

Abstract
Type I interferons induce a complex transcriptional program that leads to a generalized antiviral response against a large panel of viruses, including human immunodeficiency virus type 1 (HIV-1). However, despite the fact that interferons negatively regulate HIV-1 ex vivo, a chronic interferon state is linked to the progression of AIDS and to robust viral replication, rather than protection, in vivo. To explain this apparent contradiction, we hypothesized that HIV-1 may have evolved a partial resistance to interferon, and to test this hypothesis, we analyzed the effects of alpha interferon (IFN-α) on the infectivity of HIV-1, human immunodeficiency virus type 2 (HIV-2), and rhesus monkey simian immunodeficiency virus (SIVmac). The results we obtained indicate that HIV-1 is more resistant to an IFN-α-induced response than are HIV-2 and SIVmac. Our data indicate that the accumulation of viral DNA is more compromised following the infection of IFN-α-treated cells with HIV-2 and SIVmac than with HIV-1. This defect correlates with a faster destabilization of HIV-2 viral nucleoprotein complexes (VNCs), suggesting a link between VNC destabilization and impaired viral DNA (vDNA) accumulation. The differential susceptibilities to IFN-α of the primate lentiviruses tested here do not map to the capsid protein (CA), excluding de facto a role for human tripartite motif protein isoform 5 alpha (Trim5α) in this restriction; this also suggests that an additional restriction mechanism differentially affects primate lentivirus infection. The different behaviors of HIV-1 and HIV-2 with respect to IFN-α responses may account at least in part for the differences in pathogenesis observed between these two virus types.
INTRODUCTION
Alpha and beta interferons (IFN-α and IFN-β), collectively defined as type I interferons (IFN-I) (1), induce a complex transcriptional program involving as many as 1,000 genes that are referred to as interferon-stimulated genes (ISGs) (2). The activation of this program leads to a generalized antiviral state that, in the vast majority of cases, inhibits viral spread.
In the case of human immunodeficiency virus type 1 (HIV-1), multiple studies have clearly established that type I interferon responses inhibit its replication. The antiviral effects of interferon on HIV-1 vary in magnitude according to the experimental conditions and the cell types used, and they encompass both early and late phases of the viral life cycle. Despite the fact that the roles of a few ISGs have been clearly defined, the contributions of most ISGs to this antiviral response remain to be characterized (3–37).
Although interferons exert an inhibitory effect against HIV-1 ex vivo, the relationship between virus replication and this antiviral response appears instead to be far more complex in vivo.
The comparison of pathogenic and nonpathogenic animal models of infection has surprisingly revealed that simian immunodeficiency virus (SIV)-induced pathogenesis is markedly linked to the presence of a chronic interferon response (38–40). These findings have been confirmed in HIV-1-infected individuals, where the molecular signature of a chronic IFN response has been associated with the progression of AIDS and with the presence of high viral loads (41–43).
These observations indicate that even though the interferon response is capable of inhibiting viral replication ex vivo, a chronic IFN response does not protect infected individuals from the virus and may contribute to its pathogenesis.
Since HIV-1 replicates robustly despite strong IFN-I responses in vivo, we set out to test the hypothesis that this virus may have evolved a partial resistance to IFN-I in contrast to the responses of other primate lentiviruses. To test this hypothesis, we compared HIV-1 to the closely related and less-pathogenic human immunodeficiency virus type 2 (HIV-2) and, when possible, to its simian counterpart rhesus monkey SIV (SIVmac). HIV-2 is closely related to HIV-1, but in contrast to the latter, it is characterized by poor transmission rates (HIV-2 infection remains concentrated essentially in west Africa) and slower progression to AIDS in infected individuals. Compared to data for HIV-1-infected individuals, less data are available about the relationship between the progression toward overt disease and the presence of an IFN state in HIV-2-infected individuals at different stages of the disease. However, in marked contrast to HIV-1-infected individuals, HIV-2-infected individuals do generally display low viral loads (44–47). The reasons for these differences are unknown at present.
The results we have obtained here indicate that HIV-1 is more resistant than are members of the HIV-2/SIV from sooty mangabeys (SIVsm) lineage to the negative effects of IFN-α. The differential susceptibilities of HIV-1 and HIV-2/SIVsm toward IFN-α map to the early phases of infection and, more specifically, to reverse transcription. Our data indicate that the functional stability of HIV-2 viral nucleoprotein complexes (VNCs) is compromised more rapidly in the presence of IFN-α, suggesting a link between VNC destabilization and the accrued defect in viral DNA (vDNA) accumulation. This phenotype does not map to the capsid protein (CA) and is not sensitive to saturation, excluding de facto a role for human tripartite motif protein isoform 5 alpha (T5α) in this restriction and suggesting that an additional restriction mechanism differentially affects primate lentiviruses in human cells that are treated with IFN-α.
MATERIALS AND METHODS
Cell culture, cytokines, and antibodies.
Cells were maintained as follows: 293T and HeLa cells in Dulbecco's modified Eagle medium (DMEM), 10% fetal calf serum (FCS), and 100 U/ml of penicillin/streptomycin; THP-1 cells in RPMI 1640, 10% FCS, 10 mM HEPES, 0.05 mM β-mercaptoethanol, and 100 U/ml of penicillin/streptomycin; primary cells in RPMI 1640, 10% FCS, 10 mM HEPES, 100 U/ml of penicillin/streptomycin, and the cytokines specified below. Prior to use, THP-1 cells were treated with 100 ng/ml of phorbol 12-myristate 13-acetate (PMA) (Sigma) for 24 h to induce their differentiation into macrophage-like cells. Monocytes and peripheral blood lymphocytes (PBLs) were purified from the blood of healthy donors (Etablissement Français du Sang [EFS]-Lyon) on Ficoll and Percoll gradients, followed by negative depletion (Miltenyi Biotec). This procedure yields cell populations of >95% purity, as has been described extensively (48). Monocytes were differentiated into macrophages or immature dendritic cells (DCs) after incubation for 4 to 5 days in 100 ng/ml of monocyte colony-stimulating factor (MCSF) or 100 ng/ml of granulocyte–macrophage colony-stimulating factor (GM-CSF)/interleukin 4 (IL-4), respectively (AbCys). PBLs were stimulated with phytohemagglutinin (PHA) (1 μg/ml; Sigma) and interleukin 2 (IL-2) (150 U/ml; AIDS Reagents and Reference Program of the NIH) for 24 h prior to infection. Unless otherwise indicated, IFN-α-2A (Tebu-Bio) was used at the indicated concentration for 24 h prior to infection. Anti-Flag, antihemagglutinin (HA), and antitubulin monoclonal antibodies were purchased from Sigma. The anti-CA antibody was obtained from the AIDS Reagent Program of the NIH (clone 183-H12-5C).
DNA constructs, viral production, and primary viral strains.
Lentiviral vectors derived from HIV-1, HIV-2, and SIVmac251 have been described previously (48–52). These vectors were originally engineered from NL4-3, ROD, and SIVmac251 viral strains. They have an identical structure, bear an identical CMV-GFP expression cassette, and have been pseudotyped with the pantropic envelope vesicular stomatitis virus G glycoprotein (VSVg) to confer ample cellular tropism. The HIV-1 chimeric vector containing the SIVmac CA has been described previously, while the reciprocal SIVmac vector was constructed on the basis of previous studies (53 and 54, respectively).
