

Abstract
Coxsackievirus B3 (CVB3) is known to infect stem cells in the neonatal central nervous system. Here, we evaluated the effects of CVB3 infection on the major source and repository of stem cells, the bone marrow (BM). Viral genome was detectable in BM within 24 h of infection, and productive infection of BM cells was evident, peaking at 48 h postinfection (p.i.), when ∼1 to 2% of BM cells produced infectious virus particles. Beginning at 2 to 3 days p.i., a dramatic and persistent loss of immature erythroid cells, B and T lymphocytes, and neutrophils was observed in BM and, by day 3 to 4 p.i., the femoral BM stroma was largely destroyed. Analysis of peripheral blood revealed a modest neutrophilia, a loss of reticulocytes, and a massive lymphopenia. The abundance of multipotent progenitor cells (Lin−/c-kit+/Flt3+) in BM declined ∼10-fold during CVB3 infection and, consistent with a deficiency of primitive hematopoietic progenitors, serum levels of the hematopoietic growth factor Flt3 ligand were dramatically elevated. Therefore, we analyzed the regenerative capacity of BM from CVB3-infected mice. Granulocyte/macrophage progenitors displayed a relatively normal proliferative ability, consistent with the fact that the peripheral blood level of neutrophils—which are very short-lived cells—remained high throughout infection. However, erythroid and lymphoid hematopoietic progenitors in BM from CVB3-infected mice showed a markedly reduced colony-forming capacity, consonant with the observed loss of both lymphocytes and immature erythroid cells/reticulocytes from the BM and peripheral blood. In summary, CVB3 infects the BM and exerts differential effects on the various hematopoietic progenitor populations.
INTRODUCTION
Type B coxsackieviruses (CVB) are nonenveloped positive-strand RNA viruses of the Picornaviridae within the Enterovirus genus. The six viral serotypes (CVB1 to CVB6) cause acute infections in humans and also in mice, the most commonly used animal model, and long-term persistence of viral RNA and/or protein has frequently been reported. Frequently, an acute infection is subclinical or causes only mild disease, but in some cases the outcome may be severe or even lethal. CVB3 is among the most common causes of infectious myocarditis, a disease that can lead to dilated cardiomyopathy, a progressive ventricular dilatation (1–4); this outcome may be related to the persistence of viral RNA in the myocardium (5–7). CVB3 and CVB4 also infect acinar cells of the pancreas and can trigger profound pancreatitis (8, 9). CVB has long been known to infect the central nervous system (CNS), causing aseptic meningitis, and in immunocompromised patients, a persistent CNS infection may be established (10–12). CVB3 infects stem cells in the neonatal mouse CNS (13). These cells, which are located in the subventricular zone (SVZ), normally migrate into the olfactory bulb and cerebral cortex, proliferating as they travel, and differentiate into mature neurons at their final destination (14); CVB3 infection prevents proliferation, but some degree of migration and differentiation is maintained, perhaps contributing to viral spread within the CNS (15). In adults, the vast majority of stem cells originates in, and resides in, the bone marrow (BM). Therefore, we considered it important to extend our analyses to this tissue, by investigating the effects of CVB infection on the BM of adult animals.
The BM, a highly vascular and cellular tissue, is the primary site of hematopoiesis, a process that relies on hematopoietic stem cells (HSCs) which ultimately give rise to erythrocytes, leukocytes, and megakaryocytes. A second population of stem cells, mesenchymal stem cells, is located in the BM stroma and can differentiate to form various types of connective tissue. In addition to stem cells, the BM contains a variety of mature and immature immune cells, including T and B lymphocytes, dendritic cells, and natural killer T cells, and there is a growing appreciation that the BM lies at the intersection of hematopoiesis and immune function (16, 17); thus, the BM is both a primary hematopoietic and a primary lymphoid tissue. The importance of the BM to the host immune response may explain why several viruses have evolved to disrupt BM function; the most prominent human pathogens to do so are Epstein-Barr virus (18, 19), human cytomegalovirus (20, 21), human immunodeficiency virus (HIV) (22), parvovirus B19 (23), and the flaviviruses hepatitis C virus and dengue virus (24, 25). Here, we have investigated CVB3 infection of the BM and its consequences. We report that CVB3 productively infects ∼1 to 2% of BM cells and causes dramatic changes in BM morphology. The infection profoundly alters the relative proportions of different cell populations in the BM, depletes hematopoietic progenitor cells, and markedly reduces the restorative capacity of specific subsets of progenitor cells in the BM.
MATERIALS AND METHODS
Ethics statement.
All animal experiments were approved by the The Scripps Research Institute (TSRI) Institutional Animal Care and Use Committee and were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Mice, virus, and infections.
Adult (7- to 9-week-old) male C57BL/6 mice and mice lacking the type I interferon receptor (T1IFNRKO mice) (26) were obtained from TSRI breeding facility. C57BL/6 mice were used for three reasons. First, compared to most other mouse strains, they are relatively resistant to CVB-induced diseases and, therefore, provide a robust challenge model. Second, knockout mouse lines are most commonly available on the C57BL/6 (or other H2b) background. Third, our laboratory has a large repository of immunological regents that are on this background and that have been extensively used in prior published analyses. Naïve animals were inoculated intraperitoneally with 104 PFU of wild-type (wt) CVB3, which is a plaque-purified isolate of the myocarditic Woodruff variant of CVB3 (designated H3), known to be virulent in this murine model (27). Infectious virus stocks were generated from a cDNA clone (plasmid pH 3) (28), provided by Kirk Knowlton (University of California, San Diego).
Preparation of primary bone marrow cells.
Long bones (tibiae and femora) of infected and naïve C57BL/6 mice were isolated, and connective tissues were removed. After clipping off the ends of the bones, the BM was flushed out using a Dulbecco's modified Eagle's medium (DMEM)-filled syringe with a 26-gauge needle. Subsequently, cell clumps were carefully dispersed by resuspension, and the resulting cell suspension was passed through a 70-μm-pore-size nylon cell strainer (BD Bioscience). Unless otherwise stated, red blood cell (RBC) lysis was performed by incubation with 0.83% ammonium chloride (NH4Cl) for 5 min at room temperature. BM cells were recovered by centrifugation (1,400 rpm for 8 min).
Isolation of PBMC.
Peripheral blood was obtained by heart puncture of anesthetized mice, which were subsequently euthanized. The blood was first diluted with RPMI (1% fetal bovine serum [FBS]) and subsequently underlaid with a Ficoll-Paque Plus solution (GE Healthcare; ratio of 1:1). Centrifugation (2,000 rpm for 25 min at room temperature; without brake) separated primary peripheral blood mononuclear cells (PBMC) from dead cells and erythrocytes as well as the serum. The PBMC were harvested using a pipette, and any remaining red blood cells were lysed with 0.83% NH4Cl (5 min at room temperature), after which PBMC were washed twice with RPMI (1% FBS).
Infectious center assays (ICA).
To determine the numbers of BM cells or PBMC that were productively infected in vivo, isolated cells (after RBC lysis) were incubated with 0.05% trypsin-EDTA (Gibco) for 5 min at 37°C, to remove any infectious virus particles that were bound to the surface of the cells. After cells were washed with FBS-containing DMEM, cell numbers were assessed and serial 10-fold dilutions were prepared. A total of 100 μl of each dilution was added to HeLa cell monolayers, allowed to settle, and overlaid with 0.6% agar containing DMEM (10% FBS). After 48 h, cell cultures were stained with crystal violet and plaques were counted. Each plaque indicates a single productively infected BM cell.
Complete blood counts and reticulocyte counts (CBC/RC).
Whole peripheral blood was isolated from the hearts of anesthetized mice, which were subsequently euthanized. To maintain the single-cell suspension status, blood was transferred to an EDTA-coated collection tube (BD Vacutainer K2 EDTA, 3 ml). Complete blood counts and reticulocyte counts were carried out by a certified external laboratory.
Flow cytometry.
Equal numbers of isolated BM cells (either with or without RBC, as indicated in the text) were incubated (20 min at 4°C) in FACS buffer (phosphate-buffered saline, 2% FBS, 0.1% sodium azide) containing an anti-mouse CD16/CD32 antibody (1:100; BD Bioscience). Subsequently, fluorochrome-conjugated antibodies against various surface markers were added and incubated for 30 min on ice protected from light. The following antibodies were purchased from BioLegend: TER-119 (clone TER-119), CD71 (clone RI7217), CD19 (clone 6D5), CD8 (clone 53-6.7), CD4 (clone RM4-5), Ly6G (clone 1A8), CD11b (clone M1/70), c-kit/CD117 (clone 2B8), and CD3 (clone 145-2C11). Flt3/CD135 (clone A2F10) and B220 (clone RA3-6B2) antibodies were purchased from eBioscience. Appropriate fluorochrome-conjugated isotype controls were used for each antibody. After several wash steps with FACS buffer, cells were resuspended in 200 μl of viability dye eFluor 780 (eBioscience) diluted 1:1,000 in 1× phosphate-buffered saline (PBS) and incubated for 30 min on ice protected from light. After serial wash steps with 1× PBS following FACS buffer and fixation in FACS fix (PBS, 2% neutral buffered formalin), cells were acquired on an LSRII (BD Biosciences) and data were analyzed with FlowJo software (Tree Star). Cell doublets were excluded, and live cells were gated.
Plaque assay.
Whole blood from CVB3-infected mice was isolated by heart puncture, transferred to yellow-capped MiniCollect serum tubes (Greiner Bio One), and centrifuged for 30 min at 3,000 rpm (4°C). Collected serum was subsequently used for virus titration. Plaque assays were performed on subconfluent HeLa cell monolayers as described in reference 29, and virus titers were calculated for each sample.
