

Abstract
The skeleton provides mechanical support for stature and locomotion, protects vital organs, and controls mineral homeostasis. A healthy skeleton must be maintained by constant bone modeling to carry out these crucial functions throughout life. Bone remodeling involves the removal of old or damaged bone by osteoclasts (bone resorption) and the subsequent replacement of new bone formed by osteoblasts (bone formation). Normal bone remodeling requires a tight coupling of bone resorption to bone formation to guarantee no alteration in bone mass or quality after each remodeling cycle. However, this important physiological process can be derailed by a variety of factors, including menopause-associated hormonal changes, age-related factors, changes in physical activity, drugs, and secondary diseases, which lead to the development of various bone disorders in both women and men. We review the major diseases of bone remodeling, emphasizing our current understanding of the underlying pathophysiological mechanisms.
Keywords: osteoporosis, renal osteodystrophy, osteopetrosis, Paget’s disease, rickets
INTRODUCTION
Bone is a dynamic tissue that is constantly being remodeled to maintain a healthy skeleton, which is crucial for the efficient and lifelong execution of important skeletal functions. Bone has three vital functions: (a) It provides support and sites of attachment for muscles, (b) it protects vital organs such as bone marrow and brain, and (c) it acts as a metabolic organ with major reserves of calcium and phosphate. There are two major types of bone: (a) cortical, which provides a mechanical function and is protective, and (b) trabecular, which provides strength and, more importantly, the majority of the metabolic function. Trabecular bone is the major site of bone remodeling; it is therefore the site of diseases of bone remodeling, also termed metabolic bone diseases. Bone remodeling is a physiological process in which old or damaged bone is removed by osteoclasts (i.e., bone-resorbing cells), then replaced by new bone formed by osteoblasts (i.e., bone-forming cells).
The composition of bone is approximately 10% cells, 60% mineral crystals (crystalline hydroxyapatite), and 30% organic matrix. The organic matrix includes primarily type 1 collagen (88%); other proteins represent 10%, and lipids and glycosaminoglycans represent 1–2% (see Reference 1 for a review). In normal bone remodeling, a balance between bone resorption (mediated by osteoclasts) and bone formation (mediated by osteoblasts) is tightly regulated and maintained to ensure that, in mature healthy bone, there are no major net changes in bone mass or mechanical strength after each remodeling cycle. Proper balance is controlled by the coupling of bone formation to bone resorption, which involves a number of coordinated signaling mechanisms. Nonetheless, an imbalance between bone resorption and bone formation may occur under certain pathological conditions, which leads to abnormal bone remodeling and the development of bone disorders.
The focus of this review is on diseases of bone remodeling or metabolic bone diseases. The past two decades have seen considerable advances in our understanding of bone biology, the pathobiology of bone diseases, and more importantly, the development of novel therapies. Table 1 gives our simplified classification of diseases of bone remodeling. It omits many secondary causes of osteoporosis, instead focusing on the primary forms of the disease and two common secondary causes, glucocorticoid (GC)- and immobilization-induced osteoporosis. The last four causes of metabolic bone disease listed in Table 1 are considerably less prevalent than osteoporosis. Osteoporosis is present in at least 10 million Americans over the age of 50, and an additional 33–34 million Americans have osteopenia, a preosteoporotic condition. Twenty-six percent of women over age 65 have osteoporosis. In contrast, Paget’s disease affects approximately 1 million Americans. The incidence of renal osteodystrophy is unknown; however, almost 325,000 Americans are on dialysis therapy. The percentage of those patients who have bone disease is not known. Osteopetrosis is rare; approximately 14,000 people in the United States have the disease (2). Inherited rickets is rare, and noninherited (dietary) rickets has also become a rare disease in the United States.
Table 1.
Disorders of bone remodeling
| Osteoporosis |
|---|
| Primary |
|
| Secondary |
|
| Renal osteodystrophy |
| Paget’s disease |
| Osteopetrosis |
| Rickets |
First, we review our current understanding of bone biology and remodeling, emphasizing new developments. Second, we discuss each disease state, emphasizing what is known about its prevalence and significance, its pathophysiology, and the state of current treatment options. Because of disease prevalence, most of the review focuses on osteoporosis; we describe the other four diseases—renal osteodystrophy, Paget’s disease, osteopetrosis, and rickets—in less detail.
BASIC BIOLOGY OF BONE REMODELING
Bone remodeling is carried out by a functional and anatomic structure known as the basic multicellular unit (BMU) and requires the coordinated action of four major types of bone cells: bone-lining cells, osteocytes, osteoclasts, and osteoblasts (Figure 1) (3, 4). In a quiescent state, the bone surface is covered by a monolayer of bone-lining cells, which belong to the osteoblast lineage (5, 6). Osteocytes are the most abundant bone cells; they also differentiate from osteoblasts and are embedded within the bone during skeletal development or during previous cycles of bone remodeling (7). Osteocytes may serve as the primary mechanosensing cells, and therefore they probably play a pivotal role in the initiation of bone remodeling (8). Osteoclasts, the sole bone-resorbing cells, are multinucleated giant cells that differentiate from mononuclear cells of the monocyte/macrophage lineage upon stimulation by two essential factors: the monocyte/macrophage colony–stimulating factor (M-CSF) and the receptor activator of nuclear factor κB (NF-κB) ligand (RANKL) (9, 10). Osteoclast differentiation involves several key steps: Hematopoietic stem cells give rise to colony-forming unit granulocytes/macrophages, which further differentiate into cells of the monocyte/macrophage lineage in the bone marrow. Mononuclear cells of the monocyte/macrophage lineage in the bone marrow or in the circulation are generally considered to be osteoclast precursors, which are attracted to prospective resorption sites and then attach to the bone matrix to differentiate into osteoclasts in response to M-CSF and RANKL (10, 11). Osteoblasts, the boneforming cells, are derived from mesenchymal stem cells (MSCs) through a multistep differentiation pathway. MSCs give rise to osteoprogenitors, which differentiate into preosteoblasts and then mature osteoblasts (12). Emerging evidence indicates that MSCs and osteoprogenitors not only reside in the bone marrow but also are present in the circulation (13–15).
Figure 1.

Current model for bone remodeling. The remodeling process consists of four major distinct but overlapping phases: Phase 1: initiation/activation of bone remodeling at a specific site. Phase 2: bone resorption and concurrent recruitment of mesenchymal stem cells (MSCs) and osteoprogenitors. Phase 3: osteoblast differentiation and function (osteoid synthesis). Phase 4: mineralization of osteoid and completion of bone remodeling. In normal bone remodeling, there is no net change in bone mass and strength after each remodeling cycle. However, abnormal bone remodeling in certain pathological conditions, such as osteoporosis, causes reduced bone mass and strength. Abbreviation: BRC: bone-remodeling compartment.
The remodeling process involves four major distinct but overlapping phases:
Phase 1: initiation/activation of bone remodeling at a specific site.
Phase 2: bone resorption and concurrent recruitment of MSCs and osteoprogenitors.
Phase 3: osteoblast differentiation and function (osteoid synthesis).
Phase 4: mineralization of osteoid and completion of bone remodeling (Figure 1) (16, 17).
It has been proposed that when osteocytes either sense bone deformation caused by mechanical loading or detect microdamage in old bone, they transmit signals of an unknown nature to recruit osteoclast precursors to the specific bone site (Figure 1) (18). Emerging evidence supports the concept that bone remodeling occurs within a closed system termed the bone-remodeling compartment (BRC), which is highly vascular and characterized by the presence of a canopy formed by bone-lining cells (5, 6, 19). Thus, osteoclast precursors may be recruited either from the bone marrow, by crossing the bone-lining cell monolayer, or from capillaries that penetrate into the BRC. Osteoclast precursors then attach to the bone matrix to differentiate into osteoclasts in response to elevated concentrations of M-CSF and RANKL within the BRC. With the formation of osteoclasts, the remodeling process proceeds to Phase 2, in which bone resorption represents the predominant event, but the recruitment of MSCs and/or osteoprogenitors into the BRC is also initiated (Figure 1). Similarly, MSCs and osteoprogenitors can be recruited either directly from the bone marrow or from capillaries that penetrate into the BRC. Osteoclast formation and bone resorption continue, while recruited MSCs and osteoprogenitors differentiate into preosteoblasts and subsequently osteoblasts throughout Phase 2. The remodeling process 3 as osteoblast function (osteoid synthesis) begins to overtake bone resorptionas the predominant event (Figure 1). Phase 3 continues for some time to allow the BRC to excavate more bone by trenching the bone surface and replacing it with osteoid produced by osteoblasts. Osteoblast formation and function continue even after cessation of bone resorption to ensure a balance between bone removal and bone formation. Phase 4 involves the mineralization of osteoid and concludes the bone-remodeling cycle (Figure 1).
The human adult skeleton has approximately 1–2 million active BMUs at any given time; they are spatially and temporally separated from one another and function in an asynchronous fashion within the cortical bone and on the surfaces of the trabeculae and cortex (16). Figure 1 depicts the key steps of a BMU remodeling bone on the surface of trabecular bone (semiosteonal remodeling); similar BMUs excavate and replace tunnels in cortical bone (osteonal remodeling). Importantly, normal bone remodeling depends on the tight coupling of bone formation to bone resorption to ensure no net change in bone mass or quality after each remodeling cycle. Previously, investigators believed that two mechanisms control the coupling: First, a number of local and systemic factors regulate the formation and function of both osteoblasts and osteoclasts, resulting in concomitant changes in bone resorption and bone formation (20, 21). Second, growth factors such as transforming growth factor β (TGF-β), released from the bone matrix during the resorption process, participate in regulating osteoblast differentiation and function (Figure 1) (22, 23), thereby coupling bone formation to bone resorption. Interestingly, accumulating evidence suggests that osteoclasts play a role in the coupling of bone formation to bone resorption by producing factors that stimulate osteoblast differentiation and function (Figure 1) (24–26). Finally, the BRC may also serve as a physical mechanism for coupling bone formation to bone resorption by confining the two biological processes in a closed compartment (19).
Understanding the basic biology of bone remodeling is critical for elucidation of the molecular and cellular mechanisms underlying the pathogenesis of disorders of bone remodeling. The updated bone-remodeling model described in Figure 1 suggests that the bone-remodeling process may be derailed at different points or levels, resulting in various metabolic bone diseases. Postmenopausal osteoporosis may reflect an increase primarily in the frequency of activation of BMUs, and other bone disorders may involve the effects of many systemic and local factors on the differentiation, function, and life span of bone cells. In particular, the concept of the BRC supports the notion that systemic and local factors may alter the bone-remodeling process by affecting the recruitment of bone cells into the BRC or by disrupting the structural integrity of the BRC (27). Moreover, the deregulation of secretion of osteoclast-derived factors may contribute to the pathogenesis of bone disorders. In the following sections, we discuss new advances in our understanding of the mechanisms underlying the pathogenesis of diseases of bone turnover in the context of this model and associated concepts.
OSTEOPOROSIS
Osteoporosis is a common disorder of bone remodeling characterized by low bone mass and structural deterioration of bone; it causes bone fragility and an increased vulnerability to fractures (28). Osteoporosis represents a group of distinct pathological conditions, rather than a single entity, and is traditionally classified into primary and secondary types (28, 29). Primary osteoporosis is further divided into two subtypes: type I osteoporosis and type II osteoporosis (30, 31). Type I osteoporosis (also referred to as postmenopausal osteoporosis) is a common bone disorder in postmenopausal women and is caused primarily by estrogen deficiency resulting from menopause, whereas type II osteoporosis (also known as age-related osteoporosis or senile osteoporosis) is associated primarily with aging in both women and men. In contrast, secondary osteoporosis refers to bone disorders that are secondary complications of various other medical conditions, consequences of changes in physical activity, or adverse results of therapeutic interventions for certain disorders (28, 29). Below, we discuss postmenopausal osteoporosis, age-related osteoporosis, and two types of secondary osteoporosis: GC-induced osteoporosis and immobilization-induced osteoporosis.