Vectors were produced by the cotransfection of 293T cells with 3 plasmids encoding the packaging proteins Gag-Pro-Pol and viral nonstructural proteins, a mini viral genome bearing a CMV-GFP expression cassette, and the VSVg envelope (in a ratio of 8:8:4, for a total of 20 μg). Virions were then purified through a 25% sucrose cushion and resuspended, and the titers of the virus were determined. The number of infectious viral particles present in the vector preparations was determined by infecting HeLa cells with different viral dilutions and by quantifying the number of green fluorescent protein (GFP)-positive cells obtained 3 days postinfection through flow cytometry. For the production of noninfectious virion-like particles (VLPs), the same procedure was followed, except that the viral genome was omitted in the transfection (55). VLPs were quantified by exogenous-reverse transcriptase (exo-RT) activity against standards of known infectivity. This assay measures the ability of RT molecules present in viral particles to incorporate radioactive dTTP in an exogenous RNA-DNA substrate composed of a poly(rA) matrix and an oligo(dT) primer.
Replication-competent R5-tropic HIV-1 and HIV-2 viruses (ADA and GL-AN, respectively) were produced similarly through the transient transfection of 293T cells and then were normalized either on the basis of infectivity on HeLa P4/P5 cells (containing the CD4 receptor and the CCR5 coreceptor, as well as a β-galactosidase reporter under the control of the HIV-1 long terminal repeat [LTR]) (56) or by exogenous RT assay against standards of known infectivity. GL-AN is a chimeric virus between the HIV-2 strains ROD and GH1 and has been described previously (57). Primary viral clones were obtained directly from the AIDS Reagents and Reference Program of the NIH and were expanded for a maximum of 2 weeks on Jurkat cells to obtain sufficient material for use in our experiments. Titration was carried out as described above.
Infections.
Single-cycle infections of primary cells were carried out at a multiplicity of infection (MOI) between 2 and 5 (or 10-fold less in established cell lines) for 2 h on 105 cells that had been pretreated for 24 h with IFN-α, unless otherwise specified. After infection, cells were washed and IFN-α was replaced. The percentage of infected cells was monitored 3 to 4 days postinfection using flow cytometry.
When specified, infections were carried out in the presence of noninfectious VLPs added at the moment of infection at MOIs between 1 and 10.
To determine the kinetics of functional vDNA accumulation, infections were carried out as described above and zidovudine (AZT)/dideoxyinosine (ddI) (obtained from the AIDS Reagents and Reference Program of the NIH) was added at 20 μg/ml at different times postinfection to arrest reverse transcription. The percentage of GFP+ cells obtained at each time point (i.e., in which reverse transcription was completed prior to the addition of the RT inhibitors) was then determined 3 days postinfection, as described previously (58).
To determine the kinetics of the loss of functionality of pre-RT viral nucleoprotein complexes (VNCs), infections were carried out in the presence of a reversible RT inhibitor to freeze VNCs at their pre-RT states in a reversible manner, as we described previously (58). At different times postinfection, the inhibitor was removed, allowing for the resumption of infection, prior to flow cytometry analysis 3 days postinfection. By plotting the percentage of GFP-positive cells obtained at each time point against the control infections that were performed in the absence of the drug, we obtained a kinetic measurement of the loss of infectivity of VNCs. This loss of functionality reflects the negative effects of the cytoplasmic environment on VNC functionality (due to degradation, localization, or other causes). Nevirapine was used for HIV-1, as described previously, while low concentrations of AZT were used instead for HIV-2 (0.56 μg/ml, as nevirapine did not reversibly inhibit HIV-2).
Replicative infections were carried out as described above at MOIs between 0.01 and 0.1. Viral spread throughout the culture was monitored by exo-RT, and to this end, a fraction of the supernatant was stored and replaced every 2 to 4 days with fresh medium containing IFN-α.
Quantitative real-time PCRs.
The accumulation of vDNA produced during infection was determined at 24 h postinfection, using primers specific for minus-strand strong-stop (MSSS), full-length (FL), 2LTR, and integrated proviral DNA. The oligonucleotide sequences specific to each virus were as follows (listed as upstream and downstream sequences, from the 5′ end to the 3′ end): MSSS (HIV-1, TGGGAGCTCTCTGCTAACT and ACCAGAGTCACACAACAGACG; HIV-2, TCTCTCCAGCACTA GCAGG and GAATGACCAGGCGGCGACTAG; SIVmac CGCTTGCTTGCTTAAAGACC and GCTTCGGTTTCTCAAAGCAG); 2LTR (HIV-1, TCGTTGGGAGTGAATTAGCC and CCCACTGCTTAAGCCTCAAT; HIV-2, GTGTTCACCTGAGTAACAAGAC and GATTTTATGTCTTCTTTCACTG; SIVmac CATCCTCCTGTGCCTCATCT and GCCTGGTCAACTCGGTACTC). FL vDNA was measured with oligonucleotides specific to the gfp gene that was carried by all viruses (GAACGGCATCAAGGTGAACT and TGCTCAGGTAGTGGTTGTCG). Integrated vDNA was measured according to an already-established procedure (59). Briefly, an initial PCR was carried out for 20 cycles using the GFP upstream oligonucleotide mentioned above, along with an Alu-specific oligonucleotide (CCTCCCAAAGTGCTGGGAT). Then, 1/50 of the first PCR was used as a template for a second quantitative PCR (qPCR) using the FL primers mentioned above. In this case, the controls were provided by PCRs in which the AluI-specific primer had been omitted in the initial PCR. In addition, the control infections were carried out in the presence of RT inhibitors (AZT/ddI at 20 μg/ml). Values were first normalized for the amount of cellular DNA (actin sequences, TTTTCACGGTTGGCCTTAGG and AAGATCTGGCACCACACCTTCT) and then subtracted for the values obtained for each sample in control infections performed in the presence of RT inhibitors. Experiments were discarded if these values represented ≥10% of those obtained in the absence of RT inhibitors. Quantitative PCRs were performed on a StepOnePlus real-time PCR system (Applied Biosystems) using the FastStart universal SYBR green master mix (Roche Diagnostics).
RESULTS
HIV-1 and HIV-2 display different susceptibilities to IFN-α during replicative infection of primary human macrophages.
To determine whether primate lentivirus replication might be differently affected by type I IFNs, we incubated primary human macrophages with different concentrations of IFN-α prior to infection, with the same MOIs (as estimated after a multinuclear activation of a galactosidase indicator [MAGI] assay on HeLa P4/P5 cells) of replication-competent HIV-1 and HIV-2 viruses. After challenge, cells were washed, and viral replication was measured via the accumulation of virion-associated exogenous-RT activity in the supernatant of infected cells over time. As reported in a number of other studies, the replication of both HIV-1 and HIV-2 was completely below the levels of detection at high IFN-α concentrations (500 U/ml) (Fig. 1). However, in the presence of lower IFN-α concentrations, infected macrophages supported HIV-1 replication in a manner that was inversely proportional to the IFN-α concentration used. On the contrary, HIV-2 replication remained below the levels of detection even in the presence of very low concentrations of IFN-α (7 U/ml). The more drastic phenotype observed in IFN-α-treated macrophages upon infection with HIV-2 was not due to increased apoptosis, at least under the conditions used here (data not shown).
Fig 1.
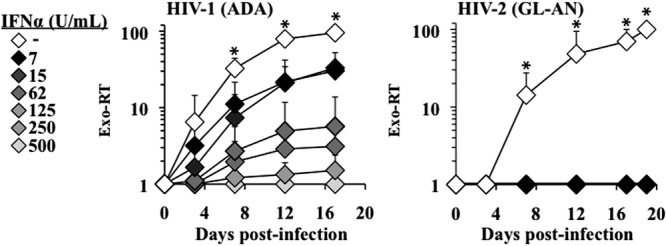
HIV-1 and HIV-2 display different susceptibilities to IFN-α. (A) Monocyte-derived macrophages were pretreated for 24 h with various concentrations of IFN-α (from 7 to 500 U/ml) and then infected with replication-competent, macrophage-tropic HIV-1 (ADA), and HIV-2 (pGL-AN) viruses in the continuous presence of IFN-α. Viral spread throughout the culture was assessed by exogenous RT activity (exo-RT) on aliquots of cell supernatants harvested at different times after infection. The graphs present data obtained from 3 independent experiments with cells of 3 different donors. Each error bar represents the standard error of the mean (SEM), and the asterisk indicates a P value of ≤0.05 according to an unpaired Student t test between treated and untreated cells.