ELISA.
Erythropoietin (Epo), Flt3 ligand, CXCL12/stromal cell-derived factor 1α (SDF-1α), CXCL2/macrophage inflammatory protein 2 (MIP-2), granulocyte colony-stimulating factor (G-CSF), and alpha interferon (IFN-α) levels of serum were measured using enzyme-linked immunosorbent assay (ELISA) kits purchased from R&D Systems (Quantikine ELISA immunoassays: mouse erythropoietin MEP00B, mouse Flt3 ligand MFK00, mouse CXCL12/SDF-1α MCX120, mouse CXCL2/MIP-2 MM200, or mouse G-CSF MCS00) or from eBioscience (Mouse IFN-α Platinum ELISA, BMS 6027) and used according to the manufacturer's instructions. For serum collection, whole blood from CVB3-infected and naïve mice was isolated by heart puncture, transferred to yellow-capped MiniCollect serum tubes (Greiner Bio One), and centrifuged for 30 min at 3,000 rpm (4°C). The clear supernatant serum was used for ELISA.
Mouse CFU assays.
Primary BM cells of uninfected and CVB3-inoculated mice were isolated as described above and subsequently resuspended in Iscove's MDM containing 2% FBS (Stemcell Technologies). CFU assays were then performed according to the manufacturer's instructions by mixing a known number of either NH4Cl-treated or untreated BM cells with methylcellulose-containing complete medium (MethoCult, Stemcell Technologies). Three types of supplemented media were used, appropriate for the development of (i) CFU-erythrocytes (CFU-E; supplemented with recombinant human [rh] Epo [03334]); (ii) CFU-pre B lymphocytes (CFU-preB; supplemented with rh IL-7 [03630]); or (iii) CFU-granulocyte-macrophage (CFU-GM; supplemented with recombinant murine [rm] SCF, rm interleukin 3 [IL-3], and rh IL-6 [03534]). Cell suspensions were dispensed into 35-mm culture dishes (Stemcell Technologies), which were pretested for optimal colony growth without supporting anchorage-dependent cells in methylcellulose-based assays by the manufacturer. Cultures were incubated at 37°C/5% CO2 for the time periods defined in the manufacturer's instructions (CFU-E, 2 days; CFU-preB, 7 days; CFU-GM, 14 days).
Quantitative real-time PCR to detect positive-sense (genomic) CVB3 RNA.
BM cells from uninfected control mice and infected animals were isolated as described above, with RBC lysis. RNA was isolated from the same number of cells per sample using the RNeasy minikit (Qiagen) according to the manufacturer's instructions. A total of 0.45 μg total RNA per sample was reverse transcribed using SuperScript III reverse transcriptase (Invitrogen) according to the manufacturer's protocol: an oligo(dT)18 primer was used for the reverse transcriptase (RT) reaction. The RT reactions were carried out in a thermocycler as follows: 65°C for 5 min, 50°C for 45 min, 70°C for 15 min. Samples were then treated with 1 μl RNase H (Invitrogen) for 20 min at 37°C to remove RNA complementary to the cDNA. Subsequently, TaqMan quantitative real-time PCR was performed using CVB3-specific primers (forward primer, 5′CACACTCCGATCAACAGTCA3′; reverse primer, 5′GAACGCTTTCTCCTTCAACC3′) and a 6-carboxyfluorescein (FAM)/ 6-carboxytetramethylrhodamine (TAMRA)-labeled probe (5′CGTGGCACACCAGCCATGTTT3′) as previously described (30). PCR amplification was done using Platinum quantitative PCR SuperMix-UDG ready-to-use cocktail (Invitrogen) as described by the manufacturer. Quantitative analysis of viral RNA was carried out using a Bio-Rad iQ5 real-time PCR system in 96-well optical reaction plates heated to 50°C for 2 min to digest dUTP-containing contaminants, 95°C for 2 min to deactivate uracyl-N-glycosylase and activate Platinum Taq DNA polymerase, followed by 40 cycles of denaturation at 95°C for 15 s and annealing and extension at 60°C for 30 s. All samples were evaluated in 4 parallel amplification reactions. In order to assign a genome copy number to the cycle threshold value, a standard curve was generated: a known quantity of in vitro-transcribed CVB genomic RNA was serially diluted, and all dilutions were subjected to the above-described reverse transcriptase and quantitative PCRs. Values are expressed as the average numbers of CVB genome copies per μg RNA.
Adoptive transfer experiments.
To evaluate the involvement of type I interferon signaling in CVB3-induced lymphopenia, an adoptive transfer experiment was carried out. Splenocytes from either naïve C57BL/6 mice (wild type [wt]) or from mice lacking the type I interferon receptor (T1IFNRKO) were isolated by mechanical disruption of the spleen over a 70-μm-pore-size nylon cell strainer (BD). RBC lysis was performed using 0.83% NH4Cl (5 min at room temperature). Immediately after, cells were stained with different concentrations of 5- and 6-carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen): wt splenocytes were stained with 5 μM CFSE, and T1IFNRKO cells were stained with a lower dose (1 μM). The splenocytes were washed with, and resuspended in, RPMI lacking FBS and then counted and mixed at a ratio of 1:1. A total of 2.6 × 107 cells were transferred into naïve C57BL/6 recipient mice via the intravenous (i.v.) route; this large number of cells was used to maximize the sensitivity of the experiment, because we anticipated dramatic lymphopenia would occur in infected mice. Twenty-four hours posttransfer, animals were inoculated intraperitoneally (i.p.) with 104 PFU wt CVB3 or remained uninfected. Three days p.i., PBMC were isolated as described above. Additionally, cells of different lymph nodes (axillary, pancreatic, and inguinal) and of the spleen were isolated by mechanical homogenization over a 70-μm cell strainer and a following RBC lysis. All obtained cells were subsequently prepared for flow cytometry and as part of this labeled with different fluorochrome-conjugated antibodies against common lymphocyte surface markers as described above.
Histological images.
Femora of infected and naïve C57BL/6 mice were isolated, connective tissues were removed, and the bones were fixed in buffered zinc formalin (Z-FIX; Anatech) at room temperature overnight. Bones were subsequently decalcified (Cal-Ex* II Fixative/Decalcifier; Fisher) for 4 to 6 h and paraffin embedded. Sections (3 μm) were prepared and stained using hematoxylin and eosin (H&E). Images were taken at a ×10 magnification with an Axiovert 200 inverted microscope (Carl Zeiss) using AxioVision (version 4.8.1) software (Carl Zeiss).
Statistics and analysis.
Statistical significance was determined by one-way analysis of variance (ANOVA) (GraphPad Prism 6). P values less than 0.05 were considered significant and are indicated in figures as follows: *, 0.05 ≥ P > 0.01; **, 0.01 ≥ P > 0.001; ***, 0.001 ≥ P > 0.0001; ****, P ≤ 0.0001.
RESULTS
The overall goal of the present study was to investigate the impact of CVB3 infection on the BM, and to this end, C57BL/6 mice were infected intraperitoneally (i.p.) with wt CVB3 at a dose of 104 PFU. Visible signs of disease (e.g., hunched posture, lack of grooming, decreased locomotor activity) were first observed 3 days postinfection (p.i.) and were accompanied by weight loss and dehydration. Between days 3 to 5 p.i., ∼1/3 of all infected mice died; consequently, the experiments reported herein were extended only until day 5 p.i.
CVB3 productively infects BM resident cells.
We first determined whether or not CVB3 productively infects BM cells by carrying out infectious center assays (ICA; see Materials and Methods). In brief, BM cells were harvested from infected mice (and from uninfected controls), reduced to a single-cell suspension, and cleared of erythrocytes. The cells were treated with trypsin to remove adherent virus and then were plated, at various dilutions, on a HeLa cell monolayer which was immediately overlaid with a semisolid matrix. Forty-eight hours later, plaques were counted; each plaque represents a single productively infected BM cell. As shown in Fig. 1A, infected BM cells were not detected on day 1 p.i., but on day 2 p.i., ∼1 to 2% of BM cells produced infectious virus particles. After this peak, the proportion of infected cells decreased gradually between days 3 to 5 p.i. Real-time PCR analyses (Fig. 1B) showed that the CVB3 genome was present in the BM at 24 h p.i., 4-fold above the limit of detection (red dashed line). As the infection progressed, CVB3 genome copy numbers increased by ∼3 logs, peaking at day 2 p.i. (Fig. 1B), concurrent with the peak of productively infected BM cells (Fig. 1A). The genome copy number fell on day 3, but the level of viral RNA then was maintained until day 5 (Fig. 1B); this contrasts with the continued decline in infected BM cells over the same time period (Fig. 1A). The BM is highly vascular, and, therefore, we considered the possibility that the productively infected cells in BM might represent infected peripheral blood mononuclear cells (PBMC) that were in transit through the BM, rather than BM resident cells. We reasoned that, if this were the case, the kinetics of productive infection of PBMC would be similar to the kinetics observed in BM. Infectious center assays showed that productively infected PBMC were first detected on day 2 p.i., increased significantly on day 3 p.i. (P < 0.01), and fell thereafter (Fig. 1C). The different ICA kinetics in cells isolated from BM and PBMC suggest that the infected cells in BM (Fig. 1A) were predominantly BM-resident cells. As expected, infectious virus was detected in serum at day 1 p.i., peaking at day 2 and remaining readily detectable over the course of the experiment (Fig. 1D). The BM is extremely well perfused, so the fact that virus was not detected by ICA on day 1 (Fig. 1A) provides further confirmation that any plaques formed during the BM ICA assay are derived from BM resident cells that are productively infected.