Postmenopausal Osteoporosis
The pathogenesis of postmenopausal osteoporosis is caused primarily by the decline in estrogen levels associated with menopause. This hypothesis was initially proposed by Albright et al. (32), whose seminal work in the 1940s demonstrated that postmenopausal women with osteoporosis exhibit negative calcium balance, a condition that could be reversed by estrogen replacement. Albright and colleagues believed that bone loss in postmenopausal women was caused by impaired bone formation resulting from estrogen deficiency, but subsequent studies (33–35) indicated that estrogen deficiency leads mainly to an increase in bone resorption rather than to impaired bone formation. Nevertheless, it is well established that both bone resorption and bone formation are increased in postmenopausal osteoporosis; however, the extent of increased bone resorption exceeds that of augmented bone formation, which causes an imbalance between bone resorption and bone formation in favor of bone resorption (34–36). Since the establishment of a central role for estrogen deficiency in the pathogenesis of postmenopausal osteoporosis, enormous effort has been focused on elucidating the mechanisms by which estrogens exert their bone-sparing effects. The past three decades have witnessed several major advances in the understanding of the molecular and cellular mechanisms underlying the role of estrogen deficiency in the pathogenesis of postmenopausal osteoporosis.
Investigations in the 1980s demonstrated that osteoclast differentiation and activity are regulated by a variety of cytokines, including interleukin (IL)-1, −6, and −7; tumor necrosis factor (TNF); granulocyte/macrophage colony–stimulating factor; and M-CSF (37, 38), which raises a question as to whether estrogen plays a protective role against osteoporosis by modulating the production of these cytokines. Indeed, subsequent in vitro and in vivo studies revealed that estrogen suppresses the expression of IL-1, TNF, and IL-6 in monocytes and/or osteoblasts and stromal cells (Figure 2) (39, 40). Human peripheral-blood monocytes from osteoporosis patients produce larger amounts of IL-1, and elevated production of IL-1 was not observed in human peripheral-blood monocytes from either estrogen-treated postmenopausal women with osteoporosis or untreated premenopausal women (41). Later studies showed that human peripheral-blood monocytes from ovariectomized premenopausal women secrete increased amounts of IL-1 and TNF (42). Interestingly, another group (43) demonstrated that estrogen also inhibits the expression of TNF and IL-1 in osteoblasts and stromal cells. The pathological role for TNF in postmenopausal osteoporosis was further supported by animal model studies demonstrating that knockout mice deficient in either TNF or TNF receptor 1 are resistant to ovariectomy-induced bone loss (44). Taken together, these data support the notion that estrogen exerts protective effects on bone via suppression of IL-1 and TNF expression. However, estrogen inhibits the expression of IL-6 indirectly through the action of IL-1 and TNF, which stimulate the expression of IL-6 in osteoblasts and stromal cells (45). Elevated levels of these three cytokines augment bone resorption by distinct mechanisms (Figure 2). Early studies showed that IL-6 exerts a primarily stimulatory effect on osteoclastogenesis (46). In contrast, IL-1 and TNF directly target mature osteoclasts to enhance osteoclast activity and indirectly stimulate osteoclastogenesis by upregulating the expression of IL-6. More recent investigations have found that these two cytokines can also directly target osteoclast precursors to promote osteoclastogenesis (Figure 2) (47–50). Although studies performed in the late 1980s and early 1990s largely established that estrogen preserves the skeleton by suppressing cytokine production, there remain numerous inconsistencies and controversies regarding the precise target cells of estrogen, the exact effects and regulatory mechanisms of the cytokines on osteoclasts and their precursors, and how these cytokines functionally interact and are coordinated in the pathogenesis of postmenopausal osteoporosis.
Figure 2.

Current understanding of the pathological mechanisms of postmenopausal osteoporosis. Dates indicate the time of the discovery of the functions. Abbreviations: FSH, follicle-stimulating hormone; IL, interleukin; M-CSF, monocyte/macrophage colony–stimulating factor; OPG, osteoprotegerin; RANK, receptor activator of nuclear factor κB; RANKL, RANK ligand; TNF, tumor necrosis factor.
The discovery of the RANKL/RANK/OPG (osteoprotegerin) system and the subsequent unraveling of an essential role for this system in osteoclast biology in the late 1990s not only represent milestones in bone biology research but have also significantly helped advance our understanding of the pathological mechanism of various bone disorders, including postmenopausal osteoporosis. RANKL (also known as OPGL, ODF, and TRANCE) was identified as a new member of the TNF superfamily independently by several groups (51–54). RANKL binds to its cognate receptor RANK (receptor activator of NF-κB), which is a member of the TNF receptor superfamily (53). Moreover, a soluble decoy receptor for RANKL, OPG, was identified and shown to antagonize RANKL functions by competing with RANK for binding of RANKL (55, 56). Although initial in vitro studies showed that the RANKL/RANK/OPG axis regulates several biological processes such as osteoclast formation and function (51, 52), dendritic cell survival and activation (57, 58), and T cell activation (59, 60), RANKL and RANK knockouts soon demonstrated that this system also plays a critical role in lymph node organogenesis (59, 61), B cell differentiation (59, 61), and mammary gland development (62). Intriguingly, a recent study revealed that in females this system is implicated in thermoregulation as well as in fever response in inflammation (63).
In the bone microenvironment and, more specifically, in normal bone remodeling, cells of the osteoblast lineage—including bone-lining cells, stromal cells (e.g., MSCs), osteoprogenitors, and osteoblasts—serve as sources of RANKL as well as of M-CSF (Figure 2) (64). Cells of the osteoblast lineage express both M-CSF and RANKL (membrane-bound RANKL and soluble RANKL), which bind to their receptors, c-fms and RANK, respectively. These receptors are expressed on osteoclast precursors and stimulate osteoclast formation. In mature osteoclasts, RANKL also stimulates osteoclast activation and survival (51). In addition, OPG is derived primarily from cells of the osteoblast lineage in normal bone remodeling. RANK activates intracellular signaling pathways to regulate osteoclast formation and function in part through recruiting TNF receptor–associated factors (TRAFs). RANK contains three functional TRAF-binding motifs (PFQEP369–373, PVQEET560–565, and PVQEQG604–609) that collectively activate six major signaling pathways [NF-κB; c-jun N-terminal kinase (JNK); extracellular signal–related kinase (ERK); p38; nuclear factor of activated T cells, cytoplasmic 1 (NFATc1); and Akt], which in turn regulate osteoclast formation, function, and/or survival (10, 65, 66). In addition, considerable experimental data support an important role for Ca2+ and calmodulin in osteoclast development and function (67, 68), and administration of calmodulin antagonists prevents ovariectomy-induced bone loss in mice (69).
Since the discovery of the RANKL/RANK/OPG axis, it has become clear that estrogen also exerts bone-sparing effects by targeting this regulatory axis (Figure 2). Specifically, estrogen stimulates the expression of OPG in mouse osteoblasts and stromal cells (70). Moreover, the expression of RANKL was elevated on the surface of bone marrow cells, such as osteoblasts and lymphocytes, from postmenopausal women with osteoporosis compared with cells from premenopausal controls (71); this finding indicates that RANKL plays an important role in the pathogenesis of postmenopausal osteoporosis. Moreover, estrogen directly inhibits the RANK-activated JNK pathway in osteoclast precursors (72, 73). As discussed above, estrogen deficiency leads to an increase in the secretion of IL-1 and TNF, which promote osteoclastogenesis by upregulating the expression of IL-6. IL-1 and TNF also stimulate RANKL gene expression in osteoblasts and stromal cells (52), which adds another level of complexity to the action of these two cytokines in the development of postmenopausal osteoporosis.
The first decade of the new century has already seen several new developments in the investigation of the molecular and cellular mechanisms that underlie the pathogenesis of postmenopausal osteoporosis (Figure 2). First, nude mice, which lack T lymphocytes, are protected from ovariectomy-induced bone loss (74), which demonstrates that circulating T cells, not monocytes, are the major source of estrogen-regulated TNF. Second, estrogen directly targets mature osteoclasts to promote apoptosis of osteoclasts via the action of the Fas/Fas ligand system (75, 76). Modulation of osteoclast life span has long been considered a potential mechanism through which estrogen exerts a protective effect against osteoporosis (77), and an earlier study showed that estrogen regulates osteoclast life span indirectly through the regulation of TGF-β (78). Finally, in 2006, Sun et al. (79) reported that the pituitary follicle-stimulating hormone (FSH) plays a role in the pathogenesis of postmenopausal osteoporosis, a finding that challenges the long-held notion that estrogen alone is central to the pathogenesis of this disorder of bone remodeling (Figure 2). This provocative notion remains controversial. Nonetheless, it has presented an intriguing new concept and created an impetus for further validation. Future studies to address this issue further will lead to a more precise understanding of the mechanism underlying the pathogenesis of postmenopausal osteoporosis.
Given that postmenopausal osteoporosis reflects an imbalance between bone resorption and bone formation in favor of the former, effective inhibition of bone resorption has long been recognized as an important therapeutic strategy for this disorder of bone remodeling. Although calcium and vitamin D remain the first line of therapy for postmenopausal osteoporosis, there are now four major classes of antiresorptive drugs (i.e., agents that can inhibit osteoclast formation and/or function) on the market: estrogen (80), selective estrogen receptor modulators (81), bisphosphonates (82), and calcitonin (83). Therapy for postmenopausal osteoporosis has changed dramatically over the past two decades. Before then, estrogen replacement therapy was the lone class of drugs used for treatment of osteoporosis, in addition to calcium and vitamin D. Since then, several classes of drugs have been developed. The most widely used are bisphosphonates, which generally shorten osteoclast life span. In addition to selective estrogen receptor antagonists, a monoclonal antibody to RANKL was recently developed (84, 85). All these drugs target osteoclasts and, therefore, primarily inhibit bone resorption. On the anabolic (as opposed to antiresorptive) side, parathyroid hormone (PTH) analogs increase bone mineral density when administered daily (86). The osteoporosis drug industry is in a state of flux, given that in 2009 the first bisphosphonate used widely for osteoporosis therapy, alendronate, became available as an inexpensive ($0.25–$0.50 per day) generic.
Age-Related Osteoporosis
Age-related bone loss affects both women and men (36). Recent studies indicate that significant trabecular bone loss begins as early as the twenties in men and women—long before any major hormonal changes (87). In women, however, bone loss accelerates for 5 to 10 years after menopause due to the rapid decline in estrogen levels; after this phase, bone loss continues at approximately the same rate as in elderly males. These observations indicate that there is an element of the aging process in bone, other than an age-associated failure of other organs or tissues, that is a common cause of bone loss in both aging women and men. Thus, the pathogenesis of osteoporosis in women consists of two components (Figure 3a). The first involves primarily osteoclasts (bone resorption) and results from changes in estrogen and FSH levels at menopause (Figure 3a), as discussed in the preceding section. The second component is age related, is centered on osteoblasts (bone formation), and engages a number of distinct factors associated with the aging process in both men and women, as we discuss below (Figure 3).
Figure 3.