IFN-α exerts a negative, but similar, effect during the late phases of HIV-1 and HIV-2 infection.
To pinpoint exactly the phases that are differentially inhibited by IFN-α, we first examined the later steps of the viral life cycle in primary macrophages. VSVg-pseudotyped replication-competent HIV-1 and HIV-2 viruses were produced through the transient transfection of 293T cells, normalized, and used to infect primary macrophages. Two days after infection, further viral spread was prevented by the addition of AZT/ddI to the macrophages. This setup allows for the rapid recovery of a relevant proportion of primary macrophages that are competent in the late phases of the viral life cycle in the absence of spreading infection, which may confound analysis of the results (Fig. 2A). To ensure that the Western blot (WB) signals were due to de novo protein synthesis, rather than from a carryover of the initial input virus, a control was added in which VSVg-pseudotyped virus infection was prevented by the addition of AZT/ddI to the macrophages. Macrophages were extensively washed and treated with up to 1,000 U/ml of IFN-α for 24 h prior to analysis. Cells were lysed and analyzed by WB to pinpoint transcriptional or posttranscriptional defects that may affect intracellular Gag levels, and supernatants were analyzed by exo-RT to determine the presence of more specific virion assembly defects. When cells were examined, no major defects were observed in the intracellular accumulation levels of HIV-1 and HIV-2 Gag proteins (Fig. 2B). Instead, a modest, but detectable, decrease in the amount of viral particles released in the cell supernatant was observed upon treatment with IFN-α. However, this defect was similar for both HIV-1 and HIV-2 (3-fold on average at the highest IFN-α concentration used; see Fig. 2C).
Fig 2.
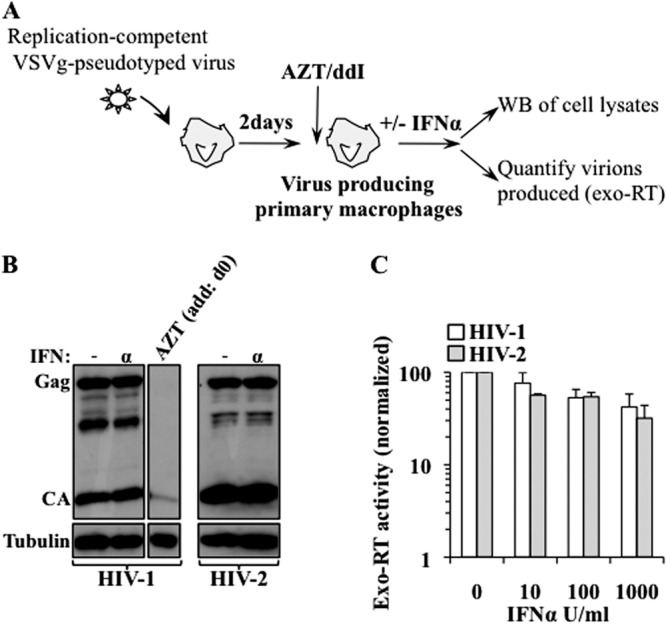
IFN-α inhibits HIV-1 and HIV-2 in a similar manner during the late phases of the viral life cycle in primary macrophages. (A) Schematic representation of the experimental setup used here. Macrophages were challenged for 2 h with replication-competent VSVg-pseudotyped HIV-1 and HIV-2 viruses (ADA and GL-AN, respectively), and 2 days after infection, further viral spread was arrested through the addition of AZT/ddI. The VSVg envelope allows for an efficient entry across the first round of infection, thus resulting in the establishment of a consistent proportion of virus-producing macrophages. These cells were then treated or not for 24 h with IFN-α prior to WB and exo-RT analysis (B and C, respectively). (B) The WB panel represents one out of 3 experiments. As a control, macrophages were infected in the continuous presence of RT inhibitors to ensure that the signals obtained by WB were mainly due to de novo protein synthesis from virus-producing macrophages, rather than from a protein that was left over from the initial infection. (C) The graph depicts the averages and SEMs obtained in 3 distinct experiments carried out with the cells of 3 different donors. No statistically significant differences were observed upon treatment with IFN-α between HIV-1- and HIV-2-producing cells, according to a Student t test.
The early phases of primate lentiviral infection are differently affected by IFN-α.
To determine the effects of IFN-α on the early phases of infection, target cells were pretreated for 24 h with IFN-α, challenged with VSVg-pseudotyped GFP-coding single-round infection-competent lentivectors, and then analyzed by flow cytometry 3 days postinfection (Fig. 3A). VSVg pseudotyping was used to bypass possible differences at the entry level and to focus only on postentry events.
Fig 3.

Primate lentiviruses exhibit distinct susceptibilities to IFN-α during the early phases of infection. (A) Schematic representation of the assay used here. (B) Established cell lines were pretreated with IFN-α for 24 h then challenged with equal amounts of single-round infection-competent VSVg-pseudotyped CMV-GFP bearing HIV-1, HIV-2, and SIVmac vectors (MOI between 0.2 and 0.5). The percentage of GFP-positive cells was determined 3 days afterward using flow cytometry. (C) Macrophages, monocyte-derived dendritic cells (DCs), and PHA-activated PBLs were treated similarly (except that vectors were used at MOIs between 2 and 5) and analyzed. The panels present averages and SEMs obtained in 3 to 10 independent experiments, each of which was carried out with cells obtained from different donors. The asterisk indicates a P value of ≤0.05, according to an unpaired Student t test, between the defects observed upon treatment with IFN-α in HIV-1 versus HIV-2 (or SIVmac).
We first analyzed the effects of IFN-α on the infectivity of a number of established cell lines (Fig. 3B). Treatment of 293T or HeLa cells with IFN-α at the highest dose used (1,000 U/ml) did not significantly modify the extent of infection for the lentiviruses tested. In contrast, a marked and differential inhibition was observed in THP-1 cells that differentiated into a macrophage-like status upon incubation with PMA. In this case, IFN-α treatment reduced HIV-1 infectivity by 8-fold and the infectivity of HIV-2 and SIVmac by 60-fold.
Next, primary macrophages, dendritic cells (DCs), and PHA/IL-2-activated peripheral blood lymphocytes (PBLs) were analyzed using various concentrations of IFN-α (Fig. 3C). Infection of primary macrophages was inhibited for all viruses upon treatment with IFN-α in a dose-dependent manner. However, HIV-2 and SIVmac were inhibited more potently than HIV-1 (3 to 5 times more on average). The differential inhibition observed between HIV-1 and HIV-2/SIVmac was also present in IFN-α-treated DCs and activated PBLs.
Of note, the effects of IFN-α were truly specific to the early phases of infection, because the addition of IFN-α 24 to 36 h after infection (i.e., after integration) did not modify the extent of infection, in line with what was reported previously (reference 16 and data not shown).
Overall, our results indicate that the different susceptibilities of primate lentiviruses to IFN-α map to the early phases of infection and are observed in multiple primary cell types, as well as in at least one established cell line of myeloid origins. HIV-2 and SIVmac exhibit very similar behaviors with respect to IFN-α, which is not surprising in light of their close phylogeny. In replicative infection, the inhibitory effect of IFN-α becomes apparent at concentrations that are lower than those used in single-round infections. We believe this is due to the fact that small defects that may be undetectable in single-round infections are amplified exponentially over multiple rounds of infection during viral replication, which yields a more appreciable phenotype.
IFN-α differentially affects vDNA accumulation during the early phases of infection.
