

Abstract
Herpes simplex virus 2 (HSV-2) infection is still one of the common causes of sexually transmitted diseases worldwide. The prevalence of HSV strains resistant to traditional nucleoside antiviral agents has led to the development of novel antiviral drugs. Human alpha-defensin 5 (HD5), a kind of endogenous antimicrobial peptide expressed in the epithelia of the small intestine and urogenital tract, displays natural antiviral activity. Based on arginine-rich features and adaptive evolution characteristics of vertebrate defensins, we conducted a screen for HD5 derivatives with enhanced anti-HSV-2 activity by a single arginine substitution at the adaptive evolution sites. Cell protection assay and temporal antiviral studies showed that HD5 and its mutants displayed affirmatory but differential anti-HSV-2 effects in vitro by inhibiting viral adhesion and entry. Inspiringly, the E21R-HD5 mutant had significantly higher antiviral activity than natural HD5, which is possibly attributed to the stronger binding affinity of the E21R-HD5 mutant with HSV-2 capsid protein gD, indicating that E21R mutation can increase the anti-HSV-2 potency of HD5. In a mouse model of lethal HSV-2 infection, prophylactic and/or therapeutic treatment with E21R-HD5 via intravaginal instillation remarkably alleviated the symptoms and delayed disease progress and resulted in about a 1.5-fold-higher survival rate than in the HD5 group. Furthermore, the E21R variant exhibited a 2-fold-higher antiviral potency against HIV-1 over parental HD5 in vitro. This study demonstrates that arginine mutagenesis at appropriate evolution sites may significantly enhance the antiviral activity of HD5, which also paves a facile way to search for potent antiviral drugs based on natural antimicrobial peptides.
INTRODUCTION
Herpes simplex virus (HSV) is highly prevalent worldwide and plays an important role in fueling the human immunodeficiency virus (HIV) epidemic (1). In the United States, the seroprevalences of HSV-1 and HSV-2 infection have been estimated to be approximately 50% and 20%, respectively (2). HSV-2 is generally associated with primary and recurrent vesicular lesions of the anogenital region. Noticeably, HSV-2 infection may result in severe outcomes in immunocompromised patients. As reported previously, in patients with HIV infection, HSV-2 incursion can lead to disseminated visceral infections, such as esophagitis, hepatitis, and pneumonia, as well as meningoencephalitis (3). The traditional drugs for the treatment of HSV-2 infection are usually derived from nucleoside analogs. Unfortunately, in recent years, reports pertinent to HSV resistance to nucleoside analogs have dramatically increased (4). Therefore, it is urgent to develop new antiherpetic agents with alternative mechanisms of action.
Defensins, a kind of endogenous antimicrobial peptide enriched with cysteine (Cys) and arginine (Arg), are widely distributed in internal organs and recognized as defenders against invasion by pathogenic organisms. Human alpha-defensin 5 (HD5) contains 32 amino acids and shares a common structural feature of the alpha-defensin family characterized by three-stranded antiparallel β-sheets stabilized by three intramolecular disulfide bridges (5). HD5 is primarily expressed in small intestinal Paneth cells and epithelial cells of the urogenital tract (6–9). In previous studies, HD5 was demonstrated to have broad-spectrum activity against Gram-positive and Gram-negative bacteria (10). Recently, it was found that HD5 also displays particular activity against various pathogenic viruses, such as HSV, human papillomavirus, adenovirus, and polyomavirus (11–13). The distribution and instinctive antiviral activity make it a potential candidate for an alternative antiviral agent to control sexually transmitted diseases, although the mechanism of action of HD5 is yet to be fully elucidated.
Notably, most alpha-defensins, including HD5, possess antiviral potency that is relatively lower than their bactericidal activity (14). Therefore, the antiviral ability of HD5 for treating virus infections is expected to be improved. As presumed, the mammalian alpha-defensins likely derive from an ancestor, and the formation of individual alpha-defensins with variable antimicrobial activities is just a result of naturally selective evolution against environmental microbes (15, 16). Except for the commonly conserved residues and structural determinants, 14 amino acid sites of mammal alpha-defensins are predicted to be subject to positive selection (16). Based on the positive Darwinian evolution, we hypothesized that the virucidal potency of HD5 may be enhanced through direct mutagenesis of the amino acids at the adaptive evolution sites. It is known that as a kind of cationic peptide, positively charged Arg residues are crucial for the function of HD5, because the antibacterial activity of HD5 could be markedly impaired or attenuated when the Arg residues are replaced with other amino acids (17). However, the effect of Arg substitution of amino acid residues at adaptive evolution sites on the antiviral activity of HD5 has not yet been studied.
