

Abstract
Objective:
The objective of this study was to validate the immunohistochemical assay for the diagnosis of nondystrophic myotonia and to provide full clarification of clinical disease to patients in whom basic genetic testing has failed to do so.
Methods:
An immunohistochemical assay of sarcolemmal chloride channel abundance using 2 different ClC1-specific antibodies.
Results:
This method led to the identification of new mutations, to the reclassification of W118G in CLCN1 as a moderately pathogenic mutation, and to confirmation of recessive (Becker) myotonia congenita in cases when only one recessive CLCN1 mutation had been identified by genetic testing.
Conclusions:
We have developed a robust immunohistochemical assay that can detect loss of sarcolemmal ClC-1 protein on muscle sections. This in combination with gene sequencing is a powerful approach to achieving a final diagnosis of nondystrophic myotonia.
We have previously reported an increased frequency of coexisting recessive CLCN1 mutations in patients with currently diagnosed myotonic dystrophy type 2 (DM2),1 because patients with DM2 heterozygous for a recessive CLCN1 mutation have more pronounced myotonia.1 With the aim of showing this modifying effect of the cosegregating CLCN1 mutations on the protein level, we developed an immunohistochemical assay for ClC-1 protein expression. The method proved to be efficient in the molecular diagnostic clarification of nondystrophic myotonias (NDMs) caused by mutations in CLCN1 and SCN4A genes.
Autosomal recessive Becker (OMIM #255700) and dominant Thomsen (OMIM #160800) congenital myotonia are NDMs caused by mutations in CLCN1 on chromosome 7q35.2 More than 100 different CLCN1 mutations have been identified.3 Some CLCN1 mutations are clearly more common than others. R894X (c.2680C>T) has an estimated carrier frequency of about 1% in the European population. In the Finnish population the mutation F413C (c.1238T>G) is almost as frequent, at least in northern Finland.4 However, these 2 mutations explain only about half of the congenital myotonias in the studied population, and many myotonia patients remain with just one mutation identified when screening for these 2 common mutations.
In this study, we focused on the validation of an immunohistochemical assay for the diagnosis of NDM. With this method combined with molecular genetics, we were able to clarify all undetermined myotonia patients, identify new recessive mutations, and verify normal protein expression with dominant CLCN1 mutations.
METHODS
Standard protocol approvals, registration, and patient consents
All blood and tissue samples were obtained with written informed consent according to the Helsinki declaration and the study was approved by the local ethical board.
Patients and controls
The study included patients for whom muscle biopsy was available: 29 patients with NDM, 15 male and 14 female, with an average age of 49 years (range 20–78 years); 8 patients with DM1; 10 patients with DM2; 5 asymptomatic carriers of recessive CLCN1 mutations; and 6 nonrelated normal controls.
Twenty-five patients with NDM had clear clinical/subclinical myotonia. Twenty-four were from sporadic/recessive pedigrees. Five of the 24 were homozygotes for R894X, one was a compound heterozygote R894X and F413C, and in the remaining 18 screening for R894X and F143C had failed to establish a final genetic diagnosis (single mutation or no mutation detected). One patient, in whom screening for the 2 common CLCN1 mutations had been negative, was from an autosomal dominant pedigree. In the patients without a final genetic diagnosis, the whole CLCN1 exome or cDNA was sequenced so that the efficacy of our assay for ClC-1 expression to detect a second mutation could be determined.
Four NDM myalgic patients had myotonia detectable by EMG but not by clinical examination.
All patients with DM1 and DM2 had been genetically diagnosed. Two of the patients with DM2 had a cosegregating CLCN1 mutation. The remaining 8 patients with DM2 and 2 patients with DM1 who had been screened were negative for R894X and F413C. None of the patients were on antimyotonic drugs. Patients are summarized in table e-1 on the Neurology® Web site at www.neurology.org.
Population controls
We screened 100 Finnish population controls for the W118G mutation. Additionally, a cohort of 65 Finnish population samples from a genetically isolated Larsmo island region was screened for both W118G and c.264G>A changes and 100 from the same population were also screened for F413C mutations.