IFN-α has been shown to inhibit reverse transcription. To determine whether the differential inhibition observed here mapped also at this step, primary macrophages were treated or not with IFN-α and then challenged with the above-mentioned vectors, prior to cell lysis and qPCR analysis of vDNA 24 h postinfection (Fig. 4). Under these conditions, and in agreement with previous results (5, 16, 32, 60, 61), the accumulation of vDNA was inhibited for all 3 viruses upon treatment with IFN-α. However, while the accumulation of MSSS vDNA was inhibited in a similar manner between HIV-1, HIV-2, and SIVmac, the accumulation of FL, 2LTR, and ultimately, proviral DNA was more severely impaired upon infection with HIV-2 and SIVmac than with HIV-1. These results confirm previous findings that treatment with IFN-α affects the accumulation of reverse transcription products, and the findings extend these results by indicating that the negative effects exerted by IFN-α become more pronounced against HIV-2 and SIVmac as reverse transcription proceeds. We believe that the apparently higher defect detected in HIV-2/SIVmac integrated proviral DNA with respect to FL and 2LTR forms may be due to the Alu-PCR amplification method, rather than to the presence of an additional defect at the integration step; however, we cannot formally exclude this hypothesis.
Fig 4.
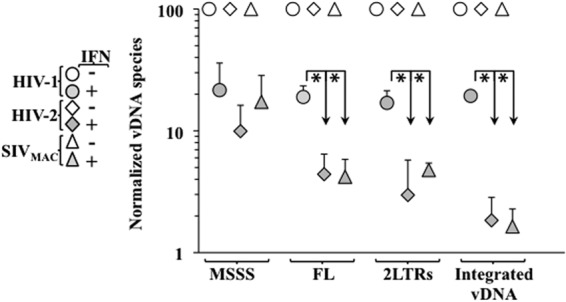
The differential defect imposed by IFN-α targets the accumulation of vDNA. Primary macrophages were treated or not with IFN-α at 1,000 U/ml for 24 h prior to infection with the virus indicated in the figure. Twenty-four hours afterward, cells were lysed and the accumulation of vDNA products was analyzed by qPCR. The values obtained in the absence of IFN-α stimulation have been set to 100 for each virus and for each vDNA product. The graph depicts averages and SEMs obtained from 3 independent experiments using cells of 3 different donors. The asterisk indicates a P value of ≤0.05 according to an unpaired Student t test between the defects observed upon treatment with IFN-α in HIV-1 versus HIV-2 (or SIVmac).
IFN-α differentially affects the functional stability of viral nucleoprotein complexes.
qPCR yields a precise measurement of the accumulation of bulk vDNA, but it does not yield information as to the functionality of neosynthesized vDNA. To gather more insights into the behaviors of the minority of functional viral genomes that are produced during viral infection, we used a method that we described previously (58). In this setup, the accumulation of functional viral genomes (defined as those capable of expressing themselves and therefore able to express the virus-encoded GFP) is followed kinetically over time by arresting reverse transcription by the addition of an RT inhibitor at different time points after infection. This inhibitor prevents infection, but only if added prior to the completion of reverse transcription. By measuring the percentage of GFP-positive cells obtained at each time point using flow cytometry 3 days postinfection, a precise kinetic measurement of the accumulation of functional viral genomes can be obtained, as we described previously (58). This experimental setup was applied to macrophages, treated or untreated with IFN-α and challenged with the indicated viruses (Fig. 5A). When examined in this manner, we noticed that the kinetics of reverse transcription of HIV-2 and SIVmac viruses were consistently faster than the kinetics measured for HIV-1. This may be expected, as these viruses encode Vpx, a protein that has been shown to speed up reverse transcription (48, 62, 63). However, no differences were observed in each virus between IFN-α-treated and -untreated samples, suggesting that the kinetics of functional vDNA accumulation are not affected by IFN-α.
Fig 5.

IFN-α affects the stability of viral nucleoprotein complexes in a CA-independent manner. (A) IFN-α-treated and -untreated macrophages were challenged with VSVg-pseudotyped HIV-1, HIV-2, and SIVmac vectors prior to the addition of the RT inhibitors AZT/ddI at the indicated times postinfection. The extent of GFP-positive cells at each time point was assessed 72 h afterward using flow cytometry (as described in reference 58) and is presented here after normalization with the percentage of GFP+ cells obtained in the absence of inhibitors. (B) Macrophages were treated or not with 250 U/ml of IFN-α, which is the maximal dose in this assay that allowed for a reliable measurement of viral infectivity in the case of HIV-2. Then, cells were challenged with VSVg-pseudotyped HIV-1 and HIV-2 vectors in the presence of a reversible RT inhibitor that was removed at different times postinfection. The extent of infection was determined for each time point 3 days postinfection, and the values are presented after normalization with the percentage of GFP+ cells obtained in the absence of viral inhibitors. (C) Macrophages were incubated in the presence or absence of 1,000 U/ml of IFN-α for 24 h. Cells were challenged afterward with various viral inputs of HIV-1 and SIVmac lentivectors prior to flow cytometry analysis 3 days postinfection. (D) Macrophages were treated as in (C) and then challenged with a constant amount of GFP-coding HIV-1 or SIVmac lentivectors (MOI, 1) in the presence of increasing amounts of noninfectious virus-like particles (VLPs), as indicated (MOI equivalents, 1 to 10). The extent of infection was determined 3 days afterward using flow cytometry. (E) GFP-coding chimeric vectors were used to identify the viral determinant of the different susceptibilities of primate lentiviruses to IFN-α. HIV-1-siv is a chimeric virus that contains an SIV-derived genome in an HIV-1 particle. The remaining two chimeras contain an exchange of CA between HIV-1 and SIVmac, as indicated here. The graphs present averages and SEMs obtained with 3 to 7 different experiments, each carried out with cells of different donors. One asterisk indicates a P value of ≤0.05 according to an unpaired Student t test between treated and untreated cells (B, C, and E), or between HIV-1 infection carried out in IFN-treated cells in the absence or presence of SIVmac VLPs (D).
Given that the overall efficiency of reverse transcription is influenced by the stability of viral nucleoprotein complexes (VNCs), we used a technique we developed previously to determine the speed at which functional viral complexes that are kept at their pre-RT state lose their functionality upon entry into target cells (see Fig. 3 and 5B). In this setup, infections are carried out in the presence of a reversible RT inhibitor that is removed at different times postinfection to allow for resumption of the infection process (i.e., functional viral genomes again will yield a GFP-positive cell). In the presence of the drug, VNCs are forcibly maintained at their pre-RT states for prolonged periods of time, during which they are submitted to various effects of the cellular environment. A negative effect on VNCs (degradation, mislocalization, etc.) is measured as a loss of infectivity (i.e., of GFP+ cells) compared to infections carried out in the absence of the drug. We have already determined that these kinetics are cell type specific and are heavily influenced by the cellular activation state. To determine whether IFN-α affected the kinetics of viral complex stability, macrophages were treated with IFN-α and then were challenged according to the experimental setup explained above. The analysis was restricted to HIV-1 and HIV-2, as we were not able to identify a reliable reversible RT inhibitor for SIVmac, and assays were performed at 250 U/ml, a concentration that allowed for a reliable measurement of infectivity in the presence of the differential phenotype. Under these conditions, no significant changes in viral complex stability were observed for HIV-1. However, a more rapid loss of functionality was observed when IFN-α-treated macrophages were challenged with HIV-2. These results indicate that IFN-α treatment results in a more rapid destabilization of functional viral complexes.
IFN-α restriction cannot be saturated and does not map to CA.
