

Abstract
Yersinia pestis is one of the most dangerous bacterial pathogens. PhoP and cyclic AMP receptor protein (CRP) are global regulators of Y. pestis, and they control two distinct regulons that contain multiple virulence-related genes. The PhoP regulator and its cognate sensor PhoQ constitute a two-component regulatory system. The regulatory activity of CRP is triggered only by binding to its cofactor cAMP, which is synthesized from ATP by adenylyl cyclase (encoded by cyaA). However, the association between the two regulatory systems PhoP/PhoQ and CRP-cAMP is still not understood for Y. pestis. In the present work, the four consecutive genes YPO1635, phoP, phoQ, and YPO1632 were found to constitute an operon, YPO1635-phoPQ-YPO1632, transcribed as a single primary RNA, whereas the last three genes comprised another operon, phoPQ-YPO1632, transcribed with two adjacent transcriptional starts. Through direct PhoP-target promoter association, the transcription of these two operons was stimulated and repressed by PhoP, respectively; thus, both positive autoregulation and negative autoregulation of PhoP/PhoQ were detected. In addition, PhoP acted as a direct transcriptional activator of crp and cyaA. The translational/transcriptional start sites, promoter −10 and −35 elements, PhoP sites, and PhoP box-like sequences were determined for these PhoP-dependent genes, providing a map of the PhoP-target promoter interaction. The CRP and PhoP regulons have evolved to merge into a single regulatory cascade in Y. pestis because of the direct regulatory association between PhoP/PhoQ and CRP-cAMP.
INTRODUCTION
Yersinia pestis, the causative agent of plague, is one of the most dangerous bacterial pathogens: it causes deadly systemic infections in mammals, including humans. Y. pestis is transmitted through flea bites, direct contact, or inhalation of aerosols that contain the bacterium (1). Y. pestis has been classified as a category A-listed pathogen by U.S. CDC because it can be used as a potential biowarfare or bioterrorism agent.
The cyclic AMP (cAMP) receptor protein (CRP) controls the transcription of more than 100 bacterial genes or operons (2). CRP is active only in the presence of cAMP, which behaves as a classic small-molecule inducer. The cAMP-CRP complex binds to a symmetrical consensus sequence, TGTGA-N6-TCACA (known as the CRP box sequence), located within the target promoter regions (3). The major control of cAMP levels in cells is at the point of synthesis catalyzed by adenylyl cyclase (encoded by cyaA) from ATP.
PhoP and PhoQ constitute a classic two-component regulatory system (4). The sensor PhoQ is activated under acidic or low-divalent-cation conditions or in response to host-secreted antimicrobial peptides, and it then phosphorylates PhoP. Phosphorylated PhoP acts as an active transcriptional regulator and recognizes an 18-bp PhoP box sequence (TGTTTAWN4TGTTTAW, where W is A or T) in Y. pestis (5) which is very similar to those established in Escherichia coli (6, 7) and Salmonella enterica (8).
During its life cycle, Y. pestis encounters distinct and complex in vivo environments, because it may transit from a flea to the dermis, macrophage, lymph, blood, and then viscera. PhoP responds to environmental low Mg2+, acidic pH, or antimicrobial peptides; the first two are frequently found in macrophages, whereas various antimicrobial peptides are secreted by hosts as a mechanism of native immunity (9). CRP senses the switch of catabolites, and glucose may be the only free sugar that the bacteria can detect in hosts, whereas the presence of glucose stops the CRP-cAMP machinery (10). Both PhoP and CRP are global regulators controlling complex cellular pathways, including multiple virulence determinants in Y. pestis (5, 11–16).
Our previous microarray expression analysis of the phoP mutant of Y. pestis showed the positive regulation of crp and cyaA by PhoP (17), indicating the potential regulatory association between PhoP/PhoQ and CRP-cAMP in Y. pestis. In this study, the combined use of primer extension analysis, LacZ fusion and β-galactosidase assay, electrophoretic mobility shift assay (EMSA), and DNase I footprinting analysis disclosed that PhoP was an activator of crp and cyaA, while it acted as both an activator and repressor of the phoPQ loci. The CRP and PhoP regulons have evolved to merge into a single regulatory cascade in Y. pestis, because of the direct regulatory association between PhoP/PhoQ and CRP-cAMP.
MATERIALS AND METHODS
Bacterial strains and growth.
The wild-type (WT) Y. pestis biovar Microtus strain 201 is avirulent to humans but highly virulent to mice (18). The nonpolar phoP mutant of Y. pestis, designated the ΔphoP strain, was previously described (19). BHI broth containing 3.7% Bacto brain heart infusion (BD Biosciences) was used for bacterial cultivation. An overnight cell culture with an optical density at 620 nm (OD620) of about 1.0 was diluted 1:20 into 18 ml of fresh medium, and bacteria were allowed to grow at 26°C with shaking at 230 rpm. Kanamycin sulfate was used at a final concentration of 50 μg/ml.
RNA isolation.
Total bacterial RNAs were extracted using the TRIzol reagent (Invitrogen). RNA quality was monitored by agarose gel electrophoresis, and RNA quantity was determined by spectrophotometry.
Reverse transcription (RT)-PCR.