The W118G change had been previously screened for in 261 unrelated patients with myotonia and in 64 unrelated population control samples from the United Kingdom as a part of the clinical genetics service of the National Hospital for Neurology & Neurosurgery, Queen Square, London (unpublished data, 2010).
ClC-1 immunohistochemistry on human skeletal muscle
Frozen sections of muscle tissue were used for immunohistochemical double staining of ClC-1 protein using 2 different antibodies pooled together, a commercial ClC-1 antibody against an extracellular domain close to the C-terminal (Alpha Diagnostic International, San Antonio, TX) and a ClC-1 antibody generated against the 15 C-terminal amino acids.5 The double immunohistochemical staining was performed on the BenchMark (Roche Tissue Diagnostics/Ventana Medical Systems Inc.) immunostainer, visualized with a peroxidase-based detection kit and the signal amplified (Roche Tissue Diagnostics/Ventana Medical Systems Inc.). The stainings were analyzed and compared to normal controls. Samples used for immunohistochemistry are listed in table e-1.
Genomic DNA and cDNA sequencing of CLCN1 gene
Genomic DNA was extracted from peripheral blood leukocytes. Primer sequences to the 23 CLCN1 exons are available upon request. All 23 exons were amplified by PCR and sequenced using bidirectional fluorescent sequencing on an ABI3130xl automatic DNA sequencer system (Applied Biosystems, Foster City, CA), with Big-Dye version 3.1 chemistry. For cDNA analysis, RNA was extracted from muscle biopsies using Trizol according to the manufacturer's suggestions (Invitrogen, Carlsbad, CA) and cDNA was generated using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). CLCN1 gene transcript was sequenced using 5 overlapping primer pairs. All sequences were analyzed with Sequencer software (Gene Codes Corporation, Ann Arbor, MI).
SDS-PAGE and Western blotting
Membrane proteins were extracted from muscle biopsies: 2 normal controls, 1 cosegregating DM2 and heterozygous F413C mutation, 1 homozygous R894X, 3 heterozygous R894X, and 2 heterozygous F413C (table e-1). The membrane protein phases were used for SDS-PAGE and Western blotting according to standard protocols. Nitrocellulose membranes with transferred proteins were immunolabeled with ClC-1 antibody.5
In vivo electroporation and expression analysis of chimeric green fluorescent protein CIC-1 constructs
Mammalian expression plasmids encoding chimeric green fluorescent protein (GFP) ClC-1 and GFP ClC-1 W118G were prepared. In vivo electroporation of plasmids encoding GFP ClC-1s into the living rat flexor digitorum brevis muscle was performed as described.6 Three to 5 days after the operation, the rats were killed, and the transfected muscles were excised, frozen in liquid nitrogen–cooled isopentane, and cryosectioned.
Patch clamp analysis
The chloride currents supported by either homodimeric W118G mutant or wild-type channels were assessed by whole cell patch clamp. The W118G point mutation was introduced into the cDNA for human CLCN1 in a mammalian expression vector (PCDH1, System Biosciences) using the QuickChange Site-directed Mutagenesis Kit (Agilent Technologies, Inc., Santa Clara, CA). HEK293T (ATCC) cells were transfected with 0.5 μg CLCN1 DNA using Lipofectamine2000 (Invitrogen) and studied by patch clamp 24–48 hours after transfection.
To obtain the voltage dependence of activation, the instantaneous current on stepping to −100 mV (tail current) was measured after pre-pulses to variable voltages from −140 mV to +120 mV. The full voltage protocol started from a holding potential of −40 mV after which the voltage was first stepped to +60 mV (which fully activates wild-type channels) before applying the variable pre-pulse voltage and then the step to −100 mV. The normalized tail current, I, was plotted against pre-pulse voltage, and was fitted with a Boltzmann function (y = Imin + [Imax–Imin]/[1 + exp([V50–Vprepulse]/slope)]) to estimate the voltage of half maximal activation (V50) and slope factor.