To determine whether the viral input dose can affect the differential phenotype displayed by primate lentiviruses, IFN-α-treated macrophages were challenged with different amounts of HIV-1 and SIVmac vectors, prior to flow cytometry analysis that occurred 3 days later (Fig. 5C). Similarly to what was observed before, both viruses were inhibited by IFN-α treatment, and the inhibition observed for SIVmac was consistently higher than that of HIV-1; this indicates that the phenotype observed here was largely independent of the viral dose. To further support this argument, IFN-α-treated macrophages were challenged with GFP-coding HIV-1 and SIVmac in the presence of increasing amounts of noninfectious virion-like particles derived from one of the two viruses (MOI of 1 for GFP-coding viruses; MOI equivalents ranging from 1 to 10 for the VLPs) (Fig. 5D). Under these conditions, the infectivity defect of HIV-1 was not rescued by the addition of HIV-1 VLPs; however, it was rescued by the addition of SIVmac VLPs. This rescue is not surprising and has been described previously by Luban's laboratory to be due to the activity of Vpx (64). In agreement with this result, SIVmac VLPs devoid of Vpx lost their positive effect on HIV-1 infectivity. On the contrary, neither HIV-1 nor SIVmac VLPs were able to rescue the infectivity defect of SIVmac.
Overall, these results indicate that the restriction induced by IFN-α may be bypassed in an artificial setup (as in the case of HIV-1 plus Vpx), but cannot be saturated in wild-type (WT) infection, at least under the conditions studied here.
Lastly, we attempted to define the viral elements that might be responsible for the higher susceptibility of HIV-2/SIVmac to IFN-α (Fig. 5E). To this end, we analyzed three chimeras: HIV-1 viruses packaging an SIVmac genome (HIV-1–siv), an HIV-1 chimera in which CA had been replaced with the SIVmac chimera (HIV-1-sCA [32]), and the reciprocal SIVmac chimera (SIVmac-hCA]). We reported previously that the HIV-1-siv chimera displays a specific infectivity defect in myeloid cells that is not displayed by WT HIV-1. This defect is particularly strong in DCs but is more moderate in macrophages; therefore, by increasing the viral input to an MOI of 10, we can easily use this chimera on IFN-α-treated macrophages (65). The loss of infectivity of HIV-1-siv was similar to that of HIV-1, indicating that the viral genome of SIVmac does not specify a cis element that is more susceptible to IFN-α. Similarly, swapping CA between HIV-1 and SIVmac did not modify the susceptibilities of chimeric viruses to IFN-α compared to those of the parental viruses.
Overall, these results indicate that CA is not the main viral target of the differential inhibition observed between HIV-1 and HIV-2/SIVmac viruses upon treatment with IFN-α. These results also indicate that human T5α, whose only known target is CA, is unlikely to be the key determinant in the phenotype observed here (66).
Kinetics of induction of the restrictive phenotype observed upon IFN-α treatment.
To characterize more precisely the defect induced by IFN-α, we sought to determine the delay that exists between the initial IFN-α stimulus and the appearance of an effective antiviral response. To this end, the times for the addition of IFN-α were between 24 h prior to infection and 72 h postinfection, and the extent of infection was determined using flow cytometry 3 days after the infection (Fig. 6). As described above, pretreatment of target macrophages for 24 h resulted in higher inhibitions of HIV-2 and SIVmac, and this phenotype was observed up to 6 h pretreatment. However, the differential inhibitions of HIV-1 and HIV-2/SIVmac were lost when cells were pretreated for shorter time periods prior to infection. IFN-α ceased to exert an antiviral effect when added ≥24 h postinfection, in agreement with a specific role during the early phases of infection.
Fig 6.
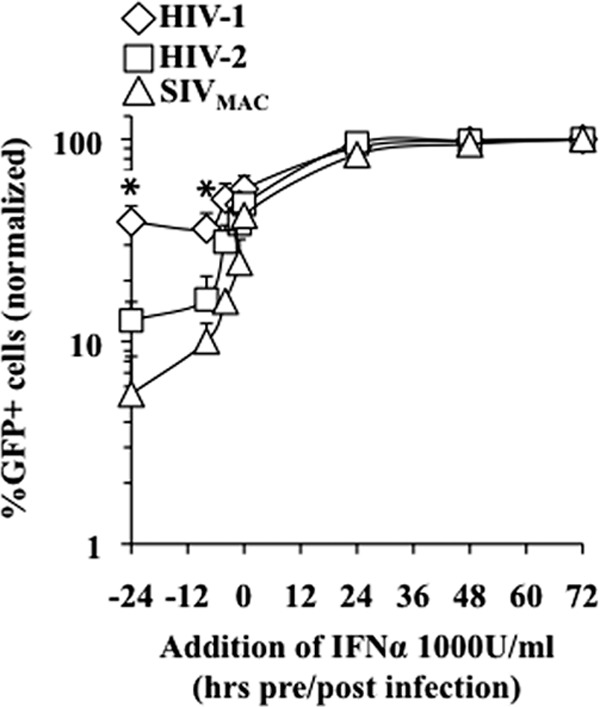
Kinetics of the induction of the differential antiviral effect of IFN-α during the early phases of infection. Macrophages were treated with 1,000 U/ml of IFN-α at the indicated times pre- and postinfection. The extent of infection was assessed by flow cytometry 3 days afterward. The graph presents data normalized to data from non-IFN-treated infections set to 100% (from 4 independent experiments and with cells of 4 different donors). The asterisk indicates a P value of ≤0.05 according to an unpaired Student t test between HIV-1-infected and HIV-2- or SIVmac-infected cells after IFN-α stimulation.
Overall, these results indicate that the restriction mechanism that more potently targets HIV-2 and SIVmac in IFN-α-treated macrophages requires a minimum of 6 h prior to infection to be functionally established. This delay may reflect the time required for the transcription of novel antiviral factors or for their posttranscriptional modification.
Evaluation of the sensitivities of different primary viral strains to IFN-α inhibition.
To determine the behaviors of different viral strains with respect to IFN-α, a limited number of experiments were carried out with a few viral strains available in the laboratory and that were able to grow on macrophages. To this end, we used 3 primary HIV-1 strains belonging to group M or O, as well as two HIV-2 strains. The two group O viruses were tested because, despite having the same genetic structure, they are phylogenetically distinct from group M viruses and their infection is concentrated essentially in west central Africa. When we assessed the ability of these viruses to replicate in macrophages treated with different amounts of IFN-α (Fig. 7), we found that even though all were inhibited at high concentrations of IFN-α, they displayed various degrees of resistance to low concentrations of IFN-α. In particular, the single group M HIV-1 strain analyzed here was the only one that replicated robustly under conditions of IFN stimulation.
Fig 7.

Susceptibility of primary viral strain replication to IFN-α. Several viral strains retrieved from the AIDS Reagents Program of the NIH were used to infect IFN-α-treated macrophages as described in the legend to Fig. 1. Viral spread throughout the culture was monitored by exo-RT. The graphs depict averages obtained from 2 independent experiments for each virus.
Overall, these results suggest, albeit within the limited analysis conducted here, that group M HIV-1 strains may replicate more robustly in the presence of IFN-α than do group O HIV-1 and HIV-2 strains.
DISCUSSION
In the present study, we determined that even though the primate lentiviruses we tested are all susceptible to a type I interferon response, HIV-1 displays a higher resistance to its negative effects than do HIV-2 and SIVmac. The increased susceptibilities displayed by HIV-2 and SIVmac toward IFN-α are exerted during the early phases of infection and, more specifically, during reverse transcription. Our data suggest that this inhibition may involve faster viral nucleoprotein complex destabilization in a manner that appears to be independent from the capsid protein. In this respect, the results presented here are in agreement with those of previous studies that reported a negative effect of IFN-α on the early phases of HIV infection, and our results extend those findings by providing an explanation as to how HIV-1 may continue to replicate in vivo in spite of the continuous presence of a persistent antiviral IFN state.
