

Abstract
T-lymphocytes promote cerebral inflammation, thus aggravating neuronal injury after stroke. Fingolimod, a sphingosine 1-phosphate receptor analog, prevents the egress of lymphocytes from primary and secondary lymphoid organs. Based on these findings, we hypothesized fingolimod treatment would reduce the number of T-lymphocytes migrating into the brain, thereby ameliorating cerebral inflammation following experimental intracerebral hemorrhage (ICH). We investigated the effects of fingolimod in two well-established murine models of ICH, implementing intrastriatal infusions of either bacterial collagenase (cICH) or autologous blood (bICH). Furthermore, we tested the long term neurological improvements by Fingolimod in a collagenase-induced rat model of ICH. Fingolimod, in contrast to vehicle administration alone, improved neurological functions and reduced brain edema at 24 and 72 hours following experimental ICH in CD-1 mice (n=103; p<0.05). Significantly fewer lymphocytes were found in blood and brain samples of treated animals when compared to the vehicle group (p<0.05). Moreover, fingolimod treatment significantly reduced the expression of intercellular adhesion molecule-1 (ICAM-1), interferon-γ (INF-γ), and interleukin-17 (IL-17) in the mouse brain at 72 hours post-cICH (p<0.05 compared to vehicle). Long-term neurocognitive performance and histopathological analysis were evaluated in Sprague-Dawley rats between 8 and 10 weeks post-cICH (n=28). Treated rats showed reduced spatial and motor learning deficits, along with significantly reduced brain atrophy and neuronal cell loss within the basal ganglia (p<0.05 compared to vehicle). We conclude that fingolimod treatment ameliorated cerebral inflammation, at least to some extent, by reducing the availability and subsequent brain infiltration of T-lymphocytes, which improved the short and long-term sequelae after experimental ICH in rodents.
Keywords: Intracerebral hemorrhage, Fingolimod, Lymphocyte, Inflammation, Neuroprotection, Brain Edema, Behavior
INTRODUCTION
Primary intracerebral hemorrhage (ICH) is a common consequence of hypertensive arteriosclerosis and amyloid angiopathy. Intracerebral bleeds associated with entities such as arteriovenous malformation, neoplasia, or coagulopathy are defined as secondary ICH (Sutherland and Auer 2006). Primary and secondary ICH account for approximately 15% of all strokes (Sutherland and Auer 2006). Although being a major cause of mortality and morbidity (Broderick et al. 1999), no effective therapy has yet been established to counteract the severity of this detrimental hemorrhagic stroke subtype (Gebel and Broderick 2000).
Cerebral inflammation plays an important role in injury progression after ICH (Aronowski and Hall 2005; Wang and Tsirka 2005; Xi et al. 2006; Wang and Dore 2007; Sansing et al. 2011). Evoked by the initial bleed, locally released chemotactic cytokines promote the migration of peripheral immune cells into the brain (Kane et al. 1992; Suzuki et al. 1995; Wang and Dore 2007; Ma et al. 2011). Furthermore, extravasated T-lymphocytes have been identified as being the driving force of neuroinflammation following experimental ischemic stroke (Yilmaz et al. 2006; Lo 2009; Shichita et al. 2009). Fingolimod (FTY720, Gilenya®), a sphingosine 1-phosphate (S1P) analog, interacts with the S1P receptor (S1PR) and regulates various cellular responses, including proliferation, apoptosis, and inflammation (Cohen and Chun 2011; Ontaneda and Cohen 2011; Soliven et al. 2011). In addition, fingolimod reduces the expression of S1PR-1 on T-lymphocytes, thus preventing their egress from primary and secondary lymphoid organs (Chiba 2005). By reducing the pool of circulating T-lymphocytes, fewer immune cells are expected to migrate into the brain after experimental ICH in rodents, thereby ameliorating cerebral inflammation and improving neurological functions. To test this theory, we used two well-established murine models of ICH, implementing intrastriatal infusions of either bacterial collagenase (cICH) or autologous blood (bICH). We further tested the long term functional improvement by Fingolimod in a collagenase-induced rat model of ICH.
MATERIAL AND METHODS
Animals
Male CD-1 mice, weighing 30–40g (Charles River Laboratory, Wilmington, MA) were used to evaluate behavioral performances, brain edema, peripheral leukocytes, brain-infiltrated T-lymphocytes, and cerebral pro-inflammatory proteins at 24 and/or 72 hours after ICH-induction (n=103). Male Sprague-Dawley rats, weighing 280–350g (Harlan, Livermore, CA) were used to evaluate short and long-term behavioral and neurocognitive performances, as well as histopathological changes at 10 weeks after ICH-induction (n=28). Animals were maintained on a 12:12 light/dark cycle and fed ad libitum. All procedures were in compliance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee at Loma Linda University.
Surgical procedures
CD-1 mice were subjected to experimental ICH, which was achieved by intrastriatal infusions of either bacterial collagenase (cICH) or autologous blood (bICH), as previously reported (Rosenberg et al. 1990; Wang et al. 2008; Ma et al. 2011). Briefly, mice (and rats) were anesthetized by intraperitoneal co-injection of ketamine (100 mg/kg) and xylazine (10mg/kg). Positioned prone, and secured to a stereotactic head frame (Kopf Instruments, Tujunga, CA), a cranial burr hole was made close to the right coronal suture, 1.7 mm lateral and 0.9 mm posterior to bregma. For cICH-induction, the 26 Gauge needle, of a glass syringe filled with bacterial collagenase (Type VII-S, Sigma-Aldrich, St Louis, MO), was lowered 3.7 mm ventrally through the burr hole and 0.075 U of bacterial collagenase dissolved in 0.5 μl PBS was infused at a rate of 2 μL/min, using a Nanomite Syringe Pump (Harvard Apparatus, Holliston, MA). A previously described double blood infusion model, with minor modifications, was utilized for bICH-induction in mice (Wang et al. 2008). A minimum of 30 μL of the rodent’s central tail artery blood was collected and transferred into a glass syringe. The 26 Gauge needle of the syringe was then lowered 3.0 mm ventrally through the burr hole and 5 μL of autologous blood was infused at a rate of 2 μL/min. Next, the needle was advanced 0.7 mm ventrally and after a waiting period of 5 minutes, 25 μL of blood was infused into the right striatum. The needle was left in place for additional 10 minutes after completed infusion in order to prevent the backflow of bacterial collagenase or blood along the needle tract. After withdrawing the needle at a rate of 1mm/min, the burr hole was sealed with bone wax and mice were allowed to recover under observation. Sham operation consisted of needle insertion only. The coordinates for the intrastriatal infusion of bacterial collagenase (0.2 U in 1 μl PBS) in Sprague-Dawley rats were 2.9 mm lateral (to the right), 0.2 mm rostral (relative to bregma), and 5.6 mm ventral (relative to the skull surface).
