

Abstract
Morphogenesis encompasses programmed changes in gene expression that lead to the development of specialized cell types. In the model fungus Aspergillus nidulans, asexual development involves the formation of characteristic cell types, collectively known as the conidiophore. With the aim of determining the transcriptional changes that occur upon induction of asexual development, we have applied massive mRNA sequencing to compare the expression pattern of 19-h-old submerged vegetative cells (hyphae) with that of similar hyphae after exposure to the air for 5 h. We found that the expression of 2,222 (20.3%) of the predicted 10,943 A. nidulans transcripts was significantly modified after air exposure, 2,035 being downregulated and 187 upregulated. The activation during this transition of genes that belong specifically to the asexual developmental pathway was confirmed. Another remarkable quantitative change occurred in the expression of genes involved in carbon or nitrogen primary metabolism. Genes participating in polar growth or sexual development were transcriptionally repressed, as were those belonging to the HogA/SakA stress response mitogen-activated protein (MAP) kinase pathway. We also identified significant expression changes in several genes purportedly involved in redox balance, transmembrane transport, secondary metabolite production, or transcriptional regulation, mainly binuclear-zinc cluster transcription factors. Genes coding for these four activities were usually grouped in metabolic clusters, which may bring regulatory implications for the induction of asexual development. These results provide a blueprint for further stage-specific gene expression studies during conidiophore development.
INTRODUCTION
Public availability of hundreds of fungal genome sequences, as well as the advent of high-throughput proteomic and transcriptomic methods, has allowed the acquisition of genome-scale data and the characterization of transcripts and proteins which have no designated function (1, 2).
In the genus Aspergillus, which includes model organisms as well as industrially and medically important species, proteomic studies have focused mainly on two-dimensional PAGE (2D-PAGE) coupled to tandem mass spectrometry (MS-MS), gaining valuable insight into the composition of the proteome under different growth and stress conditions (3). The main transcriptomic approach to the development, stress response, or secondary metabolite production of aspergilli has involved microarray analyses (see, for example, references 4–6, and 7). RNA sequencing (RNA-seq) (8) technology allows a deeper and more reproducible analysis of gene expression and regulation with a higher sensitivity than microarray analysis (8–10). It has been successfully used to elucidate transcriptomes of microbes and higher eukaryotes (see references within reference 11). Thus, this powerful technique arises as an efficient tool for transcriptomic analyses in the genus Aspergillus, as shown in 2010 by Wang and coworkers (11), who published the first RNA-seq-based transcriptomic study of this genus. In these last 2 years, the RNA-seq-based studies involving Aspergillus species analyzed the temperature effect on secondary metabolite synthesis, biofilm formation, the response to lignocellulose, or domestication in A. fumigatus, A. flavus, A. niger, or A. oryzae, respectively (11–15).
Aspergillus nidulans is the reference organism in the study of fungal asexual development, also known as conidiation or conidiophore development (16–19). The required morphological changes during conidiophore formation (Fig. 1) (20) are induced by environmental signals and arise from nonspecialized cells called vegetative hyphae. Conidiophore development starts with the formation of the foot cell, which has a thick cell wall. Then, a branch emerges from the foot cell and elongates through apical extension displaying negative geotropism, thus forming the stalk. This is followed by a swelling process of the stalk tip to form the vesicle. Then, a massive multipolar budding process at the dome of the vesicle generates a layer of approximately 60 primary sterigmata or metulae, followed by their respective apical budding to generate 120 secondary sterigmata or phialides. The vesicle, metulae, phialides, and conidia are separated by septa, the production of which is regulated by conidiophore-nonspecific and -specific bud site markers (see, for example, references 21 and 22). Finally, each phialide produces, via basipetal cell divisions, long chains of more than 100 asexual propagules called conidia (Fig. 1).
Fig 1.
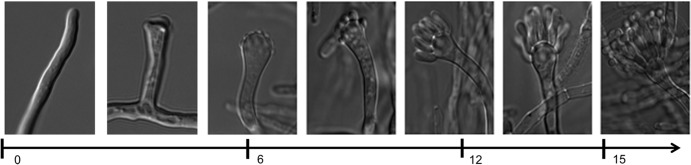
Morphogenetic transformations leading to conidium production: Time after induction of conidiophore development is indicated (in hours).
The morphological changes described above are programmed at the genetic level. For practical purposes, initial works divided the process into two genetic stages (16). Stage one included factors involved in the perception of environmental changes, transduction of these signals, and the launching of the initial morphogenetic transformations leading to vesicle formation. These are the upstream developmental activators (UDA) (17). Loss-of-function mutations in these genes yield a “fluffy” aconidial phenotype that is manifested as cotton-like masses of vegetative cells and the absence of cell differentiation (23). From the genetic point of view, the fluffy phenotype of UDA mutants is associated with the inability to induce the second stage, characterized by the control exerted by the C2H2-type transcription factor (TF) brlA, the first conidiation-specific TF (24). BrlA and downstream factors of the central developmental pathway of conidiation (CDP), such as AbaA and WetA (25, 26), regulate the spatiotemporal morphological transformations leading to spore formation.
In this simplified genetic model, the handover of control between UDA and CDP pathways is not addressed. The requirement of additional proteins other than those reported in early asexual transformations is, in addition, highly feasible. With the aim of identifying the genes and determining the cellular processes significantly altered during this transition, we used in this work a RNA-seq approach to compare the transcriptomes of submerged 19 h-old vegetative hyphae (nonspecialized cells) and 5 h-old air-exposed samples (in which asexual structures started to develop). The first time point was chosen based, on one hand, on the acquisition of the developmental competence, which is required to develop conidiophores (16) and, on the other hand, because this was the reference time point for the vegetative stage in our previous works (27, 28). The second time point was chosen based on the expression peak of UDA coding genes, which coincide with a remarkable induction of brlA and the CDP pathway (17). We found that exposure of hyphae to the air provokes a reorganization of primary metabolic pathways, a restructuring of the cell wall, and a repression of polar growth as well as sexual development. In addition, expression of transcripts predictably involved in redox balance, transmembrane transport, and transcriptional regulation is strongly affected, some of them being grouped in metabolic clusters together with polyketide synthases or nonribosomal peptide synthetases. Taken together, this work shows that asexual developmental induction involves the simultaneous activation or repression of specific pathways and cellular processes.
MATERIALS AND METHODS
Fungal strains and culture conditions.
As a reference A. nidulans strain, we used MAD2666 (kindly provided by Ane Markina-Iñarrairaegui), an isogenic strain of TN02A3 (29), where the pyrG89 mutation was eliminated by gene replacement using a wild-type (WT) fragment of the pyrG gene. Vegetative cell samples were obtained by culturing 106 spores/ml in liquid minimal medium (MMA) with the appropriate supplements (30) for 19 h. Filtered mycelia were processed for RNA extraction.
Induction of asexual development was conducted as described previously (31, 32). Briefly, after 19 h of culture in liquid MMA as described above, mycelia were filtered using nitrocellulose membranes (0.45 μm, MicronSep; GE Water and Process Technologies). These membranes were placed on solid minimal medium, and mycelia were cultured for 5 h before being collected and processed for RNA extraction. Two biological replicates were processed for each culture condition.
RNA isolation, mRNA library construction, and Illumina sequencing.
Mycelium samples (100 mg [dry weight]) were frozen in liquid nitrogen, and total RNA extraction from these samples was performed according to the Invitrogen protocol based on TRIzol reagent using 1 ml of TRIreagent (Fluka) per sample. Isolated total RNA samples were then further purified using the Qiagen RNeasy minikit, following the manufacturer's instructions. The concentration and integrity of total RNA were checked using a Nanodrop instrument (Thermo Scientific) and/or a Bioanalyzer 2100 system (Agilent Technologies).
mRNA libraries were prepared from A. nidulans total RNA samples following Illumina standard protocols (Ilumina, San Diego, CA). Briefly, each total RNA sample (20 to 50 μg) was treated with DNase and enriched for mRNA using oligo(dT) tags. Samples of poly(A) RNA (0.2 to 1 μg) were fragmented into smaller pieces (200 to 500 bp; mean for all libraries is approximately 280 bp) and used to synthesize cDNA. The cDNA library construction involved end repair, A tailing, adapter ligation, and library amplification, followed by cluster generation and sequencing. Sequencing was performed in a pair-end-read, 2× 76-base mode on a GAIIx sequencer (Illumina, San Diego, CA), running four samples per lane (multiplexing).