IFN-α has pleiotropic effects on the viral life cycle and, not surprisingly, we have found that IFN-α inhibits the late phases of infection and, in particular, virion release. This inhibition is moderate (2- to 3-fold, although one can speculate that it might increase at later time points) and not differential, so it hardly explains the replication differences observed between HIV-1 and HIV-2. Although we have not investigated this further, we believe it to be likely that tetherin mediates this inhibition by partially overcoming HIV-1 Vpu or HIV-2 Env upon IFN-α stimulation (26, 33; for a recent review see reference 67).
On the contrary, IFN-α affects differentially the early phases of infection, in that both the accumulation of the late products of reverse transcription and viral capsid stability are more severely diminished following HIV-2/SIVmac infection. These two defects lead us to propose a model in which the more rapid loss of functionality of HIV-2 viral capsids may be responsible for the more drastic defects observed in reverse transcription; this is in line with multiple evidence that links correct viral complex stability to the process of reverse transcription (68, 69).
These distinct behaviors cannot be accounted for by differences in the viral genomes, because genome swapping did not affect the outcome of infection in the presence of IFN-α. Similarly, nonstructural viral proteins are largely dispensable for completion of the early phases of infection under interferon stimulation (data not shown). Vpx may represent an exception. However, Vpx exerts a protective effect on HIV-1 infection (43), but it fails to protect its cognate virus, indicating that even this protein does not play a major role in determining resistance to IFN-α in its natural setting.
Therefore, our results indicate that the main viral determinant for the differential inhibition observed here maps to Gag-Pro-Pol. At present, we are ignoring its identity, but the use of CA-chimeric viruses seemingly exclude CA, and by extension, hT5α; this indicates the presence of additional restrictions targeting viral complexes in IFN-α-treated human cells.
One of the major differences that exists between HIV-1 and HIV-2 infections in vivo is that while the former is characterized by high viral loads in the majority of untreated patients, a high proportion of HIV-2-infected patients present with relatively low viral loads for decades (44–47). In light of the results presented here, it would be tempting to speculate that the higher susceptibility of HIV-2 to IFN-α is at least in part responsible for the presence of lower viral loads, while the partial resistance displayed by HIV-1 allows the virus to replicate despite the presence of chronic type I interferon responses. In turn, the ongoing viral replication observed in this case may entertain a persistent stimulation of interferon responses, as is observed in the case of HIV-1 patients. Multiple studies have shown that deregulated interferon production is deleterious to the functionality of the immune system and is detrimental to cell physiology (70, 71). Therefore, the ability of HIV-1 to replicate in the presence of interferon may be a key factor in the maintenance of a chronic IFN state that becomes detrimental to the functionality and homeostasis of the immune system (70, 71).
Since even in the case of HIV-1, resistance to IFN-α is partial and not total, interferon is likely to exert a negative selective pressure on the virus. We can speculate that the susceptibilities of primary viral strains to IFN-α may be highly dynamic and are possibly distinct between lineages (as shown here for HIV-1 and HIV-2), between groups (as group M or O), or even over time within a given patient (for example, between acute and chronic infection, as suggested by at least two previous studies [72, 73]). The analysis that we have carried out here is reassuring in that the two HIV-2 viruses, as well as the single HIV-1 group M isolate, reproduce the phenotype that is observed with laboratory-adapted viruses. In addition, the higher susceptibility to IFN-α displayed by group O HIV-1 strains is intriguing, as the presence of group O viruses is limited to west central Africa, indicating its suboptimal adaptation to spread among humans worldwide (74). A clear inability of group O viruses to counteract the block imposed by IFN during the late phases of infection via tetherin has been established recently (75), and it is possible that further restrictions exist also across the early phases of infection, although this remains to be established. However, it is clear that these results have been obtained with a small number of viral isolates, which warrants further studies with primary viral strains obtained from larger cohorts of HIV-1- and HIV-2-infected patients.
ACKNOWLEDGMENTS
We thank Jeanine Bernaud and Dominique Rigal at the EFS of Lyon for blood sample collection. We are indebted to Theodora Hatziioannou for the sCA-HIV chimera, to the AIDS Reagent Program of the NIH for providing the indicated material, and to the technical facilities and staff of the UMS3444/US8 in Lyon.
A.C. is a CNRS researcher, S.C. is a PhD student financed by the ANRS, S.D. is supported by a postdoctoral fellowship of Sidaction, and X.N.N. is supported by the FRM. This work received the support of the ANRS, the ENS-L, the FRM, and Sidaction.
Footnotes
Published ahead of print 19 December 2012
REFERENCES
- 1. Isaacs A, Lindenmann J. 1957. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 147:258–267 [PubMed] [Google Scholar]
- 2. Samarajiwa SA, Forster S, Auchettl K, Hertzog PJ. 2009. INTERFEROME: the database of interferon regulated genes. Nucleic Acids Res. 37:D852–D857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Agy MB, Acker RL, Sherbert CH, Katze MG. 1995. Interferon treatment inhibits virus replication in HIV-1- and SIV-infected CD4+ T-cell lines by distinct mechanisms: evidence for decreased stability and aberrant processing of HIV-1 proteins. Virology 214:379–386 [DOI] [PubMed] [Google Scholar]
- 4. Aquaro S, Perno CF, Balestra E, Balzarini J, Cenci A, Francesconi M, Panti S, Serra F, Villani N, Cali ò R. 1997. Inhibition of replication of HIV in primary monocyte/macrophages by different antiviral drugs and comparative efficacy in lymphocytes. J. Leukoc. Biol. 62:138–143 [DOI] [PubMed] [Google Scholar]
- 5. Baca-Regen L, Heinzinger N, Stevenson M, Gendelman HE. 1994. Alpha interferon-induced antiretroviral activities: restriction of viral nucleic acid synthesis and progeny virion production in human immunodeficiency virus type 1-infected monocytes. J. Virol. 68:7559–7565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barr SD, Smiley JR, Bushman FD. 2008. The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 4:e1000007 doi:10.1371/journal.ppat.1000007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bednarik DP, Mosca JD, Raj NB, Pitha PM. 1989. Inhibition of human immunodeficiency virus (HIV) replication by HIV-trans-activated alpha 2-interferon. Proc. Natl. Acad. Sci. U. S. A. 86:4958–4962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brinchmann JE, Gaudernack G, Vartdal F. 1991. In vitro replication of HIV-1 in naturally infected CD4+ T cells is inhibited by rIFN alpha 2 and by a soluble factor secreted by activated CD8+ T cells, but not by rIFN beta, rIFN gamma, or recombinant tumor necrosis factor-alpha. J. Acquir. Immune Defic. Syndr. 4:480–488 [PubMed] [Google Scholar]
- 9. Coccia EM, Krust B, Hovanessian AG. 1994. Specific inhibition of viral protein synthesis in HIV-infected cells in response to interferon treatment. J. Biol. Chem. 269:23087–23094 [PubMed] [Google Scholar]
- 10. Dolei A, Fattorossi A, D'Amelio R, Aiuti F, Dianzani F. 1986. Direct and cell-mediated effects of interferon-alpha and -gamma on cells chronically infected with HTLV-III. J. Interferon Res. 6:543–549 [DOI] [PubMed] [Google Scholar]