The contaminated DNA in the total-RNA samples was removed by using the Ambion DNA-free kit. cDNAs were generated by using 5 μg of RNA and 3 μg of random hexamer primers in a 40-μl reaction mixture. A volume of 30 μl of PCR mixture contained 50 mM KCl; 10 mM Tris-HCl (pH 8.0); 2.5 mM MgCl2; 0.001% gelatin; 0.1% bovine serum albumin (Sigma); 100 μM (each) dATP, dCTP, dGTP, and dTTP (GE Healthcare); a 0.1 μM concentration of each primer; 1 U of Taq DNA polymerase (MBI Fermentas); and 2 μl of cDNA sample. The parameters for amplification were as follows: 95°C for 3 min; 30 cycles of 94°C for 30 s, 56°C for 30 s, and 72°C for 1 min; and a final extension step of 72°C for 5 min. The PCR products were then analyzed by 1.2% agarose gel electrophoresis with ethidium bromide staining.
Primer extension assay.
For the primer extension assay (19), an oligonucleotide primer complementary to a portion of the RNA transcript of each gene was employed to synthesize cDNAs from the RNA templates. The total RNA (5 to 10 μg) from each strain was annealed with 1 pmol of reverse primer (end labeled with γ-32P) using a primer extension system (Promega) according to the manufacturer's instructions. The same labeled primer was also used for sequencing with the fmol DNA cycle sequencing system (Promega). The primer extension products and sequencing materials were concentrated and analyzed by 8 M urea–6% polyacrylamide gel electrophoresis, and the result was detected by autoradiography (Kodak film).
LacZ fusion and β-galactosidase assay.
The promoter-proximal DNA region of each indicated gene was obtained through PCR with Ex Taq DNA polymerase (TaKaRa) using Y. pestis 201 genome DNA as the template. All the primers used are listed in Table 1. PCR fragments were then directionally cloned into the EcoRI-BamHI (or BamHI-HindIII) sites of the low-copy-number transcriptional fusion vector pRW50, which harbors a tetracycline resistance gene and a promoterless lacZ reporter gene (19). Correct cloning was verified by DNA sequencing. An empty pRW50 plasmid was also introduced into each strain tested and considered the negative control. The Y. pestis strains transformed with the recombinant plasmids and the empty pRW50 plasmid were grown as previously described to measure the β-galactosidase activity in the cellular extracts using the β-galactosidase enzyme assay system (Promega). Assays were performed with at least three biological replicates.
Table 1.
Oligonucleotide primers used in this study
| Purpose and target | Primers (forward/reverse, 5′–3′) |
|---|---|
| Mutant construction, phoP | ATGCGGGTTCTGGTTGTGGAAGATAACGCGTTGTTGCGTCAGTTGTGTCTCAAAATCTCTG/CTAGTTGACGTCAAAACGATATCCCTGACCACGAATAGTCGAAAGCCGCCGTCCCGTCAAG |
| RT-PCR, operons | AAGGCATCAGCAAACTGGAAG/ACGGTTATCATTGGCAAGGC |
| CAGATATTGGCGTGAACATCG/GGAACGGTTTGGTCACATAATC | |
| GCCCTTCCAGATTGACCTTTC/ATGTGCGACTCCAGCTCAG | |
| TTGAAGTCACCACCTTACACTC/TCTACCGCCTGAACTAACAATG | |
| LacZ fusion | |
| YPO1635 | GCGGGATCCCCGACTCGACCGTGCTAC/GCGAAGCTTATGGCGACACTACAGGAACC |
| phoP | CCGGAATTCTGATGCCAGCAAAGACG/CGCGGATCCAGATGGTGACGCAACAAC |
| crp | CCGGAATTCAGACACGACATCAATGGC/CGCGGATCCTGGCAATGAGACAGGAAC |
| cyaA | CCGGAATTCCCATCCTATCACTGACCG/CGCGGATCCGTTGATCGCATCCAGTCG |
| Primer extension | |
| YPO1635 | /CAGGGCAAGGGATAATAGGG |
| phoP | /ACCCGCATACACCAATCCTT |
| crp | /GCTACAGCATCACTTTCG |
| cyaA | /CCCGTGCTCCAAATTCG |
| EMSA | |
| YPO1635 | CCGACTCGACCGTGCTAC/ATGGCGACACTACAGGAACC |
| phoP | TGATGCCAGCAAAGACG/AGATGGTGACGCAACAAC |
| crp | AGACACGACATCAATGGC/TGGCAATGAGACAGGAAC |
| cyaA | CCATCCTATCACTGACCG/GTTGATCGCATCCAGTCG |
| DNase I footprinting | |
| YPO1635 | TAATACAGGTGGCTTAGTTTGG/CAGGGCAAGGGATAATAGGG |
| phoP | TGATGCCAGCAAAGACG/GCATCGCTTGGTCACTGACT |
| GGCGAGTCAGTGACCAAGCG/CGCTCATTATGTAGGTGC | |
| crp | TGCCTTATCACCCGAATTTC/GCTACAGCATCACTTTCG |
| cyaA | CTCATTGGAGCCAGTGATTCG/GTTGATCGCATCCAGTCG |
Purification of 6×His-tagged PhoP (His-PhoP) protein.
The entire coding region of phoP of strain 201 was directionally cloned into the BamHI-HindIII sites of plasmid pET28a (Novagen), which was verified by DNA sequencing (19). The recombinant plasmid encoding the His-PhoP protein was transformed into E. coli BL21λDE3 cells. Overexpression of His-PhoP was induced by the addition of 1 mM isopropyl-β-d-thiogalactoside. The overproduced proteins were purified under native conditions using a nickel-nitrilotriacetic acid (Ni-NTA) agarose column (Qiagen). The purified protein was concentrated with the Amicon Ultra-15 centrifugal filter device (Millipore), and the protein purity was verified by sodium dodecyl sulfate-polyacryalmide gel electrophoresis.