RESULTS
Immunohistochemistry and sequencing
Patients with clinical and EMG myotonia
All 5 patients with homozygous R894X mutations showed total loss of sarcolemmal ClC-1 expression. Ten patients with clinical myotonia but just one heterozygous R894X mutation after first screening had total/subtotal loss of sarcolemmal ClC-1 protein (figure 1). Sequencing of the whole gene revealed that out of these 10 patients, 6 harbored an additional heterozygous W118G (c.352T>G) change located in exon 3 and 4 had an additional heterozygous synonymous change, c.264G>A, located in exon 2.
Figure 1. CLC-1 immunohistochemical staining of muscle biopsies.

(A) Sarcolemmal staining of ClC-1 in a normal biopsy. Total loss of sarcolemmal ClC-1 protein in (B) R894X homozygous and in (C) compound heterozygous (R894X and W118G) patient samples. (D) Subtotal loss of sarcolemmal ClC-1 protein in a compound heterozygous (F413C and W118G) patient. Patient with (E) compound heterozygous R984X and c.264G>A, (F) compound heterozygous F413C and c.264G>A, and (G) homozygous c.264G>A mutations show total loss of sarcolemmal ClC-1 protein. Arrows indicate where ClC-1 protein should be localized at the sarcolemma.
Of the 4 patients with clinical myotonia and heterozygous F413C mutation (table e-1, patients P12–P14, P20), one showed total loss of protein and was found to be compound heterozygous with a c.264G>A change. The 3 other patient biopsies showed subtotal loss of sarcolemmal ClC-1 protein and sequencing the whole gene identified compound heterozygosity with W118G.
One patient without common mutations and subtotal loss of sarcolemmal ClC-1 was compound heterozygous for 2 previously unknown mutations, V536I (c.1606G>A) and c.264G>A. In 2 other patients without common mutations and total loss of sarcolemmal ClC-1, sequencing the whole CLCN1 gene revealed a homozygous c.264G>A change only.
The c.264G>A mutation is a silent mutation with no amino acid change (p.V88V). However, cDNA sequencing of patients homozygous for c.264G>A revealed that all mRNA transcripts lacked exon 2. Exon 2 was also lacking in one allele in patients heterozygous for the c.264G>A change (figure 2). According to Human Splicing Finder7 c.264G>A breaks several potential exonic splicing enhancer sites. Nucleotides c.263–270 are markedly conserved in mammals (figure 2). To ensure that c.264G>A is the cause of exon skipping and not just linked to it, we sequenced introns 1 and 2 (apart from base pairs c.180 + 938_180 + 1,167) in patients P22 and P16, and no variants were found.
Figure 2. Effect of c.264G>A on CLCN1 cDNA.

A) CLCN1 cDNA sequences of c.264G>A carriers compared to normal control. Sequence of a patient homozygous for c.264G>A shows that exon 2 has been skipped totally so that exon 1 is followed by exon 3. Patient heterozygous for c.264G>A has 2 alleles: one normal and one lacking exon 2. (B) The nucleotide alignment of CLCN1 cDNA shows marked conservation for c.264G (in red).
One patient with dominant familial myotonia had a dominant F307S (c.920T>C) mutation located in exon 8 in compound heterozygosity with c.2284+5C>T that has been suggested to be a splice mutation8 but is known to occur in 1% of normal population (1,000 genes database). There was no loss of ClC-1 protein on the sarcolemma. One patient with clinical myotonia and heterozygous R894X mutation had more or less normal amount of sarcolemmal ClC-1. Sequencing of the whole gene and mRNA in this patient did not disclose other CLCN1 mutations. The normal ClC-1 immunohistochemistry directly suggested a different cause and an A1156T mutation in the sodium channel SCN4A gene was subsequently identified.
Asymptomatic first-degree carrier relatives of patients with Becker myotonia
In the case of the homozygous c.264G>A brothers F1-II-1 and F1-II-2, we were able to study the asymptomatic mother F1-I-1, who logically was a carrier of the mutation. Each asymptomatic parent (F2-I-1 and F2-I-2) of patient F2-II-1 compound heterozygous for c.264G>A and R894X was found to be a carrier for one of the mutations each. Furthermore, the asymptomatic mother (F3-I-2) of the compound heterozygous patient F3-II-1 harboring c.264G>A and F413C carried the F413C mutation only. On immunohistochemistry, carriership of the F413C mutation in the mother was not associated with any clear loss of sarcolemmal protein. Thus, the segregation of c.264G>A is compatible with a recessive pathogenic mutation.