In this study, we designed and synthesized a series of HD5-derived peptides based on strongly Arg-selected bias at the positive evolution sites. A comprehensive anti-HSV-2 functional investigation was performed, and the related virucidal mechanism was also explored. Remarkably, it was shown that the E21R mutation could significantly enhance the anti-HSV-2 efficacy of HD5 without increasing cytotoxcity. We also found, for the first time, that the antiviral activity of HD5 by inhibiting viral adhesion and entry to cells might be related to the interfering effect on HSV-2 capsid protein glycoprotein D (gD), and that the enhanced binding affinity of E21R-HD5 with gD might contribute to the improved anti-HSV-2 activity of E21R-HD5. Furthermore, E21R-HD5 showed a higher efficacy in blocking HSV-2 as a prophylactic or therapeutic agent in a mouse model of genital infection than did natural HD5. In addition, an enhanced anti-HIV-1 activity of E21R-HD5 was also observed in vitro. The results obtained in this study provide an alternative way to find novel antiviral drugs based on endogenous antimicrobial peptides.
MATERIALS AND METHODS
Cells, viruses, and reagents.
Vero African green monkey kidney cells (CCL 81), CaSki human cervical epithelial cells (ATCC CRL-1550), and HSV-2G (ATCC VR-734) were purchased from the China Type Culture Collection. JLTRG cells were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (catalog no. 11587), from Olaf Kutsch. C34 peptide was ordered from the NIH AIDS Reagent Program, and H9/HIVHTLV-IIIB cells were purchased from the ATCC (catalog no. CRL-8543). Glycosylated recombinant HSV-2 capsid protein D (gD) was purchased from Meridian Life Science, Inc. (Memphis, TN).
Peptide preparation.
The sequences of HD5 and the mutants (T7R-HD5, L16R-HD5, E21R-HD5, and S23R-HD5) used in this study are described in Fig. 1 (upper and middle portions). All peptides were custom synthesized, folded, and purified by Chinese Peptide Ltd. Corp. (Hangzhou, China) according to previously reported methods (18). Briefly, machine-assisted solid-phase chemical synthesis of peptides was adopted on Boc-Cys (4MeBz)-OCH2-PAM resin using an in-house chemistry tailored from the N,N-diisopropylethylamine in situ neutralization/2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate activation protocol (19). Oxidative folding and disulfide formation of crude peptides were carried out in the presence of reduced (3 mM) and oxidized (0.3 mM) glutathione, 2 M urea, and 25% N,N-dimethylformamide, pH 8.3 (18). All peptides were purified to homogeneity by preparative reverse-phase high-performance liquid chromatography (RP-HPLC), and their molecular masses were ascertained by electrospray ionization-mass spectrometry (ESI-MS). The peptides were quantified by measuring the UV absorbance at 280 nm using molar extinction coefficients calculated according to a published algorithm (20).
Fig 1.
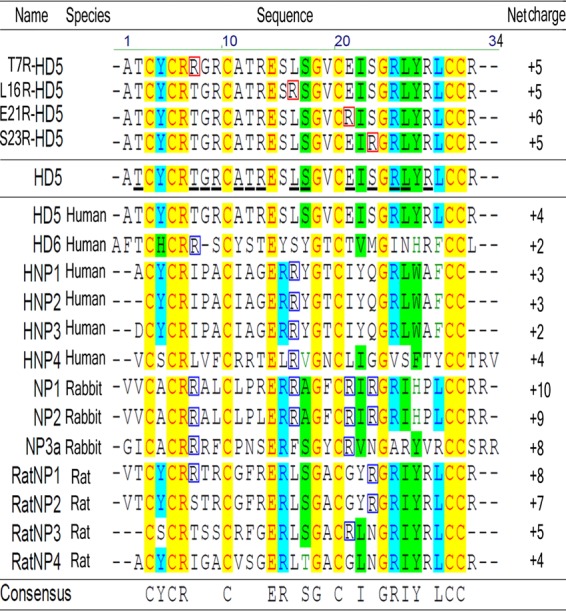
Multiple-amino-acid alignment of HD5 analogs and selected human, rabbit, and rat antiviral alpha-defensins (http://aps.unmc.edu/AP/main.php). The numbering on the top is that of HD5. Notice that all natural antiviral defensins and HD5 analogs contain conserved amino acids at special sites (in light red against a yellow background). (Upper portion) HD5 analogs contain mutated Arg residues boxed in red. (Middle portion) HD5 harbors 14 residues undergoing positive evolution (underlined in black) predicted by maximum likelihood methods (16). Lower portion, sequence characteristics of natural antiviral defensins derived from different mammals. The Arg residue distributed in at least four kinds of antiviral defensins corresponding to the variable sites in HD5 is boxed in blue.