EMSA.
For EMSA (19), the target promoter-proximal DNA fragments were prepared by PCR, and the 5′ ends of DNA were labeled using [γ-32P]ATP and T4 polynucleotide kinase. DNA binding was performed in a 10-μl reaction volume containing binding buffer [1 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol (DTT), 50 mM NaCl, 10 mM Tris-HCl (pH 7.5), and 0.05 mg/ml poly(dI-dC)], labeled DNA (1,000 to 2,000 cpm/μl), and increasing amounts of the His-PhoP protein. Three controls were included in each EMSA experiment: (i) the cold probe as the specific DNA competitor (the same promoter-proximal DNA region, unlabeled), (ii) the negative probe as the nonspecific DNA competitor (the unlabeled coding region of the 16S rRNA gene), and (iii) the nonspecific protein competitor (rabbit anti-F1 protein polyclonal antibodies). After 20 min of incubation at room temperature, the products were loaded onto a native 4% (wt/vol) polyacrylamide gel and electrophoresed in 0.5× Tris-borate-EDTA (TBE) buffer for about 1 h at 200 V. Radioactive species were detected by autoradiography after exposure to a Kodak film at −70°C.
DNase I footprinting.
For DNase I footprinting (19), the target promoter-proximal DNA regions with a single 32P-labeled end were prepared by PCR using promoter-specific primer pairs, including a primer labeled with 32P at the 5′ end (either forward or reverse) and its nonlabeled counterpart. The PCR products were purified using QiaQuick reaction cleanup columns (Qiagen). Increasing amounts of His-PhoP were incubated with the purified, labeled DNA fragment (2 to 5 pmol) for 30 min at room temperature, in a final 10-μl reaction volume containing the binding buffer used in EMSA. Before DNA digestion, 10 μl of the Ca2+-Mg2+ solution (5 mM CaCl2 and 10 mM MgCl2) was added, followed by 1 min of incubation at room temperature. The optimized RQ1 RNase-free DNase I (Promega) was added to the reaction mixture, and the mixture was incubated at room temperature for 30 to 90 s. The reaction was quenched by adding 9 μl of stop solution (200 mM NaCl, 30 mM EDTA, and 1% SDS), followed by 1 min of incubation at room temperature. The partially digested DNA samples were extracted with phenol-chloroform, precipitated with ethanol, and analyzed in a 8 M urea–6% polyacrylamide gel. Protected regions were identified by comparison with the sequence ladders. For sequencing, we used the fmol DNA cycle sequencing system (Promega). The templates for sequencing were the same as the DNA fragments in the DNase I footprinting assays. Radioactive species were detected as previously described.
cAMP determination.
Intracellular cAMP was measured using the cAMP enzyme immunoassay kit (Sigma). Briefly, 1.6 ml of cell culture at an OD620 of about 1.2 was pelleted and lysed with 400 μl of 0.1 M HCl. The unacetylated bacterial lysate samples were transferred into wells precoated with goat anti-rabbit IgG. After the addition of rabbit anti-cAMP antibody, the bound cAMP in each well was measured spectrophotometrically in a competition assay with cAMP-alkaline phosphatase conjugate and p-nitrophenyl phosphate substrate.
Experimental replicates and statistical methods.
For LacZ fusion and cAMP determination, experiments were performed with at least three independent bacterial cultures, and the values were expressed as means ± standard deviations. Paired Student's t tests were performed to determine statistically significant differences; a P value of <0.01 was considered to indicate statistical significance. For primer extension, EMSA, and DNase I footprinting, representative data from at least two independent biological replicates are shown.
RESULTS
Bacterial growth in BHI medium.
In our previous studies, the chemically defined TMH broth medium containing 10 μM Mg2+ (modified from the original 20 mM Mg2+ [20]) was used to dissect PhoP-dependent expression of genes in Y. pestis (5, 17, 19). However, Y. pestis strains, especially the ΔphoP strain, exhibited seriously inhibited growth under low-magnesium conditions (19). In the present work, the BHI broth medium was used instead of TMH to cultivate bacteria. BHI facilitated the good growth of both the ΔphoP and WT strains, and these two strains exhibited almost similar growth rates (data not shown), indicating that the phoP mutation had no effect on the bacterial growth in BHI. For all the following cell culture-related assays, the bacterial cells were harvested at an OD620 of about 1.2 (at the mid-exponential phase).
Transcriptional organization of phoPQ loci.
The phoPQ loci designated herein contained four consecutive genes—YPO1635, phoP, phoQ, and YPO1632—transcribed in the same direction. Through RT-PCR (Fig. 1) and the following primer extension experiments (Fig. 2a and 3a), these four genes were found to constitute an operon, YPO1635-phoPQ-YPO1632, transcribed as a single primary RNA. Meanwhile, the last three genes comprised another operon, phoPQ-YPO1632, that was transcribed with two adjacent transcriptional start sites located upstream of phoP (i.e., two primary RNAs).
Fig 1.