Demultiplexing, mapping, assembling and quantifying sequencing data.
Sequences were demultiplexed using the software program by Brian J. Knaus, freely accessible from his web page (http://brianknaus.com/). They are 75 nucleotides (nt) in length, since the barcodes have been removed or trimmed.
Read quality was checked using the fastQC software program, and only reads with quality values higher than Q30 were introduced for mapping. All reads were mapped using the software program Bowtie 2.0.0-beta5 (http://bowtie-bio.sourceforge.net/index.shtml), using parameters by default, which allow two mismatches per read. The version s07-m02-r07 of the Aspergillus Genome Database (http://www.aspergillusgenome.org/) provided the annotated genome of Aspergillus nidulans, which was used as the template for mapping.
Differential expression.
The volume and complexity of data from RNA-seq experiments demand mathematical analysis software. TopHat and Cuffdiff are open-source software tools for gene discovery and comprehensive expression analysis of high-throughput RNA sequencing data. The Cuffdiff program (http://cufflinks.cbcb.umd.edu/index.html) was used in order to detect genes differentially expressed between different samples. To associate with predicted genes, we used the fasta file provided by the Aspergillus Genome Database as the reference for the gtf file from previous mapping steps. As input files, we used the preformatted mapping files obtained after running the software program TopHat V1.4.1 (http://tophat.cbcb.umd.edu/). We followed the protocol described by Trapnell and collaborators (33).
Cuffdiff learns how read counts vary for each gene across the replicates and uses these variance estimates to calculate the significance of observed changes in expression. Cuffdiff calculates the P value (the uncorrected P value of the test statistic) and q value (the FDR-adjusted P value of the test statistic). The significance depends on whether P is greater than the false discovery rate (FDR) after a Benjamini-Hochberg correction for multiple testing (in our case, q values between 0 and 0.5 indicate significant changes).
Data visualization.
We used an R software application called CummeRbund to visualize the results of the RNA-seq analysis. This R program converts the different output files from TopHat or Cufflinks into a related database (CuffData.db) in order to obtain customized graphs.
Gene ontology analysis.
Gene ontology (GO) terms for each A. nidulans gene were obtained from the Aspergillus genome database (http://www.aspgd.org/download/go/gene_association.aspgd.gz) and were related with terms downloaded from OBO (http://www.geneontology.org/ontology/obo_format_1_2/gene_ontology_ext.obo). The Gene Ontology (GO) project provided a standardized set of terms describing the molecular functions of genes. We used the topGO software package from the Bioconductor project (http://www.bioconductor.org/packages/release/bioc/html/topGO.html) to identify overrepresented GO terms from a set of differentially expressed genes. The Python programming language (http://www.python.org/) was used to prepare the data, utilizing rpy2 (http://rpy.sourceforge.net/rpy2.html) to call R for the statistical analysis.
Nucleotide sequence accession number.
Our Illumina sequence reads have been submitted to the NCBI Sequence Read Archive (SRA) with the accession number SRR623029.
RESULTS AND DISCUSSION
Summary of the RNA-seq data set.
To identify genes that might be involved in the induction of asexual development and obtain a broad view on the associated cellular processes, total RNA samples from submerged 19-h-old vegetative hyphae (VG) and 5-h-old air-exposed (asexual development induction [AD]) hyphae were subjected to high-throughput Illumina sequencing. We obtained an average of 8,566,985 reads of 72 bp per sample (34,267,942 reads, for all 4 samples), representing nearly 20 A. nidulans genome lengths per sample (∼82 genome lengths with all 4 samples). Two biological replicates showed a high level of correlation (r = 0.909 for VG and r = 0,839 for AD; see Fig. S1 in the supplemental material).
RNA-seq analysis revealed that almost the whole set of genes carried in the A. nidulans genome is expressed during vegetative or early asexual stages. Of the 10,943 transcripts predicted by the Aspergillus Genome Database, 9,763 (89.2%) were expressed during VG and 10,059 (91.9%) during early AD (see Table S1 in the supplemental material). Ten thousand one hundred ninety-two genes were expressed under one or both conditions, and 751 genes were not expressed under either condition. Of the 10,192 expressed genes, 429 were uniquely expressed during VG and 113 genes at the early AD. Of the remaining 9,650 genes that were found to be expressed under both conditions, 2,222 showed a significant differential expression, of which 187 were upregulated (higher transcript levels in asexual than in vegetative samples) and 2,035 were downregulated (Fig. 2B; see also Table S2). The number of genes with a significant expression difference (2,222) is considerably higher than that described in early works on A. nidulans conidiation (34). It was estimated that 45 to 150 loci contributed specifically to spore production, while contributions by Timberlake and collaborators increased this number and suggested that approximately 1,200 unique mRNAs accumulated preferentially during conidiation (35, 36). Tables 1 and 2 show the top 20 genes with the highest significant increase (upregulated) or decrease (downregulated) in expression levels upon induction of conidiation, respectively. In order to obtain an overview of the process, an envisaged functional analysis of the top 20 genes is presented in the next section together with the rest of the significantly regulated transcripts.
Fig 2.

Summary of the RNA-seq data set. (A) Box plot (csBoxplot software program) showing the distribution of the FPKM (fragments per kilobase of exon per million fragments mapped) values. (B) Volcano graph showing differentially (in blue) and nondifferentially (in red) expressed genes. Values of >0 correspond to downregulated genes, while values of <0 correspond to upregulated genes. (C) Schematic representation showing the location of significantly upregulated (in red) and downregulated (in green) genes in each Aspergillus nidulans chromosome.
Table 1.
Top 20 upregulated genes
| Ranka | Gene | FPKM valueb |
Log2 FC | Description | |
|---|---|---|---|---|---|
| Asex | Veg | ||||
| 1 | An3227 | 24.1786 | 0.113703 | −7.73232 | Predicted monooxygenase activity |
| 2 | An7521 | 77.5662 | 0.482759 | −7.32798 | Unknown |
| 3 | An3247 | 20.8309 | 0.199484 | −6.70631 | Predicted ATP binding, ATPase activity |
| 4 | An4119 | 404.778 | 8.4472 | −5.58252 | Putative major facilitator superfamily protein |
| 5 | gelD | 341.63 | 9.37753 | −5.18708 | Putative 1,3-beta-transglycosidase |
| 6 | An8459 | 20.258 | 0.560254 | −5.17626 | Predicted role in transmembrane transport |
| 7 | An6401 | 929.352 | 28.7111 | −5.01655 | Putative hydrophobin |
| 8 | rodA | 93.0036 | 3.02418 | −4.94267 | Hydrophobin; protein involved in conidium development |
| 9 | An6477 | 110.534 | 4.10222 | −4.75194 | Predicted role in transmembrane transport |
| 10 | ivoB | 214.457 | 8.3293 | −4.68635 | Conidiophore-specific phenol oxidase |
| 11 | An3336 | 15.8021 | 0.632131 | −4.64375 | Putative enodomannanase |
| 12 | An12331 | 5.72154 | 0.241462 | −4.56653 | Putative PKS-like enzyme |
| 13 | An2841 | 12.5751 | 0.540246 | −4.54081 | Predicted role in transmembrane transport |
| 14 | An8308 | 285.408 | 12.347 | −4.5308 | Unknown |
| 15 | An7898 | 6.75941 | 0.312821 | −4.43349 | Predicted role in transmembrane transport |
| 16 | phiA | 188.471 | 9.37504 | −4.32938 | Protein required for normal phialide development |
| 17 | atrA | 37.9445 | 1.91388 | −4.30932 | Putative plasma membrane ATP-binding cassette (ABC) transporter |
| 18 | An7891 | 13.9495 | 0.732813 | −4.25062 | Putative beta-1.4-endoglucanase |
| 19 | An5370 | 21.7855 | 1.27754 | −4.09192 | Predicted role in transmembrane transport |
| 20 | apdA | 10.4226 | 0.670061 | −3.95928 | Putative hybrid PKS-NRPS |
Genes are ranked according to level of change in expression.