Fig 1.

CVB3 productively infects bone marrow cells. C57BL/6 mice were infected i.p. with 104 PFU wt CVB3. (A and C) At the indicated times p.i., mice were sacrificed and BM cells and PBMC were isolated and analyzed by infectious center assay (ICA). (B) In addition, viral genome copy numbers in BM cells were quantified by real-time PCR (red dashed line = limit of detection). (D) Finally, viral titers in the sera were determined by plaque assay. Data are combined from the following numbers of experiments: 10 (A), 1 (B), 2 (C), 4 (D). Means + standard errors are shown, and significant differences are indicated (*, 0.05 ≥ P > 0.01; **, 0.01 ≥ P > 0.001; ***, 0.001 ≥ P > 0.0001; ****, P ≤ 0.0001).
CVB3 infection leads to marked histological changes in BM.
We next evaluated the effect of CVB3 infection on BM morphology. Virus-infected mice were sacrificed daily on days 1 to 5 p.i.; their femora were harvested, decalcified, and paraffin embedded; and thin sections were stained with H&E. Representative images are shown in Fig. 2. The BM was histologically normal at 24 h p.i.; the medullary cavity was densely packed with nucleated cells and interspersed with venous sinusoids, and megakaryocytes were numerous. However, at day 2 p.i., the time point at which productive BM infection peaks (see Fig. 1A), enlargement/engorgement of the central venous sinus was observed in all mice analyzed, and morphological changes progressed rapidly thereafter; by day 3 p.i., BM architecture was severely compromised and megakaryocytes were difficult to detect. Ultimately, nucleated cells no longer predominated, having been replaced by loosely packed erythrocytes. We therefore analyzed, in more detail, the impact of CVB3 infection on (i) cells in the erythroid lineage and (ii) nucleated BM cells, focusing on lymphocytes and neutrophils.
Fig 2.

Marked disruption of BM following CVB3 infection. C57BL/6 mice were inoculated i.p. with 104 PFU wt CVB3. At the indicated times p.i., femora were removed, fixed, decalcified, and paraffin embedded. Sections were prepared and stained using hematoxylin and eosin. A representative image for each time point is shown.
CVB3 affects cells of the erythroid lineage in both BM and blood.
Single-cell suspensions (without RBC lysis) were prepared from BM at the various p.i. time points and were stained with the TER-119 antibody (31) which is specific for glycophorin A-associated antigen, an erythroid-lineage antigen that is first expressed on early proerythroblasts and is thereafter maintained throughout their development into mature erythrocytes (32). Confirming the impression obtained by histological analysis (Fig. 2), CVB3 infection led to an increase in both frequency (Fig. 3A) and number (Fig. 3B) of TER-119+ cells, beginning with a slight elevation on day 2 p.i. and gradually rising through day 5 p.i., at which time erythroid cell numbers in BM were ∼40 to 60% higher than in an uninfected animal. Next, we assessed the maturation status of these TER-119+ BM cells by examining the expression of the transferrin receptor CD71, which is critical for iron uptake and cell proliferation processes (33); all immature erythroid cells, including early precursors, proerythroblasts, and reticulocytes, express this surface protein. As shown in Fig. 3C, in uninfected C57BL/6 mice, the ratio between TER-119+ CD71+ (immature, black) and TER-119+ CD71− (mature, white) erythroid cells was roughly 1:1. This ratio was maintained until day 2 p.i., after which the proportions changed dramatically, and, by day 5 p.i., almost all erythroid cells in the BM were mature erythrocytes. Parallel analysis of the peripheral blood revealed a rapid rise in reticulocytes at day 1 p.i. (Fig. 3D, retics, blue), but this was not maintained and the reticulocyte count fell to almost undetectable levels by day 5 p.i. However, there was no anemia, with mature erythrocyte counts being stable over the course of the study (Fig. 3D, rbc, red). Consistent with the latter observation, there was no statistically significant change in the level of serum erythropoietin (EPO; Fig. 3E), suggesting that peripheral hypoxia did not occur. In summary, the paucity of CD71+ cells in BM at days 4 and 5 p.i., together with the absence of peripheral blood reticulocytes at the same time points, suggests that CVB3 infection may have compromised the restorative capacity in the erythroid lineage.
Fig 3.

CVB3 infection affects the erythroid lineage in BM and blood. Primary BM cells were isolated at the indicated times after an i.p. wt CVB3 infection with 104 PFU. No RBC lysis was carried out. Flow cytometry analysis was performed to determine the frequency (A) and number (B) of TER-119+ cells. (C) After gating on this specific population, cells were analyzed to enumerate immature (CD71+) and mature (CD71−) BM cells. (D) Blood was collected from infected and uninfected C57BL/6 mice, and RBC and reticulocyte counts were carried out. (E) Serum levels of erythropoietin were determined by ELISA. Means + standard errors are shown.
CVB3 infection affects white blood cell counts in BM and peripheral blood.
We then assessed the effects of CVB3 infection on cells of lymphoid and myeloid origin in BM and blood. In BM, there was a marked reduction in numbers of B cells (CD19+ cells; Fig. 4A) and of CD4+ and CD8+ T cells (Fig. 4B and C, respectively) beginning on day 3 p.i. On day 5 p.i., compared to uninfected control animals, all three classes of lymphocyte (B, CD4+ T, and CD8+ T) were reduced by ∼90% in BM. Neutrophils (Ly6G+ CD11b+), too, were depleted from BM, dropping to ∼15% of normal by day 2 p.i. and remaining low thereafter (Fig. 4D). In the peripheral blood, there was a pronounced lymphopenia; B cells, CD4+ T cells, and CD8+ T cells all were dramatically reduced (Fig. 4E to G) to <5% of normal. In contrast, peripheral blood neutrophils were largely spared (Fig. 4H). Finally, consistent with the observed loss of megakaryocytes from BM (Fig. 2), a substantial thrombocytopenia was present from day 3 onward (not shown).
Fig 4.

CVB3 infection affects white blood cell counts in BM and peripheral blood. C57BL/6 mice were infected i.p. with 104 PFU wt CVB3. At the indicated times p.i., BM cells (A to D) and PBMC (E to H) were isolated and treated to lyse RBC. The numbers of lymphocytes (B [CD19+], CD4+ T, and CD8+ T) and neutrophils (Ly6G+/CD11b+) were determined by flow cytometry. Means + standard errors are shown, and significant differences are indicated (*, 0.05 ≥ P > 0.01; **, 0.01 ≥ P > 0.001; ***, 0.001 ≥ P > 0.0001; ****, P ≤ 0.0001).
Type I interferons play a role in CVB3-driven lymphopenia.
Type I interferons (T1IFNs) are generally considered immunostimulatory and proinflammatory, but several studies have indicated that at least some of the several cytokines in this class can suppress the immune response (34–38), and they also have been implicated in lymphopenia induced by other viruses (39). Therefore, we investigated the involvement of T1IFNs during CVB3 infection. As shown in Fig. 5A, serum IFN-α levels rose dramatically on day 2 p.i., coinciding with the onset of the marked lymphopenia (Fig. 5B). To determine if this association was causal, splenocytes were obtained from congenic mice lacking the T1IFN receptor and from wt mice. Each population was labeled with a different concentration of CFSE, and then the cells were washed, mixed at a 1:1 ratio, and injected into nonirradiated naïve C57BL/6 mice. Twenty-four hours later, some of the mice were infected with wt CVB3, and 3 days p.i.—a time point at which CVB-induced lymphopenia is profound (Fig. 5B)—all mice were sacrificed, and their peripheral blood was analyzed by flow cytometry. The absolute numbers of both CFSE+ transferred populations were markedly reduced in the blood of CVB3-infected mice, although, as shown in Fig. 5C, lymphocytes lacking the T1IFNR (red bars) were at an advantage. The extent of the effect varied depending on the type of lymphocyte: for B cells and CD8+ T cells, there was a 24- to 37-fold loss of wt cells (blue) and an ∼8- to 15-fold reduction in T1IFNRKO cells (red), resulting in a final ratio (wt/T1IFNRKO) of ∼1:3 in both cases. Wild-type CD4+ T cells were depleted ∼200-fold by infection, while their receptor-deficient counterparts were reduced by ∼25-fold; the ratio was ∼1:8 in favor of the receptor knockout cells. We also enumerated the transferred cells in spleen and lymph nodes at 3 days p.i.; for all three lymphocyte populations, infection caused an ∼2- to 4-fold reduction in numbers, but there was minimal difference between wt and T1IFNRKO populations (not shown). Thus, T1IFN signaling contributes to CVB3-induced lymphopenia but does not absolutely control it, because the receptor-negative populations were by no means immune to the effects of infection, and the dramatic loss of transferred lymphocytes from the blood is not paralleled by similar reductions in spleen and lymph nodes.
Fig 5.