Transcriptional organization of the phoPQ loci. (a) Operon structure. The boxed arrows represent the length and direction of the coding regions of corresponding genes. The broken arrows indicate the transcriptional starts (i.e., transcribed promoters). The YPO1635-phoPQ-YPO1632 operon is transcribed as a single primary RNA, whereas another operon, phoPQ-YPO1632, is transcribed with two adjacent transcriptional start sites. The arrowheads indicate the location of primer pairs and the expected amplicons of PCR. The horizontal arrows depict the putative primary RNA transcripts. (b) Detection of transcribed RNA. The cDNA samples were generated by RT from total RNAs of the WT strain. Genomic DNA and cDNA were used as the templates for PCR and RT-PCR, respectively. To ensure that no contamination of genomic DNA in the RT reactions would occur, RT-PCR of negative controls was performed using the “cDNA” sample generated without reverse transcriptase as the template. Reaction mixtures containing primer pairs without templates were also included as blank controls. As expected, both negative and blank controls of RT-PCR did not provide an amplicon (data not shown).
Fig 2.

Regulation of YPO1635 and phoP by PhoP. Lanes C, T, A, and G represent Sanger sequencing reactions. The negative and positive numbers indicated nucleotide positions upstream and downstream of the indicated genes. (a) Primer extension. An oligonucleotide primer was designed to be complementary to the RNA transcript of each gene tested. The primer extension products were analyzed with an 8 M urea–6% acrylamide sequencing gel. The transcriptional start sites are indicated by arrows with nucleotides. (b) LacZ fusion. The target promoter-proximal DNA region was cloned into the lacZ transcriptional fusion vector pRW50 and then transformed into the WT or ΔphoP strain to determine the promoter activity, i.e., the β-galactosidase activity (Miller units) in the cellular extracts. (c) EMSA. The radioactively labeled promoter-proximal DNA fragments were incubated with increasing amounts of purified His-PhoP protein and then subjected to 4% (wt/vol) polyacrylamide gel electrophoresis. The band of free DNA disappeared with increasing amounts of His-PhoP, resulting in a retarded DNA band with decreased mobility, which presumably represented the DNA-PhoP complex. Shown also is the schematic representation of the EMSA design. (d) DNase I footprinting. Labeled coding or noncoding DNA probes were incubated with increasing amounts of purified His-PhoP and then subjected to DNase I footprinting assay. The footprint regions are indicated with vertical bars.
Fig 3.

Regulation of crp and cyaA by PhoP. For the primer extension (a), an oligonucleotide primer was designed to be complementary to the RNA transcript of each gene tested. For LacZ fusion (b), the target promoter-proximal fragment was cloned into pRW50 and then transformed into the WT or ΔphoP strain to determine the promoter activity, i.e., the β-galactosidase activity (Miller units) in the cellular extracts. For EMSA (c) and DNase I footprinting (d), the target promoter-proximal fragment was radioactively labeled and then incubated with increasing amounts of purified His-PhoP protein (lanes 1, 2, 3, and 4, containing 0, 15.8, 31.6, and 63.2 pmol, respectively). The experiments were performed as described for Fig. 4. Lanes G, A, T, and C represent Sanger sequencing reactions. The negative and positive numbers indicate nucleotide positions upstream and downstream of the relevant genes, respectively. The transcriptional start sites and footprint regions are indicated with arrows and vertical bars.
Regulation of YPO1635 and phoP by PhoP.
YPO1635 and phoP (the first genes of the above two operons, respectively) were subjected to the following gene regulation experiments. The mRNA levels of these two genes were measured in the WT and ΔphoP strains by primer extension (Fig. 2a). This assay detected a single transcriptional start site located 36 bp upstream of YPO1635. Therefore, a single PhoP-activated promoter was transcribed for YPO1635 under the tested growth conditions. Moreover, the YPO1635 mRNA levels decreased dramatically in the ΔphoP strain relative to the WT strain. Two transcriptional start sites (two primer extension products) located 118 and 90 bp upstream of phoP were detected for phoP. Both of the corresponding promoters were under the negative control of PhoP based on the abundance of the primer extension products in the ΔphoP and WT strains.
A YPO1635::lacZ or phoP::lacZ fusion vector, which contains the promoter-proximal region of YPO1635 or phoP and the promoterless lacZ, was transformed into the WT and ΔphoP strains to compare the YPO1635 or phoP promoter activities in these two strains, respectively (Fig. 2b). The LacZ fusion experiments revealed that the YPO1635 or phoP promoter activity was significantly decreased or enhanced, respectively, in the ΔphoP strain relative to the WT strain. These results further confirmed the positive and negative regulation of YPO1635 and phoP by PhoP, respectively.
The target promoter-proximal DNA region of YPO1635 or phoP was subjected to EMSA with purified His-PhoP protein (Fig. 2c). The results showed that His-PhoP bound to the target DNA fragment in a dose-dependent manner in vitro. Full DNA retardation occurred at 4.6 pmol of His-PhoP for YPO1635, whereas only a partial retardation was observed at 9 pmol for phoP. This phenomenon indicated that PhoP had a considerably higher affinity for the YPO1635 upstream region than the phoP one. The His-PhoP proteins at all tested amounts could not bind to the 16S ribosomal RNA fragment as the negative control, confirming the specificity of EMSA in this study.
As further determined by the DNase I footprinting (Fig. 2d), His-PhoP protected a single region from 95 bp to 51 bp upstream of YPO1635, as well as two distinct regions from 371 bp to 322 bp and from 186 bp to 115 bp upstream of phoP, against DNase I digestion in a dose-dependent manner. All the footprints contained PhoP box-like sequences and were considered PhoP sites. For YPO1635, 8.7 pmol of His-PhoP provided complete protection against DNase I digestion. For sites 1 and 2 of phoP, much larger amounts of His-PhoP were needed to generate clear footprints. These results further indicated that the binding affinity of PhoP for the YPO1635 upstream region is higher than that for the phoP one.