Fingolimod treatment and experimental groups
Fingolimod (2-amino-2-[2-(4-octylphenyl)ethyl]-1,3-propanediol, hydrochloride) was purchased from Cayman Chemical Co. (Ann Arbor, MI). The compound was dissolved in 5 % DMSO with 0.9 % saline and given by intraperitoneal injection at a dose of 1 mg/kg. Treatments were conducted as single dose fingolimod (1 hour after ICH-induction) or daily administrations (1 hour, 24 hours, and 48 hours after ICH-induction). Sham and vehicle animals received the same volumes of 5 % DMSO and saline. Animals were randomly divided into the following groups: Sham, Vehicle, Single Dose Fingolimod, and Daily Fingolimod.
Behavioral testing
Sensorimotor deficits were evaluated in mice at 24 and 72 hours after cICH or bICH, as well as in rats at 24 hours, 48 hours, 72 hours, and 10 days after cICH. Behavior testing was conducted in a blinded fashion during the animal’s light cycle. Behavioral scores of sensorimotor tests were converted into percentage of sham performance.
Modified Garcia Neuroscore
As previously reported, the composite modified Garcia Neuroscore, is a sensorimotor assessment, consisting of 7 individual test, which examine the rodent’s spontaneous activity, body sensation, vibrissae proprioception, fore and hind limb symmetry, ability of lateral turning, forelimb outstretching, and ability of climbing (Garcia et al. 1995; Krafft et al. 2012a). According to their performance, animals received a score for each sub-test ranging from 0 (worst performance) to 3 (best performance). The Garcia Neuroscore was calculated as the sum of all sub-tests.
Wire hang test
To perform the wire hang test (Wu et al. 2010), mice were placed midway on a wire (50 × 0.15 cm), which was connected between two platforms, approximately 40 cm above the surface. The appropriate usage of all four limbs as well as the distance moved along the wire was monitored. A score between 0 (worst performance) and 5 (best performance) was given for each trial.
Beam balance test
To perform the beam balance test (Zausinger et al. 2000), mice were placed in the middle of a horizontal rod (90 × 1 cm) that connected two platforms, approximately 40 cm above the surface. The animal’s ability to balance along the rod, and the covered distanced was recorded. A score between 0 (worst performance) and 5 (best performance) was given for each trial.
Forelimb use asymmetry test
An open Plexiglas cylinder (20 × 30 cm) was utilized to assess forelimb use asymmetry as previously described (Hua et al. 2002). Briefly, mice were placed into the cylinder for a time period of 5 minutes and exploratory movements of the forelimbs along the vertical cylinder wall were monitored. The number of contacts of the left forepaw (contralateral side to ICH) and the cylinder wall, were recorded as percentage of all forepaw movements (ipsi-, contra- and bilateral).
Corner test
To perform the corner test (Hua et al. 2002), mice were permitted to approach a 30 degree corner that was made out of two Plexiglas walls. In order to exit the corner, animals had to turn either to the right or to the left side, usually by rearing along the corner wall. The choice of turning side was recorded for 10 trials per test day, and a score was calculated as number of left turns/all turns × 100 (%).
Paw placement test
Mice and rats were evaluated for their ability to respond to a vibrissae-elicited excitation, by forward movement of their paw (Hua et al. 2002). To execute this test, animals were held by the trunk ensuring that the forelimbs were not restrained. The rodent was then oriented in parallel to a table top and slowly moved vertically until the vibrissae on one side touched the surface. Adequate forward movements of the ipsilateral paw in response to vibrissae stimulation were recorded out of 10 trials.
Brain edema measurement
Brain edema (brain water content) was measured in mice at 24 and 72 hours after cICH, as well as at 72 hours after bICH, using the wet brain weight/dry brain weight method as previously described (Tang et al. 2004). Mice were decapitated under deep isoflurane anesthesia, and brains were removed and immediately divided into five parts: ipsilateral and contralateral basal ganglia, ipsilateral and contralateral cortex, and cerebellum. All samples were weighed with an electronic analytical balance (AE100; Mettler Instrument Co, Columbus, OH) to obtain a wet weight (WW). Tissues were then dried at 100°C for 24 hours and the dry weights (DW) were determined. The brain water content (%) was calculated as (WW-DW)/WW × 100.
Peripheral leukocyte count
Peripheral leukocyte counts were conducted in mice at 24 and 72 hours post-cICH. Under deep isoflurane anesthesia, blood samples were collected via retro-ocular puncture. The samples were then transferred into 0.5 ml reaction tubes and 0.3 μl of heparin was added. Leukocytes were isolated by density centrifugation and the quantities of lymphocytes, monocytes, granulocytes, and total leukocytes were determined.
Immunofluorescence and quantification of T-lymphocytes
At 72 hours after cICH, mice under deep isoflurane anesthesia, were transcardially perfused with ice cold PBS (40ml), followed by 4 % paraformaldehyde (40 ml). Following that, brain samples were collected, formalin fixed (at 4°C for 2 days), and dehydrated with 30 % sucrose in PBS (at 4°C for 2 days). Frozen coronal brain slices (10 μm) were cut using a cryostat (CM3050S; Leica Microsystems) and placed onto poly-lysine coated glass slides (Richard Allen Scientific, Kalamazoo, MI). Immunofluorescence staining was performed as previously described (Ostrowski et al. 2005) using the following primary antibodies: anti-CD3 and anti intercellular adhesion molecule-1 (anti-ICAM-1) (Abcam, Cambridge, MA). Secondary antibodies were obtained from Jackson Immunoresearch Laboratories (West Grove, PA). Samples were evaluated using fluorescent microscopy and Magna Fire SP imaging system (Olympus). Immunoreactive cells were counted as previously described (Wang and Dore 2008). Briefly, the CD3 positive cell numbers were counted in 12 locations per mouse (three sections per mouse, four fields per section, n=5, microscopic field × 20). The results were then averaged and expressed as positive cells/mm2.
Western blot analysis of pro-inflammatory proteins
At 72 hours after cICH, mice under deep isoflurane anesthesia, were transcardially perfused with ice cold PBS (40 ml), and the ipsilateral hemispheres were collected and processed as previously described (Ma et al. 2011). Equal amounts of protein (50 μg per sample) were separated by SDS-PAGE and transferred onto nitrocellulose membranes, which were incubated with the following primary antibodies: anti-ICAM-1, anti interferon-γ (anti-IFN-γ) (Santa Cruz Biotechnology, Santa Cruz, CA), anti interleukin-17 (anti-IL-17), and anti-β-actin (Abcam, Cambridge, MA). The membranes were then incubated with the appropriate secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Immunoblots were visualized with the ECL Plus chemiluminescence reagent kit (Amersham Bioscience, Arlington Heights, IL) and semi-quantitatively analyzed using Image J software (4.0, Media Cybernetics, Silver Springs, MD). Results were expressed as relative density, normalized to mean density of sham.