Asex, asexual stage; Veg, vegetative stage.
Table 2.
Top 20 downregulated genes
| Ranka | Gene | FPKM valueb |
Log2 FC | Description | |
|---|---|---|---|---|---|
| Asex | Veg | ||||
| 1 | An2808 | 0.214378 | 11.3192 | 5.72247 | Unknown |
| 2 | An9006 | 0.467779 | 20.7339 | 5.47002 | Unknown |
| 3 | An4392 | 0.636518 | 23.7142 | 5.21941 | Unknown |
| 4 | An7200 | 0.606272 | 21.4411 | 5.14427 | Predicted role in transmembrane transport and integral to membrane localization |
| 5 | An8779 | 0.189746 | 6.34872 | 5.06433 | Predicted hydrolase activity |
| 6 | An12277 | 0.138087 | 4.50668 | 5.02842 | Predicted iron ion binding, nucleotide binding, oxidoreductase activity |
| 7 | An8159 | 0.321225 | 10.4837 | 5.02842 | Predicted DDE1 transposon-related |
| 8 | An4586 | 35.07 | 951.717 | 4.76222 | Predicted nucleic acid binding, zinc ion binding activity and intracellular localization |
| 9 | An7954 | 0.78813 | 21.1459 | 4.7458 | Unknown |
| 10 | An5332 | 11.8009 | 294.354 | 4.64059 | Predicted nutrient reservoir activity |
| 11 | An11313 | 5.67334 | 141.02 | 4.63556 | Unknown |
| 12 | An10039 | 3.54792 | 88.0903 | 4.63394 | Putative histidine acid phosphatase |
| 13 | An8733 | 1.43304 | 35.0303 | 4.61146 | Predicted oxidoreductase activity |
| 14 | An7357 | 29.2723 | 705.244 | 4.59052 | Unknown |
| 15 | An3341 | 0.351559 | 8.42835 | 4.58341 | Predicted chromate transmembrane transporter activity |
| 16 | An3175 | 1.5925 | 35.8123 | 4.49109 | Predicted transferase activity |
| 17 | An1320 | 0.233046 | 5.11311 | 4.45552 | Predicted serine-type peptidase activity and role in proteolysis |
| 18 | An8621 | 5.62032 | 122.55 | 4.44657 | Predicted role in transmembrane transport |
| 19 | mdpA | 1.12835 | 22.6884 | 4.32967 | Secondary metabolite regulatory protein |
| 20 | An5505 | 0.32594 | 6.49 | 4.31554 | Unknown |
Genes are ranked according to level of change in expression.
Asex, asexual stage; Veg, vegetative stage.
We also analyzed the distribution of significantly regulated genes along the A. nidulans chromosomes and confirmed that there was not any obvious genomic region enriched in them (Fig. 2C). However, chromosome III contained a significant increase in downregulated genes compared to the rest of the chromosomes, with a ratio (down- versus upregulated genes) of 22.6 in comparison to an average of 10.9. In contrast, chromosome VII contained the highest proportion of upregulated genes, with a down- versus upregulated ratio of 7.7.
Functional analysis of early asexual development.
To obtain a comprehensive picture of the pathways and cellular processes switched on/off as the initial stages of asexual development proceeded, we divided the list of 2,222 genes with significant altered expression (see Table S2 in the supplemental material) into two groups. On one hand, we studied those genes having a standard name, which meant that they were previously functionally annotated (274 genes; 12.3%). On the other hand, we analyzed those containing only a systematic name and not characterized to date (1,948 genes; 87.6%).
From the 274 genes in the first group, 236 (86.1%) showed lower expression levels after the induction than in VG (downregulated; log2 fold change [FC] > 0), while 38 (13.9%) showed higher expression at the AD (upregulated; log2 FC < 0). This is clearly in agreement with the Volcano graph shown in Fig. 2B.
Using the Aspergillus Genome Database (www.aspgd.org) and previously published works, we extracted all the available information on the function, localization, genetic pathway, and/or cellular process in which those genes in the first group are described or predicted to participate. This information is available in Table S3 in the supplemental material. Genes were grouped according to their participation in different cellular processes. Genes encoding putative cytochrome P450s were included in a separated group called “Electron transfer and energy metabolism.” Some genes were included in more than one group, since they have been described to participate in several cellular processes. Figure 3 shows how these cellular processes are represented, as well as the proportions of downregulated (green) and upregulated (red) genes. Genes predicted to code for proteins with miscellaneous functions are listed in the group called “Unknown/Other” (see Table S3). The most represented processes are “Primary carbon and nitrogen metabolism,” with 76 genes, 67 down- and 9 upregulated (76: 67 + 9), “Stress response” (37: 34 + 3), “Hyphal morphogenesis” (25: 24 + 1), “Conidiation” (27: 21 + 6), “Cell wall organization and biogenesis” (19: 10 + 9), “Secondary metabolism” (17: 13 + 4), “Nucleic acid assembly, organization, and integrity” (18: 18 + 0), and “Sexual development” (15: 14 + 1). Other underrepresented processes are “Fatty acid metabolism” (9: 7 + 2), “GTPase, ATPase, and channels” (11: 10 + 1), “Nuclear transport” (6: 6 + 0), “7 transmembrane domain and heterotrimeric G protein signaling” (6: 6 + 0), “Cell death” (9: 9 + 0), “Siderophore synthesis and transport” (3: 2 + 1), and “Cell cycle regulation” (3: 3 + 0).
Fig 3.
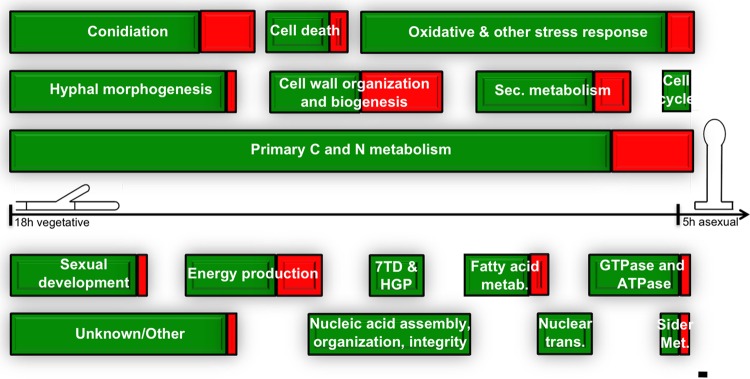
Cellular processes significantly regulated during the transition from VG (19 h) to early stages of AD (5 h after induction) in A. nidulans. Block size is determined by the number of previously known genes listed in Table S3 in the supplemental material and participating in each of these processes (bar = 1 gene). Downregulated genes are shown in green, while upregulated genes are in red. For the complete list of genes in each process and their described or putative function, see Table S3.
Induction of asexual development provokes alterations in primary metabolism pathways.
The analysis of significantly regulated genes suggests that distinct cellular processes are altered to fulfill the requirements of conidiophore development. For instance, primary metabolism appears to be strongly reoriented. Several genes coding for enzymes acting in glycolysis and gluconeogenesis, tricarboxylic acid (TCA) cycle, urea cycle, or amino acid synthesis pathways are downregulated, and few of them upregulated. We also identified downregulated transcriptional regulators that play a key role in nitrogen metabolite repression (AreA and MeaB [37, 38]) or carbon catabolite repression (CreA [39]) or are involved in the cross-pathway control of amino acid biosynthesis in response to amino acid starvation (CpcA and JlbA) (40, 41). Strong variations in the levels of primary metabolism enzymes under a wide array of growth conditions have been commonly described, including reports of early biochemical studies of fungal AD (42, 43). For example, experiments examining calcium-induced conidiation in Penicillium notatum (44) showed several regulated enzymes, such as fructose-bisphosphate aldolase, triosephosphate isomerase, pyruvate kinase, or glucose-6-phosphate dehydrogenase. Genes coding for these enzymes also appear in our analysis to be significantly regulated. This metabolic switch can be attributed to the starvation in nutrients associated with the exposition of hyphae to the aerial environment, the stimulus used in this work to induce conidiophore development (see below).