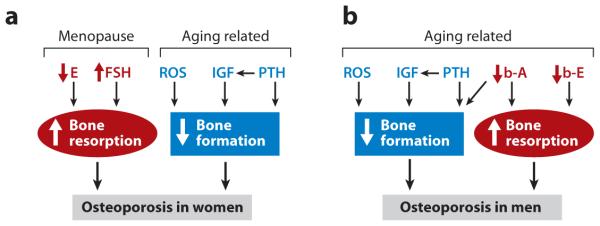
Current understanding of the pathological mechanisms of age-related osteoporosis. (a) Pathological mechanisms of osteoporosis in women. (b) Pathological mechanisms of osteoporosis in men. Up and down arrows represent increases and decreases, respectively. Abbreviations: A, androgen; b, bioavailable; E, estrogen; FSH, follicle-stimulating hormone; IGF, insulin-like growth factor; PTH, parathyroid hormone; ROS, reactive oxygen species.
Reactive oxygen species (ROS), the radical forms of oxygen, are produced in the mitochondria as by-products of respiration and oxidase activity and in cellular responses to various external stimuli ranging from inflammatory cytokines to ionizing radiation (88). ROS either cause cellular damage (proteins, lipids, and DNA), which lead to cell death, or trigger the activation of cellular signaling pathways. Cells possess intricate antioxidant defense mechanisms to counteract the adverse effects of oxidative stress. There is an age-related increase in ROS levels that arises from age-related increases in ROS production and/or a decrease in antioxidants (89, 90). Consistently, oxidative stress is inversely correlated with longevity in flies, nematodes, and mammals. As discussed above, a healthy adult skeleton is maintained by constant bone remodeling that involves osteoclasts and osteoblasts, whose activities are tightly coordinated to ensure that no net change in bone mass takes place during this process. Thus, age-related changes in the activity of either cell type may lead to bone loss. Members of the Wnt family play an important role in osteoblast differentiation and function (91). Binding of Wnts to the receptors Frizzled and LRP5/6 leads to the activation of β-catenin, which then translocates into the nucleus to associate with T cell factor (TCF) transcription factors to activate target genes. Age-dependent increases in oxidative stress antagonize Wnt signaling by diverting β-catenin from TCF to forkhead box O transcription factor–mediated transcription in osteoblasts, leading to the attenuation of osteoblastogenesis and bone formation (Figure 3) (92, 93). Calcium and its intracellular receptor, calmodulin, also are important regulators of osteoblast differentiation through both calmodulin kinase II and calcineurin pathways (94–96), which may also be a target for reactive oxygen species.
The growth hormone (GH)/insulin-like growth factor 1 (IGF-1) regulatory axis both plays a pivotal role in longitudinal bone growth and acquisition of peak bone mass during skeletal development and is critically involved in skeletal maintenance in adults (97). As discussed above (Figure 1), osteoblasts differentiate from MSCs, which can differentiate into other cell types, such as chondrocytes and adipocytes, in response to distinct differentiating stimulators. GH promotes differentiation of MSCs into osteoblasts and inhibits differentiation of MSCs into adipocytes (98, 99). GH also stimulates the proliferation of osteoblasts (100, 101). In addition to these direct effects on osteoblast proliferation and differentiation, GH regulates the differentiation and function of osteoblasts indirectly through, for instance, bone morphogenetic proteins (BMPs) and IGF-1/IGF-2. Thus, GH upregulates the expression of BMPs (102), which in turn promote osteoblast differentiation and bone formation. Moreover, GH increases the expression of IGF-1 (98), which exerts a primarily stimulatory effect on the function of osteoblasts. As such, a decline in IGF-1 production associated with the aging process has been implicated in the pathogenesis of age-related osteoporosis (Figure 3). Serum IGF-1 levels are correlated with bone mineral density in many osteoporosis patients, and polymorphisms of the IGF-1 promoter are linked to bone mass (103, 104).
An increase in serum PTH levels associated with aging has also been implicated in the pathogenesis of age-related bone loss (Figure 3). Multiple factors are probably involved in increasing serum PTH levels in the elderly. First, vitamin D deficiency is fairly common in the elderly and thus may contribute to the increase in serum PTH levels by decreasing calcium resorption from the gastrointestinal tract (105). Moreover, estrogen deficiency results in reduced intestinal calcium absorption as well as impaired renal tubular calcium reabsorption, which leads to a chronic negative calcium balance (106). If this negative calcium balance is not constantly compensated for by an increase in dietary calcium intake, it may cause an elevation in serum PTH levels. PTH acts on osteoblast precursors directly and indirectly by increasing IGF-1 production (107). Constant elevation of PTH is detrimental to bone strength, in contrast to intermittent elevation of PTH, which is anabolic.
Age-related osteoporosis in men also has a multifactorial etiology (Figure 3b). The decreased bone formation caused by changes in ROS, IGF-1, and PTH levels associated with aging plays a predominant role in the pathogenesis of age-related osteoporosis in men (87, 93). However, age-related changes in the levels of sex steroids, including both estrogen and androgen, also contribute to the pathogenesis of age-related osteoporosis in men (108). Sex steroid deficiency was not initially recognized as a cause of age-related osteoporosis in men primarily because of the failure of earlier investigations to detect significant changes in serum sex steroid levels. Subsequent studies revealed that free or bioavailable sex steroid levels change with aging (109), prompting additional studies to examine the potential role of sex steroids in the pathogenesis of age-related osteoporosis in men. Consequently, it is now appreciated that a decline in bioavailable estrogen and androgen levels also plays a role in the development of age-related osteoporosis in men by leading to an increase in osteoclast formation and function (bone resorption) (Figure 3b). Moreover, a decline in bioavailable androgen levels results in an increase in bone resorption as well as a decrease in bone formation. The decline in bioavailable estrogen and androgen levels in men may begin as early as the twenties. However, unlike the drastic decline in estrogen levels observed at menopause in women, the change in bioavailable sex steroids levels in men is gradual and related primarily to the aging process (109). The accelerated phase of bone loss observed in postmenopausal osteoporosis does not occur in male osteoporosis.
Despite the involvement of the sex steroids in the pathogenesis of age-related osteoporosis in men, hormone replacement (estrogen or testosterone) therapy and selective estrogen receptor modulators have not been approved for prevention and treatment of male osteoporosis because of safety and effectiveness concerns. Currently, bisphosphonates represent the best treatment option for bone loss in men (110).
Glucocorticoid-Induced Osteoporosis
GCs are potent immunomodulatory drugs that are commonly used to treat a variety of inflammatory conditions and autoimmune disorders, such as rheumatoid arthritis, asthma, and multiple sclerosis. However, therapeutic use of GCs often leads to numerous clinical complications, including bone loss and increased risk of fracture (111). Bone loss occurs within the first several months of initiating GC treatment, and prolonged GC-based therapy leads to a significant decrease in bone mass and an increased risk of fracture (112). Despite new advances in our understanding of GC-induced osteoporosis pathophysiology and improvements in its clinical management over the past decade, this disease still represents the most common form of drug-induced osteoporosis.
Pharmacological doses of GCs induce osteoporosis primarily by altering normal bone remodeling. GCs exert deleterious effects on the differentiation, function, and survival of multiple cell types involved in the remodeling process (Figure 4). GCs have profound effects on osteoblast differentiation and function. Although physiological levels of GCs are required for normal osteoblast differentiation, excess GCs exert an inhibitory effect on osteoblast differentiation. As discussed above, osteoblasts are derived from MSCs, which can also differentiate into a number of other cell types, including adipocytes (12, 113). Pharmacological doses of GCs inhibit differentiation of MSCs into osteoblasts and promote their differentiation into adipocytes (Figure 4a) (114). The GC-induced shift toward increased adipogenesis leads to a reduced pool of osteoprogenitors in the bone marrow. Thus, fewer osteoprogenitors are available for recruitment into the BRC during Phase 2 of bone modeling, which results in fewer osteoblasts in the BRC to adequately replace the resorbed bone. Moreover, GCs target mature osteoblasts by impairing their function and increasing apoptosis (Figure 4b). GCs inhibit the expression of bone matrix proteins such as type I collagen and osteocalcin (115, 116) and enhance the expression of two mineralization inhibitors, Dmp-1 and Phex (117). The GC-mediated impairment in osteoblast function and enhancement in osteoblast apoptosis further aggravates the reduction in bone mass and quality. Also, high GC levels promote apoptosis of osteocytes (Figure 4c) (118); GC-induced apoptosis of osteocytes is unlikely to significantly affect the ongoing bone-remodeling process (active BMUs). However, osteocytes play a primary mechanosensing role and are probably involved in the initiation of bone remodeling. Thus, GC-mediated osteocyte apoptosis may affect the prompt and efficient initiation of bone modeling.
Figure 4.

Current understanding of the pathological mechanisms of glucocorticoid (GC)-induced osteoporosis. (a) Inhibition of differentiation of mesenchymal stem cells (MSCs) and osteoprogenitors into osteoblasts. (b) Impairment of osteoblast function and enhancement of osteoblast apoptosis. (c) Induction of apoptosis of osteocytes. (d) Enhancement of receptor activator of nuclear factor κB ligand and reduction of osteoprotegerin expression in osteoblasts. (e) Prolonging of the life span of osteoclasts. (f) Inhibition of osteoclast function. (g) Suppression of osteoclast-generated osteogenic factors. Abbreviation: BRC, bone-remodeling compartment.
GCs can indirectly increase osteoclast formation and function by reducing OPG expression and increasing RANKL expression in osteoblasts (Figure 4d) (119). As discussed above and illustrated in Figure 1, bone remodeling has several distinct but overlapping phases. Bone resorption is the predominant event in Phase 2 (the bone-resorption phase), whereas osteoblast differentiation and function (osteoid synthesis) become predominant in Phase 3 (the bone-formation phase). The transition from Phase 2 to Phase 3 depends upon a critical decrease in the RANKL/OPG ratio. The GC-induced increase in the RANKL/OPG ratio changes the balance toward bone resorption or delays the reversal, both of which lead to a net negative change in bone mass. Furthermore, GCs can directly target mature osteoclasts to prolong their life span (Figure 4e) (120), which can further worsen the imbalance in favor of bone resorption. However, Kim et al. (121, 122) recently showed that GCs exert a direct inhibitory effect on osteoclast function (Figure 4f). The net effect of GCs on bone resorption is determined by their combined actions on the RANKL/OPG ratio and osteoclast survival and function. As discussed above, osteoclasts play a role in enhancing bone formation by secreting factors that promote osteoblast differentiation and function (24–26). Interestingly, the GC-induced inhibitory effect on bone formation is indirectly mediated through osteoclasts (121, 122), which suggests that GCs may also inhibit bone formation by suppressing the production of osteogenic factors by osteoclasts (Figure 4g).
In addition to affecting the bone-remodeling process, pharmacological doses of GCs contribute to the pathogenesis of osteoporosis by targeting other organs or physiological processes (123). Excess GCs decrease calcium resorption in the gut and increase loss of calcium in the kidney, which results in negative calcium balance. Moreover, GCs impair production and function of sex steroid hormones and GH, which play important roles in bone metabolism.
Clinical management of GC-induced osteoporosis currently involves reduction of GCs to minimal levels, supplementation of calcium and vitamin D, and therapeutic intervention with antiresorptive drugs at the initiation of GC therapy (111). Bisphosphonates are the most frequently used antiresorptive drugs for the prevention and treatment of GC-induced osteoporosis. PTH, an anabolic drug that has been approved for therapeutic intervention of postmenopausal osteoporosis, has the potential to become another therapeutic option for GC-induced osteoporosis, given that it exerts a stimulating effect on bone formation (124).
