

Abstract
The opportunistic human pathogen Pseudomonas aeruginosa uses two surface organelles, flagella and pili, for motility and adhesion in biotic and abiotic environments. Polar flagellar placement and number are influenced by FlhF, which is a signal recognition particle (SRP)-type GTPase. The FlhF proteins of Bacillus subtilis and Campylobacter jejuni were recently shown to have GTPase activity. However, the phenotypes associated with flhF deletion and/or mutation differ between these organisms and P. aeruginosa, making it difficult to generalize a role for FlhF in pseudomonads. In this study, we confirmed that FlhF of P. aeruginosa binds and hydrolyzes GTP. We mutated FlhF residues that we predicted would alter nucleotide binding and hydrolysis and determined the effects of these mutations on FlhF enzymatic activity, protein dimerization, and bacterial motility. Both hydrolytically active and inactive FlhF point mutants restored polar flagellar assembly, as seen for wild-type FlhF. However, differential effects on flagellar function were observed in single-cell assays of swimming motility and flagellar rotation. These findings indicate that FlhF function is influenced by its nucleotide binding and hydrolytic activities and demonstrate that FlhF affects P. aeruginosa flagellar function as well as assembly.
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative bacterium that is isolated from water and soil environments. The two surface organelles of P. aeruginosa, flagella and pili, are important for its ability to move and survive in these environments and to colonize and infect human hosts. P. aeruginosa has a single polar flagellum that powers swimming in liquid media and swarming in semisolid media. This organelle is also implicated in pathogenesis and biofilm formation (1, 2). The structure of the bacterial flagellum is well described (3). The regulation of flagellar biosynthesis is less well understood, and different models have been proposed for the hierarchy of flagellar assembly in diverse bacteria (4–6).
One protein that participates in the regulation of flagellar assembly is FlhF. In the absence of FlhF in P. aeruginosa, flagellum assembly occurs but is no longer restricted to the pole (7, 8). ΔflhF bacteria move in circular rather than straight paths when swimming in liquid, and the decline in their mean velocity under conditions of increased viscosity is more pronounced than that observed for wild-type (WT) organisms (7). FlhF is encoded by polarly flagellated bacteria (P. aeruginosa, Vibrio alginolyticus, Vibrio cholerae, and Campylobacter jejuni). With the exception of Bacillus spp., the commonly studied peritrichously flagellated bacteria do not carry flhF. In Bacillus cereus, deletion of flhF leads to a reduction in flagellar number rather than to a change in flagellar position (9).
FlhF is one of three members of the signal recognition particle (SRP)-GTPase subfamily of SIMIBI-class nucleotide binding proteins. It is involved in flagellar biosynthesis; the other two members are required for SRP protein targeting (10). The SRP-GTPases regulate global cell processes such as cell polarity, signal transduction, and cell division (11). The structure and general catalytic cycle of SRP-GTPases are known (12).
The FlhF proteins of Bacillus subtilis (BsFlhF) and C. jejuni (CjFlhF) were recently shown to have GTPase activity (13, 14). However, the phenotypes associated with flhF deletion or mutation differ for these organisms and P. aeruginosa, making it difficult to generalize a role for FlhF in P. aeruginosa based on these studies. We therefore tested whether P. aeruginosa FlhF (PaFlhF) binds and hydrolyzes GTP. Point mutations predicted to alter nucleotide binding and/or hydrolysis were introduced into FlhF. The effects of these mutations on enzymatic activity of FlhF in vitro were analyzed. In addition, we tested whether hydrolytic activity of FlhF was required for polar flagellar assembly or function.
MATERIALS AND METHODS
Bacterial strains, media, and culture conditions.
The strains and plasmids used in this study are listed in Table 1. Bacteria were cultured and propagated in Luria broth (LB) (1% tryptone, 0.5% yeast extract, 1% NaCl), in M8 medium with 0.4% glucose and 0.05% sodium glutamate, or on Vogel-Bonner minimal medium (VBM) agar plates (15). Antibiotics were added to liquid and solid media as appropriate, at the following concentrations: for Escherichia coli, 100 μg/ml ampicillin, 50 μg/ml kanamycin, and 20 μg/ml tetracycline; and for P. aeruginosa, 200 μg/ml carbenicillin, 100 μg/ml gentamicin, and 100 μg/ml tetracycline.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description or relevant genotype | Source or reference |
|---|---|---|
| Strains | ||
| E. coli strains | ||
| DH5α | Used for subcloning | Invitrogen |
| XL1-Blue | Used for subcloning | Stratagene |
| BL21(DE3) | Used for overexpression of FlhF protein | Novagen |
| S17-1 | Used for mating constructs in P. aeruginosa | 41 |
| KDZif1ΔZ | Used for bacterial two-hybrid assay | 24 |
| P. aeruginosa strains | ||
| PA103 | Virulent lung isolate of P. aeruginosa | 42 |
| PAK | Wild-type isolate | J. Mattick |
| PAK ΔpilA | Deletion of pilA | This work |
| PAK ΔflhF | In-frame deletion of amino acids 7 to 430 of FlhF | 7 |
| PAK ΔflhF + WT | flhF-His6 under the control of an arabinose promoter integrated at the attB site in the ΔflhF background [attB::pARA flhF(WT)-His6] | This work |
| PAK ΔflhF + K222A | flhF-K222A-His6 under the control of an arabinose promoter integrated at the attB site in the ΔflhF background [attB::pARA flhF(K222A)-His6] | This work |
| PAK ΔflhF + R251G | flhF-R251G-His6 under the control of an arabinose promoter integrated at the attB site in the ΔflhF background [attB::pARA flhF(R251G)-His6] | This work |
| PAK ΔflhF + D294A | flhF-D294A-His6 under the control of an arabinose promoter integrated at the attB site in the ΔflhF background [attB::pARA flhF(D294A)-His6] | This work |
| PAK ΔflhF + L298R,P299L | flhF-L298R,P299L-His6 under the control of an arabinose promoter integrated at the attB site in the ΔflhF background [attB::pARA flhF(L298R,P299L)-His6] | This work |
| Plasmids | ||
| pBAD30 | Cloning vector; Apr | 43 |
| pSW196 | Vector for introducing genes under the control of an arabinose promoter at the attB site in the P. aeruginosa chromosome; Tcr | 18 |
| pFLP2 | Source of inducible Flp recombinase; Apr | 20 |
| pEX18Gm/ΔpilA | Vector for introducing a deletion of the pilA gene into the P. aeruginosa chromosome; Gmr Tcr | 21 |
| pGEX-6P-2 | Vector for in-frame fusion with a GST tag; Apr | Amersham |
| pBRGPω | Vector for bacterial two-hybrid assay with a C-terminal domain reporter fusion of the ω subunit of E. coli RNA polymerase; Apr | 24 |
| pACTR-AP-Zif | Vector for bacterial two-hybrid assay with a C-terminal domain reporter fusion of zinc finger DNA binding domain of Zif; Tcr | 24 |
| pGEX-6P-2/flhF-WT-His6 | Vector for overexpression of GST-FlhF-WT-His6; Apr | This work |
| pGEX-6P-2/flhF-K222A-His6 | Vector for overexpression of GST-FlhF-K222A-His6; Apr | This work |
| pGEX-6P-2/flhF-R251G-His6 | Vector for overexpression of GST-FlhF-R251G-His6; Apr | This work |
| pGEX-6P-2/flhF-D294A-His6 | Vector for overexpression of GST-FlhF-D294A-His6; Apr | This work |
| pGEX-6P-2/flhF-L298R,P299L-His6 | Vector for overexpression of GST-FlhF-L298R,P299L-His6; Apr | This work |
| pBRGP/flhF-WT-ω | Vector for bacterial two-hybrid assay with FlhF-WT-ω fusion; Apr | This work |
| pBRGP/flhF-R251G-ω | Vector for bacterial two-hybrid assay with FlhF-R251G-ω fusion; Apr | This work |
| pBRGP/flhF-K222A-ω | Vector for bacterial two-hybrid assay with FlhF-K222A-ω fusion; Apr | This work |
| pBRGP/flhF-D294A-ω | Vector for bacterial two-hybrid assay with FlhF-D294A-ω fusion; Apr | This work |
| pACTR/flhF-WT-Zif | Vector for bacterial two-hybrid assay with FlhF-WT-Zif fusion; Tcr | This work |
| pACTR/flhF-R251G-Zif | Vector for bacterial two-hybrid assay with FlhF-R251G-Zif fusion; Tcr | This work |
| pACTR/flhF-K222A-Zif | Vector for bacterial two-hybrid assay with FlhF-K222A-Zif fusion; Tcr | This work |
| pACTR/flhF-D294A-Zif | Vector for bacterial two-hybrid assay with FlhF-D294A-Zif fusion; Tcr | This work |
Construction of FlhF-His6 and GST-FlhF-His6 fusion proteins.