Therefore, the PhoP regulator bound to the promoter-proximal regions of YPO1635 and phoP to stimulate and repress directly the transcription of operons YPO1635-phoPQ-YPO1632 and phoPQ-YPO1632 in Y. pestis, respectively.
Regulation of crp and cyaA by PhoP.
The primer extension experiments (Fig. 3a) detected a single transcriptional start site located 287 bp upstream of crp and two other sites located 183 and 133 bp upstream of cyaA. All three corresponding promoters were under the positive control of PhoP. The positive regulation of crp and cyaA by PhoP was further confirmed by the LacZ fusion assay (Fig. 3b), with the crp::lacZ and cyaA::lacZ fusion vectors transformed into the WT and ΔphoP strains. The EMSA (Fig. 3c) and DNase I footprinting (Fig. 3d) experiments showed that His-PhoP bound to the promoter-proximal DNA fragments of crp or cyaA in a dose-dependent manner in vitro. As determined by DNase I footprinting (Fig. 3d), His-PhoP protected a single region from 325 bp to 302 bp upstream of crp and another one from 125 bp to 66 bp upstream of cyaA against DNase I digestion. The two footprints contained PhoP box-like sequences and were considered PhoP sites. Notably, the footprinting data also indicated the low-affinity PhoP sites for crp and cyaA. Therefore, PhoP bound to the promoter-proximal regions of crp and cyaA to stimulate directly the transcription of these genes in Y. pestis.
Production of cAMP in the WT and ΔphoP strains.
The intracellular levels of cAMP were determined in the WT and ΔphoP strains. Compared with the WT strain, a significantly reduced production of cAMP was observed in the ΔphoP; as expected, the ΔcyaA mutation gave almost no detection of cAMP (Fig. 4). These results verified the PhoP-mediated transcriptional stimulation of cyaA that encoded the enzyme for cAMP synthesis in Y. pestis.
Fig 4.
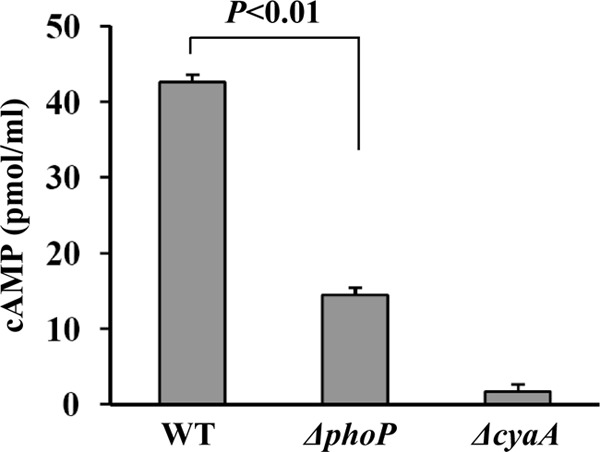
Synthesis of cAMP in different strains. Bacterial cell cultures were harvested at the mid-exponential phase (an OD620 value of about 1.2), and the cAMP content was measured as pmol of cAMP per 1 ml of cell culture.
Promoter structure of YPO1635, phoP, crp, and cyaA.
The primer extension assay was employed to map the 5′ termini (transcriptional start sites) of RNA transcripts of the target genes tested (YPO1635, phoP, crp, and cyaA). Accordingly, the −10 and −35 core promoter elements for RNA polymerase recognition were predicted. The DNase I footprinting experiments were performed to determine precisely the PhoP sites containing PhoP box-like sequences for the direct PhoP targets tested. Collection of data from the translational/transcriptional start sites, core promoter −10 and −35 elements, Shine-Dalgarno sequences for ribosomal binding, PhoP sites, and PhoP box-like sequences enabled us to depict the organization of the PhoP-dependent promoters of YPO1635, phoP, crp, and cyaA characterized herein (Fig. 5a to c).
Fig 5.

Organization of promoter-proximal DNA regions. The promoter-proximal DNA regions of direct PhoP targets YPO1635 (a), phoP (b), crp (c), and cyaA (d) are derived from Y. pestis CO92, and the PhoP box-like sequences for the above genes are aligned (e). The start codon of each gene is shown at the 3′-terminal end of each promoter-proximal DNA sequence. The bent arrows indicate transcriptional starts. The predicted core promoter −10 and −35 elements are boxed. The PhoP sites are underlined. The short consensus (TGTTTAW) sequences are highlighted in red, and the mismatch positions are shaded in gray.
The PhoP-box like sequences determined for phoP, YPO1635, crp, and cyaA were aligned with the PhoP box sequence to identify the mismatched nucleotides (Fig. 5c). Only a single mismatch was identified for YPO1635, while at least three mismatches were identified for phoP, crp, and cyaA, which was consistent with the observation that PhoP bound to the YPO1635 upstream region with an affinity higher than for the phoP, crp, and cyaA ones.
DISCUSSION
A phoP null mutant of Y. pestis exhibits a reduced ability to survive in macrophages (21, 22), but the phoPQ deletion has no effect on the virulence of Y. pestis in either the bubonic or pneumonic murine model of infection (23). Intracellular growth of Y. pestis in macrophages occurs at early stages of systemic infection, which is believed to be a shelter for this pathogen to proliferate and to synthesize virulence determinants, enabling the released bacteria to acquire the ability to annihilate the host immune response (24). Several genes such as mgtCB and udg, whose transcription is directly stimulated by PhoP, encode the factors promoting the intracellular growth of Y. pestis (5, 14).