- 11. Fernie BF, Poli G, Fauci AS. 1991. Alpha interferon suppresses virion but not soluble human immunodeficiency virus antigen production in chronically infected T-lymphocytic cells. J. Virol. 65:3968–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gendelman HE, Baca L, Turpin JA, Kalter DC, Hansen BD, Orenstein JM, Friedman RM, Meltzer MS. 1990. Restriction of HIV replication in infected T cells and monocytes by interferon-alpha. AIDS Res. Hum. Retroviruses 6:1045–1049 [DOI] [PubMed] [Google Scholar]
- 13. Gendelman HE, Baca LM, Turpin J, Kalter DC, Hansen B, Orenstein JM, Dieffenbach CW, Friedman RM, Meltzer MS. 1990. Regulation of HIV replication in infected monocytes by IFN-alpha. Mechanisms for viral restriction. J. Immunol. 145:2669–2676 [PubMed] [Google Scholar]
- 14. Gendelman HE, Friedman RM, Joe S, Baca LM, Turpin JA, Dveksler G, Meltzer MS, Dieffenbach C. 1990. A selective defect of interferon alpha production in human immunodeficiency virus-infected monocytes. J. Exp. Med. 172:1433–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gessani S, Puddu P, Varano B, Borghi P, Conti L, Fantuzzi L, Gherardi G, Belardelli F. 1994. Role of endogenous interferon-beta in the restriction of HIV replication in human monocyte/macrophages. J. Leukoc. Biol. 56:358–361 [DOI] [PubMed] [Google Scholar]
- 16. Goujon C, Malim MH. 2010. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J. Virol. 84:9254–9266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hammer SM, Gillis JM, Groopman JE, Rose RM. 1986. In vitro modification of human immunodeficiency virus infection by granulocyte-macrophage colony-stimulating factor and gamma interferon. Proc. Natl. Acad. Sci. U. S. A. 83:8734–8738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hartshorn KL, Neumeyer D, Vogt MW, Schooley RT, Hirsch MS. 1987. Activity of interferons alpha, beta, and gamma against human immunodeficiency virus replication in vitro. AIDS Res. Hum. Retroviruses 3:125–133 [DOI] [PubMed] [Google Scholar]
- 19. Hartshorn KL, Vogt MW, Chou TC, Blumberg RS, Byington R, Schooley RT, Hirsch MS. 1987. Synergistic inhibition of human immunodeficiency virus in vitro by azidothymidine and recombinant alpha A interferon. Antimicrob. Agents Chemother. 31:168–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Honda Y, Rogers L, Nakata K, Zhao BY, Pine R, Nakai Y, Kurosu K, Rom WN, Weiden M. 1998. Type I interferon induces inhibitory 16-kD CCAAT/enhancer binding protein (C/EBP)beta, repressing the HIV-1 long terminal repeat in macrophages: pulmonary tuberculosis alters C/EBP expression, enhancing HIV-1 replication. J. Exp. Med. 188:1255–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kornbluth RS, Oh PS, Munis JR, Cleveland PH, Richman DD. 1989. Interferons and bacterial lipopolysaccharide protect macrophages from productive infection by human immunodeficiency virus in vitro. J. Exp. Med. 169:1137–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mac é K, Duc Dodon M, Gazzolo L. 1989. Restriction of HIV-1 replication in promonocytic cells: a role for IFN-alpha. Virology 168:399–405 [DOI] [PubMed] [Google Scholar]
- 23. Martinand C, Montavon C, Salehzada T, Silhol M, Lebleu B, Bisbal C. 1999. RNase L inhibitor is induced during human immunodeficiency virus type 1 infection and down regulates the 2-5A/RNase L pathway in human T cells. J. Virol. 73:290–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Michaelis B, Levy JA. 1989. HIV replication can be blocked by recombinant human interferon beta. AIDS 3:27–31 [PubMed] [Google Scholar]
- 25. Mosborg-Petersen P, Toth FD, Zachar V, Villadsen JA, Nørskov-Lauritsen N, Aboagye-Mathiesen G, Chermann JC, Ebbesen P. 1991. Differential HIV replication and HIV-induced interferon production in mononuclear phagocytes: relationship to cell maturation. Res. Virol. 142:353–361 [DOI] [PubMed] [Google Scholar]
- 26. Neil SJD, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430 [DOI] [PubMed] [Google Scholar]
- 27. Poli G, Orenstein JM, Kinter A, Folks TM, Fauci AS. 1989. Interferon-alpha but not AZT suppresses HIV expression in chronically infected cell lines. Science 244:575–577 [DOI] [PubMed] [Google Scholar]
- 28. Rusconi S, Merrill DP, Hirsch MS. 1994. Inhibition of human immunodeficiency virus type 1 replication in cytokine-stimulated monocytes/macrophages by combination therapy. J. Infect. Dis. 170:1361–1366 [DOI] [PubMed] [Google Scholar]
- 29. Shirazi Y, Pitha PM. 1992. Alpha interferon inhibits early stages of the human immunodeficiency virus type 1 replication cycle. J. Virol. 66:1321–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shirazi Y, Pitha PM. 1998. Interferon downregulates CXCR4 (fusin) gene expression in peripheral blood mononuclear cells. J. Hum. Virol. 1:69–76 [PubMed] [Google Scholar]
- 31. Smith MS, Thresher RJ, Pagano JS. 1991. Inhibition of human immunodeficiency virus type 1 morphogenesis in T cells by alpha interferon. Antimicrob. Agents Chemother. 35:62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Taylor MD, Korth MJ, Katze MG. 1998. Interferon treatment inhibits the replication of simian immunodeficiency virus at an early stage: evidence for a block between attachment and reverse transcription. Virology 241:156–162 [DOI] [PubMed] [Google Scholar]
- 33. Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Woods MW, Kelly JN, Hattlmann CJ, Tong JGK, Xu LS, Coleman MD, Quest GR, Smiley JR, Barr SD. 2011. Human HERC5 restricts an early stage of HIV-1 assembly by a mechanism correlating with the ISGylation of Gag. Retrovirology 8:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yamada O, Hattori N, Kurimura T, Kita M, Kishida T. 1988. Inhibition of growth of HIV by human natural interferon in vitro. AIDS Res. Hum. Retroviruses 4:287–294 [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto JK, Kruzel ML, Louie H, Georgiades JA. 1993. Inhibition of human immunodeficiency virus type 1 replication by human interferons alpha, beta and gamma. Arch. Immunol. Ther. Exp. (Warsz.). 41:185–191 [PubMed] [Google Scholar]
- 37. Yasuda Y, Miyake S, Kato S, Kita M, Kishida T, Kimura T, Ikuta K. 1990. Interferon-alpha treatment leads to accumulation of virus particles on the surface of cells persistently infected with the human immunodeficiency virus type 1. J. Acquir. Immune Defic. Syndr. 3:1046–1051 [PubMed] [Google Scholar]
- 38. Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, Francella N, Sidahmed A, Smith AJ, Cramer EM, Zeng M, Masopust D, Carlis JV, Ran L, Vanderford TH, Paiardini M, Isett RB, Baldwin DA, Else JG, Staprans SI, Silvestri G, Haase AT, Kelvin DJ. 2009. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J. Clin. Invest. 119:3556–3572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, Dillies MA, Roques P, Butor C, Silvestri G, Giavedoni LD, Lebon P, Barré-Sinoussi F, Benecke A, Müller-Trutwin MC. 2009. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Invest. 119:3544–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lederer S, Favre D, Walters KA, Proll S, Kanwar B, Kasakow Z, Baskin CR, Palermo R, McCune JM, Katze MG. 2009. Transcriptional profiling in pathogenic and non-pathogenic SIV infections reveals significant distinctions in kinetics and tissue compartmentalization. PLoS Pathog. 5:e1000296 doi:10.1371/journal.ppat.1000296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Herbeuval JP, Shearer GM. 2007. HIV-1 immunopathogenesis: how good interferon turns bad. Clin. Immunol. 123:121–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kamat A, Misra V, Cassol E, Ancuta P, Yan Z, Li C, Morgello S, Gabuzda D. 2012. A plasma biomarker signature of immune activation in HIV patients on antiretroviral therapy. PLoS One 7:e30881 doi:10.1371/journal.pone.0030881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rotger M, Dalmau J, Rauch A, McLaren P, Bosinger SE, Martinez R, Sandler NG, Roque A, Liebner J, Battegay M, Bernasconi E, Descombes P, Erkizia I, Fellay J, Hirschel B, Mir ó JM, Palou E, Hoffmann M, Massanella M, Blanco J, Woods M, Günthard HF, de Bakker P, Douek DC, Silvestri G, Martinez-Picado J, Telenti A. 2011. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J. Clin. Invest. 121:2391–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Azevedo-Pereira JM, Santos-Costa Q, Mansinho K, Moniz-Pereira J. 2003. Identification and characterization of HIV-2 strains obtained from asymptomatic patients that do not use CCR5 or CXCR4 coreceptors. Virology 313:136–146 [DOI] [PubMed] [Google Scholar]