DNA was transformed into chemically competent E. coli or electroporated into competent P. aeruginosa (16). DNA primers used in cloning and site-directed mutagenesis are listed in Table 2. Nucleotide sequencing was performed by the Keck DNA Sequencing Facility (Yale University). The wild-type flhF gene was amplified from P. aeruginosa PA103 by use of the primers KA-1 and KA-2 and then cloned using a pCR-Blunt II-TOPO cloning kit according to the manufacturer's instructions (Invitrogen). The sequence of the cloned gene was confirmed by sequencing. The flhF-His6 gene was then subcloned into pBAD30 and pGEX-6P-2 expression vectors. These constructs were transformed into E. coli BL21(DE3) for expression and purification.
Table 2.
Primers used in this study
| Primer | Sequence (5′–3′) |
|---|---|
| KA-1 | GAA TTC AGG AGG TTC GGG CAA TGC AAG TCA A |
| KA-2 | GAG CTC TCA GTG GTG GTG GTG GTG GTG ACG CCG CGC CGG TTG CTG |
| R251G-1 | GCA TGG ACA GCT ACG GTA TCG GCG CCC AGG |
| R251G-2 | CCT GGG CGC CGA TAC CGT AGC TGT CCA TGC |
| K222A-1 | GCC CGG CAG GTG CCG GAG CGA CCA CCA CCC |
| K222A-2 | GGG TGG TGG TCG CTC CGG CAC CTG CCG GGC |
| D294A-1 | TGG TGC TGA TCG CTA CCG CCG GCC TGC |
| D294A-2 | GCA GGC CGG CGG TAG CGA TCA GCA CCA |
| L298R,P299L-1 | ATA CCG CCG GCC GGC TGG CAA GCG ACC |
| L298R,P299L-2 | GGT CGC TTG CCA GCC GGC CGG CGG TAT |
| NdeI-flhF-forw | CAT ATG CAA GTC AAA CGC TTC TTC GCC GCC GAT ATG CGC |
| NotI-flhF-rev | GCG GCC GCG CCG GCA CGC CGC GCC GGT TGC TGG TAG AG |
Protein expression and purification.
Overnight cultures prepared from single colonies were subcultured (1:100) and incubated at 37°C in LB plus ampicillin until the optical density at 600 nm (OD600) reached 0.4. Fusion protein expression was induced by the addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 3 h at 37°C. Cells were harvested by centrifugation at 8,000 × g for 20 min at 4°C. All subsequent protein purification procedures were performed either on ice or at 4°C, unless otherwise noted.
Cells were resuspended in a 1/50 culture volume in lysis buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 10 mM KCl) supplemented with 2 mM dithiothreitol (DTT), RNase, DNase, and a protease inhibitor mix (Roche). Cells were passed twice through a French press (12,000 lb/in2) and then centrifuged (20,000 × g for 30 min). The fusion protein was largely insoluble. The pellet was resuspended in 20 ml TG buffer (1% Triton X-100, 10% glycerol, 50 mM Tris, pH 8.0, 50 mM NaCl), incubated for 20 min on a rotator at room temperature, and then centrifuged at 4,000 × g for 10 min. The washed pellet was resuspended in 10 ml PreScission protease buffer (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM DTT, 1 mM EDTA) containing 6 M urea and solubilized overnight on a rotator. Insoluble protein was removed by centrifugation at 4,000 × g for 10 min. Solubilized protein was incubated with Ni-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen) overnight on a rotator. Protein-bound resin was washed and eluted under denaturing conditions according to the manufacturer's instructions, and elution fractions were analyzed by SDS-PAGE. Elution fractions containing GST-FlhF-His6 were dialyzed against PreScission protease buffer without DTT (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA) at a ratio of 1:100. To slow down refolding, dialysis was performed in a stepwise fashion against buffers containing 4 M, 2 M, 1 M, or 0 M urea. Precipitated protein (>90%) was removed by centrifugation at 4,000 × g for 10 min. To avoid freeze-thaw cycles, single-use aliquots of refolded protein containing 5% glycerol were stored at −80°C.
Aliquots of FlhF were run on SDS-PAGE together with a bovine serum albumin (BSA) standard curve. Gels were silver stained and imaged by densitometry, allowing the amount of refolded soluble FlhF to be estimated.
The glutathione S-transferase (GST) control (pGEX-6P-2 without insert) was purified via glutathione Sepharose. The empty vector control (pGEX-6P-2 without insert) was treated the same way as the cells expressing FlhF protein (with Ni-NTA under denaturing conditions, followed by refolding).
MS/MS analysis.
Purified GST-FlhF-His6 protein was submitted to the Keck Mass Spectrometry and Proteomics Resource Laboratory (Yale University) for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis following trypsin digestion. MS/MS spectra were searched against E. coli and P. aeruginosa databases using MASCOT Distiller (17).
Site-directed mutagenesis.
Site-directed mutagenesis of the flhF gene was performed using a QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The template for all mutagenesis reactions was flhF-His6 subcloned into pBAD30. flhF(K222A) was generated using primers K222A-1 and K222A-2, flhF(R251G) using primers R251G-1 and R251G-2, flhF(D294A) using primers D294A-1 and D294A-2, and flhF(L298R,P299L) using primers L298R,P299L-1 and L298R,P299L-2. The sequence of each construct was confirmed by sequencing; mutated genes were then subcloned into pGEX-6P-2 to generate amino-terminal GST fusion proteins as described above. Mutant proteins were expressed and purified using the same methods as those described for wild-type FlhF.
Construction of complemented P. aeruginosa ΔflhF strains.