Type I interferons play a role in CVB3-driven lymphopenia. Blood was isolated at each time point after wt CVB3 infection, and serum IFN-α levels were measured by ELISA (A) or total lymphocyte counts were determined (B). Means + standard errors are shown, and significant differences are indicated (*, 0.05 ≥ P > 0.01; **, 0.01 ≥ P > 0.001; ***, 0.001 ≥ P > 0.0001; ****, P ≤ 0.0001). To determine the involvement of type I interferon signaling in CVB-driven lymphopenia, wt and T1IFNRKO RBC-lysed splenocytes were labeled with different concentrations of CFSE, mixed 1:1, and adoptively transferred into wt mice. The following day, some of the mice were infected with 104 PFU wt CVB3, and 3 days later, all mice were sacrificed. CFSE+ wt and receptor-deficient cells were enumerated in spleen and lymph nodes (not shown) and in blood (C) by flow cytometry. Columns show the ratio, in peripheral blood, of wt cells (blue) to T1IFNRKO cells (red) at day 3 p.i. The white numerals show the fold reduction in abundance of each of the six populations compared to their levels in uninfected mice; e.g., in this experiment, wt CD4+ T cells (right column, blue) were 200-fold less abundant in the virus-infected mice than the uninfected animals.
Virus-triggered changes in cytokine levels accompany the depletion of neutrophils from the BM and the related neutrophilia.
Next, we sought an explanation for CVB3-driven loss of neutrophils from the BM. We found no evidence for increased apoptosis of BM resident cells during CVB3 infection (data not shown), suggesting that the reduction in cell numbers might result from their egress as a response to the infection. Neutrophil exit from the BM is controlled by at least two related mechanisms. First, egress is actively promoted by chemoattractants, such as macrophage inflammatory protein 2 (MIP-2; also known as CXCL2), that act both on the BM to mobilize neutrophils and locally to recruit the mobilized neutrophils to the site of infection and chemokine production (40); MIP-2 has been shown to be important in other virus infections (41). Granulocyte colony-stimulating factor (G-CSF) also mobilizes BM neutrophils, although it appears not to subsequently recruit the cells to the production site (40). Second, interactions that normally retain neutrophils within the BM can be downregulated during infection. Neutrophils express the chemokine receptor CXCR4, which, when bound by its agonist, the chemokine CXCL12 (also known as stromal cell-derived factor 1α [SDF-1α]), activates a signaling pathway that retains the cells in the BM. A reduction in CXCL12/CXCR4 signaling may contribute to the efflux of these cells into peripheral blood (40). Thus, we measured the concentration of MIP-2, G-CSF, and CXCL12 in the serum at days 1 to 5 p.i.; the results are shown in Fig. 6. There is a dramatic, and temporary, increase in both MIP-2 and G-CSF at day 2 p.i., correlating reasonably well with the reduction in BM neutrophils shown in Fig. 4D. There is no detectable decrease in CXCL12; indeed, a modest increase is observed on days 3 and 4 p.i., consistent with the stabilization in neutrophil abundance in BM (Fig. 4D) and parallel decrease in blood (Fig. 4H).
Fig 6.
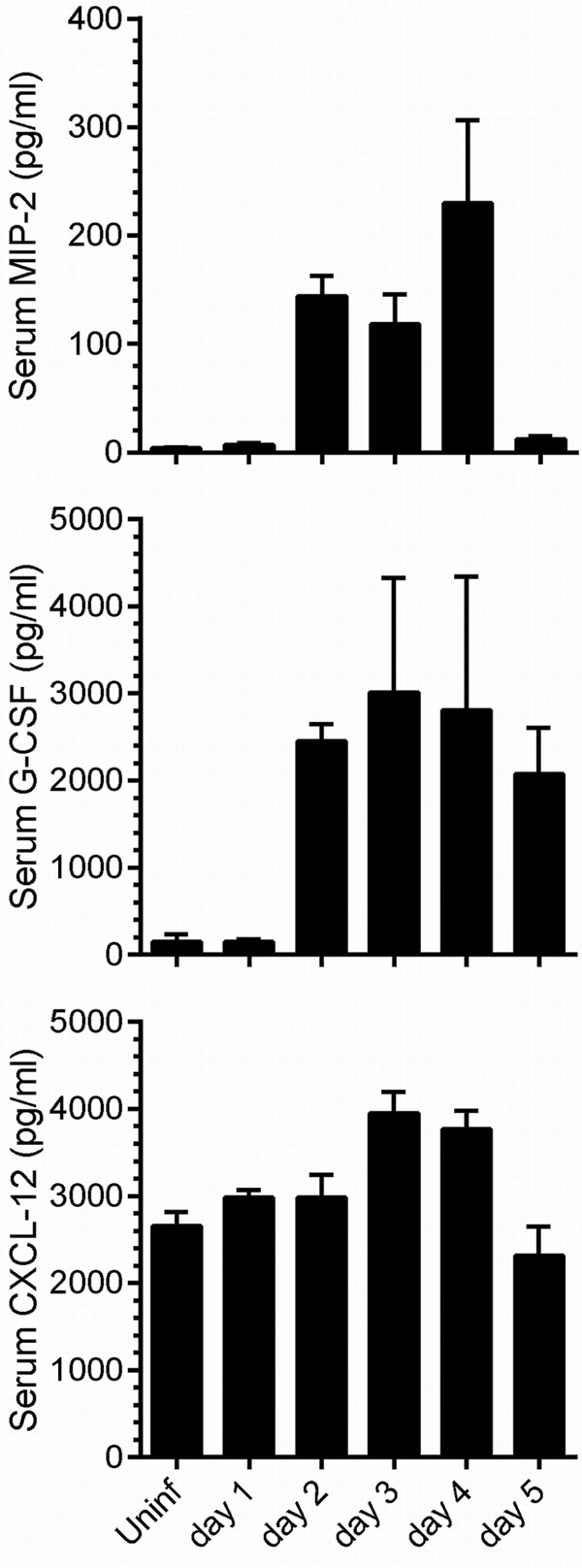
Changes to serum levels of three key cytokines occur during CVB3 infection. At the indicated time points after wt CVB3 infection, the serum levels of MIP-2, G-CSF, and CXCL-12 were measured by ELISA. Means + standard errors are indicated.
BM hematopoietic progenitor cells are depleted during CVB3 infection.
The loss of immature erythroid cells (Fig. 3C) and of lymphocytes and neutrophils (Fig. 4A to D) from the BM, coupled with the paucity of reticulocytes in the peripheral blood (Fig. 3D) and the profound lymphopenia (Fig. 4E to G), suggested that there might be a CVB3-induced defect in the restorative capacity of the BM. Therefore, we sought to determine the impact of CVB3 infection on early hematopoietic progenitor cells. All blood cells ultimately arise from hematopoietic stem cells (HSCs), and, in the adult, these multipotent and self-renewing cells are located mainly in the BM. Several cytokines regulate normal hematopoiesis (42). Two of the most important are stem cell factor (SCF) and Flt3 ligand (Flt3L), which induce the growth/differentiation of early hematopoietic progenitors that bear the appropriate receptors. The SCF receptor, c-kit, is a type III tyrosine kinase that is expressed on several cell types, including HSCs, and is central to their regulation (43). In contrast, the Flt3L receptor (Flt3/CD135) is absent from HSCs, but it is expressed by their immediate descendants, multipotent progenitor cells (MPPs), and is maintained during early maturation stages of some cells, especially of the lymphoid lineage; this appears to be true in both mouse (44) and humans (45, 46). Hence, we investigated the effect of CVB3 infection on BM MPPs, which are c-kit+/Flt3+. However, as stated above, the c-kit and Flt3 proteins also are expressed on some relatively mature cell types, so it was necessary to first identify these additional cells and exclude them from our analyses. To do so, BM cells were stained with an antibody cocktail to detect CD11b, Ly6G, B220, CD3, and TER-119. Positive cells (maturing/mature) were gated out, allowing us to focus on the remaining cells, which are termed “lineage negative” (Lin−); a large proportion of these Lin−/c-kit+/Flt3+ cells are MPPs. Representative flow cytometry dotplots, gated on Lin− cells, are shown in Fig. 7A. The frequency of MPPs (top right quadrant) drops substantially on day 2 p.i. but rises again and exceeds normal levels on day 4. MPP frequency is determined by both numerator (MPPs) and denominator (MPPs and other cell types) and can, therefore, change independently of changes in MPP abundance, so we next directly evaluated MPP numbers. MPP abundance fell by ∼50% on day 2 p.i., and by ∼90% on day 3, after which numbers rose somewhat, although not to normal levels (Fig. 7B). Thus, BM MPPs are depleted during CVB3 infection.
Fig 7.

CVB3 infection depletes BM hematopoietic progenitors and causes a rise in serum Flt3L. BM cells were isolated at the indicated times after an i.p. wt CVB3 infection, and RBC lysis was performed. Total numbers and frequencies of Lin− cells coexpressing the receptors Flt3 and c-kit were determined by flow cytometry. Dotplots shown in panel A are representative examples of each day of infection, and group data are shown (B). (C) Serum Flt3L levels were measured by ELISA. Data presented are from 2 (A and B) or 5 (C) experiments. (B and C) Means + standard errors are shown (*, 0.05 ≥ P > 0.01; **, 0.01 ≥ P > 0.001; ***, 0.001 ≥ P > 0.0001; ****, P ≤ 0.0001).
As noted above, Flt3L is a key growth factor that induces growth and differentiation of early hematopoietic progenitors that express the receptor, Flt3/CD135 (47). Flt3L is produced largely by peripheral T cells and BM stromal cells, which store it as preformed protein. In response to lymphopenia, a soluble isoform of this protein is rapidly released (48), causing the Flt3+ progenitor cells in the BM to increase in numbers; and when the peripheral blood has been repopulated, Flt3L levels fall. Thus, there should be an inverse relationship between the number of Flt3+ progenitor cells in BM and the level of serum Flt3L. Therefore, we determined the serum levels of Flt3L during CVB3 infection (Fig. 7C), and the inverse relationship is clear. On day 2 p.i., CVB3 infection causes a reduction in Flt3+ progenitor cells (Fig. 7B) and a marked lymphopenia (Fig. 4E to G), and this was accompanied by a rise in serum Flt3L at this time point. Conversely, Flt3+ cell numbers began to rise on days 4 and 5 (Fig. 7B), and there was a corresponding fall in Flt3L (Fig. 7C). Because there is no corresponding rise in lymphocytes in the peripheral blood (Fig. 5B), we considered it possible that the observed increase in MPPs did not include lymphocyte progenitors.