Immobilization-Induced Osteoporosis
One of the major functions of bone remodeling is to adapt bone material and structural properties to the mechanical demands that are placed on the skeleton, including mechanical loading and weight bearing. For ordinary individuals, the skeleton is developed in childhood and then constantly remodeled throughout adulthood to maintain mechanical strength that can sufficiently support normal weight bearing and routine physical activities. However, for individuals such as construction workers and athletes, the mechanical needs increase for certain regions of the skeleton; consequently, bone modeling results in the formation of stronger bone to replace old bone that could not adequately meet the increased mechanical demands. Conversely, prolonged bed rest or immobilization resulting from paralysis or casting of a limb reflects a decrease in mechanical requirements, which results in a disorder of bone remodeling termed immobilization-induced osteoporosis (also known as disuse osteoporosis) (125). Another example of this disorder is whole-body microgravity exposure, which is experienced by astronauts during prolonged space flight (126). Prolonged exposure to microgravity leads to bone loss of 0.5% to 2.0% per month in load-bearing bones (127, 128); both increased resorption and decreased mineralization occur (129, 130). As described above, postmenopausal osteoporosis, age-related osteoporosis, and GC-induced osteoporosis result from altered normal bone remodeling caused by a change in hormones at menopause, various aging-related factors, and therapeutic use of GCs, respectively. In contrast, immobilizationinduced osteoporosis results from a physiological response of bone remodeling to decreased mechanical demands.
Although we do not completely understand the molecular and cellular mechanisms by which the remodeling process is regulated to adjust bone material and structural properties to changes in mechanical loading or weight bearing, osteocytes have been proposed to play a crucial role in the process by serving as primary mechanosensing cells (8). Mechanical loading or weight bearing of the skeleton leads to deformation of bones (strain), shear forces, and streaming potentials within osteocyte lacunae and the canalicular network, which are detected primarily by osteocytes (131). The central role of osteocytes in adapting bone properties to mechanical loading was convincingly demonstrated by a recent study (132) showing that genetically modified mice lacking osteocytes develop osteoporosis due to defective mechanotransduction. Moreover, osteoblasts function as important mechanosensing cells (133). Furthermore, several studies (134–137) indicate that mechanical stress can exert direct and indirect effects on osteoclasts. All bone cells probably respond in some fashion to loaded mechanical stress and contribute to the pathogenesis of immobilization-induced bone loss.
Various cell-surface proteins or membrane structures, such as integrins, ion channels, connexons, gap junctions, and primary cilia, are believed to serve as mechanosensors to sense strains, flow-shear forces, and streaming potentials and to activate diverse intracellular pathways that modulate the differentiation and function of bone cells (138, 139). For instance, conditional knockout mice in which β1 integrin is deleted in cortical osteocytes are protected from disuse (hindlimb-unloading)-induced bone loss in cortical bones (140). Moreover, mechanosensitive ion channels have been identified in both osteoblasts (141) and osteocytes (139). Various types of mechanical forces directly target osteoblasts through these mechanosensors to activate intracellular pathways so as to modulate osteoblast differentiation and function (142, 143). However, although mechanical stimuli can directly affect the apoptosis of osteocytes in response to various mechanical stimuli, osteocytes regulate bone remodeling mainly by signaling to osteoblasts and osteoclasts via soluble factors and direct cell-cell contact (139).
Immobilization dramatically decreases mechanical loading of the skeleton, thereby diminishing the deformation of bones and abolishing fluid-shear forces. Consequently, the mechanosensing pathways and corresponding signaling pathways involved in promoting bone formation and modulating bone remodeling are no longer activated, which leads to a decrease in bone mass and strength. Currently, there is no better treatment for immobilization-induced osteoporosis than prophylaxis. Thus, decrease in physical activities should be minimized whenever possible, and therapeutic exercise may be prescribed to apply a certain level of mechanical stress to the affected sites of the skeleton. Pharmacological intervention with bisphosphonates or PTH analogs has not been rigorously evaluated in humans, but these agents have been effective in treating bone loss in the hindlimb-suspension model of weightlessness in rodents (144, 145).
RENAL OSTEODYSTROPHY
Renal osteodystrophy refers to a heterogeneous group of metabolic bone diseases that accompany chronic kidney disease. There is considerable evidence linking the development of renal osteodystrophy to increased cardiovascular calcification and associated morbidity and mortality (146). Renal osteodystrophy remains multidimensional both pathophysiologically and, more importantly, therapeutically. It is the only group of metabolic bone diseases whose treatment can be aided by the evaluation of bone biopsies. Quantitative histomorphometry, in conjunction with appropriate laboratory analyses, is often used to make a specific diagnosis (147). The classification of renal osteodystrophy on the basis of biopsy analysis includes assessment of three main parameters: bone turnover, mineralization, and volume (TMV) (Table 2). In this classification, turnover reflects the rate of remodeling, mineralization reflects the process of collagen calcification, and volume is the relative amount of bone per total tissue volume. In a 2008 survey of 544 bone biopsies from late-stage advanced chronic kidney disease, the TMV results were as follows: (a) for turnover, 52% were low, 21% were normal, and 27% were high; (b) for mineralization, 3% were defective and 97% were normal; and (c) for volume (trabecular), 32% were low, 30% were normal, and 38% were high (147).
Table 2.
Turnover, mineralization, and volume classification system for renal osteodystrophy
| Turnover | Mineralization | Volume |
|---|---|---|
| Low | Low | |
| Normal | ||
| Normal | Normal | |
| Abnormal | ||
| High | High |
The pathophysiology of renal osteodystrophy is complex and clearly reflects the importance of PTH and vitamin D on bone turnover and related pathological abnormalities. The classification into high and low bone-turnover groups (Table 2) reflects the disparate nature and complexity of the pathophysiology. High-turnover renal osteodystrophy can best be categorized as a secondary hyperparathyroidism state, whereas low-turnover disease is characterized by a relative hypoparathyroidism state. Thus, the pathology of high-turnover renal osteodystrophy is that of hyperparathyroidism bone disease. Sustained excess PTH increases bone turnover with increased osteoblasts, osteoclasts, and osteocytes. Disordered remodeling results in the formation of abnormal bone, increased osteoid, and fibrosis (osteitis fibrosis). Primary factors contributing to secondary hyperparathyroidism are hyperphosphatemia, hypocalcemia, and vitamin D deficiency (147, 148).
Abnormal mineralization reflects the process of calcification of collagen in bone and is reflected histologically as abnormal osteoid. This parameter can be accurately assessed only by biopsy and histomorphometric analyses. The processes that result in impaired mineralization are poorly understood but are thought to be caused by one or a combination of vitamin D and/or mineral deficiencies, aluminum toxicity, and possibly a prolonged acidotic state.
The most common form of adynamic osteodystrophy is characterized by an absolute or relative PTH deficiency and by an extremely low rate of bone formation. Aluminum toxicity has historically been associated with adynamic renal osteodystrophy incurred through dialysis. Adynamic bone disease without aluminum toxicity is common, but its pathogenesis is not well understood. Factors implicated in this disorder include increased plasma calcium; vitamin D toxicity; and altered growth factors and cytokines such as BMP, TGF-β and IGF-1, and IL-6. These are only a few components of what is a poorly understood multifactorial pathogenesis (148, 149).
The importance of making the distinction between low- and high-turnover states lies in the differences in treatment. For high-turnover disease, therapy is focused on phosphorus restriction or use of phosphate binders, correcting vitamin D deficiency, and the occasional use of calcium supplements. Calcimimetic agents that bind to the parathyroid-gland plasmamembrane Ca2+ receptor were recently used to inhibit PTH secretion (149). In contrast, low-turnover disease is managed primarily by methods designed to increase PTH levels. Reducing calcium and vitamin D is often necessary. Low-turnover disease is often the consequence of aggressive treatment of high-turnover disease, and prevention should be the therapeutic focus.
PAGET’S DISEASE OF BONE
Paget’s disease is a focal disease of high bone turnover that affects one or more bones; it becomes more prevalent with increasing age (150). In the United States, Paget’s disease is the second most common disorder of remodeling, after osteoporosis. Clinically, most patients are asymptomatic, but those who are symptomatic have variable clinical presentations. Symptoms include bone pain, bone deformities, secondary arthritis, and often neurological problems that are secondary to bone deformities. Paget’s disease is believed to be a primary disorder of increased osteoclast bone resorption with a secondary marked increase in osteoblast activity and new bone formation. The resulting trabecular bone is abnormal in that it has a “woven,” disorganized appearance. X-ray findings are highly variable because of the multiple possible locations of the disease. One scenario is shown in Figure 5, which illustrates the changes that took place over 23 years in the tibia of an untreated woman with Paget’s disease. There is also an increased frequency of osteosarcoma in the areas of the affected bone.
Figure 5.
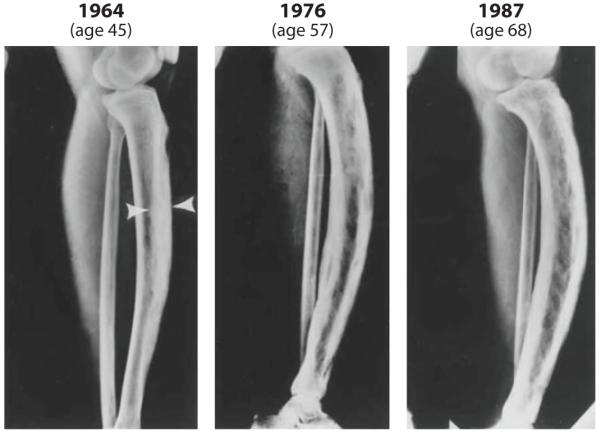
Radiographic changes in the left tibia of an untreated woman with Paget’s disease over 23 years (from age 45 to age 68). The arrows indicate the area of marked cortical thickening. The distal tibia appeared normal in 1964, but sclerotic changes progressively increased by 1987. Anterior bowing, which was mild in 1964, became progressively worse by 1987 (150). Reprinted with permission from Reference 151.
Little is known about the pathogenesis of Paget’s disease. Genetics plays a role, as the disorder occurs in a strongly familial pattern. Several genes have been implicated; however, the most commonly described mutation is a gene that encodes a ubiquitin-binding protein that plays a role in NF-κB signaling. The other pathogenic mechanism proposed to underlie this disease is associated with chronic viral infection. A paramyxoviral association is most frequently described; however, the canine distemper virus has also been implicated (152, 153).
Therapy is directed at inhibiting osteoclasts, most commonly through the use of bisphosphonates. Sometimes calcitonin is used; in addition to exhibiting antiosteoclast activity, calcitonin also has analgesic properties that often help treat patients with substantial bone pain. Additional therapy includes analgesics and surgery to correct deformities (152).
OSTEOPETROSIS
Osteopetrosis refers to a heterogeneous group of rare heritable bone-remodeling disorders that are characterized by increased bone density caused by a defect in bone resorption by osteoclasts. Subclassification of these disorders is based upon the mode of inheritance, age of onset, severity, and clinical symptoms. As a result, classification becomes rather fluid as more complex syndromes are described. The best-known forms are generally classified as (a) severe and (b) intermediate autosomal recessive types and (c) a milder autosomal dominant subtype. Importantly, molecular genetic studies have identified several gene mutations, all of which encode proteins that involve osteoclast-mediated bone resorption. The incidence of the autosomal recessive form is approximately 1 in 250,000 to 1 in 300,000 births, whereas the autosomal dominant form is more common at approximately 1 in 20,000. In addition to these three subtypes, there are several forms associated with other syndromes such as renal tubular acidosis, ectodermal dyplasias, and leukocyte adhesion deficiencies (154). For a complete summary of ostepetrosis subtypes, see Reference 155.