Wild-type flhF, flhF-His6, and mutant flhF-His6 alleles were cloned into pSW196 under the control of the pBAD promoter (18). This plasmid, which integrates at the chromosomal attB site, was transformed into E. coli S17-1 and mobilized into P. aeruginosa PAK ΔflhF by mating. Selection of integrants was carried out on VBM-tetracycline plates as previously described, and mutant constructs were confirmed by PCR and immunoblotting (19). After integration into the attB site, vector backbone sequences were excised by Flp recombinase as previously described (20).
Antibody against PAK flagellin (FliC).
The PAK ΔpilA strain was constructed by allelic recombination, using a previously described pEX18Gm pilA::aacC1 suicide vector in which the pilA gene is replaced by a Gmr cassette (21). Briefly, this construct was mobilized into PAK by mating. Exconjugants were selected on VBM-gentamicin plus 5% (wt/vol) sucrose (to select for gene replacement and loss of the vector-carried sacB gene). The mutation was confirmed by PCR, Western blotting, and the phenotype (loss of twitching motility). Flagella were then sheared from the surfaces of PAK ΔpilA cells and purified as described previously (22). Rabbits were immunized with purified flagella in complete Freund's adjuvant and then boosted with flagella in incomplete Freund's adjuvant. Antisera were adsorbed by incubation with whole PAK ΔfliC cells, and antibodies were purified using a Hi-Trap protein G column (GE Amersham). For some experiments, anti-FliC antibodies were labeled with Alexa Fluor 488 (Invitrogen) according to the manufacturer's instructions.
Antiserum against P. aeruginosa FlhF.
A P. aeruginosa FlhF peptide (amino acids [aa] 20 to 34; Ac-DELGPDAAILGNRRC-NH2) was synthesized and conjugated to keyhole limpet hemocyanin (KLH) via cysteine (LifeTein). Rabbits were immunized with KLH-peptide in complete Freund's adjuvant and boosted with KLH-peptide in incomplete Freund's adjuvant (Cocalico Biologicals).
Western blot analysis.
Western blotting was carried out as previously described (7). Membranes were probed with anti-GST–horseradish peroxidase (HRP) alone (1:4,000; GE Healthcare) or with anti-FlhF antiserum (1:4,000) followed by HRP-conjugated goat anti-rabbit antibody (1:4,000; Bio-Rad).
GTPase activity assays.
Two types of GTPase activity assay were performed. For qualitative analysis of GTPase activity, single-turnover reactions were initiated by addition of 0.2 μCi of [α-32P]GTP (50 nM) to a reaction buffer (20 mM HEPES, pH 7.5, 350 mM NaCl) containing excess FlhF-His6 protein (∼5 μM). Aliquots (10 μl) were removed from the reaction mixture at specified times, and the reaction was stopped by adding an equal volume of 200 mM MgCl2. The reaction substrate and product were separated by thin-layer chromatography (TLC) (PEI cellulose F; Merck). Saturated ammonium sulfate and 1.5 M potassium dihydrogen phosphate were mixed at a 1:1.5 ratio and used as the mobile phase. Control reactions were performed in the absence of protein.
Determination of kinetic parameters for GTPase activity of FlhF.
A steady-state kinetic analysis of wild-type and mutant proteins was performed by mixing 89 nM purified GST fusion protein with various concentrations of nonradioactive GTP, with 0.1 μCi of [γ-32P]GTP as a tracer. The reactions were initiated by addition of the appropriate 10× GTP master mix (containing different concentrations of nonradioactive GTP and MgCl2) to the protein solution. Aliquots of 10 μl were removed from the reaction mixture at specified time points (0 to 90 min) and mixed by vortexing with 750 μl quenching buffer (50 mM Tris-HCl, pH 8.0, 350 mM NaCl, and 100 mM MgCl2) containing 5% activated charcoal. Control reaction mixtures (containing no protein) were set up the same way. The quenched reaction mixtures were centrifuged at 10,000 × g for 10 min to remove unhydrolyzed [γ-32P]GTP substrate, and 500 μl of supernatant was immobilized on filter paper in scintillation vials and mixed with 3 ml scintillation fluid. In addition, an aliquot of 10 μl of control reaction mixture was removed and diluted in 490 μl quenching buffer, and the complete 500 μl was immobilized on filter paper in scintillation vials. Samples were counted using a Tri-Carb 2800TR liquid scintillation analyzer (PerkinElmer).
The data were analyzed using GraphPad Prism (GraphPad Software, San Diego, CA). The initial rate of reaction was obtained by plotting the amount of product (GDP) over time at a given GTP concentration, which then fit into a linear line. The initial rate obtained at a given GTP concentration was plotted, and nonlinear regression was used to determine the kinetic parameters by using the Michaelis-Menten equation, i.e., V0 = Vmax[S]/Km + [S], where S is the substrate, V0 is the initial rate, Vmax is the maximum velocity, and Km is [S] at half of Vmax.
DRaCALA GTP binding assay.
The differential radial capillary action of ligand assay (DRaCALA) was performed as described by Roelofs et al. (23). BL21(DE3) cells carrying GST-FlhF constructs (or empty vector controls) were induced with 1 mM IPTG, collected by centrifugation, and resuspended in reaction buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 10 mM KCl) containing DNase and protease inhibitor cocktail (Roche). Cells were disrupted by sonication, aliquoted, and stored at −80°C. Western blotting against GST was used to confirm equivalent expression of wild-type and mutant proteins in samples.
Binding of GTP to the FlhF protein was measured by mixing lysates with 2.4 μCi of [α-32P]GTP (t = 0). Aliquots of 5 μl (containing 0.06 μCi) were removed from the reaction mixture at specified time points (0 to 40 min) and spotted on a dry nitrocellulose membrane (Bio-Rad). Control reaction mixtures containing empty vector lysates or no protein were performed in the same way. The nitrocellulose was allowed to dry and then exposed to a PhosphorImager screen, which was read by a Typhoon FLA 7000 system (GE Healthcare). Competition reaction mixtures containing 400 μM cold GTP (10,000-fold excess) were set up in the same way, with cold competitor being added to the reaction ca. 30 to 60 s after lysates were mixed with [α-32P]GTP. Triplicate samples were measured at each time point, and each experiment was repeated 3 to 5 times. A representative example of the raw data generated in this assay is presented in Fig. S7 in the supplemental material.
Data were analyzed using Carestream molecular imaging software (version MI 5.0.2.28). The signal intensities (counts per pixel) of the inner and outer circles of each spot were determined, and the fraction of bound GTP was calculated according to the method of Roelofs et al. (23). The fraction of GTP bound in the absence of protein was determined for each assay and subtracted prior to plotting values for samples and empty vector controls. In experiments containing excess cold GTP, the percentage of bound radioactivity at each time point was normalized to the amount of radioactivity bound by that sample in the absence of cold competitor, as determined in parallel (% bound = fraction boundw/cold/fraction boundw/o cold × 100).
Bacterial two-hybrid assay.
Wild-type and mutant FlhF alleles were subcloned into pBRGPω and pACTR-AP-Zif to generate plasmids expressing carboxy-terminal fusions of the ω subunit of E. coli RNA polymerase and the Zn finger DNA binding domain of murine Zif268, respectively, to full-length FlhF (24). FlhF mutant alleles were amplified using the primers NdeI-flhF-forw and NotI-flhF-rev and then ligated into NdeI-NotI-restricted pACTR-AP-Zif. The sequence of each construct was confirmed by sequencing. The NdeI-NotI fragment was then subcloned into pBRGPω. Both plasmids were electroporated at the same time into the E. coli reporter strain KDZif1ΔZ, which carries a lacZ reporter gene downstream of a promoter containing the Zif binding site (24). Expression of the fusion proteins was induced by growth in the presence of 50 μM IPTG. β-Galactosidase activity was measured as described previously (25) and is reported in Miller units. Samples were assayed in duplicate in 3 to 5 independent experiments.