Air emergence and regulation of HogA/SakA MAP kinase stress response pathway.
Table S3 in the supplemental material shows that an important number of significantly up- or downregulated genes are involved in the response to stress situations. A large number of them belong to the HogA/SakA-mediated general stress response pathway (18, 45, and references therein). The expression of multiple factors from this pathway, starting with those involved in signal perception and transduction, such as histidine kinases (TcsA, involved in AD [46], and FphA, involved in reception of red light), the phosphotransfer protein YpdA, the response regulators SrrA and SrrC, the mitogen-activated protein (MAP) kinase kinase (MAPKK) PbsB, and the MAP kinase (MAPK) HogA/SakA, is downregulated. TFs that act downstream, such as NapA and AtfA, which are supposed to activate the expression of proteins involved in the detoxification of stress-causing agents, such as catalases CatA, CatC, or CpeA (47), are also downregulated.
Previous works linked elements from this pathway with different stages of AD. It was described that the loss of tcsA, the homologue of the Saccharomyces cerevisiae transmembrane osmosensor Sln1p, did not block initiation of conidiophore development but appeared to prevent the cell divisions preceding formation of conidia from phialides (46). Conidiation was also reduced in a ΔfphA strain in comparison to that in the wild type (48). The loss of SrrA or SskA activity has been linked to decreased brlA levels (49). These previous observations strongly suggested that histidine kinases and components of the phosphorelay system are required to coordinate different stages of AD and the response to ambient stimuli (18).
Although transcriptional results obtained in this work provided valuable information on how this pathway is regulated at VG and 5 h of AD, previous reports also described transcriptional and translational changes at later stages of AD. For example, we show in this work that the expression of the catalase-coding gene catA decreases 10 times in the VG-to-early-AD transition (log2 FC = 3.17) (see Table S2 in the supplemental material). However, it increases again at late stages of conidium production and in mature conidia (50, 51). Furthermore, protein interaction, as well as phosphorylation, is key in the control of the activity of proteins from the HogA/SakA pathway (52, 53). For example, the TF AtfA physically interacts with SakA and is required for its nuclear accumulation in conidia and in stressed hyphae (53). SakA is transiently phosphorylated after 20 to 60 min of air exposure (52) and is also phosphorylated in conidia (53). It has been proposed that SakA phosphorylation could be a general mechanism to regulate the transition between nongrowing and growing states in fungi (53). Thus, we suggest that the activity of this pathway may be differently regulated as conidiophores mature, with stages of transcriptional inhibition/activation and/or protein (de)phosphorylation.
Proteins involved in vegetative growth are inhibited after asexual induction, while the composition of the cell wall undergoes strong alterations.
Polar-growth-related functions active at VG are, in general terms, inhibited 5 h after the induction of conidiation. At this stage of AD, vesicles are forming or already formed, and this requires an isotropic mode of growth. In agreement with this morphological observation, we found that of a total of 25 significantly regulated genes that code for proteins involved in different aspects of polar growth (establishment of polarity and germination, endocytosis, polarisome components, proteins required for a proper branching pattern, cytoskeleton proteins, etc.), all except one are downregulated (Fig. 3; see also Table S3 in the supplemental material).
Seminal works on the morphology of conidiophores reported that one characteristic feature of the foot cell was a thicker cell wall than that of growing hyphae (20). Furthermore, it was described that cell wall modifications associated with the maturation of conidia occurred in three stages, which demanded the production of four wall layers (19, 54). In this work, we identified 19 genes involved in cell wall organization and biogenesis (see Table S3 in the supplemental material), 10 down- and 9 upregulated in the VG-to-AD transition. Both groups included genes involved in the synthesis and processing of the main constituents of the cell wall: α- and β-glucans and chitin (55). Consequently, we cannot provide a detailed description on how the down- or upregulation of these genes could affect the final cell wall composition of developing stalks and vesicles in comparison to that of vegetative hyphae. However, our and previous data strongly point to the cell wall as a target of important transformations during the initiation and progression of the synthesis of the conidiophore.
Sexual-asexual development balance.
A general inhibition of regulators of sexual development was also found in our study. It has been previously reported that the deletion of the oxylipin biosynthetic gene ppoA increases the conidium/ascospore ratio (56). In this context, here we found that ppoA levels are significantly reduced after conidiation induction, probably to favor AD. Light receptors of the velvet complex (48, 57, 58) were also downregulated, as were other TFs, such as NsdC, an activator of sexual development (59), or RosA, a repressor of sexual development under carbon starvation conditions and in submerged culture (60).
Among genes included in the group of conidiation genes, there are factors that indirectly regulate AD or regulate the balance of the asexual cycle and other morphogenetic processes, such as sexual reproduction or VG (veA, bemA, or ppoA, for example). We also found that the expression of TFs from the CDP pathway of conidiation is differently regulated. Transcripts coding for factors supposed to act at late stages of AD (metulae, phialides, and conidia) are downregulated at early stages. This occurs with TF-coding transcripts, such as vosA and abaA, and also with yA, required for the production of the pigment that provides A. nidulans spores with the characteristic green color. Previous works described no detection of these three transcripts by Northern blotting (61–63). Although abaA is significantly repressed after the induction of AD, our results also showed that its mRNA levels remained near zero at both stages (see Table S2 in the supplemental material), probably not being enough for detection by Northern blotting. vosA and yA mRNA levels are higher than those of abaA (see Table S2), and the two transcripts are expressed at similar levels. However, neither vosA nor yA was detected at VG or early AD by Northern hybridization (62, 63). This strongly suggests that the apparent disagreement between our results and those previously obtained by Northern blotting is a consequence of the differences in the detection limit between this technique and RNA sequencing.
We found six transcripts that were upregulated in the VG-to-early-AD transition. One is, as expected, brlA, but we also found transcripts required for conidium differentiation and integrity (phiA and the hydrophobin coding gene rodA [64, 65]) or conidium pigmentation (ivoB and ivoC [66, 67]). It would be interesting to know why pigmentation genes such as yA are downregulated at this stage and ivoB or ivoC is upregulated. It has been described that the expression of the phenol oxidase-coding transcript ivoB increases with the levels of its substrate N-acetyl-6-hydroxytryptophan (AHT) (67). The increase in ivoC expression is delayed approximately 4 h with respect to that of ivoB (66) (see Table S2 in the supplemental material). However, we cannot compare the expression pattern of these genes with that of yA, since it was measured in surface cultures while yA expression was analyzed after the deposition on solid plates of liquid-medium-grown mycelium pellets (63).
It is noteworthy that none of the TFs belonging to the UDA pathway was found within this group of significantly altered genes. These TFs are expressed both at VG and at early AD (17, 27, 28, 68), and some of them show remarkable differences in expression according to results of Northern blot experiments (27, 28, 68). Our RNA-seq results do not correlate with those previously described, but their transcript levels support the proposed role of UDA factors at both time points of development studied in this work.
GO analysis reveals strong alterations in oxidoreduction, transcriptional, and transmembrane transport processes.
The analysis of the 1,948 genes that were significantly altered and contained only a systematic name revealed that the expression of 149 (7.6%) genes was increased at early AD, while 1,798 (92.3%) genes were downregulated. Their GO analyses included the prediction of the cellular localization (cellular component), the function (biological function), and the cellular process in which they may participate (biological process). Figure S2 in the supplemental material shows the statistical distribution of these GO analyses, while Fig. 4 focuses on only the most-represented biological functions.
Fig 4.