- 45. Leligdowicz A, Feldmann J, Jaye A, Cotten M, Dong T, McMichael A, Whittle H, Rowland-Jones S. 2010. Direct relationship between virus load and systemic immune activation in HIV-2 infection. J. Infect. Dis. 201:114–122 [DOI] [PubMed] [Google Scholar]
- 46. Soares RS, Tendeiro R, Foxall RB, Baptista AP, Cavaleiro R, Gomes P, Camacho R, Valadas E, Doroana M, Lucas M, Antunes F, Victorino RMM, Sousa AE. 2011. Cell-associated viral burden provides evidence of ongoing viral replication in aviremic HIV-2-infected patients. J. Virol. 85:2429–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sousa AE, Carneiro J, Meier-Schellersheim M, Grossman Z, Victorino RM. 2002. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J. Immunol. 169:3400–3406 [DOI] [PubMed] [Google Scholar]
- 48. Goujon C, Rivière L, Jarrosson-Wuilleme L, Bernaud J, Rigal D, Darlix JL, Cimarelli A. 2007. SIVsm/HIV-2 Vpx proteins promote retroviral escape from a proteasome-dependent restriction pathway present in human dendritic cells. Retrovirology 4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. D'Costa J, Brown H, Kundra P, Davis-Warren A, Arya S. 2001. Human immunodeficiency virus type 2 lentiviral vectors: packaging signal and splice donor in expression and encapsidation. J. Gen. Virol. 82:425–434 [DOI] [PubMed] [Google Scholar]
- 50. Mangeot PE, Nègre D, Dubois B, Winter AJ, Leissner P, Mehtali M, Kaiserlian D, Cosset FL, Darlix JL. 2000. Development of minimal lentivirus vectors derived from simian immunodeficiency virus (SIVmac251) and their use for gene transfer into human dendritic cells. J. Virol. 74:8307–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263–267 [DOI] [PubMed] [Google Scholar]
- 52. Strappe PM, Greatorex J, Thomas J, Biswas P, McCann E, Lever AM. 2003. The packaging signal of simian immunodeficiency virus is upstream of the major splice donor at a distance from the RNA cap site similar to that of human immunodeficiency virus types 1 and 2. J. Gen. Virol. 84:2423–2430 [DOI] [PubMed] [Google Scholar]
- 53. Hatziioannou T, Princiotta M, Piatak M, Jr, Yuan F, Zhang F, Lifson JD, Bieniasz PD. 2006. Generation of simian-tropic HIV-1 by restriction factor evasion. Science 314:95. [DOI] [PubMed] [Google Scholar]
- 54. Kootstra NA, Munk C, Tonnu N, Landau NR, Verma IM. 2003. Abrogation of postentry restriction of HIV-1-based lentiviral vector transduction in simian cells. Proc. Natl. Acad. Sci. U. S. A. 100:1298–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Goujon C, Jarrosson-Wuillème L, Bernaud J, Rigal D, Darlix JL, Cimarelli A. 2006. With a little help from a friend: increasing HIV transduction of monocyte-derived dendritic cells with virion-like particles of SIV(MAC). Gene Ther. 13:991–994 [DOI] [PubMed] [Google Scholar]
- 56. Charneau P, Mirambeau G, Roux P, Paulous S, Buc H, Clavel F. 1994. HIV-1 reverse transcription. A termination step at the center of the genome. J. Mol. Biol. 241:651–662 [DOI] [PubMed] [Google Scholar]
- 57. Kawamura M, Shimano R, Ogasawara T, Inubushi R, Amano K, Akari H, Adachi A. 1998. Mapping the genetic determinants of human immunodeficiency virus type 2 for cell tropism and replication efficiency. Arch. Virol. 143:513–521 [DOI] [PubMed] [Google Scholar]
- 58. Arfi V, Lienard J, Nguyen XN, Berger G, Rigal D, Darlix JL, Cimarelli A. 2009. Characterization of the behavior of functional viral genomes during the early steps of human immunodeficiency virus type 1 infection. J. Virol. 83:7524–7535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Berger G, Goujon C, Darlix JL, Cimarelli A. 2009. SIVMAC Vpx improves the transduction of dendritic cells with nonintegrative HIV-1-derived vectors. Gene Ther. 16:159–163 [DOI] [PubMed] [Google Scholar]
- 60. Cheney KM, McKnight A. 2010. Interferon-alpha mediates restriction of human immunodeficiency virus type-1 replication in primary human macrophages at an early stage of replication. PLoS One 5:e13521 doi:10.1371/journal.pone.0013521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shirazi Y, Pitha PM. 1993. Interferon alpha-mediated inhibition of human immunodeficiency virus type 1 provirus synthesis in T-cells. Virology 193:303–312 [DOI] [PubMed] [Google Scholar]
- 62. Bergamaschi A, Ayinde D, David A, Le Rouzic E, Morel M, Collin G, Descamps D, Damond F, Brun-Vezinet F, Nisole S, Margottin-Goguet F, Pancino G, Transy C. 2009. The human immunodeficiency virus type 2 Vpx protein usurps the CUL4A-DDB1 DCAF1 ubiquitin ligase to overcome a postentry block in macrophage infection. J. Virol. 83:4854–4860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Srivastava S, Swanson SK, Manel N, Florens L, Washburn MP, Skowronski J. 2008. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 4:e1000059 doi:10.1371/journal.ppat.1000059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pertel T, Reinhard C, Luban J. 2011. Vpx rescues HIV-1 transduction of dendritic cells from the antiviral state established by type 1 interferon. Retrovirology 8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Goujon C, Jarrosson-Wuilleme L, Bernaud J, Rigal D, Darlix JL, Cimarelli A. 2003. Heterologous human immunodeficiency virus type 1 lentiviral vectors packaging a simian immunodeficiency virus-derived genome display a specific postentry transduction defect in dendritic cells. J. Virol. 77:9295–9304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848–853 [DOI] [PubMed] [Google Scholar]
- 67. Malim MH, Bieniasz PD. 2012. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2:a006940 doi:10.1101/cshperspect.a006940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76:5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hulme AE, Perez O, Hope TJ. 2011. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc. Natl. Acad. Sci. U. S. A. 108:9975–9980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Guo B, Chang EY, Cheng G. 2008. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J. Clin. Invest. 118:1680–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. 2011. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J. Exp. Med. 208:2367–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Edlin BR, St Clair MH, Pitha PM, Whaling SM, King DM, Bitran JD, Weinstein RA. 1992. In-vitro resistance to zidovudine and alpha-interferon in HIV-1 isolates from patients: correlations with treatment duration and response. Ann. Intern. Med. 117:457–460 [DOI] [PubMed] [Google Scholar]
- 73. Kunzi MS, Farzadegan H, Margolick JB, Vlahov D, Pitha PM. 1995. Identification of human immunodeficiency virus primary isolates resistant to interferon-alpha and correlation of prevalence to disease progression. J. Infect. Dis. 171:822–828 [DOI] [PubMed] [Google Scholar]
- 74. Heeney JL, Dalgleish AG, Weiss RA. 2006. Origins of HIV and the evolution of resistance to AIDS. Science 313:462–466 [DOI] [PubMed] [Google Scholar]
- 75. Vigan R, Neil SJ. 2011. Separable determinants of subcellular localization and interaction account for the inability of group O HIV-1 Vpu to counteract tetherin. J. Virol. 85:9737–9748 [DOI] [PMC free article] [PubMed] [Google Scholar]