Fluorescence microscopy.
P. aeruginosa overnight cultures grown at 37°C in LB plus 0.2% arabinose were diluted in fresh medium the next morning and incubated for 1 h at 37°C. Cells were harvested by centrifugation (3,000 × g for 3 min), fixed in 4% paraformaldehyde for 20 min at room temperature, and washed twice with phosphate-buffered saline (PBS). Flagella were stained with anti-FliC antibodies labeled with Alexa Fluor 488 (1 μg/ml in PBS) for 30 min at room temperature and then washed with PBS. Stained flagella were visualized using a Nikon Eclipse TS100 microscope (100× objective) equipped with a fluorescein isothiocyanate filter and a monochrome Spot camera (Diagnostic Instruments) running Spot 4.0.1 software.
Transmission electron microscopy (TEM).
P. aeruginosa overnight cultures grown at 37°C in LB plus 0.2% arabinose were diluted 1:100 in fresh medium the next morning and then incubated for 1.5 h at 37°C. Non-glow-discharged carbon-coated grids were incubated for 5 min with bacterial suspensions, stained with 0.1% phosphotungstic acid (PTA) for 30 s, and air dried for at least 30 min. Bacteria were visualized using a Tecnai 12 Biotwin electron microscope (Center for Cell Imaging, Yale University).
Video microscopy.
Bacteria were cultured in LB plus 0.2% arabinose, as described above for TEM, and then spotted onto glass slides and observed by dark-field microscopy with a Zeiss Axiostar Plus microscope (10× objective). Video clips were obtained using a Canon Vixia HFS200 camera and analyzed with ImageJ software, using the manual tracking function. For each strain, the movement of at least 60 bacteria was followed for 30 consecutive images (30 frames per 1 s) to obtain the average velocity (path length/s).
FliC binding assay.
P. aeruginosa was grown overnight at 37°C in LB medium containing 0.2% arabinose, diluted 1:25 in fresh medium the next morning, and incubated for 1 h at 37°C. Slides were incubated with PBS containing BSA (100 μg/ml) or BSA (100 μg/ml) plus anti-FliC antibodies (2 μg/ml), as indicated, for 30 min at room temperature (RT). Antibody- or BSA-coated slides were incubated with bacteria for 5 min at RT prior to imaging. Movies were taken immediately, using a Nikon Eclipse TS100 microscope (100× objective) equipped with a Canon Vixia HFS200 camera. One-minute video clips were analyzed manually to differentiate bacteria that were attached versus not attached (i.e., subject to Brownian motion).
Modeling of P. aeruginosa FlhF.
The Phyre2 server (http://www.sbg.bio.ic.ac.uk/phyre2/) was used to generate a structural model for PaFlhF (26). Amino acids 150 to 402 could be modeled with high confidence (100%), using FtsY (Protein Data Bank [PDB] files 2YHS, 3B9Q, 2OG2, 1VMA, and 3DMD) and BsFlhF (PDB file 2PX0) as templates. 3DLigandSite was used to predict the binding site for the Mg2+ and GTP heterogens (27). Swiss-PDBViewer v. 4.1 (http://www.expasy.org/spdbv/) was used to visualize and evaluate models and to generate images for publication (28).
Statistical analysis.
Data were analyzed using Prism 5.0 (GraphPad) software. The chi-square test was used to compare swimming velocity distributions. P values of <0.5 were considered significant.
RESULTS
P. aeruginosa FlhF is a GTPase.
FlhF from B. subtilis has been crystallized (29, 30). We threaded the P. aeruginosa FlhF sequence onto the crystal structure of the B. subtilis protein (PDB file 2PX0D) (Fig. 1A and B). Although the majority of residues that are conserved in SRP-like GTPases are easily identified in P. aeruginosa FlhF, some residues implicated in nucleotide-Mg2+ ligation and nucleophilic attack are altered in the P. aeruginosa protein, e.g., the G3 motif DXXGRL (aa 187 to 191 in E. coli Ffh) is DXXGLP. We therefore explicitly tested whether purified FlhF exhibited GTPase activity.
Fig 1.

FlhF of P. aeruginosa is a GTPase. (A) Amino acids 150 to 401 of FlhF were modeled by the Phyre2 server and are shown threaded onto the BsFlhF structure (PDB file 2PX0D). The ribbon diagram is colored by secondary structure succession (blue to red) and is shown in the same orientation as in panel B. (B) Molecular surface model of PaFlhF. Mutated residues are colored as follows: K222, aqua; R251, yellow; D294, red; L298, purple; and P299, dark plum. The positions of GTP (blue) and Mg2+ (green sphere) were predicted using 3DLigandSite. (C) Wild-type FlhF was incubated with 50 nM [α-32P]GTP. The reaction mixture was sampled at the indicated time points, and the reaction was stopped by the addition of 200 mM MgCl2. Reaction products were separated on TLC plates and visualized by autoradiography. The positions of GTP (*) and GDP (**) are indicated.
Carboxy-terminally His6-tagged FlhF was expressed in P. aeruginosa and E. coli. The protein complemented the motility and swarming defects of the P. aeruginosa ΔflhF strain, demonstrating that the tag did not interfere with biological function of the protein (data not shown and see below). The protein was purified from E. coli by metal affinity chromatography and assayed for GTP hydrolytic activity. FlhF hydrolyzed GTP to GDP and Pi (Fig. 1C).
The amount of soluble wild-type FlhF-His6 produced in E. coli was quite low and precluded careful kinetic analysis of wild-type and mutant FlhF proteins. Expression of FlhF-His6 as an amino-terminal GST fusion protein resulted in increased protein yields. The majority of GST-FlhF-His6 was insoluble, but we established a protocol for purification by metal affinity chromatography under denaturing conditions and subsequent refolding. We confirmed that wild-type GST-FlhF-His6 retained a hydrolytic activity after this purification procedure similar to that of FlhF-His6 purified under nondenaturing conditions. The refolded protein was therefore used in all subsequent assays (see Fig. S1 in the supplemental material).
Mutation of predicted FlhF active site residues alters GTP hydrolysis.
FlhF consists of an amino-terminal B domain followed by a conserved NG domain common to all SRP-GTPases (Fig. 2A). Several amino acid residues predicted to play roles in GTP binding and/or hydrolysis, based on available crystal structures of the BsFlhF, EcFfh, and EcFtsY GTPases, were mutated (29, 31–33). These included two residues within the nucleotide binding pocket (K222A and D294A) and the predicted catalytic arginine within the active site (R251G). We noted that PaFlhF lacks the positively charged residue that usually follows the D294XXG motif in nonpseudomonad FlhF homologs, and we used site-directed mutagenesis to change this sequence (DTAGLP) to the commonly observed consensus sequence DTAGRL, creating the L298R,P299L double mutant (Fig. 1B and 2B). All mutated FlhF proteins were expressed in E. coli, purified, and assayed for GTP hydrolysis in the presence of 1 μM MgCl2 (Fig. 2C; Table 3).