Heterozygosity for R894X in the asymptomatic father F4-I-2 of patient P23 produced an irregular minor reduction of sarcolemmal ClC-1 (table e-1). Altogether, these results suggest that the compound heterozygous mutations in patients were on separate alleles.
Patients with EMG myotonia and myalgia but without clinical myotonia
In patients (P26–P29) with only EMG myotonia, sarcolemmal ClC-1 protein was close to normal, slightly irregular, or just moderately reduced. These patients were found to have heterozygous R894X and F413C mutations only, even after sequencing the whole CLCN1 gene.
DM1 and DM2 patients
The sarcolemmal ClC-1 staining in DM1 and DM2 samples was variable from severe reduction to normal staining when compared to normal controls. Muscle biopsy samples were available of only 2 patients with DM2 with cosegregating recessive CLCN1 mutations. One with a cosegregating heterozygous R894X mutation showed loss of ClC-1 protein while the other patient with DM2 with a cosegregating heterozygous F413C mutation showed subtotal loss of the protein in both immunohistochemistry and Western blotting. These reductions were in the range of ClC-1 expression seen in patients with DM1 and DM2. ClC-1 immunohistochemical results and results from CLCN1 sequencing are summarized in table e-1.
Western blotting
In Western blots, we observed 80%–90% reduction of ClC-1 protein in biopsies with a homozygous R894X mutation (figure 3). Heterozygous c.264G>A mutations combined with both R894X and F413C mutation also showed 80%–90% reduction of ClC-1 when compared to muscle biopsies from normal controls (figure 3). These results correlated well with the results in ClC-1 immunohistochemistry. Patients with combined heterozygous R894X and W118G showed less clear reduction of total ClC-1 protein expression in Western blots (figure 3) in contrast to the very clear reduction of sarcolemmal expression observed with immunohistochemistry. The method should however be refined if further standardized quantification is needed.
Figure 3. CLC-1 Western blot on patient biopsies.
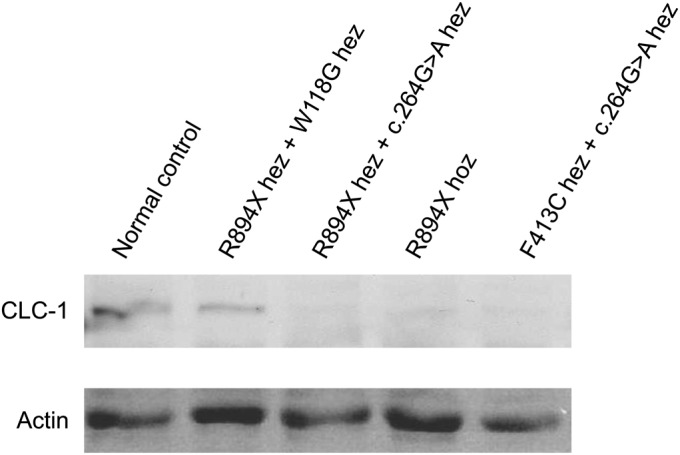
Western blot shows total amount of ClC-1 protein in muscle tissue. Total protein amount in a patient compound heterozygous for R894X and W118G is almost normal. The protein amount in heterozygous c.264G>A combined with R894X or F413C, as well as in homozygous R894X, is clearly reduced/almost absent. Ponceau stained actin as a loading control. Hez = heterozygous; hoz = homozygous.
Results from functional analysis of W118G
Both W118G and wild-type ClC-1 channels produced robust chloride currents in human embryonic kidney (HEK) cells. In contrast to currents produced by many dominantly inherited ClC-1 mutants, there was no obvious difference in current amplitudes between the wild-type and mutant clones. In addition, there was no significant difference in voltage dependence between the W118G-W118G homodimeric ClC-1 mutant and the wild-type (figure 4). Wild-type ClC-1 has been shown to localize in the sarcolemma and T-tubules of wild-type rat myofibers.5,9-12 Transfections of the chimeric GFP-ClC1 wild-type and W118G mutant into the living rat muscle fibers by means of electroporation did not reveal clear differences in the localization patterns between wild-type and mutant (data not shown).