The pathophysiology of osteopetrosis involves mutations that affect osteoclast function. For both the autosomal recessive and autosomal dominant forms, the most common mutations are listed in Table 3. Our current knowledge of osteoclast function suggests that each of these mutations impairs osteoclast function. The location of these mutations in the osteoclast is shown in Figure 6. The three most important mutations—carbonic anhydrase II, proton pump, and chloride channel—are highlighted. The proton pump, which is abundantly present in the ruffled membrane, transports protons (H+) into the resorption lacuna to create and maintain a low-pH (~4.5) environment. The low-pH condition deposits high concentrations of acid onto a strongly basic mineral to dissolve the inorganic component of bone hydroxyapatite: [Ca3(PO4)2]3Ca(OH)2 + 8 H+ ↔ 10 Ca3 + 6 HPO42− + 2 H2O. The cytoplasmic protons are generated from carbon dioxide and water by carbonic anhydrase II (CO2 + H2O ↔ H2CO3 ↔ H+ + HCO3−). Importantly, the chloride channel, which is also present in the ruffled membrane, transports Cl− into the resorption lacuna to maintain electron neutrality. The removal of bicarbonates from the cell and the continuous supply of Cl− ions into the cell for secretion into the resorption lacuna are accomplished by a passive chloride-bicarbonate exchanger in the basolateral membrane. Also shown in Figure 6 is a rare mutational defect in cathepsin K, which plays a crucial role in degrading the bone matrix proteins such as type I collagen. The classification of these rare disorders is continually being updated as new mutations and clinical syndromes are identified. A recent review (155) extensively discusses the various clinical subtypes.
Table 3.
Genetic causes of osteopetrosis
| Genetic subtype | Gene | Function | Frequency | Disease severity |
|---|---|---|---|---|
| Autosomal recessive |
CAII | Produces H+ in cytoplasm | < 5% of patients with severe osteopetrosis |
Somewhat more severe than the other types |
| Autosomal recessive |
TCIRG1 | Proton pump | 60% of patients with severe osteopetrosis |
Severe |
| Autosomal recessive |
CLCN7 | Chloride channel | 15% of patients with severe osteopetrosis |
Either severe or intermediate |
| Autosomal recessive |
g1/g1 | Unknown | < 5% of patients with severe osteopetrosis |
Extremely severe |
| Autosomal recessive |
TNFSF1 1−/− | RANKL | Rare | Severe |
| Autosomal dominant |
CLCN7 | Chloride channel | Rare; 1 in 100,000 to 1 in 500,000 |
Mild delayed onset |
Figure 6.

Defects in osteoclast function in osteopetrosis. The sites of the four main genetic defects in osteoporosis—CAII (carbonic anhydrase), V-ATPase (proton pump), CLCN7 (chloride channel), and CTSK (cathespin K)—are highlighted in blue.
Osteopetrosis is diagnosed through the use of a combination of clinical and radiological parameters. Increased serum concentrations of creatine kinase BB isozyme and tartrate-resistant acid phosphatase (both derived from osteoclasts) are often observed. The prognosis as outlined in Table 3 is highly variable: The severe infantile forms are lethal, as untreated patients may die from bone marrow suppression in the first or second decade of life, whereas life expectancy in the delayed-onset form is normal.
Therapy is largely supportive. For severe lethal forms, hematopoietic stem cell transplantation can prolong lives for five years or more (156). Various treatment regimens with interferon-γ, calcium restriction, vitamin D, steroids, and PTH have all been tried without consistent documented success. For the delayed-onset, benign form of disease with normal life expectancy, supportive therapy involves managing complications such as fractures, arthritis, hypocalcemia, and various levels of bone marrow failure.
RICKETS
The most common form of rickets is nutritional vitamin D deficiency, which may be either primary or secondary to other diseases such as malabsorption, severe liver disease, impaired renal function, and heritable abnormalities of vitamin D metabolism. The consequences of vitamin D deficiency in bone are both direct inhibition of osteoblast progenitors and, more prominently, an increase in RANKL and a decrease in OPG. The major effects of the last two features are enhanced bone resorption and increased bone turnover (157).
It is crucial to place the pathophysiology of decreased vitamin D in the context of systemic calcium and PTH. Low vitamin D is secondary either to low sunshine exposure and low vitamin D intake or to decreased kidney function (vitamin D 1-α-hydoxylase is in the kidney). The combination of low calcium and high PTH contributes to bone loss. If rickets occurs in children before the growth plates heal, the bones may become markedly deformed. If it occurs in adults, osteopenia or osteoporosis occurs, along with an increased risk of fracture. There are additional, nonskeletal effects of vitamin D that may cause muscle weakness and subsequent falls.
Treatment is focused on repairing the deficiency in vitamin D, whether by increased sunshine (enhancing the skin’s ability to make vitamin D) and/or through dietary supplements. Repairing the deficiency results in reversal of most of the bone changes, except for major changes that occur during childhood. Given all the controversies about nonbone benefits of vitamin D, the normal range of vitamin D levels is unknown. There may soon be a generation of individuals whose serum levels of vitamin D are at least double the values found in the current generation. The fact that some uncommon forms of rickets could not be corrected by supplemental doses of vitamin D resulted in the discovery of rare, inheritable forms of the disease, including abnormalities in the vitamin D receptor, the rendering of cells insensitive to vitamin D, and abnormalities in vitamin D metabolism.
CONCLUSIONS
The mysteries of bone biology are being unraveled at an unprecedented pace, and our understanding of bone-remodeling diseases and targeted therapies is rapidly evolving. The concept of the BMU as the basic functional anatomic unit of bone remodeling and turnover permits us to predict and describe the underlying pathophysiology of several bone diseases. Of the eight forms of bone remodeling discussed herein, primary osteoporosis—both postmenopausal and age related—is by far the most common. Breakthroughs in basic biology that are being translated into clinical practice include the discovery of the roles of RANK/RANKL and OPG in osteoclastogenesis. Elucidation of the signals that regulate MSC differentiation—not only into osteoblasts but also into adipocytes, chondrocytes, and muscle cells—could further our understanding of mineralization and bone-formation disorders and may allow us to develop new anabolic therapies to build bone. Although the molecular defects in osteoclast function in osteopetrosis are rare, they have confirmed the vital components and molecular regulation of osteoclast function. In the future, we can expect the development of even more new therapies to evolve from a better understanding of the complex molecular aspects of bone turnover.
SUMMARY POINTS.
Bone remodeling is the result of a complex interaction among multiple cells in a tightly controlled environment (BMU). Bone remodeling occurs in four phases: (a) initiation/activation, (b) bone resorption and recruitment of MSCs and osteoprogenitors, (c) osteoblast differentiation and function, and (d) mineralization of osteoid.
Osteoporosis, which affects more than 10 million Americans, is by far the most prevalent disorder of bone remodeling.
Pathogenesis of osteoporosis in women involves two major components: (a) an increase in bone resorption resulting from changes in estrogen and FSH levels at menopause and (b) a decrease in bone formation caused by a number of distinct factors associated with the aging process.
Development of osteoporosis in men involves primarily a decrease in bone formation caused by multiple factors associated with the aging process, but an increase in bone resorption caused by a decline in bioavailability of sex steroids may also contribute.
GC-induced osteoporosis is caused by adverse effects of excess GCs on differentiation, function, and survival of the major bone cell types involved in bone modeling. It represents the most common form of drug-induced osteoporosis.
Immobilization-induced osteoporosis results from a physiological response of bone remodeling to decreased mechanical demands.
Classification of renal osteodystrophy is the only disorder of bone remodeling that can be accurately assessed only by bone biopsy.
Osteopetrosis is caused by inheritable specific defects in osteoclast function.
FUTURE ISSUES.
What is the relative contribution of estrogen deficiency and an increase in FSH to the pathogenesis of postmenopausal osteoporosis?
What is the precise mechanism underlying the rapid bone loss that occurs within 5 to 10 years of menopause?
Do ROS play a role in the pathogenesis of osteoporosis by regulating the differentiation, function, and survival of osteoclasts?
Can the BRC serve as a mechanosensing unit?
ACKNOWLEDGMENTS
The original work in the authors’ laboratory is supported by grant numbers AR47830 (to X.F.) and R01 AR050235 (to J.M.M.) from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS); grant number NS061677 from National Institute of Neurological Disorders and Stroke (to X.F.); a Within Our Reach Innovative Basic Research grant from the Research and Education Foundation of the American College of Rheumatology (to X.F.); grant number 5P30 AR0406031, University of Alabama Core Center for Basic Skeletal Research, from NIAMS (to J.M.M.); and grant number R01 CA109119 from the National Cancer Institute (to J.M.M.).
Glossary
- Glucocorticoid (GC)-induced osteoporosis
characterized by bone loss and increased risk of fracture; occurs in patients treated with GCs
- Immobilization-induced osteoporosis
characterized by bone loss and increased risk of fracture; secondary to immobilization of all or part of the skeleton
- Paget’s disease
focal disease of high bone turnover that results in abnormal bone architecture
- Renal osteodystrophy
refers to a heterogeneous group of metabolic bone diseases that accompany chronic renal failure
- Osteopetrosis
refers to a rare heterogeneous group of genetic bone diseases; characterized by a defect in bone resorption that causes increased bone density
- Rickets
bone disease caused by absolute or relative vitamin D deficiency
- Basic multicellular unit (BMU)
the functional and anatomic site of bone remodeling; composed of bone-lining cells, osteocytes, osteoclasts, and osteoblasts
- M-CSF
monocyte/macrophage colony–stimulating factor
- RANKL
receptor activator of nuclear factor κB ligand
- MSCs
mesenchymal stem cells
- Bone-remodeling compartment (BRC)
the anatomic compartment in which bone turnover occurs; composed of BMUs
- Postmenopausal osteoporosis
occurs secondary to loss of estrogen at menopause
- Age-related osteoporosis
affects both men and women equally; increases with increasing age
- IL
interleukin
- TNF
tumor necrosis factor
- OPG
osteoprotegerin
- PTH
parathyroid hormone
- ROS
reactive oxygen species
- IGF-1
insulin-like growth factor 1
Footnotes
DISCLOSURE STATEMENT The authors are not aware of any affiliations, memberships, funding, or financial holdings that might affect the objectivity of this review.