Fig 2.

Enzymatic characterization of FlhF point mutants. (A) Domain structure of FlhF from P. aeruginosa. N-TD and C-TD, N-terminal and C-terminal domains, respectively. (B) Amino acids within G-domain motifs of FlhF implicated in catalytic activity and/or GTP binding are aligned with the E. coli FtsY sequence. Residues mutated in FlhF are shown in bold. (C) Kinetic parameters of wild-type and mutant FlhF in the presence of 1 μM MgCl2. Symbols indicate means ± standard deviations (SD) for wild-type (black circles), L298R,P299L (black triangles), R251G (open squares), K222A (open inverted triangles), and D294A (black inverted triangles) FlhF. (D) Analysis of negative controls (GST [gray circles] and empty vector [open squares]) compared to K222A (open triangles) and D294A (black triangles) FlhF.
Table 3.
GTP hydrolysis by wild-type and mutant FlhF proteins
| FlhF construct | Km (μM) | Vmax (μmol/min) | kcat (min−1) | kcat/Km (M−1 min−1) |
|---|---|---|---|---|
| Wild type | 36.65 ± 18.01 | 0.42 ± 0.07 | 0.06 ± 0.01 | 1,637 |
| R251G | 9.13 ± 0.90 | 0.50 ± 0.01 | 0.07 ± 0.01 | 7,667 |
| L298R,P299L | 18.04 ± 3.67 | 0.85 ± 0.04 | 0.13 ± 0.01 | 7,206 |
| K222A | 130.70 ± 141.90 | 0.15 ± 0.09 | 0.02 ± 0.01 | 153 |
| D294A | 87.65 ± 95.69 | 0.06 ± 0.03 | 0.01 ± 0.01 | 114 |
The L298R,P299L mutant of FlhF had both a lower Michaelis constant (Km) and a higher rate of substrate turnover (kcat) than wild-type FlhF, resulting in a 4-fold increase in enzymatic efficiency. FlhF(R251G) had a lower Km for GTP than that of wild-type FlhF; surprisingly, estimates of kcat for this enzyme were similar to those obtained for the wild-type protein, despite mutation of the residue implicated in stabilizing the transition state geometry of the substrate undergoing nucleophilic attack (Table 3). The FlhF variants with mutations in the binding pocket, i.e., the K222A and D294A mutants, generated substrate at rates similar to those observed for negative controls, and thus appeared hydrolytically inactive (Fig. 2C and D). We had a difficult time fitting a curve to these data points, as reflected in the error associated with the estimates of Km for these reactions (Table 3).
We carried out several controls to confirm that the GTP hydrolysis that we observed could be ascribed to FlhF. We purified lysates from cells carrying the empty vector pGEX-6P-2, using the same protocol as that for our FlhF-expressing cells, and analyzed these preparations along with a second negative control, purified GST (Fig. 2D). Neither of these negative-control preparations showed GTP hydrolytic activity. In addition, an aliquot of the same FlhF protein used for the enzymatic assays was analyzed by MS/MS to confirm that no other nucleotidase had copurified with FlhF (see Table S1 in the supplemental material).
Nucleotide binding characteristics of wild-type and mutant FlhF proteins.
DRaCALA was used to compare nucleotide binding properties of wild-type and mutant FlhF proteins (23). This assay, which exploits the ability of dry nitrocellulose to separate protein-ligand complexes and free ligand, was used to measure [α-32P]GTP binding over time to wild-type and mutant FlhF alleles in the absence and presence of excess cold GTP competitor. Nonspecific binding was assessed using samples prepared from cells expressing GST alone. All alleles tested, with the exception of the D294A mutant, showed comparable initial binding of [α-32P]GTP; the D294A mutant reproducibly showed no specific binding above background (Fig. 3A). In the absence of excess cold competitor, ligand binding by wild-type FlhF slowly decayed, while the K222A and L298R,P299L proteins consistently showed more rapid dissociation of ligand. As GTP is labeled on the α phosphate, this experiment does not distinguish between bound substrate (GTP) and product (GDP). The R251G mutant consistently exhibited an increase in the fraction of ligand bound over the initial period of the assay and did not show appreciable dissociation in the absence of cold competitor (Fig. 3A).
Fig 3.

FlhF mutants show differences in GTP binding. Whole-cell lysates expressing GST-FlhF constructs were incubated with [α-32P]GTP. (A) The bound fraction of GTP is plotted as a function of incubation time. (B) Bound radioactivity measured after the addition of an excess of cold GTP competitor (10,000-fold excess) at time zero. Symbols indicate means ± SD for wild-type (gray circles), R251G (black squares), L298R,P299L (gray open squares), K222A (black open circles), and D294A (black X's) FlhF. Binding by lysates carrying the empty vector is indicated by a dashed line.
Dissociation of bound [α-32P]GTP/GDP was also measured after the addition of a 10,000-fold excess of cold GTP (Fig. 3B). Again, R251G ligand binding was seen to decay at a lower rate than that with wild-type FlhF. Since the wild-type and R251G FlhF proteins hydrolyzed GTP at comparable rates (as measured by release of Pi), this result likely reflects a slower dissociation of [α-32P]GDP from the R251G mutant.
Mutations in FlhF alter protein homodimerization.
GTP binding by SRP- and SRP receptor (SR)-GTPases precedes their heterodimerization and enzymatic activation, while hydrolysis of GTP is followed by heterodimer dissociation, as determined by the work of many laboratories and as recently reviewed (34). Because several of the mutant FlhF alleles showed altered nucleotide binding, we explicitly tested whether their ability to dimerize was altered by these mutations. We used a bacterial two-hybrid assay to measure dimerization, as soluble full-length FlhF and mutant variants could not be purified at high enough concentrations to allow biophysical methods to be employed. The DNA binding domain of the murine Zif268 protein (Zif) or residues 1 to 90 of the ω subunit of E. coli RNA polymerase (ω) were fused to the carboxy terminus of full-length FlhF (24). Expression of the fusion proteins was confirmed by probing Western blots of E. coli lysates with antiserum generated against FlhF (see Fig. S2 in the supplemental material). Interaction between the fusion proteins FlhF-ω and FlhF-Zif activated transcription of a lacZ reporter gene located downstream of a promoter containing the Zif binding site and could be measured by increased β-galactosidase activity (Fig. 4A). No increase in β-galactosidase activity was seen in control cells expressing ω (without fused FlhF) and FlhF-Zif or expressing Zif and FlhF-ω (Fig. 4A). This analysis was repeated with ω and Zif fusions to the R251G, K222A, and D294A alleles of FlhF. The R251G mutant formed homodimers, but both the K222A and D294A mutants were significantly attenuated for dimerization (Fig. 4A). We also assayed the ability of each mutant protein to interact with wild-type FlhF. The R251G mutant interacted strongly with wild-type FlhF (Fig. 4B). The K222A mutant appeared to interact more strongly with WT FlhF than the D294A mutant did.
Fig 4.

Bacterial two-hybrid analysis of FlhF protein-protein interactions. Cells harboring the indicated plasmids were grown in presence or absence of IPTG and then analyzed for β-galactosidase activity. (A) Homodimer formation by wild-type and mutant FlhF. (B) Heterodimer formation between wild-type FlhF and point mutants.