Data presented indicate that YPO1635, phoP, phoQ, and YPO1632 cotranscribe into a single primary RNA transcript to constitute a four-gene operon, YPO1635-phoPQ-YPO1632, in Y. pestis, while the last three genes constitute another operon with two determined promoters, P1phoP and P2phoP. Potential roles for YPO1635 and YPO1632 in Y. pestis survival in macrophages need to be elucidated, since genes in the same operon tend to have the same physiological functional class (25).
The crp deletion leads to a huge attenuation of the virulence of Y. pestis after subcutaneous infection of mice (11). Expression of plasminogen activator (Pla), pesticin (Pst), and type III YOP secretion components is dependent on CRP in Y. pestis (11–13). Specifically, CRP directly stimulates the expression of Pla (11, 13), a virulence factor essential for bubonic and primary pneumonic plague (26, 27). Given that Pla specifically promotes Y. pestis to disseminate from peripheral infection routes, the defective expression of Pla in the crp mutant will greatly contribute to the huge loss of virulence of this mutant strain after subcutaneous infection (11).
Published reports are mainly focused on the roles of CRP, PhoP, and their target genes in the virulence of Y. pestis in mammals. However, this bacterium also replicates in the flea gut at ambient temperatures. Potential regulatory cascades controlled by CRP or PhoP under these conditions need to be evaluated.
PhoP not only directly regulates the expression of specific genes, including virulence-related ones, but also controls various indirect targets by acting on a set of regulators in Y. pestis (5, 15, 16). PhoP recognizes the promoter region of rovA to repress its transcription in Y. pestis (19). The global regulator RovA is required for bubonic plague through directly regulating psaEF, psaABC, and CUS-2 prophage loci (28). As shown in this study, PhoP recognizes the promoter regions of crp and cyaA (encoding the two components of CRP-cAMP) to stimulate these genes in Y. pestis. Hence, cellular pathways governed by PhoP, CRP, and RovA have evolved to merge into a single global regulatory circuit because of the direct regulatory action of PhoP on its target genes rovA, crp, and cyaA. This phenomenon probably contributes to the tightly controlled expression of the factors responsible for infection in mammals and those for transmission via fleas.
Transcriptional autostimulation of phoPQ through direct binding of PhoP to the upstream region of phoPQ operon has been detected in E. coli and S. enterica; in addition, phoP employs two distinct promoters (P1 and P2), and only P1 is dependent on PhoP (6, 8, 29). Thus, relevant environmental signals stimulate PhoQ to phosphorylate PhoP that is produced at the basal levels from the P2 promoter in E. coli and S. enterica, and then, the phosphorylated PhoP stimulates its own expression by acting on the P1 one (6, 8, 29).
As shown in this study, PhoP binds to a high-affinity site within the upstream region of the YPO1635-phoPQ-YPO1632 operon to stimulate the transcription of the above operon, achieving the autostimulation of phoPQ; this is consistent with the previous observation in Y. pestis KIM (16). PhoP bound to two low-affinity sites within the upstream region of the phoPQ-YPO1632 operon to repress the activity of its two promoters P1phoP and P2phoP, leading to the autorepression of phoPQ. The binding of PhoP to the phoP upstream region is consistent with our previous results (5).
PhoP binds to DNA regions at various positions from the RNA polymerase binding site (−10 and −35 core promoter elements) (5, 15, 30). For YPO1635 or crp, a single PhoP site overlaps with the −35 element that usually contacts the σ subunit C-terminal domain (σCTD) of RNA polymerase. Thus, a class II stimulation of YPO1635 or crp by PhoP may be found, requiring the σCTD of the RNA polymerase for function (31). PhoP recognizes two distinct sites (sites 1 and 2) within the phoP upstream region. Site 2 is located more than 130 bp upstream of the −35 elements, whereas site 1 overlaps the −10 and −35 regions for P2phoP and the −35 one for P1phoP. The binding of PhoP to these sites possibly leads to the formation of a loop of the phoP upstream DNA region, which will block the entry of the RNA polymerase and thereby lead to PhoP-mediated repression of phoPQ-YPO1632. For cyaA, a single PhoP site is located downstream of the transcriptional start, which is highly unusual for a regulator that stimulates the transcription of its target gene. However, this finding is expected because similar promoters have been found in S. enterica (32), Bacillus subtilis (33), and E. coli (34). For instance, the Salmonella transcriptional activator SlyA footprinted the ugtL promoter at a region located downstream of the transcription start that is necessary for gene transcription (32).
Since the genetic contents (including both coding and promoter regions) of phoP, phoQ, YPO1635, crp, and cyaA are identical in Y. pestis (35, 36) and its closely related progenitor Yersinia pseudotuberculosis (37), the regulatory circuit characterized herein will be conserved in these two bacterial species.
ACKNOWLEDGMENTS
Financial support was provided by the National Natural Science Foundation of China (30930001) and by the National Basic Research Program of China (2009CB522600).
The English writing of the manuscript was polished by EnPapers.