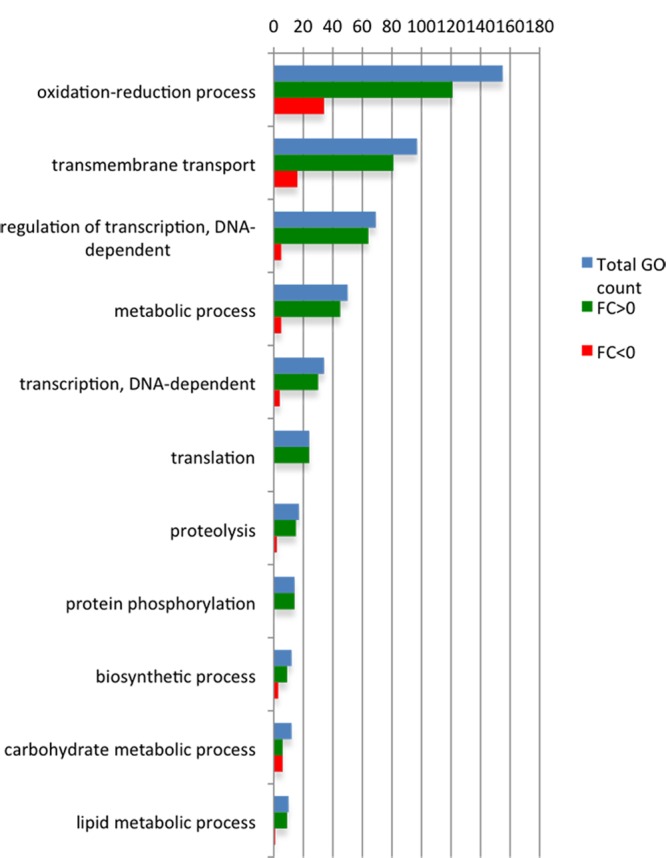
GO functional enrichment analysis of significantly regulated genes between noninducing and inducing conditions.
Figure 4 indicates that oxidation-reduction reactions are the most represented, including oxidoreductases of different types, each one requiring specific cofactors (Fig. 4; see also Fig. S2 in the supplemental material). The second and third groups include those genes with a predicted transmembrane transporter activity or transcriptional regulators. The modifications observed in the expression pattern may be linked to changes in nutrient availability and environmental conditions that occur upon induction of AD. The lower availability of nutrients on emergence to the atmosphere likely activates autophagy to sustain the energetic requirements of developmental changes (69, 70). This would involve a reorganization of carbon and nitrogen metabolism (see above) (42–44, 71, 72) and a dramatic change in nutrient compartmentalization and transport mechanisms. Furthermore, the highly oxidative air environment might require a higher potential for the detoxification of reactive oxygen species (ROS). The idea suggesting that ROS play important physiological roles was already known (73). It is tempting to suggest that these changes may be controlled by a new genetic or functional relationship of transcriptional regulators.
Role of secondary metabolism in asexual development signaling.
In our analysis, the presence of transcripts predicted to be involved in secondary metabolic and biosynthetic pathways is noteworthy. This includes polyketide synthases (PKS) and nonribosomal peptide synthetases (NRPS) but also enzymes and TFs acting on known secondary metabolite pathways (see Table S3 in the supplemental material). Besides, the three most represented biological functions in our GO analysis, oxidoreduction, transmembrane transport, and transcriptional regulation, are usually involved in the control of secondary metabolite biosynthetic processes and are grouped in metabolic clusters in fungal genomes (see, for example, references 13, 74, 75, and 76).
Secondary metabolite production is tightly linked with development in fungi, in terms of both signaling and toxin biosynthesis (77). Specific molecules are required to induce the asexual cycle (78–80). One of these metabolites has recently been identified in A. nidulans as the meroterpenoid dehydroaustinol (81), but there are additional extracellular and diffusible compounds whose structure has not been elucidated yet (see, for example, references 32, 82, and 80).
Thus, we searched for secondary metabolic enzymes within the list of genes with significantly altered expression when comparing VG and early AD. First, we confirmed that the genes An1594, An3252, and An9314, coding for diterpene synthases (83), and the genes xptA, tdiB, An11080, An11194, and An11202 (84), coding for aromatic prenyltransferases, were absent from our list of significantly regulated genes. Second, we searched for PKS- or NRPS-coding genes, following the work of Von Döhren on one hand and Nielsen and coworkers on the other hand (84, 85) (Fig. 5; see also Table S4 in the supplemental material). Yellow squares in Fig. 5 designate significantly altered PKS- or NRPS-coding genes (Fig. 5 and 6; see below). Ten genes belong to this first group, 5 being upregulated at the early AD and 5 downregulated. In this group are the PKS-coding genes An2032 (also known as pkhA), related to benzaldehyde derivative biosynthesis, An6791, An8910, An9005, and An12331 (= An7838), the NRPS-coding genes An2064, An5318, and An6236 (also known as sidD), related to fusarinine-type siderophore biosynthesis, and An9129, and the hybrid PKS-NRPS-coding gene An8412 (also known as apdA), involved in aspyridone synthesis (74, 84–87).
Fig 5.

Schematic representation of Aspergillus nidulans chromosomes showing the location of genes coding for secondary metabolite producer polyketide synthases (PKSs) (in blue), nonribosomal peptide synthetases (NRPS) (in orange), and dimethyl allyltryptophan prenyltransferases (DMAT) (in pink). Those genes significantly regulated during the morphological transition analyzed in this work are in yellow squares. In black squares are those which, being nonsignificantly regulated, belong to secondary metabolite gene clusters in which at least three genes are significantly regulated.
Fig 6.

Expression patterns of those A. nidulans secondary metabolism gene clusters in which at least three genes are significantly regulated. The position of PKSs or NRPSs in each cluster is indicated, as are the first and last gene numbers. Nonsignificantly regulated genes are in black, those upregulated are in red, and those downregulated are in green.
However, an analysis based exclusively on the PKS and NRPS coding transcripts would lead to a biased point of view. It has been shown that the transcriptional control of each metabolic cluster and thus the concentration of the secondary metabolite linked to their activity depends on mechanisms exerted on various cluster functions, such as oxidoreduction, transcriptional regulation, or transport (13, 74, 76). Thus, we decided to study the genomic flanking regions of specific PKS- or NRPS-coding genes shown in Fig. 5. We included three more PKS- or NRPS-coding genes in this analysis, since although they were not significantly regulated, they defined genomic regions where contiguous genes were significantly regulated (see black squares in Fig. 5) (74, 84): An2035 (also known as pkhB), located in the same metabolic cluster as An2032 (pkhA) (see above), An3230 (also known as pkfA), involved in orsellinaldehyde derivative synthesis, and the NRPS gene An11820 (= An9291). Based on these criteria, we focused on seven clusters in which at least three genes were significantly regulated, one of them being the PKS- or NRPS-coding gene or not (Fig. 6). The extension of each cluster in Fig. 6 was delimited according to the SMURF program, a web-based application for systematically predicting clustered secondary metabolism genes based on their genomic context and domain content (www.jcvi.org/smurf/) (88). The extension of the cluster defined by the PKS genes An2032 (pkhA) and An2035 (pkhB) was modified with respect to that delimited by SMURF based on our blast, synteny, and evolutionary analyses.
This last cluster is clearly upregulated. According to our synteny analyses (not shown), it is not conserved in the genus Aspergillus. However, it maintains the position and orientation of the genes comparing to a cluster found in Metarhizium robertsii, an endophytic insect-parasitic fungus that translocates nitrogen directly from insects to plants (89). This suggests that the activity of this cluster is not directly required for the induction of conidiation or could be required at morphological stages that occur exclusively during A. nidulans conidiophore development but not in the rest of Aspergillus spp. included in the synteny analysis.
The cluster defined by the An6236 (sidD) NRPS is also upregulated. Most genes from this cluster maintain their position in the genome of Aspergillus spp. included in the synteny analysis (not shown), suggesting that products related to fusarinine C and tryacetylfusarinine C from A. fumigatus (90) are induced during A. nidulans conidiation. Siderophore biosynthesis requires l-ornithine as the starting product. Thus, it is plausible that conidiation defects caused by mutations in the ornithine transcarbamylase coded by argB (16) could be related to alterations in the siderophore biosynthetic pathways.