Long-term behavioral testing
Sensorimotor deficits in rats were evaluated via the paw placement test (see above) at day 1 to 3, and at 10 weeks after cICH-induction. Furthermore, the rotarod test was performed at 8 weeks post-cICH to assess sensorimotor coordination and balance. To perform this test, rats were placed onto a rotarod (Columbus Instruments, Columbus, OH) and rotations were started at either 4 or 10 RPM with an acceleration of 2 RPM for every 5 seconds. The latency of falling from the rotarod was recorded using a photobeam circuit.
The Morris water maze test was additionally performed at 8 weeks post-cICH to assess learning and memory abilities in rats (Hartman et al. 2008; Lekic et al. 2010). This test required the finding of a slightly submerged platform in a pool of water (diameter: 110 cm). An overhead camera recorded the swim path of each rat, which allowed for quantification of swim distance as well as spatial distance from the platform, by a computerized tracking system (Noldus Ethovision, Tacoma, WA). Water maze testing was done over a 4 day period. Following the cued trials (visible platform) on day 1, the platform was submerged for all subsequent testing (day 2–4). The location of the platform was changed at the beginning of every testing day and the rats were released into the water on the several sites around the platform for 5 blocks of testing. At the end of day 2–4, each animal was subjected to a “probe” trial, in which the platform was removed from the water maze. The distance between the rat and the probe (where the platform used to be) was recorded for 60 seconds.
Histopathological analysis
At 10 weeks after cICH, rats under deep isoflurane anesthesia, were transcardially perfused with ice cold PBS (80 ml), followed by 4% paraformaldehyde (80 ml). Next, brain samples were collected, formalin fixed (at 4°C for 3 days), and dehydrated with 30 % sucrose in PBS (at 4°C for 3 days). Frozen coronal brain slices (10 μm) were cut between 1 mm anterior and posterior from the center of the hemorrhage, using a cryostat (CM3050S; Leica Microsystems) and placed onto poly-lysine coated glass slides (Richard Allen Scientific, Kalamazoo, MI). All brain samples were stained with cresyl violet as previously described (Kusaka et al. 2004; Ostrowski et al. 2006). Morphometric analysis was conducted using computer-assisted (Image J 4.0, Media Cybernetics, Silver Spring, MD) hand delineation of the following areas: caudate putamen, cerebral cortex, ventricle and corpus callosum. The borders of cerebral structures were based on criteria previously defined from a stereologic study using optical dissector principles (Oorschot 1996). The volumetric lesion area (cavity and cellular debris) and brain atrophy (ventriculomegaly) were calculated using the following equations: Volume of tissue lost= Volume of uninjured hemisphere − Volume of injured hemisphere, Volume of hemisphere= Average [(area of coronal section of the hemisphere)−(lesion area + ventricle area)] × section interval × number of sections (MacLellan et al. 2006; MacLellan et al. 2008).
Neuronal density was estimated in accordance to established methods (Peeling et al. 2001a; Felberg et al. 2002; MacLellan et al. 2008). Striatal neurons were counted at 400 × magnification in five areas (250 μm × 250 μm grids) per hemisphere of the section with maximal lesion diameter. The relative loss of neurons was calculated by subtracting the average neuronal counts from ipsilateral and contralateral sides. Loss of white matter was assessed by measuring the corpus callosum area in each hemisphere and comparing the opposite sides.
Statistical analysis
All data were evaluated with the statistical software Sigma Plot 10.0 (Systat Software, Inc., San Jose, CA). Data were expressed as mean ± SEM and analyzed with One-way ANOVA and Tukey post hoc test. Behavior data, expressed as mean percentage ± SEM of sham performance were statistically analyzed using Kruskal-Wallis One-way ANOVA on Ranks, followed by the Student-Newman-Keuls Method. For long-term behavioral analysis, repeated-measures ANOVA was used when appropriate. A P-value of < 0.05 was considered statistically significant.
RESULTS
Fingolimod improved sensorimotor function and reduced brain edema after experimental ICH
Behavioral performances and brain water content were evaluated in mice at 24 and 72 hours after ICH-induction. Significant sensorimotor deficits were observed at 24 hours after cICH in all conducted behavioral test (Garcia Neuroscore, wire hang, beam balance, forelimb use asymmetry, corner, and paw placements tests), (p<0.05, Fig. 1A). However, a single dose of fingolimod (1 mg/kg) improved behavioral performances in all mentioned tests (p<0.05, compared to vehicle), with the exception of the forelimb use asymmetry test (p>0.05, compared to vehicle). Improved behavior was paralleled with significantly decreased brain edema in the ipsilateral basal ganglia of treated animals when compared to vehicle administration (p<0.05, Fig. 1B).
Figure 1.

Effects of single fingolimod treatment on behavioral performances (A) and brain edema (B) at 24 hours after collagenase-induced intracerebral hemorrhage (cICH). Effects of single and daily fingolimod treatments on behavioral performances (C) and brain edema (D) at 72 hours after cICH. Effects of daily fingolimod treatment on behavioral performances (E) and brain edema (F) at 72 hours after intrastriatal blood infusion (bICH). Behavior data are expressed as mean percentage of sham values ± SEM. Brain edema values are expressed as mean ± SEM. N=7 mice per group. * p<0.05 compared to sham, † p<0.05 compared to vehicle. Contra: contralateral, Ipsi: ipsilateral, Garcia: Garcia Neuroscore, WH: wire hang test, BB: beam balance test, FUA: forelimb use asymmetry test, CT: corner test, PP: paw placement test.
The battery of behavioral tests (see above) was also conducted in mice at 72 hours post-cICH (Fig. 1C). At this time point, single dose as well as daily fingolimod treatment significantly improved the Garcia Neuroscore as well as performances on the corner and paw placement test (p<0.05, compared to vehicle). Moreover, daily fingolimod treatment showed a significantly greater improvement on the corner test performance than single dose fingolimod (p<0.05). Both treatment groups reduced brain edema in the ipsilateral cortex and basal ganglia at 72 hours post-cICH (p<0.05, compared to vehicle); however, only daily fingolimod treatment significantly reduced brain edema in the contralateral basal ganglia (p<0.05, compared to vehicle, Fig. 1D). To verify these results in a second model of experimental ICH, we subjected mice to bICH and administered the more effective, daily fingolimod treatment regimen. Sensorimotor function was then examined using the behavioral tests that showed improved performances in treated mice at 72 hours post-cICH, including the Garcia Neuroscore, corner, and paw placement test. Daily fingolimod treatment significantly improved behavioral performances at 72 hours post-bICH in all aforementioned tests (p<0.05, compared to vehicle, Fig. 1E). Furthermore, daily fingolimod treatment significantly reduced brain edema in the ipsilateral basal ganglia at 72 hours after induction of bICH (P<0.05, compared to vehicle, Fig. 1F).
Fingolimod reduced the number of circulating lymphocytes
Blood samples were collected from mice at 24 and 72 hours post-cICH to quantify the amount of peripheral lymphocytes, monocytes, and granulocytes. The number of all mentioned white blood cells did not change significantly at 24h after experimental ICH (p>0.05, compared to sham, Fig. 2A). However, administration of a single dose fingolimod reduced the number peripheral lymphocytes at 24 hours post-cICH (p<0.05, compared to sham and vehicle). Interestingly, a significant rise in the quantity of lymphocytes and total leukocytes was observed at 72 hours following cICH (p<0.05, compared to sham, Fig. 2B), which was reversed following single and daily fingolimod treatment (p<0.05, compared to vehicle). Moreover, daily fingolimod treatment caused a significant reduction in peripheral lymphocytes when compared to sham animals (p<0.05).