CVB3 infection is accompanied by a loss of colony-forming activity for certain hematopoietic lineages.
The reduced abundance of Lin−/Flt3+/c-kit+ cells in BM between days 1 to 3 p.i. (Fig. 7) led us to investigate the effects of CVB3 infection on primitive hematopoietic progenitors. Briefly, BM cells were harvested from CVB3-infected mice at days 1 to 5 p.i. and were mixed with each of three semiliquid matrices consisting of a rich cell medium and methylcellulose, supplemented by cytokines that promote growth of cells of either erythroid, myeloid, or B cell lineages. As shown in Fig. 8A, infection had only a marginal effect on myeloid progenitors (CFU-GM), but from day 3 p.i., there was a substantial reduction in the numbers of both lymphoid (CFU-preB) and erythroid (CFU-E) progenitors (for each, P values of <0.05 compared to CFU-GM). CFU-E counts were diminished by 10-fold, and CFU-preB counts diminished by between 30- and 130-fold. To determine if the observed CVB3-driven depletion of CFU-E and CFU-preB might be mediated directly, i.e., by virus infection of the progenitor cells, BM cells were harvested from uninfected mice and were exposed in vitro to CVB3 (multiplicity of infection [MOI] of 20). Control BMCs were mock infected. After a 1-h incubation, the BM cells were washed and analyzed using the same three CFU assays. The number of CFU-E was reduced by ∼40% in cultures infected with CVB3, consistent with their being directly infected by the virus (Fig. 8B). In contrast, the CFU-GM and CFU-preB lineages were not significantly affected by incubation with CVB3, suggesting that the dramatic loss of B cell progenitors (Fig. 8A) may be mediated indirectly.
Fig 8.

CVB3 infection has differing effects on lineage-specific hematopoietic progenitor cells. (A) C57BL/6 mice were inoculated i.p. with 104 PFU wt CVB3. At the indicated times p.i., BM cells were isolated and, if necessary for the desired cell population, RBC lysis was performed. Thereafter, CFU assays were carried out for each sample, as described in Materials and Methods. CFU-erythrocytes, CFU-E; CFU-pre B lymphocytes, CFU-preB; and CFU-granulocyte-macrophage, CFU-GM. The fold difference in colony number, compared to the number of colonies generated from uninfected BM, is shown for each day p.i. (B) BM cells were isolated from uninfected C57BL/6 mice, and, if necessary for the desired cell population, RBC lysis was performed. A defined number of BM cells was then incubated in the presence of wt CVB3 (MOI of 20) for 1 h at 37°C. Control BM cells suspensions were mock infected. Subsequently, different CFU assays were carried out for each sample. The data are presented as the percentage of CFU compared to the appropriate uninfected controls (i.e., uninfected controls were set at 100%). Means ± standard errors are shown.
DISCUSSION
CVB3 is known to infect several solid tissues, such as heart, pancreas, and liver, and numerous publications have documented the resulting pathogenic effects. Herein, we report the effects of CVB3 on the BM. We focused on this tissue for two reasons. First, CVB3 infects stem cells in the CNS of neonatal mice (13, 15, 49, 50). The BM is the primary repository, and source, of stem cells in the adult animal, so we considered it interesting to determine the effects of the virus on these cells and their immediate progeny. Second, CVB3 is known to infect a small proportion of B and T lymphocytes in the peripheral blood and spleen (51–54). In adults, essentially all lymphocytic progenitors come from the BM, which is an important primary immune organ that also harbors CD8+ effector and memory T cells (55); thus, we sought to characterize the impact of CVB3 infection on BM lymphocytes and their progenitors.
We first determined whether or not BM cells could be infected by CVB3. Productively infected BM cells were not detected at 24 h p.i. (Fig. 1A), indicating that fewer than ∼1 in 105 to 106 BM cells (the approximate limit of detection for our ICA) was productively infected at this time point. This cannot be attributed to the failure of virus to reach the BM, because (i) the serum titer at 24 h p.i. was ∼106 PFU/ml (Fig. 1D) and (ii) the CVB genome was detectable in BM cells (Fig. 1B). The BM cells had been trypsinized and washed prior to RNA extraction; thus, it is unlikely that these CVB3 genomes came from cell-bound virus, and we propose that, by 24 h p.i., some of the BM cells may have internalized the virus. Nevertheless, the cells did not generate plaques by ICA, but only 24 h later, there was an at least ∼10,000-fold increase in productively infected BM cells as measured by ICA (∼1 to 2% of BM cells scored positive at 48 h p.i.; Fig. 1A). We speculate that, between 24 to 48 h p.i., the activation status of infected BM cells changes in vivo, facilitating the completion of a productive CVB3 infection, and that this fails to occur in vitro, when the cells have been removed from their normal anatomical microenvironment. This proposal is consistent with prior observations from our group, showing that CVB3 protein synthesis and productive infection are profoundly affected by the cell cycle status and that CVB3 may lie quiescent within cells, beginning to replicate only when the cell status is changed (15, 56–59); for example, neural stem cells in the SVZ are infected by CVB3 but still can migrate, and we have proposed that productive infection ensues only when the cells reach their final destination and complete their differentiation into mature neurons (13, 15). Interestingly, hematopoietic progenitor cells in the BM are usually in a quiescent state and are roused from their dormancy by type I interferon (60), a cytokine that is produced early in response to CVB3 infections (Fig. 5A) (61). Therefore, day 1 after infection may represent an eclipse state of the virus infection of BM cells, which transitions to productive infection only when the BM cells receive the appropriate external stimuli. In agreement with previous studies (54, 62, 63), we also identified productively infected PBMC, the frequency of which peaked on day 3 p.i., 24 h later than in the BM (Fig. 1C). We speculate that at least some of the infected PBMC might represent cells that had been infected while in the BM and were subsequently released into the peripheral circulation. This would, potentially, accelerate viral dissemination throughout the infected host, in the form of infected cells that, as we have previously proposed, may act as “Trojan horses” (63). At later times p.i., the number of productively infected cells in BM fell sharply, although viral RNA levels were maintained (Fig. 1A and B). This is consistent with the hypothesis that the virus establishes a persistent (nonproductive) infection in some cells, depending on cell status. This also may explain earlier observations from in vitro infections of primary human PBMC and BM cells, in which viral protein was detected but infectious particles were not produced (64).
The productive CVB3 infection of BM cells, described above, was accompanied by a dramatic disruption of BM tissue. Beginning at 2 days p.i., the normally densely packed tissue structure became less compact, and nucleated hematopoietic cells were progressively replaced by RBC (Fig. 2); by day 5 p.i., ∼80 to 90% of all BM cells were positive for the cell surface marker TER-119+, which characterizes mostly all cells of the erythroid lineage (Fig. 3A). Interestingly, the developmental stage of cells within this population was also affected during CVB3 infection. The ratio between immature TER-119+ cells (CD71+, capable of dividing) and mature TER-119+ cells (CD71−, nondividing) was ∼1:1 in naïve mice and remained thus until day 3 p.i., after which almost all BM TER-119+ cells were mature (Fig. 3C). Several nonmutually exclusive mechanisms may contribute to this shift toward mature erythroid cells. (i) CD71+ erythroid precursors are released from BM during the infection, as demonstrated by the rise in the reticulocyte count early in infection (Fig. 3D), which reflects the transfer of a substantial number of immature cells from BM to the peripheral blood. (ii) There may be accelerated maturation of CD71+ cells within the infected BM. This would alter the ratio of immature to mature erythroid cells in the BM but would not alter the total number of erythroid (TER-119+) cells. However, this number does rise over the course of infection (Fig. 3B), so we postulate that (iii) there is accumulation of peripheral blood erythrocytes in the damaged BM—in effect, a virus-induced BM hemorrhage; this is consistent with the venous distension observed histologically on day 2 p.i. (Fig. 2).
Consistent with the loss of nucleated cells observed by histology, B and T lymphocyte numbers in BM also fell markedly, beginning on days 2 to 3 p.i. (Fig. 4A to C). The loss of lymphocytes from BM coincided with an even larger reduction in peripheral blood; CVB3-infected mice suffered a massive and persistent lymphopenia starting at day 2 p.i. and affecting both B and T lymphocytes (Fig. 4E to G). Several viruses induce lymphopenia, by a variety of mechanisms. In some virus infections—for example, in HIV and measles virus—lymphocytes are directly infected, and this contributes to their ultimate demise (65–67). In other cases—for example, vesicular stomatitis virus (39, 68) and lymphocytic choriomeningitis virus (LCMV) (69)—lymphopenia appears to be mediated indirectly, by cytokines such as type I interferon. However, the effects of type I IFNs on immune cells are not readily predictable. These cytokines, which are produced early and in abundance in response to many virus infections, are thought to drive the rapid and transient attrition of lymphocytes that occurs in the first 1 to 3 days following many viral infections (34–38). Furthermore, they are instrumental in the depletion of plasmacytoid dendritic cells that occurs concurrently with the above-described lymphocyte attrition (70). In contrast, they are thought to exert positive effects on BM HSCs (60) and to confer protection upon virus-specific T cells in the BM, rendering them resistant to virus infection and, thereby, more capable of fulfilling their in vivo role in combatting infection (71). Furthermore, both CD4+ and CD8+ T cell responses are dramatically reduced if the cells cannot receive T1IFN signals (72, 73). We found that the lymphopenia during CVB3 infection was concurrent with an abrupt rise in serum IFN-α levels (Fig. 5A and B) and that, compared to wt cells, receptor-negative cells (B, CD4+, and CD8+ lymphocytes) were better represented in the peripheral blood; however, the effect was far from absolute, because even the T1IFNRKO cells were markedly reduced by CVB3 infection (Fig. 5C). We do not know if the selective loss of wt lymphocytes from the blood reflects their death or merely their redistribution to other organs; we favor the latter explanation, because no selective reduction of wt cells was evident in the spleen or lymph nodes. Thus, we speculate that the major effect of T1IFNs on blood lymphocytes during CVB infection is to regulate their distribution to solid tissues, as has previously been proposed for another virus (39).