The three clusters defined by the NRPS genes An2064, An5318, and An11820 are clearly downregulated, suggesting that the unknown metabolites linked to their activity are preferentially required at VG. Finally, some genes (mainly oxidoreductases and membrane transporters) from clusters defined by the PKS genes An3230 (pkfA), involved in orsellinaldehyde derivative synthesis (74), or An9005 are upregulated while others are downregulated. This strongly suggests that the availability, concentration, and/or final structure of the related secondary intermediates are finely tuned through complex regulatory mechanisms.
Overall, the results presented in this section suggest that secondary metabolism is transcriptionally reoriented during the initial stages of conidiophore development, while cluster analysis reveals the existence of multiple regulatory mechanisms for those metabolic pathways.
Supplementary Material
ACKNOWLEDGMENTS
This work has been supported by the Basque Government through grant IT393-10 and the Ministerio de Economía y Competitividad (formerly Ministerio de Ciencia e Innovación) through grant BFU2010-17528 to U.U., grant BFU2009-08701 to E.A.E., and grants from the German Science Foundation (DFG Fi 459), the Fonds der Chemischen Industrie, the Baden-Württemberg Stiftung, and the Centre for Functional Nanostructures to R.F. A.G. is now a contract researcher from The University of The Basque Country. J.R.-R. was a postdoctoral fellow of the Ministerio de Ciencia e Innovación. O.E. is a contract researcher associated with grant BFU2010-17528.
We thank Vladimir Benes and his GeneCore service at the EMBL (Heidelberg, Germany) for their help in RNA sequencing and Francisco Codoñer from LifeSequencing (University of Valencia Scientific Park, Paterna, Spain) for assistance in data processing. We also thank Ane Markina-Iñarrairaegui for strain MAD2666.
Footnotes
Published ahead of print 21 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00274-12.
REFERENCES
- 1. Hawkins RD, Hon GC, Ren B. 2010. Next-generation genomics: an integrative approach. Nat. Rev. Genet. 11:476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nitsche BM, Crabtree J, Cerqueira GC, Meyer V, Ram AFJ, Wortman JR. 2011. New resources for functional analysis of omics data for the genus Aspergillus. BMC Genomics 12:486 doi:10.1186/1471-2164-12-486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kniemeyer O. 2011. Proteomics of eukaryotic microorganisms: the medically and biotechnologically important fungal genus Aspergillus. Proteomics 11:3232–3243 [DOI] [PubMed] [Google Scholar]
- 4. Pusztahelyi T, Klement E, Szajli E, Klem J, Miskei M, Karányi Z, Emri T, Kovács S, Orosz G, Kovács KL, Medzihradszky KF, Prade RA, Pócsi I. 2011. Comparison of transcriptional and translational changes caused by long-term menadione exposure in Aspergillus nidulans. Fungal Genet. Biol. 48:92–103 [DOI] [PubMed] [Google Scholar]
- 5. Ruger-Herreros C, Rodriguez-Romero J, Fernandez-Barranco R, Olmedo M, Fischer R, Corrochano LM, Canovas D. 2011. Regulation of conidiation by light in Aspergillus nidulans. Genetics 188:809–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Terabayashi Y, Sano M, Yamane N, Marui J, Tamano K, Sagara J, Dohmoto M, Oda K, Ohshima E, Tachibana K, Higa Y, Ohashi S, Koike H, Machida M. 2010. Identification and characterization of genes responsible for biosynthesis of kojic acid, an industrially important compound from Aspergillus oryzae. Fungal Genet. Biol. 47:953–961 [DOI] [PubMed] [Google Scholar]
- 7. Twumasi-Boateng K, Yu Y, Chen D, Gravelat FN, Nierman WC, Sheppard DC. 2009. Transcriptional profiling identifies a role for BrlA in the response to nitrogen depletion and for StuA in the regulation of secondary metabolite clusters in Aspergillus fumigatus. Eukaryot. Cell 8:104–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621–628 [DOI] [PubMed] [Google Scholar]
- 9. Shendure J. 2008. The beginning of the end for microarrays? Nat. Methods 5:585–587 [DOI] [PubMed] [Google Scholar]
- 10. Wang Z, Gerstein M, Snyder M. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10:57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang B, Guo G, Wang C, Lin Y, Wang X, Zhao M, Guo Y, He M, Zhang Y, Pan L. 2010. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 38:5075–5087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Delmas S, Pullan ST, Gaddipati S, Kokolski M, Malla S, Blythe MJ, Ibbett R, Campbell M, Liddell S, Aboobaker A, Tucker GA, Archer DB. 2012. Uncovering the genome-wide transcriptional responses of the filamentous fungus Aspergillus niger to lignocellulose using RNA sequencing. PLoS Genet. 8:e1002875 doi:10.1371/journal.pgen.1002875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gibbons JG, Beauvais A, Beau R, McGary KL, Latge JP, Rokas A. 2012. Global transcriptome changes underlying colony growth in the opportunistic human pathogen Aspergillus fumigatus. Eukaryot. Cell 11:68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gibbons JG, Salichos L, Slot JC, Rinker DC, McGary KL, King JG, Klich MA, Tabb DL, McDonald WH, Rokas A. 2012. The evolutionary imprint of domestication on genome variation and function of the filamentous fungus Aspergillus oryzae. Curr. Biol. 22:1403–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu J, Fedorova ND, Montalbano BG, Bhatnagar D, Cleveland TE, Bennett JW, Nierman WC. 2011. Tight control of mycotoxin biosynthesis gene expression in Aspergillus flavus by temperature as revealed by RNA-Seq. FEMS Microbiol. Lett. 322:145–149 [DOI] [PubMed] [Google Scholar]
- 16. Adams TH, Wieser JK, Yu J-H. 1998. Asexual sporulation in Aspergillus nidulans. Microbiol. Mol. Biol. Rev. 62:35–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Etxebeste O, Garzia A, Espeso EA, Ugalde U. 2010. Aspergillus nidulans asexual development: making the most of cellular modules. Trends Microbiol. 18:569–576 [DOI] [PubMed] [Google Scholar]
- 18. Etxebeste O, Ugalde U, Espeso EA. 2010. Adaptative and developmental responses to stress in Aspergillus nidulans. Curr. Protein Pept. Sci. 11:704–718 [DOI] [PubMed] [Google Scholar]
- 19. Ni M, Gao N, Kwon N-J, Shin K-S, Yu JH. 2010. Regulation of Aspergillus conidiation in cellular and molecular biology of filamentous fungi. ASM Press, Washington, DC [Google Scholar]
- 20. Mims CW, Richardson E, Timberlake WE. 1988. Ultrastructural analysis of conidiophore development in the fungus Aspergillus nidulans using freeze-substitution. Protoplasma 144:132–141 [Google Scholar]
- 21. Hernández-Rodríguez Y, Hastings S, Momany M. 2012. The septin AspB in Aspergillus nidulans forms bars and filaments and plays roles in growth emergence and conidiation. Eukaryot. Cell 11:311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Si H, Rittenour WR, Xu K, Nicksarlian M, Calvo AM, Harris SD. 2012. Morphogenetic and developmental functions of the Aspergillus nidulans homologues of the yeast bud site selection proteins Bud4 and Axl2. Mol. Microbiol. 85:252–270 [DOI] [PubMed] [Google Scholar]
- 23. Wieser JK, Lee BN, Fondon JW, III, Adams TH. 1994. Genetic requirements for initiating asexual development in Aspergillus nidulans. Curr. Genet. 27:62–69 [DOI] [PubMed] [Google Scholar]