Figure 2.
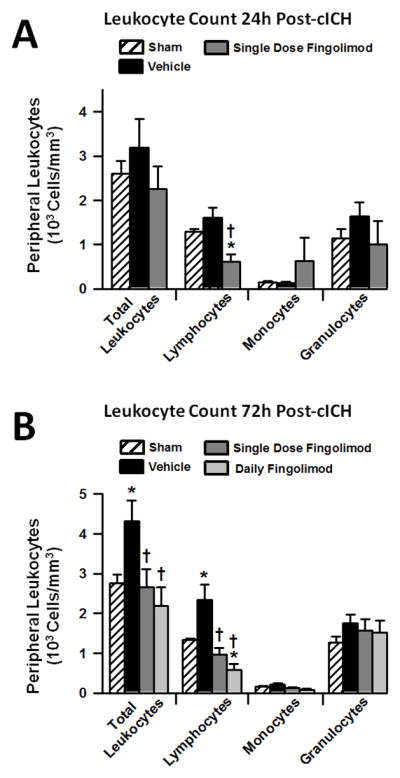
Effects of single fingolimod treatment on peripheral blood leukocyte counts at 24 hours (A) and effects of single and daily fingolimod treatments on peripheral blood leukocyte counts at 72 hours (B) after cICH in mice. Values are expressed as mean ± SEM. N=7 mice per group. * p<0.05 compared to sham, † p<0.05 compared to vehicle.
Fingolimod reduced the number of brain-infiltrated T-lymphocytes after experimental ICH
Immunofluorescence staining and quantification of CD3 positive cells within the brain was performed in sham, vehicle, and daily fingolimod treated mice, which were euthanatized at 72 hours after surgery. At this time point CD3 positive cells were present in the perihematomal region (Fig. 3A). Significantly more CD3 positive cells were found in both cICH groups (p<0.05, compared to sham); however, daily fingolimod treatment reduced their number within the brain, when compared to vehicle administration (p<0.05, Fig. 3B). Additionally, intercellular adhesion molecule-1 (ICAM-1) was co-expressed in CD3 positive cells, and daily fingolimod treatment seemed to reduce the expression of ICAM-1 positive/CD3 positive cells (Fig. 3C).
Figure 3.
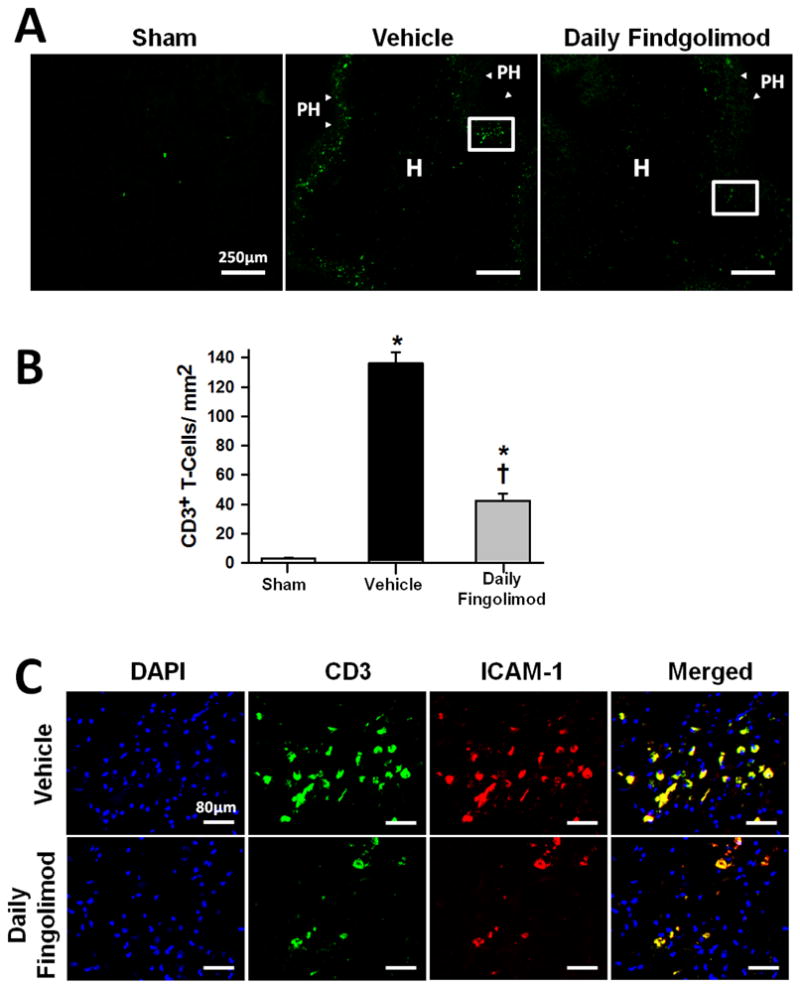
Effect of daily fingolimod treatment on brain infiltration of T-lymphocytes at 72 hours after cICH-induction or sham surgery in mice (A). Representative photomicrographs showing CD3 positive cells (arrow heads) infiltrating the perihematomal (PH) area, and to a lesser extent the hemorrhage (H) site. White box depicts the area magnified below. Bar graph showing CD3 positive cell quantification (B). Representative photomicrographs showing CD3 positive cells (green) with ICAM-1 (red) co-localization. DAPI nucleic acid stain (blue) marked DNA (C). Data in bar graphs are expressed as mean ± SEM. N=5 mice per group. * p<0.05 compared to sham, † p<0.05 compared to vehicle.
Fingolimod reduced the expression of pro-inflammatory mediators within the brain after experimental ICH
ICAM-1, interferon-γ (IFN-γ), and interleukin-17 (IL-17) expressions, within the ipsilateral hemispheres, of sham, vehicle, and daily fingolimod treated animals were quantified via Western blot at 72 hours after cICH-induction or sham surgery. IFN-γ expression was increased at 72 hours after cICH (Fig. 4A); however, fingolimod treatment reduced the expression of IFN-γ effectively (p<0.05, compared to vehicle, Fig. 4B). No significant increase of ICAM-1 was seen after cICH-induction in vehicle animals (p>0.05, compared to sham, Fig. 4C); however, daily fingolimod treatment reduced ICAM-1 expression after cICH when compared to vehicle animals (p<0.05). IL-17 expression was significantly increased after cICH-induction (p<0.05, compared to sham) and daily fingolimod treatment reversed this increase effectively (p<0.05 compared to vehicle, Fig. 4 D).
Figure 4.