FlhF mutations are associated with altered flagellar function in single-cell assays.
Bacteria expressing FlhF mutant variants were analyzed for restoration of flagellar motility by video microscopy. P. aeruginosa ΔflhF strains carrying wild-type or mutant alleles of FlhF integrated as a single copy at the chromosomal attB site and under the control of the pBAD promoter were used for these and subsequent phenotypic assays.
Swimming of individual bacteria was characterized by measuring the distance traveled by a bacterium in 1 s (path length, equal to average velocity). When grown in 0.2% arabinose, ΔflhF bacteria complemented with wild-type FlhF swam with a distribution of path lengths indistinguishable from that observed for wild-type PAK (Fig. 5A). This concentration of inducer was therefore used for all subsequent phenotypic analyses. Under these induction conditions, steady-state levels of wild-type and mutant FlhF alleles (as estimated by Western blotting) were comparable, with the exception of that of FlhF(R251G), which appeared to be present in greater amounts (see Fig. S3 in the supplemental material).
Fig 5.

Distribution of path lengths for FlhF mutants. Bacteria were grown in LB plus 0.2% arabinose to an OD600 of ∼0.5 and then spotted onto glass slides and imaged. For each strain, the movement of at least 60 bacteria was followed for 30 consecutive images (1 s each). Distributions of average velocity (μm/s) were compared using the chi-square test. (A) For PAK versus PAK ΔflhF + WT, P > 0.5; and for PAK ΔflhF + WT versus PAK ΔflhF + L298R,P299L, P > 0.5. (B) For PAK ΔflhF + R251G versus PAK ΔfliC, P < 0.5. (C) For PAK ΔflhF + K222A versus PAK ΔflhF + D294A, P < 0.5. The data set for PAK ΔflhF + WT is reproduced in panels B and C to facilitate comparison with mutant strains. (D) Slides coated with BSA or anti-FliC antibodies were incubated with bacteria as indicated and then imaged for 1 min. Percentages of attached (white) versus nonattached (black) cells are shown for PAK ΔflhF + R251G on anti-FliC-coated slides (n = 124) and BSA-coated slides (n = 43) and for PAK ΔfliC on anti-FliC-coated slides (n = 29).
The FlhF(L298R,P299L) allele supported swimming motility indistinguishable from that seen with bacteria expressing wild-type FlhF (Fig. 5A). In contrast, most bacteria expressing FlhF(R251G) did not swim, as seen by comparing the distributions of swimming velocity for this strain and the aflagellate negative control, PAK ΔfliC (Fig. 5B). Bacteria expressing the two proteins that were hydrolytically inactive in vitro, i.e., the D294A and K222A mutants, showed a distribution of path lengths intermediate to those observed for WT FlhF versus FlhF(R251G) (Fig. 5C).
FlhF mutants assemble unipolar flagella.
In C. jejuni, deletion of flhF results in absent rather than mislocalized flagella, and complementation with an allele in which the G2 motif Arg is mutated (R324A) does not restore wild-type levels of flagellar assembly (14); a similar result has been reported for V. cholerae (35). In P. aeruginosa, flhF deletion results in flagellar assembly at random locations on the bacterial envelope (7). We used several imaging techniques to determine whether polar flagellar assembly was restored by mutant variants of FlhF. Every point mutant of FlhF that we constructed restored polar flagellar assembly; no bacterium with a nonpolar flagellum was seen (Fig. 6; see Fig. S4 in the supplemental material). Thus, neither wild-type nucleotide binding nor hydrolytic activity by PaFlhF is necessary for unipolar positioning of the flagellum. Transmission electron microscopy allowed us to visualize the origin of flagella at the cell envelope. The majority of flagellated cells had a single polar flagellum, with occasional bacteria exhibiting 2 or 3 unipolar flagella when the ΔflhF strain was complemented with either wild-type or mutant FlhF alleles (see Fig. S4). Only single unipolar flagella were observed for the wild-type PAK strain under these growth conditions.
Fig 6.
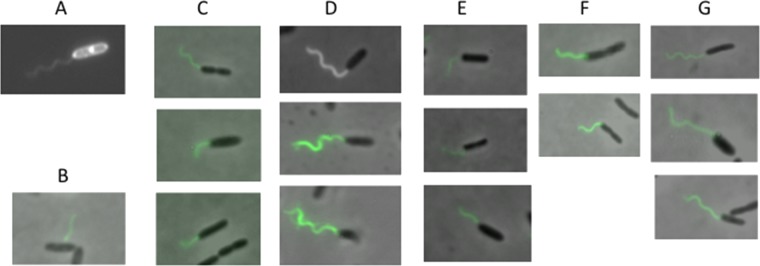
All FlhF mutants support the assembly of polar flagella. Bacteria were grown to an OD600 of ∼0.5 in LB plus 0.2% arabinose, fixed, and labeled with anti-FliC conjugated to Alex Fluor 488 as described in Materials and Methods to visualize flagella. (A) PAK; (B) PAK ΔflhF; (C) PAK ΔflhF + WT; (D) PAK ΔflhF + R251G; (E) PAK ΔflhF + L298R,P299L; (F) PAK ΔflhF + K222A; (G) PAK ΔflhF + D294A.
FlhF affects flagellar rotation of immobilized bacteria.
R251G bacteria assembled polar flagella, but most bacteria did not swim when examined by video microscopy. In order to assess flagellar presence and function in the same assay, we examined rotation of the ΔflhF deletion mutant complemented with the R251G allele (the ΔflhF + R251G strain) 5 min after binding to anti-FliC antibody-coated slides. A substantial proportion (89.5%) of bacteria showed attachment to the slide surface (and thus were no longer subject to Brownian motion) and could be observed to pivot (<360°) around a polar attachment point during the 1-min movie acquisition (Fig. 5D). Very few bacteria made a similar attachment to a BSA-coated slide or when the aflagellate ΔfliC strain was incubated with antibody-coated slides (Fig. 5D). Although many R251G cells attached to antibody-coated slides, fewer than 5% of these attached cells rotated (>360°) (see Movie S5 in the supplemental material). No rotating bacteria were observed on BSA-coated slides (see Movie S6) or when the ΔfliC strain was imaged (data not shown). This experiment confirmed that bacteria expressing the R251G allele assembled surface flagella but that the majority of these flagella did not rotate.
DISCUSSION
Enzymatic characteristics of P. aeruginosa FlhF.
In this study, we showed that purified full-length P. aeruginosa FlhF is a GTPase with a high Km (∼30 μM), a low turnover rate (kcat = 0.06/s), and a low Vmax (25 μmol/h). These kinetic parameters are similar to those reported for another SRP-GTPase, B. subtilis FlhF (20 μmol/h) (13).
FlhF proteins belong to the SRP-GTPase subfamily of SIMIBI-class nucleotide binding proteins. The other two members of this subfamily, the signal sequence binding protein Ffh (SRP54 in the Archaea and Eukarya) and the SRP receptor FtsY (SRα in the Eukarya), together constitute the universally conserved machinery that targets ribosomes in complex with nascent proteins to membrane channels for translocation. When bound to GTP, Ffh and FtsY proteins form a heterodimer with a composite active site. Elegant work has revealed that Ffh-FtsY dimerization and conformational activation of GTP hydrolysis are integrated with binding of “cargo” (complexes of ribosome and nascent chain protein) and its delivery to the membrane translocation machinery (34, 36).