CVB3 infection causes a loss of Ly6G+/CD11b+ BM neutrophils as early as day 2 p.i. (Fig. 4D) and a corresponding modest rise in the peripheral blood (Fig. 4D and H), suggesting that there was minimal change in the total number of these cells in the infected mouse, and there are parallel changes in the serum level of cytokines that are known to regulate these changes (Fig. 6). What might be the evolutionary reason for this, given that neutrophils are usually associated with responses to bacterial, rather than viral, infections? A role for neutrophils in controlling at least some virus infections has been proposed from studies of individuals with a congenital neutropenia termed WHIM syndrome (warts, hypogammaglobulinemia, bacterial infections, and myelokathexis) (74). Many WHIM patients also show a B cell lymphopenia, and most show a decrease—often quite modest—in blood immunoglobulin levels; together, these three deficiencies (neutrophils, B cells, Ig) contribute to an increased susceptibility to bacterial infection (75). WHIM syndrome results from a dominant mutation in CXCR4 (76) which enhances CXCL12 signaling into neutrophils, causing them to be retained in the BM, although they are released in relatively normal numbers in response to infection. The sufferers show normal resistance to most virus infections, with the exception of papillomaviruses, which cause extensive and severe cutaneous verrucosis (74, 77). To date, no immunological explanation for this highly specific viral susceptibility has been identified, and a likelier mechanism is that skin keratinocytes, an important target of papillomavirus infection and pathogenesis, also express CXCR4 (78). Thus, the biological significance of the very rapid mobilization of neutrophils during CVB3 infection is unknown. However, it is significant that neutrophil counts in peripheral blood are not only maintained but are ∼2-fold elevated throughout CVB3 infection (Fig. 4H). Following their release from the BM into the periphery, neutrophils have a remarkably short half-life of ∼6.5 h and are subsequently destroyed in the liver, spleen, and bone marrow; the ongoing neutrophilia therefore suggests that CVB infection does not markedly affect the capacity of the BM to constantly replenish the periphery.
In summary, lymphocytes and platelets are markedly depleted from the peripheral blood during CVB3 infection, while erythrocyte and neutrophil numbers are largely maintained. These data suggest that some degree of bone marrow function is retained, contrasting with a report that neonatal echoviral infection may cause pancytopenia and bone marrow failure (79). To directly assess BM function, we evaluated the effect of CVB3 infection on hematopoietic progenitor cells in the BM. The number of Lin−/c-kit+/Flt3+ cells in BM fell on day 2 p.i., reached its nadir on day 3, and thereafter showed a modest recovery (Fig. 7A and B). This was mirrored by complementary changes in serum Flt3L (Fig. 7C), which is released by T cells and BM stromal cells in response to lymphopenia and stimulates the Lin−/c-kit+/Flt3+ progenitor cells of all hematopoietic lineages (80, 81). Flt3L levels do not increase in diseases affecting single hematopoietic lineages (82, 83), suggesting that CVB3 might affect several of the lineages in BM. We next investigated whether or not CVB3 infection has an impact on the BM's ability to form new immature hematopoietic cells. To this end, we performed CFU assays, using methylcellulose-containing medium supplemented with different growth factors/cytokines that allowed us to assess the impact of infection upon three hematopoietic lineages. Despite the (presumably positive) influence of high Flt3L levels between days 2 to 5 p.i., we found significantly lower numbers of erythroid and lymphoid progenitor CFU in cultures gathered at or after days 2 to 3 p.i. (Fig. 8A). This may contribute to the shift in the mature/immature erythroid cell ratio in BM (Fig. 3C), the short-lived reticulocytosis (Fig. 3D), and the loss of lymphocytes from both BM and blood (Fig. 4). Interestingly, others have reported transient erythroblastopenia in a 3-year-old girl with an echoviral infection (84). The reduction in erythroid and pre B CFU may result from the transient depletion of progenitor cells from BM (Fig. 7); however, we cannot exclude the possibility that the virus infection also reduces the proliferative activity of the remaining progenitor cells, thus amplifying the effects on colony formation. In contrast to the findings with CFU-E and CFU-preB, CFU-GM counts were very similar in BM harvested from uninfected mice and from mice at each of the 5 p.i. time points (Fig. 8A), suggesting that the restorative capacity of this population was not dramatically altered by CVB3 infection. For reasons discussed above, this is consistent with the ongoing elevation of these short-lived cells in the peripheral blood. Thus, the loss of neutrophils from BM (Fig. 4D) probably reflects their transfer to the peripheral blood rather than paralysis of production within the BM, and the retained functionality of the CFU-GM explains the stabilization of neutrophil counts in BM after day 2 p.i. (Fig. 4D). The lack of effect on CFU-GM following both in vivo and in vitro infection (Fig. 8) is consonant with our findings that neutrophil levels in vivo are less markedly impacted by CVB3 infection but contrasts with a previous report suggesting that CVB3 infects human BM CFU-GM in vitro (85); this may reflect a species difference, but at present, we have no evidence to support such a conclusion. To begin to address the mechanism(s) by which CVB3 infection constrains the restorative capacity of CFU-E and CFU-preB, primary BM cells from naïve C57BL/6 mice were incubated with the virus in vitro, washed, and analyzed using each of the three different CFU assays. The numbers of CFU-E were ∼50% reduced in cultures that were established using CVB3-infected BM cells, compared with mock-infected BM cells (Fig. 8B), suggesting that erythroid precursors may be directly infected with virus. In contrast, neither CFU-GM nor CFU-preB showed any negative effect of in vitro exposure to wt CVB3. The apparent discrepancy for CFU-preB—markedly reduced during in vivo infection but relatively unaffected following in vitro exposure—indicates that the in vivo changes may be driven by something other than direct infection. There is evidence suggesting a critical role of the host's immune response in causing BM suppression in some virus infections (19, 86). Moreover, Binder et al. reported a predominantly type I IFN-mediated suppressive effect on hematopoietic precursors during acute LCMV infection (69), although this observation contrasts with a recent report that IFN-α activates dormant HSCs in vivo (60).
In conclusion, the present study reveals a profound impact of a CVB3 infection on morphology, composition, and activity of the BM of C57BL/6 mice. Future efforts are focused on the underlying mechanisms of this complex host-virus interaction.
ACKNOWLEDGMENTS
We are grateful to Sheila Silverstein for excellent secretarial support and Claudia Flynn and Stephanie Harkins for assistance with real-time PCR.
This work was supported by NIH grants AI042314 and HL093177 (to J.L.W.).
This is manuscript number 21785 from the Scripps Research Institute.