Cytotoxicity.
Cell Counting Kit-8 (CCK-8), based on 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfopheny l)-2H-tetrazolium (WST-8) (Dojindo Laboratories, Kumamoto, Japan), was used according to the manufacturer's instructions. Briefly, CaSki cells, JLTRG cells, or H9/HIVHTLV-IIIB cells (5,000 cells/well) were added to 96-well tissue culture plates (Costar, USA) and incubated in 100 μl of Dulbecco's modified Eagle's medium (DMEM) containing 2% heat-inactivated fetal bovine serum (FBS) for 12 h at 37°C. Serial 2-fold dilutions of peptides (10 μl), diluted stock solution in 0.01% acetic acid-deionized water with phosphate-buffered saline (PBS) (pH 7.4), were added to 90 μl of DMEM. Cells were maintained in the mixed media with final peptide concentrations ranging from 100 to 6.24 μg/ml for 36 h. Thereafter, 10 μl of CCK-8 solution was added to each well, and cells were continuously incubated for 3 h. Then, the absorbance of each cell well was measured at 450 nm with a microplate reader. Optical density values were calculated with SOFTmax PRO software (Molecular Devices).
Assays of cell protection against HSV-2.
Viral titers were tested by infecting CaSki cell monolayer with 10-fold serially diluted viral inocula and incubating the cells in medium supplemented with 2% heat-inactivated FBS for 72 h at 37°C. A viral dilution which caused about 75% cell death as measured by the CCK-8 kit described for the cytotoxicity assay was used in the inactivation experiments. The corresponding multiplicity of infection (MOI) and PFU/well values were approximately 0.04 and 1 × 103, respectively, for HSV-2. CaSki target cells were seeded in 96-well plates at 2 × 104 cells/well and incubated in DMEM with 10% heat-inactivated FBS for 12 h at 37°C. In the screening assays, the virus was preincubated in serum-free DMEM with different concentrations of peptides for 2 h at 37°C before being added to the target cells. Virus-free (peptide alone) or peptide-free (virus alone) cell wells were used as controls. To initiate infection, the original medium was replaced with 100 μl/well of the peptide-treated viral inoculum or serum-free medium. The cells were maintained for 3 h at 37°C and then transferred into 2% heat-inactivated FBS-DMEM for another 72-h incubation. Finally, the cell viability was measured with the CCK-8 kit. The cell protection rate was calculated to assess antiviral activity of peptides as previously described (21).
Anti-HSV assays.
All peptides were evaluated for their antiviral potential. Peptides were tested in four different stages of HSV-2 infection: preinfection, during adhesion, during entry, and postentry. Viral infection without peptide treatment was taken as an infection control; heparin (100 μg/ml) and acyclovir (100 μg/ml) were included as positive controls for adhesion and postentry inhibition, respectively.
Pre-HSV-2 infection.
To explore whether the HD5 mutants act on HSV-2 and/or cells, both the virus and CaSki cells were treated individually with each peptide prior to infection and assessed for viral titers. The effect of peptides on virus was determined by mixing 100 μl virus solution containing ∼200 PFU of virus with individual peptides made up to a final volume of 1 ml with DMEM and by incubating the mixture at 37°C for 2 h. The final concentration of each peptide was 50 μg/ml. The inoculation was then added to CaSki cells cultured to 75% confluence in 12-well plates and maintained at 37°C for 3 h in a humidified atmosphere containing 5% CO2. Next, the virus inoculum was aspirated and washed twice with fresh medium. After that, 2% methylcellulose medium containing 2% heat-inactivated FBS was added to block spreading of virus through the cell culture medium. Seventy-two hours later, plates were washed and stained with crystal violet for enumeration of plaques. Similarly, the effect of peptides on CaSki cells was analyzed by treating cells with each of these peptides for 2 h at the same concentrations as specified above. Following incubation, the peptide mixture was aspirated and cells were washed twice with fresh DMEM. These treated cells were then inoculated with a 1-ml mixture of HSV-2 (∼200 PFU) for 3 h at 37°C. Viral titers were measured by plaque assay as previously described. By reference to the number of plaques observed in virus control monolayers without addition of peptides, the concentration of test peptide which inhibited plaque numbers by 50% (IC50) was determined from dose-response curves (22).