LITERATURE CITED
- 1.Robey PG, Boskey AL. The composition of bone. In: Rosen CJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Am. Soc. Bone Miner. Res; Washington, DC: 2008. pp. 32–38. [Google Scholar]
- 2.McGowen JA, Raisz LG, Noonan AS, Elderkin AL. Bone Health and Osteoporosis: A Report of the Surgeon General. US Dep. Health Hum. Serv; Rockville, MD: 2004. The frequency of bone diseases; pp. 69–87. [Google Scholar]
- 3.Parfitt AM. Osteonal and hemi-osteonal remodeling: the spatial and temporal framework for signal traffic in adult human bone. J. Cell Biochem. 1994;55:273–86. doi: 10.1002/jcb.240550303. [DOI] [PubMed] [Google Scholar]
- 4.Seeman E. Bone modeling and remodeling. Crit. Rev. Eukaryot. Gene Expr. 2009;19:219–33. doi: 10.1615/critreveukargeneexpr.v19.i3.40. [DOI] [PubMed] [Google Scholar]
- 5.Hauge EM, Qvesel D, Eriksen EF, Mosekilde L, Melsen F. Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblastic markers. J. Bone Miner. Res. 2001;16:1575–82. doi: 10.1359/jbmr.2001.16.9.1575. [DOI] [PubMed] [Google Scholar]
- 6.Parfitt AM. The bone remodeling compartment: a circulatory function for bone lining cells. J. Bone Miner. Res. 2001;16:1583–85. doi: 10.1359/jbmr.2001.16.9.1583. [DOI] [PubMed] [Google Scholar]
- 7.Bonewald LF. Osteocytes as dynamic multifunctional cells. Ann. N.Y. Acad. Sci. 2007;1116:281–90. doi: 10.1196/annals.1402.018. [DOI] [PubMed] [Google Scholar]
- 8.Santos A, Bakker AD, Klein-Nulend J. The role of osteocytes in bone mechanotransduction. Osteoporos. Int. 2009;20:1027–31. doi: 10.1007/s00198-009-0858-5. [DOI] [PubMed] [Google Scholar]
- 9.Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–8. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 10.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–42. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 11.Ross FP, Teitelbaum SL. Osteoclast biology. In: Marcus R, Feldman D, Kelsey J, editors. Osteoporosis. Academic; San Diego: 2001. pp. 73–106. [Google Scholar]
- 12.Ducy P, Schinke T, Karsenty G. The osteoblast: a sophisticated fibroblast under central surveillance. Science. 2000;289:1501–4. doi: 10.1126/science.289.5484.1501. [DOI] [PubMed] [Google Scholar]
- 13.Kuznetsov SA, Mankani MH, Gronthos S, Satomura K, Bianco P, Robey PG. Circulating skeletal stem cells. J. Cell Biol. 2001;153:1133–40. doi: 10.1083/jcb.153.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eghbali-Fatourechi G, Lamsam J, Fraser D, Nagel D, Riggs BL, Khosla S. Circulating osteoblast-lineage cells in humans. N. Engl. J. Med. 2005;352:1959–66. doi: 10.1056/NEJMoa044264. [DOI] [PubMed] [Google Scholar]
- 15.Modder UI, Khosla S. Skeletal stem/osteoprogenitor cells: current concepts, alternate hypotheses, and relationship to the bone remodeling compartment. J. Cell Biochem. 2008;103:393–400. doi: 10.1002/jcb.21423. [DOI] [PubMed] [Google Scholar]
- 16.Parfitt AM. Skeletal heterogeneity and the purposes of bone remodeling: implications for the understanding of osteoporosis. In: Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. Elsevier; San Diego: 2008. pp. 71–92. [Google Scholar]
- 17.Martin TJ, Seeman E. New mechanisms and targets in the treatment of bone fragility. Clin. Sci. (Lond.) 2007;112:77–91. doi: 10.1042/CS20060046. [DOI] [PubMed] [Google Scholar]
- 18.Parfitt AM. Targeted and nontargeted bone remodeling: relationship to basic multicellular unit origination and progression. Bone. 2002;30:5–7. doi: 10.1016/s8756-3282(01)00642-1. [DOI] [PubMed] [Google Scholar]
- 19.Andersen TL, Sondergaard TE, Skorzynska KE, Dagnaes-Hansen F, Plesner TL, et al. A physical mechanism for coupling bone resorption and formation in adult human bone. Am. J. Pathol. 2009;174:239–47. doi: 10.2353/ajpath.2009.080627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raisz LG. Hormonal regulation of bone growth and remodelling. Ciba Found. Symp. 1988;136:226–38. doi: 10.1002/9780470513637.ch14. [DOI] [PubMed] [Google Scholar]
- 21.Mohan S, Baylink DJ. Insulin-like growth factor system components and the coupling of bone formation to resorption. Horm. Res. 1996;45(Suppl. 1):59–62. doi: 10.1159/000184833. [DOI] [PubMed] [Google Scholar]
- 22.Mundy GR. The effects of TGF-β on bone. Ciba Found. Symp. 1991;157:137–43. [PubMed] [Google Scholar]
- 23.Tang Y, Wu X, Lei W, Pang L, Wan C, et al. TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009;15:757–65. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Falany ML, Thames AM, III, McDonald JM, Blair HC, McKenna MA, et al. Osteoclasts secrete the chemotactic cytokine mim-1. Biochem. Biophys. Res. Commun. 2001;281:180–85. doi: 10.1006/bbrc.2001.4307. [DOI] [PubMed] [Google Scholar]
- 25.Martin T, Gooi JH, Sims NA. Molecular mechanisms in coupling of bone formation to resorption. Crit. Rev. Eukaryot. Gene Expr. 2009;19:73–88. doi: 10.1615/critreveukargeneexpr.v19.i1.40. [DOI] [PubMed] [Google Scholar]
- 26.Matsuo K, Irie N. Osteoclast-osteoblast communication. Arch. Biochem. Biophys. 2008;473:201–9. doi: 10.1016/j.abb.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 27.Andersen TL, Soe K, Sondergaard TE, Plesner T, Delaisse JM. Myeloma cell–induced disruption of bone remodelling compartments leads to osteolytic lesions and generation of osteoclast-myeloma hybrid cells. Br. J. Haematol. 2009;48:551–61. doi: 10.1111/j.1365-2141.2009.07980.x. [DOI] [PubMed] [Google Scholar]
- 28.Natl. Inst. Health Consens. Dev. Panel Osteoporosis prevention, diagnosis, and therapy. J. Am. Med. Assoc. 2001;285:785–95. doi: 10.1001/jama.285.6.785. [DOI] [PubMed] [Google Scholar]
- 29.Marcus R, Bouxsein M. The nature of osteoporosis. In: Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. Academic; San Diego: 2008. pp. 27–36. [Google Scholar]
- 30.Riggs BL, Wahner HW, Seeman E, Offord KP, Dunn WL, et al. Changes in bone mineral density of the proximal femur and spine with aging. Differences between the postmenopausal and senile osteoporosis syndromes. J. Clin. Investig. 1982;70:716–23. doi: 10.1172/JCI110667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riggs BL, Khosla S, Melton LJ. The type I/type II model for involutional osteoporosis. In: Marcus R, Feldman D, Kelsey J, editors. Osteoporosis. Academic; San Diego: 2001. pp. 49–58. [Google Scholar]
- 32.Albright F, Smith PH, Richardson AM. Postmenopausal osteoporosis. J. Am. Med. Assoc. 1941;116:2465–74. [Google Scholar]
- 33.Heaney RP, Recker RR, Saville PD. Menopausal changes in bone remodeling. J. Lab. Clin. Med. 1978;92:964–70. [PubMed] [Google Scholar]
- 34.Eriksen EF, Hodgson SF, Eastell R, Cedel SL, O’Fallon WM, Riggs BL. Cancellous bone remodeling in type I (postmenopausal) osteoporosis: quantitative assessment of rates of formation, resorption, and bone loss at tissue and cellular levels. J. Bone Miner. Res. 1990;5:311–9. doi: 10.1002/jbmr.5650050402. [DOI] [PubMed] [Google Scholar]
- 35.Garnero P, Sornay-Rendu E, Chapuy MC, Delmas PD. Increased bone turnover in late postmenopausal women is a major determinant of osteoporosis. J. Bone Miner. Res. 1996;11:337–49. doi: 10.1002/jbmr.5650110307. [DOI] [PubMed] [Google Scholar]
- 36.Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J. Clin. Investig. 2005;115:3318–25. doi: 10.1172/JCI27071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horowitz MC. Cytokines and estrogen in bone: antiosteoporotic effects. Science. 1993;260:626–27. doi: 10.1126/science.8480174. [DOI] [PubMed] [Google Scholar]
- 38.Manolagas SC, Jilka RL. Bone marrow, cytokines, and bone remodeling. Emerging insights into the pathophysiology of osteoporosis. N. Engl. J. Med. 1995;332:305–11. doi: 10.1056/NEJM199502023320506. [DOI] [PubMed] [Google Scholar]
- 39.Jilka RL. Cytokines, bone remodeling, and estrogen deficiency—a 1998 update. Bone. 1998;23:75–81. doi: 10.1016/s8756-3282(98)00077-5. [DOI] [PubMed] [Google Scholar]
- 40.Pacifici R. Cytokines, estrogen, and postmenopausal osteoporosis—the second decade. Endocrinol. 1998;139:2659–61. doi: 10.1210/endo.139.6.6087. [DOI] [PubMed] [Google Scholar]
- 41.Pacifici R, Rifas L, Teitelbaum S, Slatopolsky E, Miller R, et al. Spontaneous release of interleukin 1 from human blood monocytes reflects bone formation in idiopathic osteoporosis. Proc. Natl. Acad. Sci. USA. 1987;84:4616–20. doi: 10.1073/pnas.84.13.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pacifici R, Brown C, Puscheck E, Friedrich E, Slatopolsky E, et al. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc. Natl. Acad. Sci. USA. 1991;88:5134–38. doi: 10.1073/pnas.88.12.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Komm BS, Terpening CM, Benz DJ, Graeme KA, Gallegos A, et al. Estrogen binding, receptor mRNA, and biologic response in osteoblast-like osteosarcoma cells. Science. 1988;241:81–83. doi: 10.1126/science.3164526. [DOI] [PubMed] [Google Scholar]
- 44.Roggia C, Gao Y, Cenci S, Weitzmann MN, Toraldo G, et al. Up-regulation of TNF-producing T cells in the bone marrow: a key mechanism by which estrogen deficiency induces bone loss in vivo. Proc. Natl. Acad. Sci. USA. 2001;98:13960–65. doi: 10.1073/pnas.251534698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Girasole G, Jilka RL, Passeri G, Boswell S, Boder G, et al. 17 β-estradiol inhibits interleukin-6 production by bone marrow–derived stromal cells and osteoblasts in vitro: a potential mechanism for the antiosteoporotic effect of estrogens. J. Clin. Investig. 1992;89:883–91. doi: 10.1172/JCI115668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jilka RL, Hangoc G, Girasole G, Passeri G, Williams DC, et al. Increased osteoclast development after estrogen loss: mediation by interleukin-6. Science. 1992;257:88–91. doi: 10.1126/science.1621100. [DOI] [PubMed] [Google Scholar]
- 47.Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor αinduces differentiation of and bone resorption by osteoclasts. J. Biol. Chem. 2000;275:4858–64. doi: 10.1074/jbc.275.7.4858. [DOI] [PubMed] [Google Scholar]
- 48.Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, et al. Tumor necrosis factor α stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000;191:275–85. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000;106:1481–88. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li P, Schwarz EM, O’Keefe RJ, Ma L, Boyce BF, Xing L. RANK signaling is not required for TNFα-mediated increase in CD11hi osteoclast precursors but is essential for mature osteoclast formation in TNFα-mediated inflammatory arthritis. J. Bone Miner. Res. 2004;19:207–13. doi: 10.1359/JBMR.0301233. [DOI] [PubMed] [Google Scholar]
- 51.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–76. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 52.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA. 1998;95:3597–602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–79. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- 54.Wong BR, Rho J, Arron J, Robinson E, Orlinick J, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J. Biol. Chem. 1997;272:25190–94. doi: 10.1074/jbc.272.40.25190. [DOI] [PubMed] [Google Scholar]
