronically programmable attenuator over a 90-dB range. The digitized electrical responses were amplified (100,000×) and filtered via a two-step process. Initial amplification of 1000× was made using a preamplifier with a 10- to 10,000-Hz filter (DAM-6A Differential Pre-amp, World Precision Instruments, Sarasota, FL, USA) with a secondary amplification of 100× and a 0.3- to 3-kHz filter by the TDT System II spike conditioner. Responses were recorded over a 10-ms interval, which began at the onset of the tone burst and had a 20-kHz sampling rate. Single traces whose voltage exceeded a normal level were rejected as artifact derived from muscle and cardiac activity. Approximately 500 non-artifact responses were collected and averaged at each stimulus intensity. Thresholds were defined as half the interval between the lowest stimulus intensity producing a replicable response waveform and the stimulus intensity below which no reproducible response was elicited.
4.2. Statistical analysis
Statistical analysis was performed with Origin 7.5 software. Pearson correlations indicated no significant covariance for left and right ears of the same animal within the group. Two sample independent t–test was applied for statistical analysis. Corresponding statistical values (df, t and p) are indicated in Table 1. Averaging was done over 16 ears in each group of mice for every measured frequency except 32 kHz where two P3H1 null ears (from two different mice) were unresponsive at 100 dB SPL and excluded from the statistics.
4.3. Micro-CT
Mice used for ABR measurements were euthanized and the heads were separated. Images were collected using a Scanco μCT 35 micro-CT using a tungsten target x-ray tube and a 0.5mm Al filter. The system was operated at 70kVp with an intensity of 114μA resulting a <9μm spot size. Scan parameters were set with a voxel size of 10μm and a 20.5mm field of view. 939 slices were collected over a total height of 20.5mm with a 400ms integration time and data averaging at 6x. Total scan time was 12 hours. Image collections were evaluated using software provided with the Scanco micro-CT using the “3D Segmentation of VOI, 1 solid object” script.
4.4. Animals
P3H1 null mice were purchased from Deltagen (San Mateo, CA). Directed knockouts were created in which exons 1–3 (nucleotides 15–817) of the mouse P3H1 or leprecan 1 gene (NCBI Reference sequence number: NM_019782.2) were deleted. Then a LacZ-Neo cassette was inserted into the area of the target gene that was deleted. P3H1 or leprecan gene inactivation was verified in mice by RNA preparation from tissues of null mice, followed by reverse transcription and PCR with primer sets spanning the length of the target gene. No P3H1 transcripts and gene expression were detected, as compared with normal levels of the target gene expressed in wild-type animals (Vranka et al., 2010). All procedures were performed in adherence with institutional and national guidelines and were approved by the Oregon Health & Science University Institutional Animal Care and Use Committee. Mice were bred multiple generations into a C57Bl6 background prior to analysis to verify phenotype effects. Auditory brain-stem response testing was done for wild type (C57/BL6 strain) and P3H1 knockout mice at P28.
Highlights.
Prolyl 3-hydroxylase1 (P3H1) is a collagen modifying enzyme which hydroxylates proline in the Xaa position of conventional GlyXaaYaa triple helical sequence. Recent investigations have revealed that mutations in the gene encoding for prolyl 3-hydroxylase-1 cause severe osteogenesis imperfecta (OI) in humans. Similarly P3H1 knockout mice display an OI-like phenotype. Significant hearing loss is a common problem for people with osteogenesis imperfecta. Here we report for the first time that hearing of the P3H1 null mice is substantially affected. Auditory brainstem responses (ABRs) of the P3H1 null mice show an average increase of 20–30 dB in auditory thresholds. Three dimensional reconstructions of the P3H1 null middle ear bones by Micro-scale X-ray computed tomography (Micro-CT) demonstrate abnormal morphology of the incudostapedial and incudomalleal joints. We establish the P3H1 knockout mouse as a valuable model system to investigate the mechanism of hearing loss in recessive OI.
Acknowledgments
This work was supported by a grant from the Shriners Hospitals for Children (to H.P.B.) and grants from the National Institute on Deafness and Other Communication Disorders: DC R01 008595 (JVB) and the Oregon Hearing Research Center Core Grant DC005983.