Figure 4. Functional expression of wild-type CLC-1 and W118G in cells.
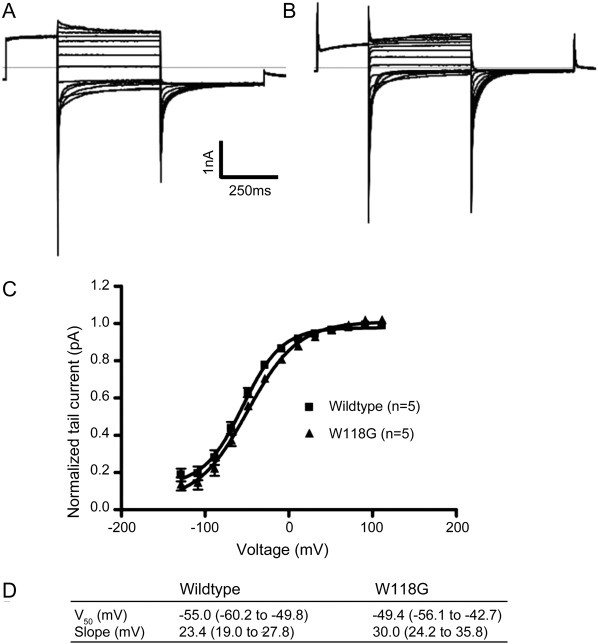
Functional expression of wild-type ClC-1 and the W118G mutation by whole cell patch clamp of transfected HEK293T cells. (A) Representative wild-type recording. (B) Representative mutant recording. (C) Boltzmann fits of the normalized tail current from 6 wild-type (squares) and 4 mutant (triangles) recordings to show the similar voltage dependence of activation. Error bars are obscured by symbol. (D) V50 and slope (with 95% confidence intervals) from the Boltzmann fits.
Population screening
In the cohort of 100 samples from Larsmo population, we found 3 heterozygous F413C mutations corresponding to a carrier frequency of 3%. In 65 individuals from the same population, the W118G mutation was found in a carrier frequency of 7.7%, whereas no carriers of the c.264G>A mutation were found. In a cohort of 100 controls from central Finland, the W118G mutation was found in a frequency of 3%, and in a cohort of 64 controls from the United Kingdom with a frequency of 4.7%.
Of 261 UK myotonia patients, 31 were found to have the W118G mutation (10 of whom were homozygotes for this mutation), corresponding to a frequency of 12%.
DISCUSSION
We developed an immunohistochemical assay for ClC-1 in muscle fibers using 2 different antibodies that proved to be a robust method for the detection of presence or absence of sarcolemmal ClC-1 protein on muscle sections. In our total cohort of 74 patients with sporadic/recessive NDM, 23% had remained without genetic diagnosis after screening for the 2 common CLCN1 mutations in Finland, R894X and F413C. Using this method we were able to establish diagnosis in all and identified new CLCN1 mutations that can cause or exacerbate low chloride conductance myotonia.
The previously unreported c.264G>A mutation was found in 4 different combinations. The silent c.264G>A is the apparent cause of exon 2 skipping on mRNA subsequently leading to frame shift and expected to cause nonsense-mediated mRNA decay, which is supported by the absent protein in c.264C>A homozygotes.