HSV-2 adhesion stage.
A total of 100 μl of virus (∼200 PFU) was mixed with an individual peptide (final concentration of 50 μg/ml) and made up to 1 ml with fresh medium, and the mixture was added directly to CaSki cell monolayers precooled to 4°C in 12-well plates. The cells were maintained for 3 h at 4°C, a temperature permissive for viral adhesion but not entry. Unbound virus was removed by washing three times with fresh medium. The cell culture plates were then incubated in 2% heat-inactivated FBS-DMEM at 37°C for 72 h. The cell monolayer was stained with crystal violet for viral plaque counting as described above.
HSV-2 entry stage.
Cells were bound in a synchronized fashion by virus as described above and washed three times in PBS at 4°C. One milliliter of medium with 50 μg of an individual peptide was added to the cells. For penetration of virus, the cell culture plates were instantly shifted to 37°C for 30 min. Then, the cells were washed twice with fresh medium and incubated in 2% heat-inactivated FBS-DMEM at 37°C for 72 h. Similarly, virus infectivity was assessed by plaque assay as described above.
Postentry stage of HSV-2.
The CaSki cell monolayer which had undergone simultaneous penetration of virus as described above was washed with a citrate buffer (pH 3.0) to inactivate any nonpenetrant virus. The cells in 12-well plates were then incubated in 1 ml of DMEM containing 50 μg of peptides at 37°C for 2 h. Afterward, the cells were washed in PBS and maintained in 2% heat-inactivated FBS-DMEM at 37°C for another 72 h. Virus particles in each well were then quantified by the plaque assay as described above.
HSV-2 adhesion studies.
Purified HSV-2 was pretreated with peptides (50 μg/ml), heparin (100 μg/ml), acyclovir (100 μg/ml), or PBS (as a control) at 37°C for 2 h and then inoculated to precooled CaSki cells for 3 h at 4°C (MOI, about 1 PFU/cell). Cells were washed twice with PBS to remove nonadhered virus. Cell-bound virus was analyzed by Western blotting, using cell lysates and an anti-gD monoclonal antibody (Meridian Life Science, Memphis, TN). Western blots were scanned and analyzed using the GELDOC 2000 Bio-Rad system.
VP16 and ICP27 analysis.
Synchronized infectivity assays were performed as described above, using an MOI equivalent to 1 PFU/cell. Certain final concentration of peptide (50 μg/ml), heparin (100 μg/ml), or acyclovir (100 μg/ml) was added either when cells were shifted from 4°C to 37°C (to initiate the viral entry period) or immediately after cells were washed with citrate buffer (postpenetration). The cells were maintained for 2 h and then overlaid with fresh medium. Viral infection was monitored by detecting transport of VP16 or expression of an infected cell protein (ICP27). Nuclear extracts were prepared 3 h after citrate treatment to examine transport of VP16 to the nucleus; alternatively, cell lysates were prepared 24 h after citrate treatment to detect ICP27 expression. Western blotting was performed as described for adhesion gels.
In vivo efficacy studies against HSV-2.
Female BALB/c mice (weight, 18 to 20 g; 7 weeks old) were subjected to infection and/or treatment with the approval of the ethics committee for animal experiments of the Third Military Medical University. All the mice were pretreated with progesterone (Pharmacia) subcutaneously (2 mg/mouse in a volume of 50 μl). Five days later, each mouse was anesthetized and inoculated with 20 μl of virus solution containing a lethal dose of HSV-2 at 1 × 105 PFU (23). Prior to or after viral inoculation, mouse received an intravaginal instillation of 20 μl of peptide solution (final peptide concentration, 10 mg/ml in PBS) as a prophylactic or therapeutic treatment. The control mice were treated with PBS alone. Genital pathology was monitored daily after viral challenge. The severity of disease was graded from 0 to 5 on the basis of no apparent abnormality (0), slight redness of the external vagina (1), redness and swelling of the external vagina (2), severe redness and swelling of the external vagina with hair loss of surrounding tissue (3), genital ulceration with severe redness (4), and severe genital ulceration extending to surrounding tissue (5) (24). The mice of parallel experiment groups were sacrificed at day 7 postinfection, and their spinal cords were collected and frozen in 0.5 ml of PBS.