Footnotes
Published ahead of print 26 December 2012
REFERENCES
- 1. Carthy CM, Yang D, Anderson DR, Wilson JE, McManus BM. 1997. Myocarditis as systemic disease: new perspectives on pathogenesis. Clin. Exp. Pharmacol. Physiol. 24:997–1003 [DOI] [PubMed] [Google Scholar]
- 2. O'Connell JB. 1987. The role of myocarditis in end-stage dilated cardiomyopathy. Tex. Heart Inst. J. 14:268–275 [PMC free article] [PubMed] [Google Scholar]
- 3. Sole MJ, Liu P. 1993. Viral myocarditis: a paradigm for understanding the pathogenesis and treatment of dilated cardiomyopathy. J. Am. Coll. Cardiol. 22:99A–105A [DOI] [PubMed] [Google Scholar]
- 4. Tam PE. 2006. Coxsackievirus myocarditis: interplay between virus and host in the pathogenesis of heart disease. Viral Immunol. 19:133–146 [DOI] [PubMed] [Google Scholar]
- 5. Kandolf R, Klingel K, Zell R, Selinka HC, Raab U, Schneider-Brachert W, Bultmann B. 1993. Molecular pathogenesis of enterovirus-induced myocarditis: virus persistence and chronic inflammation. Intervirology 35:140–151 [DOI] [PubMed] [Google Scholar]
- 6. Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R. 1992. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc. Natl. Acad. Sci. U. S. A. 89:314–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martino TA, Liu P, Sole MJ. 1994. Viral infection and the pathogenesis of dilated cardiomyopathy. Circ. Res. 74:182–188 [DOI] [PubMed] [Google Scholar]
- 8. Mena I, Fischer C, Gebhard JR, Perry CM, Harkins S, Whitton JL. 2000. Coxsackievirus infection of the pancreas: evaluation of receptor expression, pathogenesis, and immunopathology. Virology 271:276–288 [DOI] [PubMed] [Google Scholar]
- 9. Ramsingh AI. 1997. Coxsackieviruses and pancreatitis. Front. Biosci. 2:e53–e62 [DOI] [PubMed] [Google Scholar]
- 10. Palomba E, Tovo PA. 1999. Persistent fever as the only manifestation of chronic coxsackievirus B4 infection in the brain of a human immunodeficiency virus type 1-infected child. Clin. Infect. Dis. 28:912–913 [DOI] [PubMed] [Google Scholar]
- 11. Geller TJ, Condie D. 1995. A case of protracted coxsackie virus meningoencephalitis in a marginally immunodeficient child treated successfully with intravenous immunoglobulin. J. Neurol. Sci. 129:131–133 [DOI] [PubMed] [Google Scholar]
- 12. Hertel NT, Pedersen FK, Heilmann C. 1989. Coxsackie B3 virus encephalitis in a patient with agammaglobulinaemia. Eur. J. Pediatr. 148:642–643 [DOI] [PubMed] [Google Scholar]
- 13. Feuer R, Mena I, Pagarigan RR, Harkins S, Hassett DE, Whitton JL. 2003. Coxsackievirus B3 and the neonatal CNS: the roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am. J. Pathol. 163:1379–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Menezes JR, Smith CM, Nelson KC, Luskin MB. 1995. The division of neuronal progenitor cells during migration in the neonatal mammalian forebrain. Mol. Cell Neurosci. 6:496–508 [DOI] [PubMed] [Google Scholar]
- 15. Feuer R, Pagarigan RR, Harkins S, Liu F, Hunziker IP, Whitton JL. 2005. Coxsackievirus targets proliferating neuronal progenitor cells in the neonatal CNS. J. Neurosci. 25:2434–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mercier FE, Ragu C, Scadden DT. 2012. The bone marrow at the crossroads of blood and immunity. Nat. Rev. Immunol. 12:49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao E, Xu H, Wang L, Kryczek I, Wu K, Hu Y, Wang G, Zou W. 2012. Bone marrow and the control of immunity. Cell Mol. Immunol. 9:11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baranski B, Armstrong G, Truman JT, Quinnan GV, Jr, Straus SE, Young NS. 1988. Epstein-Barr virus in the bone marrow of patients with aplastic anemia. Ann. Intern. Med. 109:695–704 [DOI] [PubMed] [Google Scholar]
- 19. Shadduck RK, Winkelstein A, Zeigler Z, Lichter J, Goldstein M, Michaels M, Rabin B. 1979. Aplastic anemia following infectious mononucleosis: possible immune etiology. Exp. Hematol. 7:264–271 [PubMed] [Google Scholar]
- 20. Apperley JF, Dowding C, Hibbin J, Buiter J, Matutes E, Sissons PJ, Gordon M, Goldman JM. 1989. The effect of cytomegalovirus on hemopoiesis: in vitro evidence for selective infection of marrow stromal cells. Exp. Hematol. 17:38–45 [PubMed] [Google Scholar]
- 21. Reddehase MJ, Dreher-Stumpp L, Angele P, Balthesen M, Susa M. 1992. Hematopoietic stem cell deficiency resulting from cytomegalovirus infection of bone marrow stroma. Ann. Hematol. 64:A125–A127 [DOI] [PubMed] [Google Scholar]
- 22. Molina JM, Scadden DT, Sakaguchi M, Fuller B, Woon A, Groopman JE. 1990. Lack of evidence for infection of or effect on growth of hematopoietic progenitor cells after in vivo or in vitro exposure to human immunodeficiency virus. Blood 76:2476–2482 [PubMed] [Google Scholar]
- 23. Young N. 1988. Hematologic and hematopoietic consequences of B19 parvovirus infection. Semin. Hematol. 25:159–172 [PubMed] [Google Scholar]
- 24. La Russa VF, Innis BL. 1995. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin. Haematol. 8:249–270 [DOI] [PubMed] [Google Scholar]
- 25. Young NS. 1990. Flaviviruses and bone marrow failure. JAMA 263:3065–3068 [PubMed] [Google Scholar]
- 26. Muller U, Steinhoff U, Reis LFL, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921 [DOI] [PubMed] [Google Scholar]
- 27. van Houten N, Bouchard PE, Moraska A, Huber SA. 1991. Selection of an attenuated coxsackievirus B3 variant, using a monoclonal antibody reactive to myocyte antigen. J. Virol. 65:1286–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Knowlton KU, Jeon ES, Berkley N, Wessely R, Huber SA. 1996. A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J. Virol. 70:7811–7818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hunziker IP, Cornell CT, Whitton JL. 2007. Deletions within the 5′UTR of coxsackievirus B3: consequences for virus translation and replication. Virology 360:120–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tabor-Godwin JM, Ruller CM, Bagalso N, An N, Pagarigan RR, Harkins S, Gilbert PE, Kiosses WB, Gude NA, Cornell CT, Doran KS, Sussman MA, Whitton JL, Feuer R. 2010. A novel population of myeloid cells responding to coxsackievirus infection assists in the dissemination of virus within the neonatal CNS. J. Neurosci. 30:8676–8691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ikuta K, Kina T, MacNeil I, Uchida N, Peault B, Chien YH, Weissman IL. 1990. A developmental switch in thymic lymphocyte maturation potential occurs at the level of hematopoietic stem cells. Cell 62:863–874 [DOI] [PubMed] [Google Scholar]
- 32. Kina T, Ikuta K, Takayama E, Wada K, Majumdar AS, Weissman IL, Katsura Y. 2000. The monoclonal antibody TER-119 recognizes a molecule associated with glycophorin A and specifically marks the late stages of murine erythroid lineage. Br. J. Haematol. 109:280–287 [DOI] [PubMed] [Google Scholar]
- 33. Trowbridge IS, Lesley J, Schulte R. 1982. Murine cell surface transferrin receptor: studies with an anti-receptor monoclonal antibody. J. Cell. Physiol. 112:403–410 [DOI] [PubMed] [Google Scholar]
- 34. Selin LK, Lin MY, Kraemer KA, Pardoll DM, Schneck JP, Varga SM, Santolucito PA, Pinto AK, Welsh RM. 1999. Attrition of T cell memory: selective loss of LCMV epitope-specific memory CD8 T cells following infections with heterologous viruses. Immunity 11:733–742 [DOI] [PubMed] [Google Scholar]
- 35. McNally JM, Zarozinski CC, Lin MY, Brehm MA, Chen HD, Welsh RM. 2001. Attrition of bystander CD8 T cells during virus-induced T-cell and interferon responses. J. Virol. 75:5965–5976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McNally JM, Welsh RM. 2002. Bystander T cell activation and attrition. Curr. Top. Microbiol. Immunol. 263:29–41 [DOI] [PubMed] [Google Scholar]
- 37. Peacock CD, Kim SK, Welsh RM. 2003. Attrition of virus-specific memory CD8+ T cells during reconstitution of lymphopenic environments. J. Immunol. 171:655–663 [DOI] [PubMed] [Google Scholar]
- 38. Welsh RM, Selin LK. 2009. Attrition of memory CD8 T cells. Nature 459:E3–E4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kamphuis E, Junt T, Waibler Z, Forster R, Kalinke U. 2006. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood 108:3253–3261 [DOI] [PubMed] [Google Scholar]
- 40. Furze RC, Rankin SM. 2008. Neutrophil mobilization and clearance in the bone marrow. Immunology 125:281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yan XT, Tumpey TM, Kunkel SL, Oakes JE, Lausch RN. 1998. Role of MIP-2 in neutrophil migration and tissue injury in the herpes simplex virus-1-infected cornea. Invest. Ophthalmol. Vis. Sci. 39:1854–1862 [PubMed] [Google Scholar]
- 42. Lotem J, Sachs L. 2002. Cytokine control of developmental programs in normal hematopoiesis and leukemia. Oncogene 21:3284–3294 [DOI] [PubMed] [Google Scholar]
- 43. Ronnstrand L. 2004. Signal transduction via the stem cell factor receptor/c-Kit. Cell. Mol. Life Sci. 61:2535–2548 [DOI] [PubMed] [Google Scholar]
- 44. Buza-Vidas N, Woll P, Hultquist A, Duarte S, Lutteropp M, Bouriez-Jones T, Ferry H, Luc S, Jacobsen SE. 2011. FLT3 expression initiates in fully multipotent mouse hematopoietic progenitor cells. Blood 118:1544–1548 [DOI] [PubMed] [Google Scholar]