Representative Western blots (A) and bar graphs showing densitometric quantifications of the inflammatory markers interferon gamma (IFN-γ) (B), inter-cellular adhesion molecule-1 (ICAM-1) (C), and interleukin 17 (IL-17) (D) at 72 hours after cICH-induction or sham surgery. The band density values were calculated as a ratio of β-actin, normalized to sham. Data are expressed as mean fold change ± SEM. N=6 mice per group.* p<0.05 compared to sham, † p<0.05 compared to vehicle.
Fingolimod improved short-term sensorimotor function and long-term motor coordination in rats after experimental ICH
Within the first 2 days after surgery, cICH rats in vehicle as well as in both treatment groups showed significant sensorimotor impairments evaluated with the paw placement test (p<0.05, compared to sham, Fig. 5A). Single and daily fingolimod treatment significantly improved performance on the paw placement test 1 and 2 days after cICH (p<0.05, compared to vehicle). Some recovery of sensorimotor deficits was observed at day 3 after cICH. At this time point no significant differences in test performance was found between the vehicle and both treatment groups (p>0.05). Continuing sensorimotor recovery in rats was observed at 8 weeks after cICH-induction (Fig. 5A).
Figure 5.

Effects of single or daily fingolimod treatment on behavioral performances of the paw placement test at 1, 2, and 3 days and at 10 weeks after cICH-induction or sham surgery in rats (A). Effects of single or daily fingolimod treatment on rotarod performances at 8 weeks after cICH-induction or sham surgery in rats (B). Behavior data of the paw placement test are expressed as mean percentage of sham values ± SEM. Behavior data of the rotarod performance are expressed as mean ± SEM. N=7 rats per group. * p<0.05 compared to sham, † p<0.05 compared to vehicle.
Motor coordination in rats was evaluated via the rotarod test at 8 weeks after cICH-induction or sham surgery. At a starting rotation velocity of 4 RPM with a continuous acceleration of 2 RPM for every 5 seconds, all cICH rats demonstrated earlier falls from the rotating cylinder when compared to sham animals (p<0.05, Fig. 5B). Interestingly, both single and daily treated rats showed a significantly improved performance (p<0.05, compared to vehicle) during the more difficult trials starting at a rotation velocity of 10 RPM with continuous accelerations of 2 RPM for every 5 seconds.
Fingolimod improved long-term cognitive function in rats after experimental ICH
Cognitive function in rats was evaluated using the water maze test starting at 8 weeks after induction of cICH or sham surgery. All animals subjected to cICH showed significant spatial learning and memory deficits when compared to the sham group (p<0.05, Fig. 6A, B, C). Although there were notable differences between treated and vehicle animals on individual blocks of testing, a significance was not reached (p>0.05, Fig. 6A). However, when overall performances were analyzed across the blocks of testing, single and daily fingolimod treated rats showed significantly reduced distances swam to find the submerged platform, which indicates an improvement in spatial learning ability (p<0.05, compared to vehicle, Fig. 6B).
Figure 6.

Effects of single or daily fingolimod treatment on cognitive performance during the water maze test at 8 weeks after cICH-induction or sham surgery in rats. Bar graph showing the distances swam before finding the submerged platform for each the 5 blocks of daily trials (A). Bar graph showing the overall average distance swam before finding the submerged platform (B). Bar graph showing the average proximity to the previous target location during the probe trials (C). Values are expressed as mean ± SEM. N=7 rats per group.* p<0.05 compared to sham, † p<0.05 compared to vehicle.
At the end of each testing day, the platform was removed from the water maze, and the average distance between each rat in the water maze and the prior site of the platform (probe) was used to evaluate memory ability. Vehicle animals exhibited significant memory deficits, as they were further away from the target during the 1 minute trial (p<0.05, compared to sham, Fig. 6C). Daily fingolimod treated rats tended to be closer to the target site (p=0.079, compared to vehicle), indicating tendentially improved spatial memory function.
Fingolimod ameliorated brain atrophy in rats after experimental ICH
At 10 weeks after cICH-induction, brain samples were prepared for volumetric analysis of tissue loss. Single and daily fingolimod treatments tended to improve the gross tissue loss in the ipsilateral hemisphere, compared to vehicle (p>0.05, Fig. 7A). However, significant reductions in brain atrophy were found in the caudate putamen of both treatment groups (p<0.05, compared to vehicle, Fig. 7B). White matter loss tended to be reduced in daily fingolimod treated animals (P>0.05, Fig. 7C). Furthermore, a significant reduction in neuronal density loss was observed in the caudate putamen of those rats that received the daily fingolimod treatment regime (p<0.05, compared to vehicle, Fig. 7D). Additionally, ventriculomegaly seemed to be reduced in both treatment groups when compared to ventricle size of vehicle animals (P>0.05, Fig. 7E).
Figure 7.

Effects of single or daily fingolimod treatment on cerebral histopathology at 10 weeks after cICH-induction in rats. Bar graph showing total tissue loss of the ipsilateral brain hemisphere (A). Bar graph showing percent atrophy of the caudate putamen and cerebral cortex based on the volumetric difference of the contralateral side (B). Bar graph showing white-matter loss measured as percent volumetric difference of the corpus callosum between the ipsilateral and contralateral hemispheres (C). Bar graph depicting loss of neuronal density in the ipsilateral caudate putamen reported as the difference in cell counts between ipsilateral and contralateral regions (D). Representative microphotographs of Nissl stained brain sections illustrating ventriculomegaly at 10 weeks following cICH-induction (E). Values are expressed as mean ± SEM. N=7 rats per group. † p<0.05 compared to vehicle.
DISCUSSION
In this present study, we hypothesized fingolimod treatment would reduce the number of T-lymphocytes migrating into the brain following experimental ICH, thereby ameliorating cerebral inflammation and result in improved neurobehavioral and cognitive outcomes in rodents. Aiming to follow the recommendations of the Stroke Therapy Academic Industry Roundtable (STAIR) for preclinical investigations, we implemented 2 different experimental models (cICH, bICH), 2 species (mice, rats), 2 treatment regimes (single dose, daily), as well as multiple behavioral and histopathological evaluations (Fisher et al. 2009).
Primary ICH most commonly affects the basal ganglia, and injuries to this brain region have been shown to cause considerable sensorimotor deficits as well as learning and memory problems in patients (Bhatia and Marsden 1994; Hochstenbach et al. 1998; Chung et al. 2000; Werring et al. 2004; Su et al. 2007). To achieve a translational approach, we induced reproducible hemorrhagic lesions within the basal ganglia of rodents through intrastriatal infusions of either bacterial collagenase or autologous (arterial) blood. The collagenase infusion model mimics a spontaneous bleed that gradually develops over several hours, as seen in 14–20% of all ICH patients (Rosenberg et al. 1990; MacLellan et al. 2008). Collagenase, a protease, degrades the basement membrane of cerebral capillaries, causing vessel rupture and consequent blood extravasation into the surrounding brain tissue (Rosenberg et al. 1990). Although collagenase, itself, has been thought to exaggerate cerebral inflammation, recent studies failed to prove this theory (Matsushita et al. 2000; Wang et al. 2003; Chu et al. 2004). The forte of the intrastriatal blood infusion model is that no exogenous factors are used to induce experimental ICH in rodents. This model mirrors a rapidly developing bleed without provoking an actual vessel rupture (Krafft et al. 2012b).