The W118G mutation has been considered a polymorphism.13 However, it occurred with an unexpectedly high frequency among myotonia patients from Finland and the United Kingdom. Nine of 19 Finnish myotonia patients with inconclusive results by screening for the 2 common Finnish myotonia mutations harbored W118G, which corresponds to 12% of the total myotonia patient cohort. Also in the United Kingdom, 12% of patients with confirmed or suspected myotonia congenita harbored the mutation, compared to 5% in the general population. This highly significant overrepresentation (p < 0.001, Fisher test) in both patient cohorts suggests a functional defect of muscle chloride conductance. When expressed in HEK cells, homodimeric W118G mutant channels yielded robust chloride currents with the same voltage dependence as wild-type channels; thus the defect is not at the level of ClC-1 protein function. However, on muscle immunohistochemistry the combination W118G/R894X or W118G/F413C causes subtotal loss of sarcolemmal ClC-1 protein clearly distinct from the expression of heterozygous R894X or F413C alone. By Western blotting, which measures the combined ClC-1 content of intracellular and surface plasma membranes, the total amount of ClC-1 protein in patients with a combined heterozygous W118G mutation is less abnormal. This discrepancy between sarcolemmal staining and blotting the total protein suggests mutant W118G is not correctly transported and integrated into the sarcolemma, which is in accordance with recent results reported for CLCN1 mutations Q43R, Y137D, and Q160H.14 The W118G mutation has been reported to occur in healthy controls with a frequency of 2.9%–3.5% (database of single nucleotide polymorphisims).13 However, it affects a highly conserved amino acid in the first transmembrane region of the protein. Our population studies comparing the isolated Larsmo population to a cohort of central Finland show that the frequency of a certain mutation may be highly variable even within a population considered to be genetically homogeneous. The carrier frequency of the pathogenic F413C mutation varied in these geographic cohorts from 0.6% to 3% and the W118G showed frequencies of 3% and 7.7%, respectively.
Low chloride conductance myotonia occurs when the summated loss of function of the 2 ClC-1 alleles is greater than 60%15–17 owing to mutation of both alleles (Becker disease), a dominant negative interaction between a single mutant allele and the normal one (Thomsen disease), or a wider mRNA spliceopathy affecting both alleles (DM1 and DM2). Based on the high frequency in normal population controls and the absence of symptomatic homozygous W118G patients in our cohort, one explanation is that W118G causes a moderate loss of function (i.e., 40%–50% in a homozygote) that is insufficient to cause myotonia by itself, but sufficient to cause myotonia when the other allele shows loss of function.
Expression of ClC-1 is stimulated by action potentials18 in the muscle cell; robust ClC-1 expression in the patient harboring the known dominant F307S mutation19 is consistent with the notion of positive feedback between myotonia and expression of the dominant allele. Furthermore, this is the first confirmation in muscle from a patient with a dominant mutation that the dominant negative interaction must occur at the level of channel function and not by disrupted expression.
The marked variability of ClC-1 expression detected by our assay in muscle from patients with DM1 and DM2 is consistent with the highly variable phenotype of these diseases. While the exacerbation of the DM2 phenotype by cosegregating recessive CLCN1 mutations is detectable as a selection bias in a large population,1 in our 2 patients with DM2 we were not able to reliably distinguish the effect of a cosegregating CLCN1 mutation from inherent variability in the DM2 phenotype at the level of ClC-1 sarcolemmal expression.
A fully normal ClC-1 protein expression in a patient with myotonia may suggest Thomsen disease or a different genetic background such as sodium channel myotonia, as was the case in some of our patients. The assay is most useful when screening for common CLCN1 mutations fails to establish a genetic diagnosis in patients with sporadic or recessive myotonia; absence of protein indicates the presence of a second CLCN1 mutation, and results also assist in the classification of novel sequence variants as pathogenic or benign.