- 45. Sitnicka E, Buza-Vidas N, Larsson S, Nygren JM, Liuba K, Jacobsen SE. 2003. Human CD34+ hematopoietic stem cells capable of multilineage engrafting NOD/SCID mice express flt3: distinct flt3 and c-kit expression and response patterns on mouse and candidate human hematopoietic stem cells. Blood 102:881–886 [DOI] [PubMed] [Google Scholar]
- 46. Kikushige Y, Yoshimoto G, Miyamoto T, Iino T, Mori Y, Iwasaki H, Niiro H, Takenaka K, Nagafuji K, Harada M, Ishikawa F, Akashi K. 2008. Human Flt3 is expressed at the hematopoietic stem cell and the granulocyte/macrophage progenitor stages to maintain cell survival. J. Immunol. 180:7358–7367 [DOI] [PubMed] [Google Scholar]
- 47. Shurin MR, Esche C, Lotze MT. 1998. FLT3: receptor and ligand. Biology and potential clinical application. Cytokine Growth Factor Rev. 9:37–48 [DOI] [PubMed] [Google Scholar]
- 48. Chklovskaia E, Jansen W, Nissen C, Lyman SD, Rahner C, Landmann L, Wodnar-Filipowicz A. 1999. Mechanism of flt3 ligand expression in bone marrow failure: translocation from intracellular stores to the surface of T lymphocytes after chemotherapy-induced suppression of hematopoiesis. Blood 93:2595–2604 [PubMed] [Google Scholar]
- 49. Ruller CM, Tabor-Godwin JM, Van Deren D, Robinson SM, Maciejewski S, Gilbert PE, An N, Gude NA, Sussman MA, Whitton JL, Feuer R. 2012. Neural stem cell depletion following a viral infection leads to CNS developmental defects. Am. J. Pathol. 180:1107–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Feuer R, Ruller CM, An N, Tabor-Godwin JM, Rhoades RE, Maciejewski S, Pagarigan RR, Cornell CT, Crocker SJ, Kiosses WB, Pham-Mitchell N, Campbell IL, Whitton JL. 2009. Viral persistence and chronic immunopathology in the adult CNS following coxsackievirus infection during the neonatal period. J. Virol. 83:9356–9369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jarasch-Althof N, Wiesener N, Schmidtke M, Wutzler P, Henke A. 2010. Antibody-dependent enhancement of coxsackievirus B3 infection of primary CD19+ B lymphocytes. Viral Immunol. 23:369–376 [DOI] [PubMed] [Google Scholar]
- 52. Klingel K, McManus BM, Kandolf R. 1995. Enterovirus-infected immune cells of spleen and lymph nodes in the murine model of chronic myocarditis: a role in pathogenesis? Eur. Heart J. 16(Suppl O):42–45 [DOI] [PubMed] [Google Scholar]
- 53. Anderson DR, Wilson JE, Carthy CM, Yang D, Kandolf R, McManus BM. 1996. Direct interactions of coxsackievirus B3 with immune cells in the splenic compartment of mice susceptible or resistant to myocarditis. J. Virol. 70:4632–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jarasch N, Martin U, Zell R, Wutzler P, Henke A. 2007. Influence of pan-caspase inhibitors on coxsackievirus B3-infected CD19+ B lymphocytes. Apoptosis 12:1633–1643 [DOI] [PubMed] [Google Scholar]
- 55. Slifka MK, Whitmire JK, Ahmed R. 1997. Bone marrow contains virus-specific cytotoxic T lymphocytes. Blood 90:2103–2108 [PubMed] [Google Scholar]
- 56. Feuer R, Mena I, Pagarigan RR, Slifka MK, Whitton JL. 2002. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J. Virol. 76:4430–4440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Feuer R, Mena I, Pagarigan RR, Hassett DE, Whitton JL. 2004. Coxsackievirus replication and the cell cycle: a potential regulatory mechanism for viral persistence/latency. Med. Microbiol. Immunol. (Berl.) 193:83–90 [DOI] [PubMed] [Google Scholar]
- 58. Whitton JL, Cornell CT, Feuer R. 2005. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 3:765–776 [DOI] [PubMed] [Google Scholar]
- 59. Feuer R, Whitton JL. 2008. Preferential coxsackievirus replication in proliferating/activated cells: implications for virus tropism, persistence, and pathogenesis. Curr. Top. Microbiol. Immunol. 149–173 [DOI] [PubMed] [Google Scholar]
- 60. Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. 2009. IFNa activates dormant haematopoietic stem cells in vivo. Nature 458:904–908 [DOI] [PubMed] [Google Scholar]
- 61. Wessely R, Klingel K, Knowlton KU, Kandolf R. 2001. Cardioselective infection with coxsackievirus B3 requires intact type I interferon signaling: implications for mortality and early viral replication. Circulation 103:756–761 [DOI] [PubMed] [Google Scholar]
- 62. Klingel K, Stephan S, Sauter M, Zell R, McManus BM, Bultmann B, Kandolf R. 1996. Pathogenesis of murine enterovirus myocarditis: virus dissemination and immune cell targets. J. Virol. 70:8888–8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mena I, Perry CM, Harkins S, Rodriguez F, Gebhard JR, Whitton JL. 1999. The role of B lymphocytes in coxsackievirus B3 infection. Am. J. Pathol. 155:1205–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vuorinen T, Vainionpaa R, Kettinen H, Hyypia T. 1994. Coxsackievirus B3 infection in human leukocytes and lymphoid cell lines. Blood 84:823–829 [PubMed] [Google Scholar]
- 65. Crowe SM, Carlin JB, Stewart KI, Lucas CR, Hoy JF. 1991. Predictive value of CD4 lymphocyte numbers for the development of opportunistic infections and malignancies in HIV-infected persons. J. Acquir. Immune. Defic. Syndr. 4:770–776 [PubMed] [Google Scholar]
- 66. Okada H, Kobune F, Sato TA, Kohama T, Takeuchi Y, Abe T, Takayama N, Tsuchiya T, Tashiro M. 2000. Extensive lymphopenia due to apoptosis of uninfected lymphocytes in acute measles patients. Arch. Virol. 145:905–920 [DOI] [PubMed] [Google Scholar]
- 67. Wesley A, Coovadia HM, Henderson L. 1978. Immunological recovery after measles. Clin. Exp. Immunol. 32:540–544 [PMC free article] [PubMed] [Google Scholar]
- 68. Schattner A, Meshorer A, Wallach D. 1983. Involvement of interferon in virus-induced lymphopenia. Cell Immunol. 79:11–25 [DOI] [PubMed] [Google Scholar]
- 69. Binder D, Fehr J, Hengartner H, Zinkernagel RM. 1997. Virus-induced transient bone marrow aplasia: major role of interferon-a/b during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J. Exp. Med. 185:517–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. 2011. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J. Exp. Med. 208:2367–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hermesh T, Moltedo B, Moran TM, Lopez CB. 2010. Antiviral instruction of bone marrow leukocytes during respiratory viral infections. Cell Host Microbe 7:343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. 2005. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202:637–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Havenar-Daughton C, Kolumam GA, Murali-Krishna K. 2006. Cutting edge: the direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J. Immunol. 176:3315–3319 [DOI] [PubMed] [Google Scholar]
- 74. Wetzler M, Talpaz M, Kleinerman ES, King A, Huh YO, Gutterman JU, Kurzrock R. 1990. A new familial immunodeficiency disorder characterized by severe neutropenia, a defective marrow release mechanism, and hypogammaglobulinemia. Am. J. Med. 89:663–672 [DOI] [PubMed] [Google Scholar]
- 75. Kawai T, Malech HL. 2009. WHIM syndrome: congenital immune deficiency disease. Curr. Opin. Hematol. 16:20–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, Klotman ME, Diaz GA. 2003. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat. Genet. 34:70–74 [DOI] [PubMed] [Google Scholar]
- 77. Gorlin RJ, Gelb B, Diaz GA, Lofsness KG, Pittelkow MR, Fenyk JR., Jr 2000. WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am. J. Med. Genet. 91:368–376 [PubMed] [Google Scholar]
- 78. Chow KY, Brotin E, Ben KY, Carthagena L, Teissier S, Danckaert A, Galzi JL, Arenzana-Seisdedos F, Thierry F, Bachelerie F. 2010. A pivotal role for CXCL12 signaling in HPV-mediated transformation of keratinocytes: clues to understanding HPV-pathogenesis in WHIM syndrome. Cell Host Microbe 8:523–533 [DOI] [PubMed] [Google Scholar]
- 79. Tarcan A, Ozbek N, Gurakan B. 2001. Bone marrow failure with concurrent enteroviral infection in a newborn. Pediatr. Infect. Dis. J. 20:719–721 [DOI] [PubMed] [Google Scholar]
- 80. Lyman SD, Jacobsen SE. 1998. c-kit ligand and Flt3 ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood 91:1101–1134 [PubMed] [Google Scholar]
- 81. Brasel K, McKenna HJ, Morrissey PJ, Charrier K, Morris AE, Lee CC, Williams DE, Lyman SD. 1996. Hematologic effects of flt3 ligand in vivo in mice. Blood 88:2004–2012 [PubMed] [Google Scholar]
- 82. Lyman SD, Seaberg M, Hanna R, Zappone J, Brasel K, Abkowitz JL, Prchal JT, Schultz JC, Shahidi NT. 1995. Plasma/serum levels of flt3 ligand are low in normal individuals and highly elevated in patients with Fanconi anemia and acquired aplastic anemia. Blood 86:4091–4096 [PubMed] [Google Scholar]
- 83. Wodnar-Filipowicz A, Lyman SD, Gratwohl A, Tichelli A, Speck B, Nissen C. 1996. Flt3 ligand level reflects hematopoietic progenitor cell function in aplastic anemia and chemotherapy-induced bone marrow aplasia. Blood 88:4493–4499 [PubMed] [Google Scholar]
- 84. Elian JC, Frappaz D, Pozzetto B, Freycon F. 1993. Transient erythroblastopenia of childhood presenting with echovirus 11 infection. Acta Paediatr. 82:492–494 [DOI] [PubMed] [Google Scholar]
- 85. Vuorinen T, Vainionpaa R, Vanharanta R, Hyypia T. 1996. Susceptibility of human bone marrow cells and hematopoietic cell lines to coxsackievirus B3 infection. J. Virol. 70:9018–9023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liang DC, Lin KH, Lin DT, Yang CP, Hung KL, Lin KS. 1990. Post-hepatitic aplastic anaemia in children in Taiwan, a hepatitis prevalent area. Br. J. Haematol. 74:487–491 [DOI] [PubMed] [Google Scholar]