Abbreviations
- P3H1
Prolyl 3-hydroxylase 1
- OI
osteogenesis imperfecta
- ABR
auditory brainstem response
- CypB
cyclophilin B
- CRTAP
cartilage associated protein
- wt
wild type
- Micro-CT
Micro-scale X-ray computed tomography
- COL1A1
collagen type 1 alpha 1 chain
- COL1A2
collagen type 1 alpha 2 chain
- FKBP65
65-kDa FK506-binding protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D, Alikasifoglu M, Tuncbilek E, Orhan D, Bakar FT, Zabel B, Superti-Furga A, Bruckner-Tuderman L, Curry CJ, Pyott S, Byers PH, Eyre DR, Baldridge D, Lee B, Merrill AE, Davis EC, Cohn DH, Akarsu N, Krakow D. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuler RA, Dolan DF, Ptok M, Gholizadeh G, Bonadio J, Hawkins JE. An evaluation of otopathology in the MOV-13 transgenic mutant mouse. Ann N Y Acad Sci. 1991;630:249–252. doi: 10.1111/j.1749-6632.1991.tb19595.x. [DOI] [PubMed] [Google Scholar]
- Amin S, Matalova E, Simpson C, Yoshida H, Tucker AS. Incudomalleal joint formation: the roles of apoptosis, migration and downregulation. BMC Dev Biol. 2007;7:134. doi: 10.1186/1471-213X-7-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S, Pepin MG, Weis MA, Eyre DR, Byers PH. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet. 86:389–398. doi: 10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret G. Development of absolute auditory thresholds in the house mouse (Mus musculus) J Am Audiol Soc. 1976;1:179–184. [PubMed] [Google Scholar]
- Homan EP, Rauch F, Grafe I, Lietman C, Doll JA, Dawson B, Bertin T, Napierala D, Morello R, Gibbs R, White L, Miki R, Cohn DH, Crawford S, Travers R, Glorieux FH, Lee B. Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J Bone Miner Res. 26:2798–2803. doi: 10.1002/jbmr.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa Y, Vranka J, Wirz J, Nagata K, Bachinger HP. The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J Biol Chem. 2008;283:31584–31590. doi: 10.1074/jbc.M802535200. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Wirz J, Vranka JA, Nagata K, Bachinger HP. Biochemical characterization of the prolyl 3-hydroxylase 1.cartilage-associated protein. cyclophilin B complex. J Biol Chem. 2009;284:17641–17647. doi: 10.1074/jbc.M109.007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison JR, Allen PD, O’Neill WE. Age-related hearing loss in C57BL/6J mice has both frequency-specific and non-frequency-specific components that produce a hyperacusis-like exaggeration of the acoustic startle reflex. J Assoc Res Otolaryngol. 2007;8:539–550. doi: 10.1007/s10162-007-0098-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BP, Malfait F, Bonafe L, Baldridge D, Homan E, Symoens S, Willaert A, Elcioglu N, Van Maldergem L, Verellen-Dumoulin C, Gillerot Y, Napierala D, Krakow D, Beighton P, Superti-Furga A, De Paepe A, Lee B. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J Bone Miner Res. 2011;26:666–672. doi: 10.1002/jbmr.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuurila K, Kaitila I, Johansson R, Grenman R. Hearing loss in Finnish adults with osteogenesis imperfecta: a nationwide survey. Ann Otol Rhinol Laryngol. 2002;111:939–946. doi: 10.1177/000348940211101014. [DOI] [PubMed] [Google Scholar]
- Marini JC, Cabral WA, Barnes AM. Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res. 2010;339:59–70. doi: 10.1007/s00441-009-0872-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna MJ, Kristiansen AG, Bartley ML, Rogus JJ, Haines JL. Association of COL1A1 and otosclerosis: evidence for a shared genetic etiology with mild osteogenesis imperfecta. Am J Otol. 1998;19:604–610. [PubMed] [Google Scholar]
- McKenna MJ, Kristiansen AG, Tropitzsch AS. Similar COL1A1 expression in fibroblasts from some patients with clinical otosclerosis and those with type I osteogenesis imperfecta. Ann Otol Rhinol Laryngol. 2002;111:184–189. doi: 10.1177/000348940211100214. [DOI] [PubMed] [Google Scholar]
- Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, Castagnola P, Rauch F, Glorieux FH, Vranka J, Bachinger HP, Pace JM, Schwarze U, Byers PH, Weis M, Fernandes RJ, Eyre DR, Yao Z, Boyce BF, Lee B. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- Paterson CR, Monk EA, McAllion SJ. How common is hearing impairment in osteogenesis imperfecta? J Laryngol Otol. 2001;115:280–282. doi: 10.1258/0022215011907442. [DOI] [PubMed] [Google Scholar]
- Pedersen U, Melsen F, Elbrond O, Charles P. Histopathology of the stapes in osteogenesis imperfecta. J Laryngol Otol. 1985;99:451–458. doi: 10.1017/s0022215100097036. [DOI] [PubMed] [Google Scholar]
- Pillion JP, Shapiro J. Audiological findings in osteogenesis imperfecta. J Am Acad Audiol. 2008;19:595–601. doi: 10.3766/jaaa.19.8.3. [DOI] [PubMed] [Google Scholar]
- Schwarze U, Cundy T, Pyott SM, Christiansen HE, Hegde MR, Bank RA, Pals G, Ankala A, Conneely K, Seaver L, Yandow SM, Raney E, Babovic-Vuksanovic D, Stoler J, Ben-Neriah Z, Segel R, Lieberman S, Siderius L, Al-Aqeel A, Hannibal M, Hudgins L, McPherson E, Clemens M, Sussman MD, Steiner RD, Mahan J, Smith R, Anyane-Yeboa K, Wynn J, Chong K, Uster T, Aftimos S, Sutton VR, Davis EC, Kim LS, Weis MA, Eyre D, Byers PH. Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum Mol Genet. doi: 10.1093/hmg/dds371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Al-Owain M, Faqeih E, Al-Hashmi N, Awaji A, Al-Zayed Z, Alkuraya FS. Mutations in FKBP10 cause both Bruck syndrome and isolated osteogenesis imperfecta in humans. Am J Med Genet A. 2011;155A:1448–1452. doi: 10.1002/ajmg.a.34025. [DOI] [PubMed] [Google Scholar]
- Shapiro JR, Pikus A, Weiss G, Rowe DW. Hearing and middle ear function in osteogenesis imperfecta. Jama. 1982;247:2120–2126. [PubMed] [Google Scholar]
- Sillence DO, Rimoin DL. Classification of osteogenesis imperfect. Lancet. 1978;1:1041–1042. doi: 10.1016/s0140-6736(78)90763-8. [DOI] [PubMed] [Google Scholar]
- Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankovic KM, Kristiansen AG, Bizaki A, Lister M, Adams JC, McKenna MJ. Studies of otic capsule morphology and gene expression in the Mov13 mouse--an animal model of type I osteogenesis imperfecta. Audiol Neurootol. 2007;12:334–343. doi: 10.1159/000104789. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Kanebayashi H, Kawano A, Hagiwara A, Furuse H, Yamaguchi T, Shimizu M. Involvement of the incudostapedial joint anomaly in conductive deafness. Acta Otolaryngol. 2008;128:515–519. doi: 10.1080/00016480701596062. [DOI] [PubMed] [Google Scholar]
- Swinnen FK, De Leenheer EM, Goemaere S, Cremers CW, Coucke PJ, Dhooge IJ. Association between bone mineral density and hearing loss in osteogenesis imperfecta. Laryngoscope. 2012a;122:401–408. doi: 10.1002/lary.22408. [DOI] [PubMed] [Google Scholar]
- Swinnen FK, Dhooge IJ, Coucke PJ, D’Eufemia P, Zardo F, Garretsen TJ, Cremers CW, De Leenheer EM. Audiologic phenotype of osteogenesis imperfecta: use in clinical differentiation. Otol Neurotol. 2012b;33:115–122. doi: 10.1097/MAO.0b013e31823e28e9. [DOI] [PubMed] [Google Scholar]
- Tsuprun V, Santi P. Ultrastructure and immunohistochemical identification of the extracellular matrix of the chinchilla cochlea. Hear Res. 1999;129:35–49. doi: 10.1016/s0378-5955(98)00219-6. [DOI] [PubMed] [Google Scholar]
- van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PG, Piersma SR, Fratantoni SA, Jimenez CR, Huizer M, Morsman AC, Cobben JM, van Roij MH, Elting MW, Verbeke JI, Wijnaendts LC, Shaw NJ, Hogler W, McKeown C, Sistermans EA, Dalton A, Meijers-Heijboer H, Pals G. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet. 2009;85:521–527. doi: 10.1016/j.ajhg.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranka JA, Pokidysheva E, Hayashi L, Zientek K, Mizuno K, Ishikawa Y, Maddox K, Tufa S, Keene DR, Klein R, Bachinger HP. Prolyl 3-hydroxylase 1 null mice display abnormalities in fibrillar collagen-rich tissues such as tendons, skin, and bones. J Biol Chem. 2010;285:17253–17262. doi: 10.1074/jbc.M110.102228. [DOI] [PMC free article] [PubMed] [Google Scholar]