Supplementary Material
Glossary
- DM1
myotonic dystrophy type 1
- DM2
myotonic dystrophy type 2
- GFP
green fluorescent protein
- HEK
human embryonic kidney
- NDM
nondystrophic myotonia
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
O. Raheem: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, acquisition of data. S. Penttilä: drafting/revising the manuscript, analysis or interpretation of data, acquisition of data. T. Suominen: drafting/revising the manuscript, analysis or interpretation of data, acquisition of data. M. Kaakinen: drafting/revising the manuscript, analysis or interpretation of data, acquisition of data. J. Burge: drafting/revising the manuscript, analysis or interpretation of data, acquisition of data. A. Haworth: drafting/revising the manuscript, analysis or interpretation of data, acquisition of data. R. Sud: drafting/revising the manuscript, analysis or interpretation of data, acquisition of data. S. Schorge: drafting/revising the manuscript, analysis or interpretation of data. H. Haapasalo: drafting/revising the manuscript. S. Sandell: drafting/revising the manuscript, contribution of vital reagents/tools/patents. K. Metsikkö: drafting/revising the manuscript, analysis or interpretation of data, acquisition of data. M. Hanna: drafting/revising the manuscript, analysis or interpretation of data, acquisition of data. B. Udd: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, contribution of vital reagents/tools/patents, acquisition of data, study supervision, obtaining funding.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Suominen T, Schoser B, Raheem O, et al. High frequency of co-segregating CLCN1 mutations among myotonic dystrophy type 2 patients from Finland and Germany. J Neurol 2008;255:1731–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koch MC, Steinmeyer K, Lorenz C, et al. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science 1992;257:797–800 [DOI] [PubMed] [Google Scholar]
- 3.Matthews E, Fialho D, Tan SV, et al. The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain 2010;133:9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papponen H. The muscle specific chloride channel CLC-1 and myotonia congenita in Northern Finland [thesis], University of Oulu; 2008. [DOI] [PubMed]
- 5.Papponen H, Kaisto T, Myllyla VV, Myllyla R, Metsikko K. Regulated sarcolemmal localization of the muscle-specific ClC-1 chloride channel. Exp Neurol 2005;191:163–173 [DOI] [PubMed] [Google Scholar]
- 6.Kaakinen M, Papponen H, Metsikkö K. Microdomains of endoplasmic reticulum within the sarcoplasmic reticulum or skeletal myofibers. Exp Cell Res 2008;314:237–245 [DOI] [PubMed] [Google Scholar]
- 7.Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009;37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun C, Tranebjaerg L, Torbergsen T, Holmgren G, Van Ghelue M. Spectrum of CLCN1 mutations in patients with myotoniacongenita in Northern Scandinavia. Eur J Hum Genet 2001;9:903–909 [DOI] [PubMed] [Google Scholar]
- 9.Lueck J, Rossi A, Thorton C, Cambell K, Dirksen T. Sarcolemmal-restricted localization of functional ClC-1 channels in mouse skeletal muscle. J Gen Physiol 2010;136:597–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiFranco M, Herrera A, Vergara J. Chloride currents from the transverse tubular system in adult mammalian skeletal muscle fibers. J Gen Physiol 2010;137:21–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamb G, Murphy R, Stephenson G. On the localization of ClC-1 in skeletal muscle fibers. J Gen Physiol 2011;137:327–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papponen H, Toppinen T, Baumann P, et al. Founder mutations and the high prevalence of myotoniacongenita in northern Finland. Neurology 1999;53:297–302 [DOI] [PubMed] [Google Scholar]
- 13.Lehmann-Horn F, Mailander V, Heine R, George AL. Myotonialevior is a chloride channel disorder. Hum Mol Genet 1995;4:1397–1402 [DOI] [PubMed] [Google Scholar]
- 14.Peter K, Sternberg D, Fischer M, Fahlke C. Mutations of HCLC-1 channel without functional defects cause myotoniacongenita by impaired surface membrane insertion: a new approach combining electrophysiology with single cell fluorescence measurements. Acta Physiol 2011;201(suppl 682). The Annual Meeting of the German Physiological Society. [Google Scholar]
- 15.Kwieciński H, Lehmann-Horn F, Rüdel R. Drug-induced myotonia in human intercostal muscle. Muscle Nerve 1988;11:576–581 [DOI] [PubMed] [Google Scholar]
- 16.Colding-Jørgensen E. Phenotypic variability in myotonia congenita. Muscle Nerve 2005;32:19–34 [DOI] [PubMed] [Google Scholar]
- 17.Furman R, Barchi R. The pathophysiology of myotonia produced by aromatic carboxylic acids. Ann Neurol 1978;4:357–365 [DOI] [PubMed] [Google Scholar]
- 18.Klocke R, Steinmeyer K, Jentsch T, Jockusch H. Role of innervation, excitability and myogenic factors in the expression of the muscular chloride channel ClC-1. J Biol Chem 1994;269:27635–27639 [PubMed] [Google Scholar]
- 19.Kubisch C, Schmidt-Rose T, Fontaine B, Bretag AH, Jentsch TJ. ClC-1 chloride channel mutations in myotoniacongenita: variable penetrance of mutations shifting the voltage dependence. Hum Mol Genet 1998;7:1753–1760 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.