HSV-2 viral load analysis.
The mice spinal cords were homogenized and DNA was extracted using an ultrapure DNA isolation kit according to the manufacturer's protocol (TaKaRa, Dalian, China). Quantitative PCR was performed as previously described (25) to analyze the HSV-2 DNA load in each infected mouse.
DPI.
The dual-polarization interferometry (DPI) instrument (Farfield AnaLight BIO200) used for the experiments consists of a dual-slab waveguide sensor chip illuminated with alternately polarized light. All measurements were performed according to the AnaLight protocol (version 0.1). Briefly, an unmodified AnaChip was silanized with amino silanes and activated using the amino-to-amine linker BS3 as described previously (26). Then, to immobilize HSV-2 gD protein on the sensor surface, 250 μl of gD protein, at 0.5 mg/ml in phosphate-buffered saline supplemented with 0.01% Tween 20 (PBS-T, pH 7.4), was injected at 50 μl/min over the sample channel, followed by washing with PBS (pH 7.4) and blocking of any unreacted linker or surface groups with 100 mM ethanolamine in PBS-T (pH 8.0). Once the response of chip had stabilized, 125 μl of each peptide on 2-fold-increasing concentrations from 0.3906 to 100 nM in PBS-T was injected over the sensor surface at 25 μl/min consecutively, with intervening 125-μl injections of PBS-T washing buffer at a flow rate of 25 μl/min. The sensor response was recorded and the results were analyzed using the DPI explorer software.
Anti-HIV-1 assay.
Jurkat T cell-derived JLTRG cells which express CD4 and CXCR4 (27), and H9 cells chronically infected with HIVHTLV-IIIB, which gradually release HIV-1 particles into the culture medium, were individually maintained at an average density of 0.5 × 106 to 1× 106 cells/ml in RPMI 1640 (Invitrogen, USA) supplemented with 10% heat-inactivated FBS. To evaluate the anti-HIV activity of test peptides, JLTRG cells were seeded into 24-well tissue culture plates at a cell density of 1.0 × 105/well in RPMI 1640 containing 2% heat-inactivated FBS (27) and were preincubated with serially 2-fold-diluted HD5, E21R-HD5 (0.39063 to 50 μg/ml), or C34 (0.00064 to 3.2 μg/ml) peptide for 1 h, followed by addition of 1.0 × 104 H9/HIVHTLV-IIIB cells/well and further coculture for 3 days. As JLTRG cells harbor a stably integrated enhanced green fluorescent protein (EGFP) gene driven by an HIV-1 long terminal repeat (LTR) promoter, the levels of viral infection and propagation can be determined by flow cytometry using EGFP expression as a readout (28).
Statistical analysis.
Each experiment was performed at least three times in triplicate, and the data, expressed as means ± standard deviations (SD), were calculated with SPSS software (SPSS for Windows, version 11.0, Chicago, IL). Statistical analyses were conducted using a two-tailed Student t test, and differences were considered to be significant when the P value was <0.05.
RESULTS
Characteristics of synthesized HD5 analogs.