We found fingolimod treatment effectively ameliorated brain edema as well as behavioral deficits in rodents subjected to either cICH or bICH. Previous studies reported a close relationship between the degree of perihematomal brain edema and poor outcome in ICH patients. Accordingly, pharmacological therapies that effectively reduce cerebral edema formation are expected to improve the neurological deficits in ICH patients (Aronowski and Hall 2005). Wei et al. found that fingolimod administered at 30 minutes after transient ischemic stroke in rats, reduced brain edema at 24 hours after vessel occlusion (Wei et al. 2011). Our results also indicated daily fingolimod treatment (given at 1 hour, 24 hours, and 48 hours post-cICH) reduced brain edema and improved sensorimotor deficits to a greater extent than single dose administration. For that reason we used the daily treatment regime for all subsequent investigations (leukocyte counts, immunohistochemistry, Western blot, and long-term behavior).
The S1P analog, fingolimod (Gilenya®), has recently been approved by the FDA for oral treatment of relapsing/remitting multiple sclerosis. This compound is rapidly phosphorylated within the body before interfering with its S1PR (S1PR-1, -3, -4, or -5) (Brinkmann et al. 2002; Meno-Tetang et al. 2006). Fingolimod has also been shown to prevent the egress of lymphocytes from primary and secondary lymphoid organs (Chiba 2005). More specifically, fingolimod, in contrast to endogenous S1P ligands, causes a prolonged downregulation of the S1PR-1, a receptor required for adequate egress of lymphocytes from peripheral storages into circulation (Chiba 2005; Oo et al. 2007). The time course of S1P production following experimental stroke has been measured following photochemical induced brain ischemia where, Kimura et al. reported that S1P initially decreases up to day 3, after which it increases and peaks at day 14, remaining elevated thereafter (Kimura et al. 2008). As anticipated, our results demonstrated a significant reduction of circulating lymphocytes following fingolimod treatment.
Recent studies have identified a number of T-lymphocyte subtypes, such as Th1, Th17, gamma-delta T-cells, and natural killer cells, as important mediators of cerebral inflammation following experimental ischemic stroke (Yilmaz et al. 2006; Lo 2009; Shichita et al. 2009). These cells secrete pro-inflammatory cytokines, such as INF-γ and IL-17, thus enhancing the immunostimulatory response following experimental stroke (Neumann et al. 1997). The number of cytotoxic T-lymphocytes (CD8 positive cells) in the rat’s brain significantly increased as early as 24 hours after ICH-induction, with a maximum reached at 3–7 days (Xue and Del Bigio 2003). For that reason, we chose to evaluate the amount of circulating and perihematomal lymphocytes, as well as the expression of pro-inflammatory markers, at 72 hours after ICH-induction. In patients, however, peripheral immune cells start to infiltrate the brain between 6 and 12 hours after occurrence of symptoms, and a positive correlation between the amount of brain-infiltrated immune cells and neuronal apoptosis following ICH has been established (Guo et al. 2006). Interestingly, mice deficient in either CD4 positive and/or CD8 positive T-lymphocytes exhibited protection against brain inflammation and secondary neuronal injury following ischemic stroke (Yilmaz et al. 2006). Furthermore, inhibition of T-lymphocyte activation with the immunosuppressive drug tacolimus resulted in decreased cell death and improved functional outcome in rats subjected to experimental ICH (Peeling et al. 2001b).
We assumed fingolimod, by trapping immune cells in their peripheral storage sites, would also reduce the amount of brain-infiltrated T-lymphocytes after experimental ICH. Our results showed not only a reduced number of perihematomal T-lymphocytes, which were identified with the specific immunohistochemical marker CD3, but also decreased expressions of INF-γ and IL-17 within the brain of fingolimod treated ICH animals. This present study is the first to show an increased expression of IL-17 within the brain of rodents following experimental ICH. The importance of this particular cytokine needs to be further investigated in patients following hemorrhagic stroke. Fingolimod also reduced the expression of ICAM-1 within the brain of ICH animals. In a previous study, fingolimod was found to decrease the expression of ICAM-1 in brain endothelial cells (Wei et al. 2011). Based on this finding we suggest that a reduced capacity for immune cell adhesion may play an additional role in decreasing the migration of T-lymphocytes into the injured brain. ICAM-1 was co-expressed on CD3 positive cells, which was also observed in an experimental model of Parkinson’s disease (Brochard et al. 2009).
When evaluating the efficiency of potential therapeutic agents in preclinical stroke research, long-term functional recovery must be conducted in order to demonstrate a stable protection rather than just a short-acting amelioration of the lesion progression (Freret et al. 2011). This present study demonstrated the long-term efficiency of fingolimod, which improved delayed sensorimotor and cognitive deficits in rats subjected to experimental ICH. The improvement of long-term behavior was paralleled with reduced atrophy and increased neuronal density within the basal ganglia.
Several potential limitations of this study need to be discussed. First, although inhibition of the immune response following experimental ICH has been linked to improved outcomes in rodents (Kane et al. 1992; Del Bigio et al. 1999; Peeling et al. 2001b; Titova et al. 2008), inflammatory cells are also needed for phagocytosis of cellular debris and for induction of diverse repair mechanisms (Zhao et al. 2007). These potentially protective effects of the adaptive immune system following hemorrhagic stroke have not been evaluated in this present study. Second, while this study focuses mainly on the immunomodulatory effects of fingolimod, several other neuroprotective mechanisms have been linked to this pharmacological agent. For instance, fingolimod treatment resulted in reduced neuronal apoptosis in a rat model of middle cerebral occlusion (Hasegawa et al. 2010). Additionally, fingolimod demonstrated endothelial-barrier enhancement capacity, through S1PR activation, which stimulated the transcription of adherens junction proteins, thereby enhancing blood-brain barrier integrity (Sanchez et al. 2003; Brinkmann et al. 2004). The observed improvements in our experimental animals were most likely due to a combined protective effect involving a reduced immune response, anti-apoptotic effects and blood-brain barrier stabilization. Thirdly, a previous investigation by Liesz et al. found that fingolimod reduced post-ischemic lymphocyte brain infiltration in a murine model of permanent cerebral ischemia; however, an improved outcome was not observed in experimental animals (Liesz et al. 2011). The latter finding suggests variations, possibly due to experimental conditions. Lastly, fingolimod has been extensively studied in animal models of ischemic stroke (Czech et al. 2009; Shichita et al. 2009; Hasegawa et al. 2010; Wei et al. 2011); nonetheless, to date, we are the only group investigating the effects of fingolimod following experimental ICH.