- 24. Adams TH, Boylan MT, Timberlake WE. 1988. brlA is necessary and sufficient to direct conidiophore development in Aspergillus nidulans. Cell 54:353–362 [DOI] [PubMed] [Google Scholar]
- 25. Andrianopoulos A, Timberlake WE. 1994. The Aspergillus nidulans abaA gene encodes a transcriptional activator that acts as a genetic switch to control development. Mol. Cell. Biol. 14:2503–2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marshall MA, Timberlake WE. 1991. Aspergillus nidulans wetA activates spore-specific gene expression. Mol. Cell. Biol. 11:55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garzia A, Etxebeste O, Herrero-GarcíA E, Fischer R, Espeso EA, Ugalde U. 2009. Aspergillus nidulans FlbE is an upstream developmental activator of conidiation functionally associated with the putative transcription factor FlbB. Mol. Microbiol. 71:172–184 [DOI] [PubMed] [Google Scholar]
- 28. Garzia A, Etxebeste O, Herrero-GarcíA E, Ugalde U, Espeso EA. 2010. The concerted action of bZip and cMyb transcription factors FlbB and FlbD induces brlA expression and asexual development in Aspergillus nidulans. Mol. Microbiol. 75:1314–1324 [DOI] [PubMed] [Google Scholar]
- 29. Nayak T, Szewczyk E, Oakley CE, Osmani A, Ukil L, Murray SL, Hynes MJ, Osmani SA, Oakley BR. 2006. A versatile and efficient gene-targeting system for Aspergillus nidulans. Genetics 172:1557–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pontecorvo G, Roper JA, Chemmons LM, Macdonald KD, Bufton AWJ. 1953. The genetics of Aspergillus nidulans. Adv. Genet. 5:141–238 [DOI] [PubMed] [Google Scholar]
- 31. Aguirre J. 1993. Spatial and temporal controls of the Aspergillus brIA developmental regulatory gene. Mol. Microbiol. 8:211–218 [DOI] [PubMed] [Google Scholar]
- 32. Etxebeste O, Ni M, Garzia A, Kwon N-J, Fischer R, Yu J-H, Espeso EA, Ugalde U. 2008. Basic-zipper-type transcription factor FlbB controls asexual development in Aspergillus nidulans. Eukaryot. Cell 7:38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7:562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martinelli SD, Clutterbuck AJ. 1971. A quantitative survey of conidiation mutants in Aspergillus nidulans. J. Gen. Microbiol. 69:261–268 [DOI] [PubMed] [Google Scholar]
- 35. Timberlake WE. 1980. Developmental gene regulation in Aspergillus nidulans. Dev. Biol. 78:497–510 [DOI] [PubMed] [Google Scholar]
- 36. Zimmermann CR, Orr WC, Leclerc RF, Barnard EC, Timberlake WE. 1980. Molecular cloning and selection of genes regulated in Aspergillus development. Cell 21:709–715 [DOI] [PubMed] [Google Scholar]
- 37. Wilson RA, Arst HN. 1998. Mutational analysis of AREA, a transcriptional activator mediating nitrogen metabolite repression in Aspergillus nidulans and a member of the “streetwise” GATA family of transcription factors. Microbiol. Mol. Biol. Rev. 62:586–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wong KH, Hynes MJ, Todd RB, Davis MA. 2007. Transcriptional control of nmrA by the bZIP transcription factor MeaB reveals a new level of nitrogen regulation in Aspergillus nidulans. Mol. Microbiol. 66:534–551 [DOI] [PubMed] [Google Scholar]
- 39. Dowzer CEA, Kelly JM. 1991. Analysis of the creA gene, a regulatory of carbon catabolite repression in Aspergillus nidulans. Mol. Cell. Biol. 11:5701–5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoffmann B, Valerius O, Andermann M, Braus GH. 2001. Transcriptional autoregulation and inhibition of mRNA translation of amino acid regulator gene cpcA of filamentous fungus Aspergillus nidulans. Mol. Biol. Cell 12:2846–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Strittmatter AW, Irniger S, Braus GH. 2001. Induction of jlbA mRNA synthesis for a putative bZIP protein of Aspergillus nidulans by amino acid starvation. Curr. Genet. 39:327–334 [DOI] [PubMed] [Google Scholar]
- 42. Lloyd G, Anderson J, Smith J, Morris E. 1972. Conidiation and esterase synthesis in Aspergillus niger. Trans. Br. Mycol. Soc. 59:63–70 [Google Scholar]
- 43. Pitt D, Mosley MJ. 1985. Enzymes of gluconate metabolism and glycolysis in Penicillium notatum. Antonie Van Leeuwenhoek 51:353–364 [DOI] [PubMed] [Google Scholar]
- 44. Pitt D, Mosley MJ. 1985. Pathways of glucose catabolism and the origin and metabolism of pyruvate during calcium-induced conidiation of Penicillium notatum. Antonie Van Leeuwenhoek 51:365–384 [DOI] [PubMed] [Google Scholar]
- 45. Hagiwara D, Asano Y, Marui J, Yoshimi A, Mizuno T, Abe K. 2009. Transcriptional profiling for Aspergillus nidulans HogA MAPK signaling pathway in response to fludioxonil and osmotic stress. Fungal Genet. Biol. 46:868–878 [DOI] [PubMed] [Google Scholar]
- 46. Virginia M, Appleyard CL, McPheat WL, Stark MJ. 2000. A novel “two-component” protein containing histidine kinase and response regulator domains required for sporulation in Aspergillus nidulans. Curr. Genet. 37:364–372 [DOI] [PubMed] [Google Scholar]
- 47. Kawasaki L, Aguirre J. 2001. Multiple catalase genes are differentially regulated in Aspergillus nidulans. J. Bacteriol. 183:1434–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodriguez-Romero J, Hedtke M, Kastner C, Müller S, Fischer R. 2010. Fungi, hidden in soil or up in the air: light makes a difference. Annu. Rev. Microbiol. 64:585–610 [DOI] [PubMed] [Google Scholar]
- 49. Vargas-Pérez I, Sánchez O, Kawasaki L, Georgellis D, Aguirre J. 2007. Response regulators SrrA and SskA are central components of a phosphorelay system involved in stress signal transduction and asexual sporulation in Aspergillus nidulans. Eukaryot. Cell 6:1570–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Navarro RE, Aguirre J. 1998. Posttranscriptional control mediates cell type-specific localization of catalase A during Aspergillus nidulans development. J. Bacteriol. 180:5733–5738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Navarro RE, Stringer MA, Hansberg W, Timberlake WE, Aguirre J. 1996. catA, a new Aspergillus nidulans gene encoding a developmentally regulated catalase. Curr. Genet. 29:352–359 [PubMed] [Google Scholar]
- 52. Kawasaki L, Sánchez O, Shiozaki K, Aguirre J. 2002. SakA MAP kinase is involved in stress signal transduction, sexual development and spore viability in Aspergillus nidulans. Mol. Microbiol. 45:1153–1163 [DOI] [PubMed] [Google Scholar]
- 53. Lara-Rojas F, Sánchez O, Kawasaki L, Aguirre J. 2011. Aspergillus nidulans transcription factor AtfA interacts with the MAPK SakA to regulate general stress responses, development and spore functions. Mol. Microbiol. 80:436–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sewall TC, Mims CW, Timberlake WE. 1990. Conidium differentiation in Aspergillus nidulans wild-type and wet-white (wetA) mutant strains. Dev. Biol. 138:499–508 [DOI] [PubMed] [Google Scholar]
- 55. de Groot PWJ, Brandt BW, Horiuchi H, Ram AFJ, de Koster CG, Klis FM. 2009. Comprehensive genomic analysis of cell wall genes in Aspergillus nidulans. Fungal Genet. Biol. 46(Suppl 1):S72–S81 [DOI] [PubMed] [Google Scholar]
- 56. Tsitsigiannis DI, Kowieski TM, Zarnowski R, Keller NP. 2005. Three putative oxylipin biosynthetic genes integrate sexual and asexual development in Aspergillus nidulans. Microbiology 151:1809–1821 [DOI] [PubMed] [Google Scholar]
- 57. Bayram O, Braus GH, Fischer R, Rodriguez-Romero J. 2010. Spotlight on Aspergillus nidulans photosensory systems. Fungal Genet. Biol. 47:900–908 [DOI] [PubMed] [Google Scholar]