The alignment of human, rabbit, and rat antiviral alpha-defensin sequences (http://aps.unmc.edu/AP/main.php) indicated ∼30% similarity of these peptides among different animals, with the conserved residues of Cys at positions 3, 5, 10, 20, 30, and 31, Arg at position 6, Glu at position 14, and Gly at positions 18 and 24 and the feature of strongly selected Arg (Fig. 1, lower portion). Except for these conserved residues, which are of crucial importance for the structural stability of the molecules (5, 17), the other residues remain free to vary. In fact, 14 positive evolution sites (Fig. 1, middle portion) in HD5 were predicted by maximum likelihood methods (16). Each variable residue at these evolutional sites should be related to bioactivity of the peptide. Interestingly, we found that among the selected antiviral defensins, Arg at a high frequency (at least 23%) was selectively distributed at 4 out of the 14 variable sites corresponding to HD5 (Fig. 1, lower portion). To evaluate the antiviral activities, four mutants were designed by replacing Thr-7, Leu-16, Glu-21, and Ser-23 in HD5 with Arg and were successfully synthesized and prepared according to the previously described protocol (18). The five peptides used in this study, i.e., HD5, T7R-HD5, L16R-HD5, E21R-HD5, and S23R-HD5, were analyzed and identified by RP-HPLC and ESI-MI (see Fig. S1 in the supplemental material). The determined molecular mass of each peptide was within experimental error of the theoretical value calculated on the basis of the average isotopic compositions of folded peptides. It is worth noting that T7R-HD5, L16R-HD5, and S23R-HD5 are remarkably less hydrophobic (more polar) than HD5 and E21R-HD5, as indicated by their relative retention on C18 RP-HPLC (see Fig. S1).
Potential for cell protection from HSV-2 infection.
As expected, none of the peptides displayed marked cytotoxicity to CaSki cells, even when tested at 100 μg/ml (data not shown). Afterward, screening assays of cell protection were performed by a recently described CCK-8 method. Peptides were tested at serial concentrations of 5, 10, 25, 50, and 100 μg/ml, and the viral inoculum was preincubated for 2 h with individual peptides. The results indicated that all five peptides were substantially active in terms of inactivating HSV-2 in a dose-dependent manner (Fig. 2). The mutants of T7R-HD5 and S23R-HD5 were approximately as effective as HD5. Intriguingly, the variant of E21R-HD5 was significantly more active than HD5, showing the cell protection rates of 61.5% ± 7.1% and 90.2% ± 4.9%, while HD5 had values of 47.8% ± 3.9% and 69.4% ± 5.4% at 25 and 50 μg/ml, respectively (P < 0.05). In contrast, the L16R-HD5 mutant was less effective against HSV-2, affording only 51.2% ± 7.2% protection rates even at 100 μg/ml.
Fig 2.
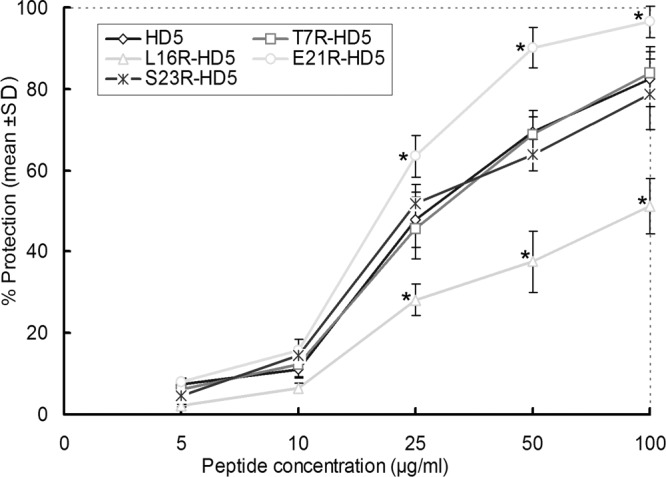
Protection of CaSki cells from HSV-2 infection. Different peptides at 5, 10, 25, 50, and 100 μg/ml were incubated with the virus for 2 h and then added to CaSki cell monolayers. Plates were incubated at 37°C for 72 h, and cell viability was measured with a CCK-8 kit. Data are presented as means ± SD of three to five experiments at each concentration of individual peptide. Asterisks identify the peptide whose activity against HSV-2 differed significantly from that of HD5 at the indicated concentration (P < 0.05).
HSV-2 neutralization activity during the preinfection stage.
To evaluate the virucidal activity of the peptides, the virus inactivation potential of all five peptides was tested. The results of these experiments revealed that preincubation of 50 μg/ml of peptide with HSV-2 was able to significantly decrease the virus titers by 40 to 80%, compared to the virus control without peptide (Fig. 3). Inspiringly, the E21R-HD5 mutant possessed obviously enhanced antiviral activity, with about 40% viral plaque reduction relative to that by natural HD5, whereas the L16R-HD5 peptide displayed weakened virucidal potential (Fig. 3). In contrast, none of the five peptides preincubated only with cells significantly reduced the virus titer compared to the control groups (P > 0.05; data not shown). Thus, the strategy of 2 h of preincubation of peptides with viral particles was adopted to evaluate the neutralization efficacy of each peptide at sequential 2-fold dilutions with the initial concentration of 100 μg/ml. HSV was neutralized by HD5 mutants in a dose-dependent manner, with 50% inhibitory concentrations (IC50) of 35, 40, 67, 25, and 36 μg/ml for HD5, T7R-HD5, L16R-HD5, E21R-HD5, and S23R-HD5, respectively.