In conclusion, this present study demonstrated that fingolimod treatment (a) reduced acute sensorimotor deficits and perihematomal brain edema in two experimental models of ICH in mice, (b) decreased the amount of peripheral and brain-infiltrated lymphocytes, as well as the protein expressions of IFN-γ, IL-17, and ICAM-1, and (c) ameliorated long-term neurocognitive deficits and brain atrophy following experimental ICH.
Highlights.
We applied Fingolimod, an S1P agonist, in two murine experimental stroke models.
Fingolimod reduced short-term brain edema and neurological deficits after ICH.
T-lymphocytes and cerebral ICAM-1, IFN-γ, and IL-17 were reduced by Fingolimod.
Fingolimod reduced long-term behavioral deficits and brain tissue damage.
Acknowledgments
Source of funding
This work was supported by NIH grant NS053407 to Dr. John H. Zhang
Footnotes
Disclosures
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res. 2005;27:268–279. doi: 10.1179/016164105X25225. [DOI] [PubMed] [Google Scholar]
- Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain. 1994;117(Pt 4):859–876. doi: 10.1093/brain/117.4.859. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Cyster JG, et al. FTY720: sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am J Transplant. 2004;4:1019–1025. doi: 10.1111/j.1600-6143.2004.00476.x. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Davis MD, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–21457. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- Brochard V, Combadiere B, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick JP, Adams HP, Jr, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: A statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke. 1999;30:905–915. doi: 10.1161/01.str.30.4.905. [DOI] [PubMed] [Google Scholar]
- Chiba K. FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol Ther. 2005;108:308–319. doi: 10.1016/j.pharmthera.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Chu K, Jeong SW, et al. Celecoxib induces functional recovery after intracerebral hemorrhage with reduction of brain edema and perihematomal cell death. J Cereb Blood Flow Metab. 2004;24:926–933. doi: 10.1097/01.WCB.0000130866.25040.7D. [DOI] [PubMed] [Google Scholar]
- Chung CS, Caplan LR, et al. Striatocapsular haemorrhage. Brain. 2000;123(Pt 9):1850–1862. doi: 10.1093/brain/123.9.1850. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Chun J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69:759–777. doi: 10.1002/ana.22426. [DOI] [PubMed] [Google Scholar]
- Czech B, Pfeilschifter W, et al. The immunomodulatory sphingosine 1-phosphate analog FTY720 reduces lesion size and improves neurological outcome in a mouse model of cerebral ischemia. Biochem Biophys Res Commun. 2009;389:251–256. doi: 10.1016/j.bbrc.2009.08.142. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR, Yan HJ, et al. Effect of fucoidan treatment on collagenase-induced intracerebral hemorrhage in rats. Neurol Res. 1999;21:415–419. doi: 10.1080/01616412.1999.11740953. [DOI] [PubMed] [Google Scholar]
- Felberg RA, Grotta JC, et al. Cell death in experimental intracerebral hemorrhage: the “black hole” model of hemorrhagic damage. Ann Neurol. 2002;51:517–524. doi: 10.1002/ana.10160. [DOI] [PubMed] [Google Scholar]
- Fisher M, Feuerstein G, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freret T, Schumann-Bard P, et al. On the importance of long-term functional assessment after stroke to improve translation from bench to bedside. Exp Transl Stroke Med. 2011;3:6. doi: 10.1186/2040-7378-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JH, Wagner S, et al. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. discussion 635. [DOI] [PubMed] [Google Scholar]
- Gebel JM, Broderick JP. Intracerebral hemorrhage. Neurol Clin. 2000;18:419–438. doi: 10.1016/s0733-8619(05)70200-0. [DOI] [PubMed] [Google Scholar]
- Guo FQ, Li XJ, et al. Study of relationship between inflammatory response and apoptosis in perihematoma region in patients with intracerebral hemorrhage. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2006;18:290–293. [PubMed] [Google Scholar]
- Hartman RE, Rojas H, et al. Long-term behavioral characterization of a rat model of intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:125–126. doi: 10.1007/978-3-211-09469-3_25. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Suzuki H, et al. Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke. 2010;41:368–374. doi: 10.1161/STROKEAHA.109.568899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstenbach J, van Spaendonck KP, et al. Cognitive deficits following stroke in the basal ganglia. Clin Rehabil. 1998;12:514–520. doi: 10.1191/026921598666870672. [DOI] [PubMed] [Google Scholar]
- Hua Y, Schallert T, et al. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002;33:2478–2484. doi: 10.1161/01.str.0000032302.91894.0f. [DOI] [PubMed] [Google Scholar]
- Kane PJ, Modha P, et al. The effect of immunosuppression on the development of cerebral oedema in an experimental model of intracerebral haemorrhage: whole body and regional irradiation. J Neurol Neurosurg Psychiatry. 1992;55:781–786. doi: 10.1136/jnnp.55.9.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Ohmori T, et al. Antagonism of sphingosine 1-phosphate receptor-2 enhances migration of neural progenitor cells toward an area of brain. Stroke. 2008;39:3411–3417. doi: 10.1161/STROKEAHA.108.514612. [DOI] [PubMed] [Google Scholar]
- Krafft PR, Altay O, et al. alpha7 nicotinic acetylcholine receptor agonism confers neuroprotection through GSK-3beta inhibition in a mouse model of intracerebral hemorrhage. Stroke. 2012a;43:844–850. doi: 10.1161/STROKEAHA.111.639989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krafft PR, Rolland WB, et al. Modeling intracerebral hemorrhage in mice: injection of autologous blood or bacterial collagenase. J Vis Exp. 2012b doi: 10.3791/4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusaka I, Kusaka G, et al. Role of AT1 receptors and NAD(P)H oxidase in diabetes-aggravated ischemic brain injury. Am J Physiol Heart Circ Physiol. 2004;286:H2442–2451. doi: 10.1152/ajpheart.01169.2003. [DOI] [PubMed] [Google Scholar]
- Lekic T, Hartman R, et al. Protective effect of melatonin upon neuropathology, striatal function, and memory ability after intracerebral hemorrhage in rats. J Neurotrauma. 2010;27:627–637. doi: 10.1089/neu.2009.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Sun L, et al. FTY720 reduces post-ischemic brain lymphocyte influx but does not improve outcome in permanent murine cerebral ischemia. PLoS One. 2011;6:e21312. doi: 10.1371/journal.pone.0021312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH. T time in the brain. Nat Med. 2009;15:844–846. doi: 10.1038/nm0809-844. [DOI] [PubMed] [Google Scholar]