- 55.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–68. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–19. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 57.Wong BR, Josien R, Lee SY, Sauter B, Li HL, et al. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell–specific survival factor. J. Exp. Med. 1997;186:2075–80. doi: 10.1084/jem.186.12.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Josien R, Wong BR, Li HL, Steinman RM, Choi Y. TRANCE, a TNF family member, is differentially expressed on T cell subsets and induces cytokine production in dendritic cells. J. Immunol. 1999;162:2562–68. [PubMed] [Google Scholar]
- 59.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–23. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 60.Bachmann MF, Wong BR, Josien R, Steinman RM, Oxenius A, Choi Y. TRANCE, a tumor necrosis factor family member critical for CD40 ligand–independent T helper cell activation. J. Exp. Med. 1999;189:1025–31. doi: 10.1084/jem.189.7.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–24. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, et al. The osteoclast differentiation factor osteoprotegerin ligand is essential for mammary gland development. Cell. 2000;103:41–50. doi: 10.1016/s0092-8674(00)00103-3. [DOI] [PubMed] [Google Scholar]
- 63.Hanada R, Leibbrandt A, Hanada T, Kitaoka S, Furuyashiki T, et al. Central control of fever and female body temperature by RANKL/RANK. Nature. 2009;462:505–9. doi: 10.1038/nature08596. [DOI] [PubMed] [Google Scholar]
- 64.Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 1999;20:345–57. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- 65.Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature. 2002;416:744–49. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- 66.Feng X. Regulatory roles and molecular signaling of TNF family members in osteoclasts. Gene. 2005;350:1–13. doi: 10.1016/j.gene.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 67.Williams JP, McDonald JM, McKenna MA, Jordan SE, Radding W, Blair HC. Differential effects of tamoxifen-like compounds on osteoclastic bone degradation, H+-ATPase activity, calmodulin-dependent cyclic nucleotide phosphodiesterase activity, and calmodulin binding. J. Cell Biochem. 1997;66:358–69. [PubMed] [Google Scholar]
- 68.Williams JP, Micoli K, McDonald JM. Calmodulin—an often-ignored signal in osteoclasts. Ann. N.Y. Acad. Sci. 2010;1192:358–64. doi: 10.1111/j.1749-6632.2009.05242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang L, Feng X, McDonald JM. The role of calmodulin in the regulation of osteoclastogenesis. Endocrinol. 2003;144:4536–43. doi: 10.1210/en.2003-0147. [DOI] [PubMed] [Google Scholar]
- 70.Saika M, Inoue D, Kido S, Matsumoto T. 17β-estradiol stimulates expression of osteoprotegerin by a mouse stromal cell line, ST-2, via estrogen receptor α. Endocrinology. 2001;142:2205–12. doi: 10.1210/endo.142.6.8220. [DOI] [PubMed] [Google Scholar]
- 71.Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J. Clin. Investig. 2003;111:1221–30. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shevde NK, Bendixen AC, Dienger KM, Pike JW. Estrogens suppress RANK ligand–induced osteoclast differentiation via a stromal cell independent mechanism involving c-Jun repression. Proc. Natl. Acad. Sci. USA. 2000;97:7829–34. doi: 10.1073/pnas.130200197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Srivastava S, Toraldo G, Weitzmann MN, Cenci S, Ross FP, Pacifici R. Estrogen decreases osteoclast formation by down-regulating receptor activator of NF-κB ligand (RANKL)-induced JNK activation. J. Biol. Chem. 2001;276:8836–40. doi: 10.1074/jbc.M010764200. [DOI] [PubMed] [Google Scholar]
- 74.Cenci S, Weitzmann MN, Roggia C, Namba N, Novack D, et al. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-α. J. Clin. Investig. 2000;106:1229–37. doi: 10.1172/JCI11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, et al. Estrogen prevents bone loss via estrogen receptor α and induction of Fas ligand in osteoclasts. Cell. 2007;130:811–23. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 76.Wu X, McKenna MA, Feng X, Nagy TR, McDonald JM. Osteoclast apoptosis: the role of Fas in vivo and in vitro. Endocrinology. 2003;144:5545–55. doi: 10.1210/en.2003-0296. [DOI] [PubMed] [Google Scholar]
- 77.Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000;21:115–37. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- 78.Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-β. Nat. Med. 1996;2:1132–36. doi: 10.1038/nm1096-1132. [DOI] [PubMed] [Google Scholar]
- 79.Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, et al. FSH directly regulates bone mass. Cell. 2006;125:247–60. doi: 10.1016/j.cell.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 80.Stefanick ML. Estrogens and progestins: background and history, trends in use, and guidelines and regimens approved by the US Food and Drug Administration. Am. J. Med. 2005;118(Suppl. 12B):64–73. doi: 10.1016/j.amjmed.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 81.Prince R, Muchmore DB, Siris ES. Estrogen analogues: selective estrogen receptor modulators and phytoestrogens. In: Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. Academic; San Diego: 2008. pp. 1705–23. [Google Scholar]
- 82.Miller P. Bisphosphonates: pharmacology and use in the treatment of osteoporosis. In: Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. Academic; San Diego: 2008. pp. 1725–42. [Google Scholar]
- 83.Gruber HE, Ivey JL, Baylink DJ, Matthews M, Nelp WB, et al. Long-term calcitonin therapy in postmenopausal osteoporosis. Metabolism. 1984;33:295–303. doi: 10.1016/0026-0495(84)90187-2. [DOI] [PubMed] [Google Scholar]
- 84.Lewiecki EM. Current and emerging pharmacologic therapies for the management of postmenopausal osteoporosis. J. Womens Health. 2009;18:1615–26. doi: 10.1089/jwh.2008.1086. [DOI] [PubMed] [Google Scholar]
- 85.Miller PD. Denosumab: anti-RANKL antibody. Curr. Osteoporos. Rep. 2009;7:18–22. doi: 10.1007/s11914-009-0004-5. [DOI] [PubMed] [Google Scholar]
- 86.Girotra M, Rubin MR, Bilezikian JP. The use of parathyroid hormone in the treatment of osteoporosis. Rev. Endocr. Metab Disord. 2006;7:113–21. doi: 10.1007/s11154-006-9007-z. [DOI] [PubMed] [Google Scholar]
- 87.Khosla S, Riggs BL. Pathophysiology of age-related bone loss and osteoporosis. Endocrinol. Metab. Clin. N. Am. 2005;34:1015–30. xi. doi: 10.1016/j.ecl.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 88.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 89.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 90.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007;8:722–28. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- 91.Baron R, Rawadi G. Wnt signaling and the regulation of bone mass. Curr. Osteoporos. Rep. 2007;5:73–80. doi: 10.1007/s11914-007-0006-0. [DOI] [PubMed] [Google Scholar]
- 92.Almeida M, Han L, Martin-Millan M, O’Brien CA, Manolagas SC. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting β-catenin from T cell factor– to forkhead box O–mediated transcription. J. Biol. Chem. 2007;282:27298–305. doi: 10.1074/jbc.M702811200. [DOI] [PubMed] [Google Scholar]
- 93.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010;31:266–300. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zayzafoon M, Fulzele K, McDonald JM. Calmodulin and calmodulin-dependent kinase II α regulate osteoblast differentiation by controlling c-fos expression. J. Biol. Chem. 2005;280:7049–59. doi: 10.1074/jbc.M412680200. [DOI] [PubMed] [Google Scholar]
- 95.Yeo H, McDonald JM, Zayzafoon M. NFATcl: A novel anabolic therapeutic target for osteoporosis. Ann. N.Y. Acad. Sci. 2006;1068:564–67. doi: 10.1196/annals.1346.053. [DOI] [PubMed] [Google Scholar]
- 96.Yeo H, Beck LH, McDonald JM, Zayzafoon M. Cyclosporin A elicits dose-dependent biphasic effects on osteoblast differentiation and bone formation. Bone. 2007;40:1502–16. doi: 10.1016/j.bone.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Giustina A, Mazziotti G, Canalis E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr. Rev. 2008;29:535–59. doi: 10.1210/er.2007-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.DiGirolamo DJ, Mukherjee A, Fulzele K, Gan Y, Cao X, et al. Mode of growth hormone action in osteoblasts. J. Biol. Chem. 2007;282:31666–74. doi: 10.1074/jbc.M705219200. [DOI] [PubMed] [Google Scholar]
- 99.Gevers EF, Loveridge N, Robinson IC. Bone marrow adipocytes: a neglected target tissue for growth hormone. Endocrinol. 2002;143:4065–73. doi: 10.1210/en.2002-220428. [DOI] [PubMed] [Google Scholar]
- 100.Barnard R, Ng KW, Martin TJ, Waters MJ. Growth hormone (GH) receptors in clonal osteoblast-like cells mediate a mitogenic response to GH. Endocrinology. 1991;128:1459–64. doi: 10.1210/endo-128-3-1459. [DOI] [PubMed] [Google Scholar]
- 101.Kassem M, Blum W, Ristelli J, Mosekilde L, Eriksen EF. Growth hormone stimulates proliferation and differentiation of normal human osteoblast-like cells in vitro. Calcif. Tissue Int. 1993;52:222–26. doi: 10.1007/BF00298723. [DOI] [PubMed] [Google Scholar]
- 102.Li H, Bartold PM, Zhang CZ, Clarkson RW, Young WG, Waters MJ. Growth hormone and insulin-like growth factor I induce bone morphogenetic proteins 2 and 4: a mediator role in bone and tooth formation? Endocrinology. 1998;139:3855–62. doi: 10.1210/endo.139.9.6211. [DOI] [PubMed] [Google Scholar]
- 103.Rivadeneira F, Houwing-Duistermaat JJ, Vaessen N, Vergeer-Drop JM, Hofman A, et al. Association between an insulin-like growth factor I gene promoter polymorphism and bone mineral density in the elderly: the Rotterdam Study. J. Clin. Endocrinol. Metab. 2003;88:3878–84. doi: 10.1210/jc.2002-021813. [DOI] [PubMed] [Google Scholar]
- 104.Langlois JA, Rosen CJ, Visser M, Hannan MT, Harris T, et al. Association between insulin-like growth factor I and bone mineral density in older women and men: the Framingham Heart Study. J. Clin. Endocrinol. Metab. 1998;83:4257–62. doi: 10.1210/jcem.83.12.5308. [DOI] [PubMed] [Google Scholar]
- 105.Riggs BL. Role of the vitamin D–endocrine system in the pathophysiology of postmenopausal osteoporosis. J. Cell Biochem. 2003;88:209–15. doi: 10.1002/jcb.10345. [DOI] [PubMed] [Google Scholar]
- 106.Kalu DN, Orhii PB. Calcium absorption and bone loss in ovariectomized rats fed varying levels of dietary calcium. Calcif. Tissue Int. 1999;65:73–77. doi: 10.1007/s002239900660. [DOI] [PubMed] [Google Scholar]
- 107.McCarthy TL, Centrella M, Canalis E. Cyclic AMP induces insulin-like growth factor I synthesis in osteoblast-enriched cultures. J. Biol. Chem. 1990;265:15353–56. [PubMed] [Google Scholar]
- 108.Riggs BL, Khosla S, Melton LJ., III Sex steroids and the construction and conservation of the adult skeleton. Endocr. Rev. 2002;23:279–302. doi: 10.1210/edrv.23.3.0465. [DOI] [PubMed] [Google Scholar]