Fig 3.
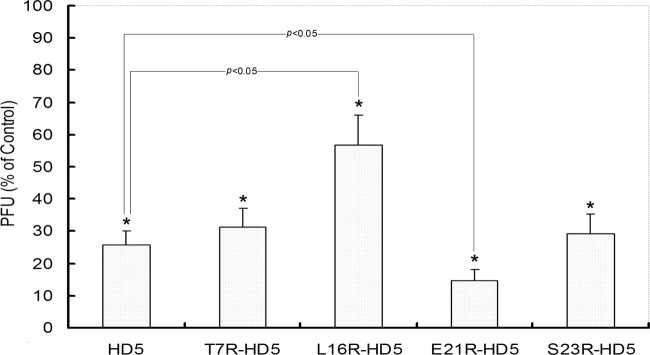
HD5 mutants block HSV-2 infection during the preinfection stage. HSV-2 particles (∼200 PFU) were preincubated with individual peptide (50 μg/ml) for 2 h at 37°C and then inoculated with CaSki cells. After another 72 h of culture, titers were measured by counting plaques. HSV-2 infection without the peptides was included as a control. All peptides demonstrated significant virus-inactivating potential (indicated by the asterisks) compared to the control (P < 0.05). Compared to natural HD5, E21R-HD5 exhibited obviously enhanced antiviral activity, while L16R-HD5 showed reduced potential (P < 0.05).
Temporal characterization of anti-HSV-2 activity.
To further assess in which stage of the virus infection the peptides exert antiviral activity, we performed synchronized experiments to analyze the antiviral activity in the viral infection course. The HSV infection process can be experimentally controlled and divided discriminately into adhesion stage (occurs at 4°C), entry stage (occurs after a shift from 4°C to 37°C), and postentry stage (begins when washing cells with acidic buffer to inactivate any bound but nonpenetrant virus) (13). Accordingly, the peptides were added during adhesion (for 3 h at 4°C), entry (for 30 min at the time of temperature shift), or instantly postentry for 3 h. All the peptides tested significantly blocked adhesion and penetration (Fig. 4). Curiously, the E21R-HD5 mutant demonstrated obviously increased potency to inhibit viral infection during either the adhesion or penetration period, compared to natural HD5. In contrast, heparin inhibited significant HSV plaque formation only when present during the adhesion period, whereas acyclovir was effective only if present postentry.
Fig 4.
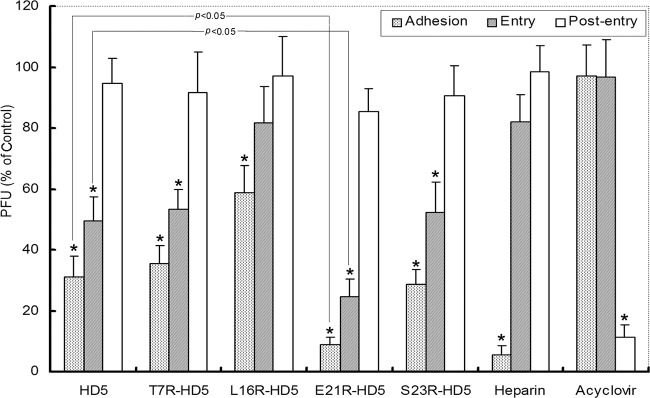
Synchronized infectivity assays demonstrated that HD5 analogs inhibited viral adhesion and entry to target cells in the viral life cycle. CaSki cells were cooled to 4°C and incubated with ∼200 PFU/well at 4°C for 3 h. After the unbound viruses were washed off, the cultures were shifted to 37°C for 30 min to admit viral entry. Adhered but nonpenetrant viruses were inactivated by washing the monolayer with a pH 3.0 buffer, after which the cells were overlaid with 2% methylcellulose-DMEM. Plaques were numerated 72 h postinfection. Peptides (50 μg/ml), heparin (100 μg/ml), acyclovir (100 μg/ml), or control buffer was added at the onset of each infection step. The bars display PFU formed in the presence of peptides as a percentage of PFU formed in the control wells and represent the means ± SD of three independent experiments. All five peptides significantly inhibited viral adhesion and/or entry (indicated by the asterisks) (P < 0.05). E21R-HD5 showed significantly enhanced antiviral efficacy compared to that of natural HD5 (P < 0.05). Heparin only blocked adhesion, and acyclovir inhibited infection only when added in the postentry stage.