- Ma Q, Manaenko A, et al. Vascular adhesion protein-1 inhibition provides antiinflammatory protection after an intracerebral hemorrhagic stroke in mice. J Cereb Blood Flow Metab. 2011;31:881–893. doi: 10.1038/jcbfm.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLellan CL, Auriat AM, et al. Gauging recovery after hemorrhagic stroke in rats: implications for cytoprotection studies. J Cereb Blood Flow Metab. 2006;26:1031–1042. doi: 10.1038/sj.jcbfm.9600255. [DOI] [PubMed] [Google Scholar]
- MacLellan CL, Silasi G, et al. Intracerebral hemorrhage models in rat: comparing collagenase to blood infusion. J Cereb Blood Flow Metab. 2008;28:516–525. doi: 10.1038/sj.jcbfm.9600548. [DOI] [PubMed] [Google Scholar]
- Matsushita K, Meng W, et al. Evidence for apoptosis after intercerebral hemorrhage in rat striatum. J Cereb Blood Flow Metab. 2000;20:396–404. doi: 10.1097/00004647-200002000-00022. [DOI] [PubMed] [Google Scholar]
- Meno-Tetang GM, Li H, et al. Physiologically based pharmacokinetic modeling of FTY720 (2-amino-2[2-(-4-octylphenyl)ethyl]propane-1,3-diol hydrochloride) in rats after oral and intravenous doses. Drug Metab Dispos. 2006;34:1480–1487. doi: 10.1124/dmd.105.009001. [DOI] [PubMed] [Google Scholar]
- Neumann H, Schmidt H, et al. Interferon gamma gene expression in sensory neurons: evidence for autocrine gene regulation. J Exp Med. 1997;186:2023–2031. doi: 10.1084/jem.186.12.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ontaneda D, Cohen JA. Potential mechanisms of efficacy and adverse effects in the use of fingolimod (FTY720) Expert Rev Clin Pharmacol. 2011;4:567–570. doi: 10.1586/ecp.11.46. [DOI] [PubMed] [Google Scholar]
- Oo ML, Thangada S, et al. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem. 2007;282:9082–9089. doi: 10.1074/jbc.M610318200. [DOI] [PubMed] [Google Scholar]
- Oorschot DE. Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: a stereological study using the cavalieri and optical disector methods. J Comp Neurol. 1996;366:580–599. doi: 10.1002/(SICI)1096-9861(19960318)366:4<580::AID-CNE3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Ostrowski RP, Colohan AR, et al. Mechanisms of hyperbaric oxygen-induced neuroprotection in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;25:554–571. doi: 10.1038/sj.jcbfm.9600048. [DOI] [PubMed] [Google Scholar]
- Ostrowski RP, Tang J, et al. Hyperbaric oxygen suppresses NADPH oxidase in a rat subarachnoid hemorrhage model. Stroke. 2006;37:1314–1318. doi: 10.1161/01.STR.0000217310.88450.c3. [DOI] [PubMed] [Google Scholar]
- Peeling J, Del Bigio MR, et al. Efficacy of disodium 4-[(tert-butylimino)methyl]benzene-1,3-disulfonate N-oxide (NXY-059), a free radical trapping agent, in a rat model of hemorrhagic stroke. Neuropharmacology. 2001a;40:433–439. doi: 10.1016/s0028-3908(00)00170-2. [DOI] [PubMed] [Google Scholar]
- Peeling J, Yan HJ, et al. Effect of FK-506 on inflammation and behavioral outcome following intracerebral hemorrhage in rat. Exp Neurol. 2001b;167:341–347. doi: 10.1006/exnr.2000.7564. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Mun-Bryce S, et al. Collagenase-induced intracerebral hemorrhage in rats. Stroke. 1990;21:801–807. doi: 10.1161/01.str.21.5.801. [DOI] [PubMed] [Google Scholar]
- Sanchez T, Estrada-Hernandez T, et al. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem. 2003;278:47281–47290. doi: 10.1074/jbc.M306896200. [DOI] [PubMed] [Google Scholar]
- Sansing LH, Harris TH, et al. Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann Neurol. 2011;70:646–656. doi: 10.1002/ana.22528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichita T, Sugiyama Y, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- Soliven B, Miron V, et al. The neurobiology of sphingosine 1-phosphate signaling and sphingosine 1-phosphate receptor modulators. Neurology. 2011;76:S9–14. doi: 10.1212/WNL.0b013e31820d9507. [DOI] [PubMed] [Google Scholar]
- Su CY, Chen HM, et al. Neuropsychological impairment after hemorrhagic stroke in basal ganglia. Arch Clin Neuropsychol. 2007;22:465–474. doi: 10.1016/j.acn.2007.01.025. [DOI] [PubMed] [Google Scholar]
- Sutherland GR, Auer RN. Primary intracerebral hemorrhage. J Clin Neurosci. 2006;13:511–517. doi: 10.1016/j.jocn.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Kelley RE, et al. Acute leukocyte and temperature response in hypertensive intracerebral hemorrhage. Stroke. 1995;26:1020–1023. doi: 10.1161/01.str.26.6.1020. [DOI] [PubMed] [Google Scholar]
- Tang J, Liu J, et al. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–1145. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- Titova E, Ostrowski RP, et al. Reduced brain injury in CD18-deficient mice after experimental intracerebral hemorrhage. J Neurosci Res. 2008;86:3240–3245. doi: 10.1002/jnr.21762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- Wang J, Dore S. Heme oxygenase 2 deficiency increases brain swelling and inflammation after intracerebral hemorrhage. Neuroscience. 2008;155:1133–1141. doi: 10.1016/j.neuroscience.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Fields J, et al. The development of an improved preclinical mouse model of intracerebral hemorrhage using double infusion of autologous whole blood. Brain Res. 2008;1222:214–221. doi: 10.1016/j.brainres.2008.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Rogove AD, et al. Protective role of tuftsin fragment 1–3 in an animal model of intracerebral hemorrhage. Ann Neurol. 2003;54:655–664. doi: 10.1002/ana.10750. [DOI] [PubMed] [Google Scholar]
- Wang J, Tsirka SE. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005;128:1622–1633. doi: 10.1093/brain/awh489. [DOI] [PubMed] [Google Scholar]
- Wei Y, Yemisci M, et al. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann Neurol. 2011;69:119–129. doi: 10.1002/ana.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werring DJ, Frazer DW, et al. Cognitive dysfunction in patients with cerebral microbleeds on T2*-weighted gradient-echo MRI. Brain. 2004;127:2265–2275. doi: 10.1093/brain/awh253. [DOI] [PubMed] [Google Scholar]
- Wu B, Ma Q, et al. Ac-YVAD-CMK Decreases Blood-Brain Barrier Degradation by Inhibiting Caspase-1 Activation of Interleukin-1beta in Intracerebral Hemorrhage Mouse Model. Transl Stroke Res. 2010;1:57–64. doi: 10.1007/s12975-009-0002-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi G, Keep RF, et al. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- Xue M, Del Bigio MR. Comparison of brain cell death and inflammatory reaction in three models of intracerebral hemorrhage in adult rats. J Stroke Cerebrovasc Dis. 2003;12:152–159. doi: 10.1016/S1052-3057(03)00036-3. [DOI] [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, et al. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- Zausinger S, Hungerhuber E, et al. Neurological impairment in rats after transient middle cerebral artery occlusion: a comparative study under various treatment paradigms. Brain Res. 2000;863:94–105. doi: 10.1016/s0006-8993(00)02100-4. [DOI] [PubMed] [Google Scholar]
- Zhao X, Sun G, et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol. 2007;61:352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]