Footnotes
Published ahead of print 21 December 2012
REFERENCES
- 1. Perry RD, Fetherston JD. 1997. Yersinia pestis—etiologic agent of plague. Clin. Microbiol. Rev. 10:35–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zheng D, Constantinidou C, Hobman JL, Minchin SD. 2004. Identification of the CRP regulon using in vitro and in vivo transcriptional profiling. Nucleic Acids Res. 32:5874–5893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gunasekera A, Ebright YW, Ebright RH. 1992. DNA sequence determinants for binding of the Escherichia coli catabolite gene activator protein. J. Biol. Chem. 267:14713–14720 [PubMed] [Google Scholar]
- 4. Groisman EA. 2001. The pleiotropic two-component regulatory system PhoP-PhoQ. J. Bacteriol. 183:1835–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Y, Gao H, Qin L, Li B, Han Y, Guo Z, Song Y, Zhai J, Du Z, Wang X, Zhou D, Yang R. 2008. Identification and characterization of PhoP regulon members in Yersinia pestis biovar Microtus. BMC Genomics 9:143 doi:10.1186/1471-2164-9-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Minagawa S, Ogasawara H, Kato A, Yamamoto K, Eguchi Y, Oshima T, Mori H, Ishihama A, Utsumi R. 2003. Identification and molecular characterization of the Mg2+ stimulon of Escherichia coli. J. Bacteriol. 185:3696–3702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamamoto K, Ogasawara H, Fujita N, Utsumi R, Ishihama A. 2002. Novel mode of transcription regulation of divergently overlapping promoters by PhoP, the regulator of two-component system sensing external magnesium availability. Mol. Microbiol. 45:423–438 [DOI] [PubMed] [Google Scholar]
- 8. Lejona S, Aguirre A, Cabeza ML, Garcia Vescovi E, Soncini FC. 2003. Molecular characterization of the Mg2+-responsive PhoP-PhoQ regulon in Salmonella enterica. J. Bacteriol. 185:6287–6294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Groisman EA, Mouslim C. 2006. Sensing by bacterial regulatory systems in host and non-host environments. Nat. Rev. Microbiol. 4:705–709 [DOI] [PubMed] [Google Scholar]
- 10. Brückner R, Titgemeyer F. 2002. Carbon catabolite repression in bacteria: choice of the carbon source and autoregulatory limitation of sugar utilization. FEMS Microbiol. Lett. 209:141–148 [DOI] [PubMed] [Google Scholar]
- 11. Zhan L, Han Y, Yang L, Geng J, Li Y, Gao H, Guo Z, Fan W, Li G, Zhang L, Qin C, Zhou D, Yang R. 2008. The cyclic AMP receptor protein, CRP, is required for both virulence and expression of the minimal CRP regulon in Yersinia pestis biovar microtus. Infect. Immun. 76:5028–5037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhan L, Yang L, Zhou L, Li Y, Gao H, Guo Z, Zhang L, Qin C, Zhou D, Yang R. 2009. Direct and negative regulation of the sycO-ypkA-ypoJ operon by cyclic AMP receptor protein (CRP) in Yersinia pestis. BMC Microbiol. 9:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee HY, Cho SA, Lee IS, Park JH, Seok SH, Baek MW, Kim DJ, Lee SH, Hur SJ, Ban SJ, Lee YK, Han YK, Cho YK, Park JH. 2007. Evaluation of phoP and rpoS mutants of Salmonella enterica serovar Typhi as attenuated typhoid vaccine candidates: virulence and protective immune responses in intranasally immunized mice. FEMS Immunol. Med. Microbiol. 51:310–318 [DOI] [PubMed] [Google Scholar]
- 14. Grabenstein JP, Fukuto HS, Palmer LE, Bliska JB. 2006. Characterization of phagosome trafficking and identification of PhoP-regulated genes important for survival of Yersinia pestis in macrophages. Infect. Immun. 74:3727–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perez JC, Groisman EA. 2009. Transcription factor function and promoter architecture govern the evolution of bacterial regulons. Proc. Natl. Acad. Sci. U. S. A. 106:4319–4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perez JC, Shin D, Zwir I, Latifi T, Hadley TJ, Groisman EA. 2009. Evolution of a bacterial regulon controlling virulence and Mg(2+) homeostasis. PLoS Genet. 5:e1000428 doi:10.1371/journal.pgen.1000428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou D, Han Y, Qin L, Chen Z, Qiu J, Song Y, Li B, Wang J, Guo Z, Du Z, Wang X, Yang R. 2005. Transcriptome analysis of the Mg2+-responsive PhoP regulator in Yersinia pestis. FEMS Microbiol. Lett. 250:85–95 [DOI] [PubMed] [Google Scholar]