Much less is known about the role that FlhF plays in flagellar assembly, though several groups have demonstrated that the NG domains of this protein exhibit GTPase activity in vitro. The BsFlhF NG construct has also been crystallized, and the observed homodimer shares features with the Ffh-FtsY heterodimer, e.g., a composite active site formed by a dimer interface between G domains and conserved nucleotide- and magnesium ion-binding residues. Bange and colleagues, however, also noted potentially significant differences between BsFlhF and EcFtsY, such as the lack of N-N contacts within the dimer interface and the replacement of a conserved P-loop asparagine in FtsY (N302) with a threonine (T184) that fails to contact the ribose of bound nucleotides (29). In PaFlhF, an alanine is found at this position (A218); interestingly, an Asn-to-Ala substitution in EcFtsY (N302A) results in defective heterodimer GTPase activation (37).
Our analysis of full-length PaFlhF suggests that several aspects of the Ffh-FtsY reaction cycle may be conserved between PaFlhF and the SRP homologs. The K222A and D294A mutations rendered purified FlhF hydrolytically inactive, as might be anticipated from the important roles played by G1 (P loop) and G3 motif residues in binding and orienting nucleotide in the enzyme active site. The D294A mutation also abrogated GTP binding, and the mutant protein showed minimal dimerization with itself or with wild-type FlhF. The K222A mutant bound GTP, but it also dimerized poorly and was hydrolytically inactive. We note that the homologous mutations in E. coli FtsY have been shown to impair FtsY-Ffh heterodimer formation (the D382A mutation more so than the K306A mutation) and subsequent GTPase activation (37).
We still observed GTP hydrolysis by the R251G mutant, with kcat and Vmax values similar to those noted for the purified wild-type protein. Arginine 251 is predicted to participate in stabilizing the transition state geometry of GTP as it undergoes nucleophilic attack, and we expected the R251G mutation to have a more pronounced effect on FlhF hydrolytic activity in vitro. Instead, the R251G protein showed marked differences in ligand binding that could reflect delayed dissociation of the FlhF-GDP dimer and/or altered release of GDP. The R251G allele dimerized as well as wild-type FlhF, which is consistent with observations that the homologous E. coli FtsY mutation, R333A, does not inhibit FtsY-Ffh complex formation (37).
FlhF and polar flagellar assembly.
FlhF plays distinct roles in flagellar assembly and positioning in bacteria with polar flagella. In C. jejuni and V. alginolyticus, flhF deletion results in nonflagellated cells that are nonmotile (14, 38). In contrast, P. aeruginosa ΔflhF bacteria assemble flagella at nonpolar sites and swim, albeit via circular rather than straight paths (7). We found that all of the alleles of FlhF that we constructed restored polar flagellar assembly to the ΔflhF strain. Thus, the flagellar positioning function of PaFlhF appears to be independent of nucleotide binding or hydrolysis. Because PaFlhF is not required for flagellar assembly per se, it is difficult to compare our findings to those of Green et al., who reported that mutations analogous to K222A and D294A (VcFlhF K295A and D367A) resulted in the absence of flagellar assembly in V. cholerae (35). These mutations were also introduced into V. alginolyticus FlhF (K305A and D377A), where they partially restored swarming motility and supported flagellar assembly in a small proportion of bacterial cells (38). It is unclear whether the differences between these Vibrio sp. studies result from differences in the experimental methods used to express FlhF alleles (from an integrated chromosomal copy versus a high-copy-number plasmid) and assess flagellar assembly (direct visualization versus functional assays) or reflect distinct roles played by FlhF in regulating flagellar gene expression, as well as flagellar placement, in various organisms (5, 39, 40).
FlhF and flagellar motility.
Although all tested mutant alleles of FlhF were able to restore polar flagellar placement, only FlhF(L298R,P299L) restored wild-type swimming motility. The three mutants that altered aspects of FlhF nucleotide binding and/or hydrolysis all exhibited impaired swimming motility. FlhF(R251G)-expressing bacteria could attach to anti-FliC antibody-coated slides and pivot (<360°) around a polar point of attachment; however, fewer than 5% of these attached bacteria rotated >360°, confirming that assembled flagella were deficient for rotation. It is unlikely that gross differences in protein expression or stability account for the altered flagellar function of the mutant proteins, as all were detected at levels comparable to or greater than that of the wild-type allele in lysates prepared from cells grown exactly as for motility assays. We did not detect endogenous PaFlhF by Western blotting, suggesting that the native protein is expressed at lower levels than our chromosomally integrated pBAD construct. Overexpression of FlhF has been associated with an increased number of polar flagella in Pseudomonas putida (8), and we also observed occasional bacteria with two or three polar flagella in our TEM analysis of the complemented ΔflhF strains. We do not think that alteration of flagellar number could account for the different swimming behaviors that we observed, however, as similar proportions of bacteria with >1 flagellum were observed for strains that showed wild-type swimming [i.e., PAK ΔflhF + WT and PAK ΔflhF + FlhF(L298R,P299L)] and those that showed reduced swimming velocities. The mechanism by which FlhF influences flagellar rotation remains unknown at this time.
Supplementary Material
ACKNOWLEDGMENTS
We thank Simon Dove for providing strains and plasmids for bacterial two-hybrid assays. We thank Morven Graham (Center for Cell Imaging, Yale University) for assistance with transmission electron microscopy and Aidan McKinlay and Ming-Jie Wu for excellent technical assistance with plasmid and strain constructions. We acknowledge Nathan Suek for excellent technical assistance with bacterial two-hybrid assays. We thank Ruchi Jain for helpful discussions.
This work was supported by an Investigator in Pathogenesis of Infectious Diseases award from the Burroughs Wellcome Fund and by award R01 AI054920 from the National Institute of Allergy and Infectious Diseases (B.I.K.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Footnotes
Published ahead of print 21 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02013-12.