- 58. Calvo AM. 2008. The VeA regulatory system and its role in morphological and chemical development in fungi. Fungal Genet. Biol. 45:1053–1061 [DOI] [PubMed] [Google Scholar]
- 59. Kim H-R, Chae K-S, Han K-H, Han D-M. 2009. The nsdC gene encoding a putative C2H2-type transcription factor is a key activator of sexual development in Aspergillus nidulans. Genetics 182:771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vienken K, Scherer M, Fischer R. 2005. The Zn(II)2Cys6 putative Aspergillus nidulans transcription factor repressor of sexual development inhibits sexual development under low-carbon conditions and in submersed culture. Genetics 169:619–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Boylan MT, Mirabito PM, Willett CE, Zimmerman CR, Timberlake WE. 1987. Isolation and physical characterization of three essential conidiation genes from Aspergillus nidulans. Mol. Cell. Biol. 7:3113–3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ni M, Yu J-H. 2007. A novel regulator couples sporogenesis and trehalose biogenesis in Aspergillus nidulans. PLoS One 2:e970 doi:10.1371/journal.pone.0000970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. O'Hara EB, Timberlake WE. 1989. Molecular characterization of the Aspergillus nidulans yA locus. Genetics 121:249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Melin P, Schnürer J, Wagner EGH. 2003. Characterization of phiA, a gene essential for phialide development in Aspergillus nidulans. Fungal Genet. Biol. 40:234–241 [DOI] [PubMed] [Google Scholar]
- 65. Stringer MA, Dean RA, Sewall TC, Timberlake WE. 1991. Rodletless, a new Aspergillus developmental mutant induced by directed gene inactivation. Genes Dev. 5:1161–1171 [DOI] [PubMed] [Google Scholar]
- 66. Birse CE, Clutterbuck AJ. 1991. Isolation and developmentally regulated expression of an Aspergillus nidulans phenol oxidase-encoding gene, ivoB. Gene 98:69–76 [DOI] [PubMed] [Google Scholar]
- 67. Clutterbuck AJ. 1990. The genetics of conidiophore pigmentation in Aspergillus nidulans. J. Gen. Microbiol. 136:1731–1738 [DOI] [PubMed] [Google Scholar]
- 68. Kwon N-J, Garzia A, Espeso EA, Ugalde U, Yu J-H. 2010. FlbC is a putative nuclear C2H2 transcription factor regulating development in Aspergillus nidulans. Mol. Microbiol. 77:1203–1219 [DOI] [PubMed] [Google Scholar]
- 69. Emri T, Molnár Z, Pusztahelyi T, Varecza Z, Pócsi I. 2005. The fluG-BrlA pathway contributes to the initialisation of autolysis in submerged Aspergillus nidulans cultures. Mycol. Res. 109:757–763 [DOI] [PubMed] [Google Scholar]
- 70. White S, McIntyre M, Berry DR, McNeil B. 2002. The autolysis of industrial filamentous fungi. Crit. Rev. Biotechnol. 22:1–14 [DOI] [PubMed] [Google Scholar]
- 71. Galbraith JC, Smith JE. 1969. Changes in activity of certain enzymes of the tricarboxylic acid cycle and the glyoxylate cycle during the initiation of conidiation of Aspergillus niger. Can. J. Microbiol. 15:1207–1212 [DOI] [PubMed] [Google Scholar]
- 72. Galbraith JC, Smith JE. 1969. The glyoxylate cycle as an important pathway in fungal morphogenesis. J. Gen. Microbiol. 58:12–13 [PubMed] [Google Scholar]
- 73. Aguirre J, Ríos-Momberg M, Hewitt D, Hansberg W. 2005. Reactive oxygen species and development in microbial eukaryotes. Trends Microbiol. 13:111–118 [DOI] [PubMed] [Google Scholar]
- 74. Ahuja M, Chiang Y-M, Chang S-L, Praseuth MB, Entwistle R, Sanchez JF, Lo H-C, Yeh H-H, Oakley BR, Wang CCC. 2012. Illuminating the diversity of aromatic polyketide synthases in Aspergillus nidulans. J. Am. Chem. Soc. 134:8212–8221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Davison J, al Fahad A, Cai M, Song Z, Yehia SY, Lazarus CM, Bailey AM, Simpson TJ, Cox RJ. 2012. Genetic, molecular, and biochemical basis of fungal tropolone biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 109:7642–7647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lim FY, Hou Y, Chen Y, Oh JH, Lee I, Bugni TS, Keller NP. 2012. Genome-based cluster deletion reveals an endocrocin biosynthetic pathway in Aspergillus fumigatus. Appl. Environ. Microbiol. 78:4117–4125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bok JW, Keller NP. 2004. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot. Cell 3:527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Herrero-GarcíA E, Garzia A, Cordobés S, Espeso EA, Ugalde U. 2011. 8-Carbon oxylipins inhibit germination and growth, and stimulate aerial conidiation in Aspergillus nidulans. Fungal Biol. 115:393–400 [DOI] [PubMed] [Google Scholar]
- 79. Leeder AC, Palma-Guerrero J, Glass NL. 2011. The social network: deciphering fungal language. Nat. Rev. Microbiol. 9:440–451 [DOI] [PubMed] [Google Scholar]
- 80. Ugalde U. 2006. Autoregulatory signals in mycelial fungi, p 203–214 In Kües U, Fischer R. (ed), Growth, differentiation and sexuality: the Mycota, vol 1 Springer, Berlin, Germany [Google Scholar]
- 81. Rodríguez-Urra AB, Jimenez C, Nieto MI, Rodríguez J, Hayashi H, Ugalde U. 2012. Signaling the induction of sporulation involves the interaction of two secondary metabolites in Aspergillus nidulans. ACS Chem. Biol. 7:599–606 [DOI] [PubMed] [Google Scholar]
- 82. Soid-Raggi G, Sánchez O, Aguirre J. 2006. TmpA, a member of a novel family of putative membrane flavoproteins, regulates asexual development in Aspergillus nidulans. Mol. Microbiol. 59:854–869 [DOI] [PubMed] [Google Scholar]
- 83. Bromann K, Toivari M, Viljanen K, Vuoristo A, Ruohonen L, Nakari-SetälÄ T. 2012. Identification and characterization of a novel diterpene gene cluster in Aspergillus nidulans. PLoS One 7:e35450 doi:10.1371/journal.pone.0035450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. von Döhren, H 2009. A survey of nonribosomal peptide synthetase (NRPS) genes in Aspergillus nidulans. Fungal Genet. Biol. 46:S45–S52 [DOI] [PubMed] [Google Scholar]
- 85. Nielsen ML, Nielsen JB, Rank C, Klejnstrup ML, Holm DMK, Brogaard KH, Hansen BG, Frisvad JC, Larsen TO, Mortensen UH. 2011. A genome-wide polyketide synthase deletion library uncovers novel genetic links to polyketides and meroterpenoids in Aspergillus nidulans. FEMS Microbiol. Lett. 321:157–166 [DOI] [PubMed] [Google Scholar]
- 86. Bergmann S, Schümann J, Scherlach K, Lange C, Brakhage AA, Hertweck C. 2007. Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans. Nat. Chem. Biol. 3:213–217 [DOI] [PubMed] [Google Scholar]
- 87. Power T, Ortoneda M, Morrissey JP, Dobson ADW. 2006. Differential expression of genes involved in iron metabolism in Aspergillus fumigatus. Int. Microbiol. 9:281–287 [PubMed] [Google Scholar]
- 88. Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, Fedorova ND. 2010. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Biol. 47:736–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Behie SW, Zelisko PM, Bidochka MJ. 2012. Endophytic insect-parasitic fungi translocate nitrogen directly from insects to plants. Science 336:1576–1577 [DOI] [PubMed] [Google Scholar]
- 90. Schrettl M, Beckmann N, Varga J, Heinekamp T, Jacobsen ID, Jöchl C, Moussa TA, Wang S, Gsaller F, Blatzer M, Werner ER, Niermann WC, Brakhage AA, Haas H. 2010. HapX-mediated adaption to iron starvation is crucial for virulence of Aspergillus fumigatus. PLoS Pathog. 6:e1001124 doi:10.1371/journal.ppat.1001124 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.