- 18. Zhou D, Tong Z, Song Y, Han Y, Pei D, Pang X, Zhai J, Li M, Cui B, Qi Z, Jin L, Dai R, Du Z, Wang J, Guo Z, Huang P, Yang R. 2004. Genetics of metabolic variations between Yersinia pestis biovars and the proposal of a new biovar, microtus. J. Bacteriol. 186:5147–5152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y, Gao H, Wang L, Xiao X, Tan Y, Guo Z, Zhou D, Yang R. 2011. Molecular characterization of transcriptional regulation of rovA by PhoP and RovA in Yersinia pestis. PLoS One 6:e25484 doi:10.1371/journal.pone.0025484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Straley SC, Bowmer WS. 1986. Virulence genes regulated at the transcriptional level by Ca2+ in Yersinia pestis include structural genes for outer membrane proteins. Infect. Immun. 51:445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oyston PC, Dorrell N, Williams K, Li SR, Green M, Titball RW, Wren BW. 2000. The response regulator PhoP is important for survival under conditions of macrophage-induced stress and virulence in Yersinia pestis. Infect. Immun. 68:3419–3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hitchen PG, Prior JL, Oyston PC, Panico M, Wren BW, Titball RW, Morris HR, Dell A. 2002. Structural characterization of lipo-oligosaccharide (LOS) from Yersinia pestis: regulation of LOS structure by the PhoPQ system. Mol. Microbiol. 44:1637–1650 [DOI] [PubMed] [Google Scholar]
- 23. Bozue J, Mou S, Moody KL, Cote CK, Trevino S, Fritz D, Worsham P. 2011. The role of the phoPQ operon in the pathogenesis of the fully virulent CO92 strain of Yersinia pestis and the IP32953 strain of Yersinia pseudotuberculosis. Microb. Pathog. 50:314–321 [DOI] [PubMed] [Google Scholar]
- 24. Lukaszewski RA, Kenny DJ, Taylor R, Rees DG, Hartley MG, Oyston PC. 2005. Pathogenesis of Yersinia pestis infection in BALB/c mice: effects on host macrophages and neutrophils. Infect. Immun. 73:7142–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Price MN, Arkin AP, Alm EJ. 2006. The life-cycle of operons. PLoS Genet. 2:e96 doi:10.1371/journal.pgen.0020096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marceau M, Sebbane F, Ewann F, Collyn F, Lindner B, Campos MA, Bengoechea JA, Simonet M. 2004. The pmrF polymyxin-resistance operon of Yersinia pseudotuberculosis is upregulated by the PhoP-PhoQ two-component system but not by PmrA-PmrB, and is not required for virulence. Microbiology 150:3947–3957 [DOI] [PubMed] [Google Scholar]
- 27. Lathem WW, Price PA, Miller VL, Goldman WE. 2007. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315:509–513 [DOI] [PubMed] [Google Scholar]
- 28. Cathelyn JS, Crosby SD, Lathem WW, Goldman WE, Miller VL. 2006. RovA, a global regulator of Yersinia pestis, specifically required for bubonic plague. Proc. Natl. Acad. Sci. U. S. A. 103:13514–13519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Soncini FC, Vescovi EG, Groisman EA. 1995. Transcriptional autoregulation of the Salmonella typhimurium phoPQ operon. J. Bacteriol. 177:4364–4371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zwir I, Shin D, Kato A, Nishino K, Latifi T, Solomon F, Hare JM, Huang H, Groisman EA. 2005. Dissecting the PhoP regulatory network of Escherichia coli and Salmonella enterica. Proc. Natl. Acad. Sci. U. S. A. 102:2862–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Niu W, Kim Y, Tau G, Heyduk T, Ebright RH. 1996. Transcription activation at class II CAP-dependent promoters: two interactions between CAP and RNA polymerase. Cell 87:1123–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shi Y, Latifi T, Cromie MJ, Groisman EA. 2004. Transcriptional control of the antimicrobial peptide resistance ugtL gene by the Salmonella PhoP and SlyA regulatory proteins. J. Biol. Chem. 279:38618–38625 [DOI] [PubMed] [Google Scholar]
- 33. Qi Y, Hulett FM. 1998. PhoP-P and RNA polymerase sigmaA holoenzyme are sufficient for transcription of Pho regulon promoters in Bacillus subtilis: PhoP-P activator sites within the coding region stimulate transcription in vitro. Mol. Microbiol. 28:1187–1197 [DOI] [PubMed] [Google Scholar]
- 34. Munson GP, Scott JR. 2000. Rns, a virulence regulator within the AraC family, requires binding sites upstream and downstream of its own promoter to function as an activator. Mol. Microbiol. 36:1391–1402 [DOI] [PubMed] [Google Scholar]
- 35. Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MT, Prentice MB, Sebaihia M, James KD, Churcher C, Mungall KL, Baker S, Basham D, Bentley SD, Brooks K, Cerdeno-Tarraga AM, Chillingworth T, Cronin A, Davies RM, Davis P, Dougan G, Feltwell T, Hamlin N, Holroyd S, Jagels K, Karlyshev AV, Leather S, Moule S, Oyston PC, Quail M, Rutherford K, Simmonds M, Skelton J, Stevens K, Whitehead S, Barrell BG. 2001. Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413:523–527 [DOI] [PubMed] [Google Scholar]
- 36. Song Y, Tong Z, Wang J, Wang L, Guo Z, Han Y, Zhang J, Pei D, Zhou D, Qin H, Pang X, Zhai J, Li M, Cui B, Qi Z, Jin L, Dai R, Chen F, Li S, Ye C, Du Z, Lin W, Yu J, Yang H, Huang P, Yang R. 2004. Complete genome sequence of Yersinia pestis strain 91001, an isolate avirulent to humans. DNA Res. 11:179–197 [DOI] [PubMed] [Google Scholar]
- 37. Chain PS, Carniel E, Larimer FW, Lamerdin J, Stoutland PO, Regala WM, Georgescu AM, Vergez LM, Land ML, Motin VL, Brubaker RR, Fowler J, Hinnebusch J, Marceau M, Medigue C, Simonet M, Chenal-Francisque V, Souza B, Dacheux D, Elliott JM, Derbise A, Hauser LJ, Garcia E. 2004. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U. S. A. 101:13826–13831 [DOI] [PMC free article] [PubMed] [Google Scholar]