REFERENCES
- 1. Hogan DA, Kolter R. 2002. Pseudomonas-Candida interactions: an ecological role for virulence factors. Science 296:2229–2232 [DOI] [PubMed] [Google Scholar]
- 2. O'Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30:295–304 [DOI] [PubMed] [Google Scholar]
- 3. Smith TG, Hoover TR. 2009. Deciphering bacterial flagellar gene regulatory networks in the genomic era. Adv. Appl. Microbiol. 67:257–295 [DOI] [PubMed] [Google Scholar]
- 4. Dasgupta N, Wolfgang MC, Goodman AL, Arora SK, Jyot J, Lory S, Ramphal R. 2003. A four-tiered transcriptional regulatory circuit controls flagellar biogenesis in Pseudomonas aeruginosa. Mol. Microbiol. 50:809–824 [DOI] [PubMed] [Google Scholar]
- 5. Correa NE, Peng F, Klose KE. 2005. Roles of the regulatory proteins FlhF and FlhG in the Vibrio cholerae flagellar transcription hierarchy. J. Bacteriol. 187:6324–6332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wilhelms M, Molero R, Shaw JG, Tomas JM, Merino S. 2011. Transcriptional hierarchy of Aeromonas hydrophila polar-flagellum genes. J. Bacteriol. 193:5179–5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murray TS, Kazmierczak BI. 2006. FlhF is required for swimming and swarming in Pseudomonas aeruginosa. J. Bacteriol. 188:6995–7004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pandza S, Baetens M, Park CH, Au T, Keyhan M, Matin A. 2000. The G-protein FlhF has a role in polar flagellar placement and general stress response induction in Pseudomonas putida. Mol. Microbiol. 36:414–423 [DOI] [PubMed] [Google Scholar]
- 9. Salvetti S, Ghelardi E, Celandroni F, Ceragioli M, Giannessi F, Senesi S. 2007. FlhF, a signal recognition particle-like GTPase, is involved in the regulation of flagellar arrangement, motility behaviour and protein secretion in Bacillus cereus. Microbiology 153:2541–2552 [DOI] [PubMed] [Google Scholar]
- 10. Carpenter PB, Hanlon DW, Ordal GW. 1992. flhF, a Bacillus subtilis flagellar gene that encodes a putative GTP-binding protein. Mol. Microbiol. 6:2705–2713 [DOI] [PubMed] [Google Scholar]
- 11. Bulyha I, Hot E, Huntley S, Sogaard-Andersen L. 2011. GTPases in bacterial cell polarity and signalling. Curr. Opin. Microbiol. 14:726–733 [DOI] [PubMed] [Google Scholar]
- 12. Luirink J, Sinning I. 2004. SRP-mediated protein targeting: structure and function revisited. Biochim. Biophys. Acta 1694:17–35 [DOI] [PubMed] [Google Scholar]
- 13. Bange G, Kummerer N, Grudnik P, Lindner R, Petzold G, Kressler D, Hurt E, Wild K, Sinning I. 2011. Structural basis for the molecular evolution of SRP-GTPase activation by protein. Nat. Struct. Mol. Biol. 18:1376–1380 [DOI] [PubMed] [Google Scholar]
- 14. Balaban M, Joslin SN, Hendrixson DR. 2009. FlhF and its GTPase activity are required for distinct processes in flagellar gene regulation and biosynthesis in Campylobacter jejuni. J. Bacteriol. 191:6602–6611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vogel HJ, Bonner DM. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218:97–106 [PubMed] [Google Scholar]
- 16. Choi K-H, Kumar A, Schweizer HP. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64:391–397 [DOI] [PubMed] [Google Scholar]
- 17. Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551–3567 [DOI] [PubMed] [Google Scholar]
- 18. Baynham PJ, Ramsey DM, Gvozdyev BV, Cordonnier EM, Wozniak DJ. 2006. The Pseudomonas aeruginosa ribbon-helix-helix DNA-binding protein AlgZ (AmrZ) controls twitching motility and biogenesis of type IV pili. J. Bacteriol. 188:132–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Laskowski MA, Osborn E, Kazmierczak BI. 2004. A novel sensor kinase-response regulator hybrid regulates type III secretion and is required for virulence in Pseudomonas aeruginosa. Mol. Microbiol. 54:1090–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86 [DOI] [PubMed] [Google Scholar]
- 21. de Kerchove AJ, Elimelech M. 2007. Impact of alginate conditioning film on deposition kinetics of motile and nonmotile Pseudomonas aeruginosa strains. Appl. Environ. Microbiol. 73:5227–5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Totten PA, Lara JC, Lory S. 1990. The rpoN gene product of Pseudomonas aeruginosa is required for expression of diverse genes, including the flagellin gene. J. Bacteriol. 172:389–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roelofs KG, Wang J, Sintim HO, Lee VT. 2011. Differential radial capillary action of ligand assay for high-throughput detection of protein-metabolite interactions. Proc. Natl. Acad. Sci. U. S. A. 108:15528–15533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vallet-Gely I, Donovan KE, Fang R, Joung JK, Dove SL. 2005. Repression of phase-variable cup gene expression by H-NS-like proteins in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 102:11082–11087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dove SL, Hochschild A. 1998. Conversion of the omega subunit of Escherichia coli RNA polymerase into a transcriptional activator or an activation target. Genes Dev. 12:745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 27. Wass MN, Kelley LA, Sternberg MJ. 2010. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 38:W469–W473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723 [DOI] [PubMed] [Google Scholar]
- 29. Bange G, Petzold G, Wild K, Parlitz RO, Sinning I. 2007. The crystal structure of the third signal-recognition particle GTPase FlhF reveals a homodimer with bound GTP. Proc. Natl. Acad. Sci. U. S. A. 104:13621–13625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bange G, Petzold G, Wild K, Sinning I. 2007. Expression, purification and preliminary crystallographic characterization of FlhF from Bacillus subtilis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 63:449–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Egea PF, Shan SO, Napetschnig J, Savage DF, Walter P, Stroud RM. 2004. Substrate twinning activates the signal recognition particle and its receptor. Nature 427:215–221 [DOI] [PubMed] [Google Scholar]
- 32. Focia PJ, Shepotinovskaya IV, Seidler JA, Freymann DM. 2004. Heterodimeric GTPase core of the SRP targeting complex. Science 303:373–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Montoya G, Svensson C, Luirink J, Sinning I. 1997. Crystal structure of the NG domain from the signal-recognition particle receptor FtsY. Nature 385:365–368 [DOI] [PubMed] [Google Scholar]
- 34. Saraogi I, Akopian D, Shan SO. 2011. A tale of two GTPases in cotranslational protein targeting. Protein Sci. 20:1790–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Green JCD, Kahramanoglou C, Rahman A, Pender AMC, Charbonnel N, Fraser GM. 2009. Recruitment of the earliest component of the bacterial flagellum to the old cell division pole by a membrane-associated signal recognition particle family GTP-binding protein. J. Mol. Biol. 391:679–690 [DOI] [PubMed] [Google Scholar]
- 36. Peluso P, Shan SO, Nock S, Herschlag D, Walter P. 2001. Role of SRP RNA in the GTPase cycles of Ffh and FtsY. Biochemistry 40:15224–15233 [DOI] [PubMed] [Google Scholar]
- 37. Shan SO, Stroud RM, Walter P. 2004. Mechanism of association and reciprocal activation of two GTPases. PLoS Biol. 2:e320 doi:10.1371/journal.pbio.0020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kusumoto A, Nishioka N, Kojima S, Homma M. 2009. Mutational analysis of the GTP-binding motif of FlhF which regulates the number and placement of the polar flagellum in Vibrio alginolyticus. J. Biochem. 146:643–650 [DOI] [PubMed] [Google Scholar]
- 39. Hendrixson DR, DiRita VJ. 2003. Transcription of sigma54-dependent but not sigma28-dependent flagellar genes in Campylobacter jejuni is associated with formation of the flagellar secretory apparatus. Mol. Microbiol. 50:687–702 [DOI] [PubMed] [Google Scholar]
- 40. Balaban M, Hendrixson DR. 2011. Polar flagellar biosynthesis and a regulator of flagellar number influence spatial parameters of cell division in Campylobacter jejuni. PLoS Pathog. 7:e1002420 doi:10.1371/journal.ppat.1002420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Biotechnology 1:784–791 [Google Scholar]
- 42. Liu PV. 1966. The roles of various fractions of Pseudomonas aeruginosa in its pathogenesis: identity of the lethal toxins produced in vitro and in vivo. J. Infect. Dis. 116:481–489 [DOI] [PubMed] [Google Scholar]
- 43. Guzman L, Belin D, Carlson MJ, Beckwith J. 1995. Tight regulation, modulation and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.