- 109.Khosla S, Melton LJ, III, Atkinson EJ, O’Fallon WM, Klee GG, Riggs BL. Relationship of serum sex steroid levels and bone turnover markers with bone mineral density in men and women: a key role for bioavailable estrogen. J. Clin. Endocrinol. Metab. 1998;83:2266–74. doi: 10.1210/jcem.83.7.4924. [DOI] [PubMed] [Google Scholar]
- 110.Khosla S. Update in male osteoporosis. J. Clin. Endocrinol. Metab. 2010;95:3–10. doi: 10.1210/jc.2009-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Adler RA, Curtis J, Weinstein RS, Saag KG. Glucocorticoid-induced osteoporosis. In: Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. Academic; San Diego: 2008. pp. 1135–66. [Google Scholar]
- 112.Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Use of oral corticosteroids and risk of fractures. J. Bone Miner. Res. 2000;15:993–1000. doi: 10.1359/jbmr.2000.15.6.993. [DOI] [PubMed] [Google Scholar]
- 113.Liu ZJ, Zhuge Y, Velazquez OC. Trafficking and differentiation of mesenchymal stem cells. J. Cell. Biochem. 2009;106:984–91. doi: 10.1002/jcb.22091. [DOI] [PubMed] [Google Scholar]
- 114.Vande Berg BC, Malghem J, Lecouvet FE, Devogelaer JP, Maldague B, Houssiau FA. Fat conversion of femoral marrow in glucocorticoid-treated patients: a cross-sectional and longitudinal study with magnetic resonance imaging. Arthritis Rheum. 1999;42:1405–11. doi: 10.1002/1529-0131(199907)42:7<1405::AID-ANR14>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 115.Canalis E. Effect of glucocorticoids on type I collagen synthesis, alkaline phosphatase activity, and deoxyribonucleic acid content in cultured rat calvariae. Endocrinol. 1983;112:931–39. doi: 10.1210/endo-112-3-931. [DOI] [PubMed] [Google Scholar]
- 116.Peretz A, Praet JP, Bosson D, Rozenberg S, Bourdoux P. Serum osteocalcin in the assessment of corticosteroid induced osteoporosis. Effect of long and short term corticosteroid treatment. J. Rheumatol. 1989;16:363–67. [PubMed] [Google Scholar]
- 117.Yao W, Cheng Z, Pham A, Busse C, Zimmermann EA, et al. Glucocorticoid-induced bone loss in mice can be reversed by the actions of parathyroid hormone and risedronate on different pathways for bone formation and mineralization. Arthritis Rheum. 2008;58:3485–97. doi: 10.1002/art.23954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J. Clin. Investig. 1998;102:274–82. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, et al. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinol. 1999;140:4382–89. doi: 10.1210/endo.140.10.7034. [DOI] [PubMed] [Google Scholar]
- 120.Jia D, O’Brien CA, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoclasts to increase their life span and reduce bone density. Endocrinol. 2006;147:5592–99. doi: 10.1210/en.2006-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, et al. Glucocorticoids suppress bone formation via the osteoclast. J. Clin. Investig. 2006;116:2152–60. doi: 10.1172/JCI28084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, et al. Glucocorticoids and the osteoclast. Ann. N.Y. Acad. Sci. 2007;1116:335–39. doi: 10.1196/annals.1402.057. [DOI] [PubMed] [Google Scholar]
- 123.Canalis E, Mazziotti G, Giustina A, Bilezikian JP. Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos. Int. 2007;18:1319–28. doi: 10.1007/s00198-007-0394-0. [DOI] [PubMed] [Google Scholar]
- 124.Saag KG, Shane E, Boonen S, Marin F, Donley DW, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N. Engl. J. Med. 2007;357:2028–39. doi: 10.1056/NEJMoa071408. [DOI] [PubMed] [Google Scholar]
- 125.Takata S, Yasui N. Disuse osteoporosis. J. Med. Investig. 2001;48:147–56. [PubMed] [Google Scholar]
- 126.Zayzafoon M, Meyers VE, McDonald JM. Microgravity: the immune response and bone. Immunol. Rev. 2005;208:267–80. doi: 10.1111/j.0105-2896.2005.00330.x. [DOI] [PubMed] [Google Scholar]
- 127.Collet P, Uebelhart D, Vico L, Moro L, Hartmann D, et al. Effects of 1- and 6-month spaceflight on bone mass and biochemistry in two humans. Bone. 1997;20:547–51. doi: 10.1016/s8756-3282(97)00052-5. [DOI] [PubMed] [Google Scholar]
- 128.Lang T, LeBlanc A, Evans H, Lu Y, Genant H, Yu A. Cortical and trabecular bone mineral loss from the spine and hip in long-duration spaceflight. J. Bone Miner. Res. 2004;19:1006–12. doi: 10.1359/JBMR.040307. [DOI] [PubMed] [Google Scholar]
- 129.Smith SM, Nillen JL, LeBlanc A, Lipton A, Demers LM, et al. Collagen cross-link excretion during space flight and bed rest. J. Clin. Endocrinol. Metab. 1998;83:3584–91. doi: 10.1210/jcem.83.10.5169. [DOI] [PubMed] [Google Scholar]
- 130.Smith SM, Wastney ME, O’Brien KO, Morukov BV, Larina IM, et al. Bone markers, calcium metabolism, and calcium kinetics during extended-duration space flight on the Mir space station. J. Bone Miner. Res. 2005;20:208–18. doi: 10.1359/JBMR.041105. [DOI] [PubMed] [Google Scholar]
- 131.You J, Yellowley CE, Donahue HJ, Zhang Y, Chen Q, Jacobs CR. Substrate deformation levels associated with routine physical activity are less stimulatory to bone cells relative to loading-induced oscillatory fluid flow. J. Biomech. Eng. 2000;122:387–93. doi: 10.1115/1.1287161. [DOI] [PubMed] [Google Scholar]
- 132.Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–75. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 133.Papachroni KK, Karatzas DN, Papavassiliou KA, Basdra EK, Papavassiliou AG. Mechanotransduction in osteoblast regulation and bone disease. Trends Mol. Med. 2009;15:208–16. doi: 10.1016/j.molmed.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 134.Rubin J, Fan X, Biskobing DM, Taylor WR, Rubin CT. Osteoclastogenesis is repressed by mechanical strain in an in vitro model. J. Orthop. Res. 1999;17:639–45. doi: 10.1002/jor.1100170504. [DOI] [PubMed] [Google Scholar]
- 135.Rubin J, Murphy TC, Fan X, Goldschmidt M, Taylor WR. Activation of extracellular signal-regulated kinase is involved in mechanical strain inhibition of RANKL expression in bone stromal cells. J. Bone Miner. Res. 2002;17:1452–60. doi: 10.1359/jbmr.2002.17.8.1452. [DOI] [PubMed] [Google Scholar]
- 136.Wiltink A, Nijweide PJ, Scheenen WJ, Ypey DL, Van Duijn B. Cell membrane stretch in osteoclasts triggers a self-reinforcing Ca2+ entry pathway. Pflüg. Arch. 1995;429:663–71. doi: 10.1007/BF00373987. [DOI] [PubMed] [Google Scholar]
- 137.Saxena R, Pan G, Dohm EK, McDonald JM. Modeled microgravity and hindlimb unloading sensitize osteoclast precursors to RANKL-mediated osteoclastogenesis. J. Bone Miner. Metab. 2010 doi: 10.1007/s00774-010-0201-4. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rubin J, Rubin C, Jacobs CR. Molecular pathways mediating mechanical signaling in bone. Gene. 2006;367:1–16. doi: 10.1016/j.gene.2005.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chen JH, Liu C, You L, Simmons CA. Boning up on Wolff’s Law: mechanical regulation of the cells that make and maintain bone. J. Biomech. 2010;43:108–18. doi: 10.1016/j.jbiomech.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 140.Phillips JA, Almeida EA, Hill EL, Aguirre JI, Rivera MF, et al. Role for β1 integrins in cortical osteocytes during acute musculoskeletal disuse. Matrix Biol. 2008;27:609–18. doi: 10.1016/j.matbio.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 141.Davidson RM, Tatakis DW, Auerbach AL. Multiple forms of mechanosensitive ion channels in osteoblast-like cells. Pflüg. Arch. 1990;416:646–51. doi: 10.1007/BF00370609. [DOI] [PubMed] [Google Scholar]
- 142.Meyers VE, Zayzafoon M, Gonda SR, Gathings WE, McDonald JM. Modeled microgravity disrupts collagen I/integrin signaling during osteoblastic differentiation of human mesenchymal stem cells. J. Cell Biochem. 2004;93:697–707. doi: 10.1002/jcb.20229. [DOI] [PubMed] [Google Scholar]
- 143.Meyers VE, Zayzafoon M, Douglas JT, McDonald JM. RhoA and cytoskeletal disruption mediate reduced osteoblastogenesis and enhanced adipogenesis of human mesenchymal stem cells in modeled microgravity. J. Bone Miner. Res. 2005;20:1858–66. doi: 10.1359/JBMR.050611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bikle DD, Morey-Holton ER, Doty SB, Currier PA, Tanner SJ, Halloran BP. Alendronate increases skeletal mass of growing rats during unloading by inhibiting resorption of calcified cartilage. J. Bone Miner. Res. 1994;9:1777–87. doi: 10.1002/jbmr.5650091115. [DOI] [PubMed] [Google Scholar]
- 145.Turner RT, Evans GL, Lotinun S, Lapke PD, Iwaniec UT, Morey-Holton E. Dose-response effects of intermittent PTH on cancellous bone in hindlimb unloaded rats. J. Bone Miner. Res. 2007;22:64–71. doi: 10.1359/jbmr.061006. [DOI] [PubMed] [Google Scholar]
- 146.Block GA, Cunningham J. Morbidity and mortality associated with abnormalities in bone and mineral metabolism in CKD. In: Olgaard K, editor. Clinical Guide to the Basics of Bone and Mineral Metabolism in CKD. Natl. Kidney Found; New York: 2006. pp. 77–92. [Google Scholar]
- 147.Moe S, Drueke T, Cunningham J, Goodman W, Martin K, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO) Kidney Int. 2006;69:1945–53. doi: 10.1038/sj.ki.5000414. [DOI] [PubMed] [Google Scholar]
- 148.Malluche HH, Monier-Faugere MC, Herberth J. Bone disease after renal transplantation. Nat. Rev. Nephrol. 2010;6:32–40. doi: 10.1038/nrneph.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hruska KA. Chronic kidney disease mineral bone disorder (CKD-MBD) In: Rosen CJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Am. Soc. Bone Miner. Res.; Washington, DC: 2008. pp. 343–49. [Google Scholar]
- 150.Singer FR. Paget disease: when to treat and when not to treat. Nat. Rev. Rheumatol. 2009;5:483–89. doi: 10.1038/nrrheum.2009.149. [DOI] [PubMed] [Google Scholar]
- 151.Siris ES, Feldman F. Natural history of untreated Paget’s disease of the tibia. J. Bone Miner. Res. 1997;12:691–92. doi: 10.1359/jbmr.1997.12.4.691. [DOI] [PubMed] [Google Scholar]
- 152.Siris ES. Extensive personal experience: Paget’s disease of bone. J. Clin. Endocrinol. Metab. 1995;80:335–38. doi: 10.1210/jcem.80.2.7852484. [DOI] [PubMed] [Google Scholar]
- 153.Michou L, Chamoux E, Couture J, Morissette J, Brown JP, Roux S. Gene expression profile in osteoclasts from patients with Paget’s disease of bone. Bone. 2010;46:598–603. doi: 10.1016/j.bone.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tolar J, Teitelbaum SL, Orchard PJ. Osteopetrosis. N. Engl. J. Med. 2004;351:2839–49. doi: 10.1056/NEJMra040952. [DOI] [PubMed] [Google Scholar]
- 155.Stark Z, Savarirayan R. Osteopetrosis. Orphanet J. Rare. Dis. 2009;4:5. doi: 10.1186/1750-1172-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Driessen GJ, Gerritsen EJ, Fischer A, Fasth A, Hop WC, et al. Long-term outcome of haematopoietic stem cell transplantation in autosomal recessive osteopetrosis: an EBMT report. Bone Marrow Transplant. 2003;32:657–63. doi: 10.1038/sj.bmt.1704194. [DOI] [PubMed] [Google Scholar]
- 157.Lips P, van Schoor NM, Bravenboer N. Vitamin D–related disorders. In: Rosen CJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Am. Soc. Bone Miner. Res.; Washington, DC: 2008. pp. 329–35. [Google Scholar]