To more specifically delineate the steps in HSV infection inhibited by the peptides, we conducted synchronized experiments and monitored infection by comparing the relative numbers of viral particles adhered (binding), the nuclear transport of HSV tegument protein VP16 (an important marker of entry), and the expression of ICP27 in the absence or presence of each peptide. Consistent with the plaque assays (Fig. 4), each of the peptides tested reduced viral adhesion and entry, whereas heparin and acyclovir only, individually, inhibited viral binding, replication, and expression of viral genes (Fig. 5). Inspiringly, the results obtained from the synchronized plaque and the Western blot assays supported the contention that E21R-HD5 had greater virus-blocking effects than natural HD5 in either the adhesion or entry stage.
Fig 5.

Effects of peptides (50 μg/ml) on adhesion (a), nuclear transport of VP16 (b), and expression of the ICP27 gene (c). Heparin (100 μg/ml), acyclovir (100 μg/ml), and PBS (pH 7.4) were used as controls. (a) CaSki cells were cooled to 4°C and exposed to HSV-2 preincubated with peptide for 2 h at 37°C (MOI, 1 PFU/cell) and then maintained at 4°C for 3 h. Bound virus was detected by analyzing Western blots of cell lysates for gD. (b) CaSki cells were cooled to 4°C and inoculated with virus (1 PFU/cell) at 4°C for 3 h. The cultures were washed with PBS to remove the unbound viruses and incubated with peptides at 37°C for 3 h. Then, nuclear extracts were eluted and analyzed for nuclear VP16. (c) CaSki cells with synchronized infectivity and citrate buffer treatment were incubated with peptides for 5 h and cultured in fresh medium supplemented with 2% heat-inactivated FBS. Cell lysates were prepared 24 h after entry of virus. Expression of ICP27 was determined by Western blotting.
Interaction of gD with HD5 analogs.
All the HD5 analogs tested blocked viral binding and penetration (Fig. 4). To obtain insight into the mechanism, we studied the binding of peptides to HSV-2 glycoprotein D (gD). Glycoprotein D is one of the essential molecules for HSV binding to and penetration of cells, especially for viral entry (1). Therefore, we examined the ability of HD5 analogs to interact with recombinant gD from HSV-2 using DPI. The experiments showed that each of the five HD5 analogs bound to gD, with increases in thickness and mass and a decrease in density with addition of 2-fold-increased concentrations from 0.3906 nM to 100 nM on a gD protein-functionalized sensor chip surface (Fig. 6a to e). Based on the mass change characteristics absorbed on the chip surface, the equilibrium dissociation constant (KD) for each HD5 analog and gD was calculated using the DPI explorer software (29, 30). The values of KD were 5.14 ± 0.51 nM (HD5), 6.69 ± 0.48 nM (T7R-HD5), 25.05 ± 3.23 nM (L16R-HD5), 2.09 ± 0.26 nM (E21R-HD5), and 8.52 ± 1.87 nM (S23R-HD5) (Fig. 6a to e). Surprisingly, E21R-HD5 possessed 2-fold-stronger binding affinity for gD than natural HD5, while L16R-HD5 had a 5-fold-weaker affinity (Fig. 6f). The efficiencies in inhibiting HSV-2 binding and entry of HD5 analogs may be at least partly dependent on their affinity for the capsid protein gD.
Fig 6.

Analysis of the deposition of HD5 mutant overlay on the HSV-2 gD protein-functionalized sensor surface (a to e) and comparative analysis of equilibrium dissociation constant (KD) for HSV-2 gD and HD5 analogs (f). The DPI graph contains nine steps, where each step contains one uphill association curve and one downhill dissociation curve. The downhill curves are not very apparent due to the slow dissociation process. The nine steps correspond to nine different concentrations of each HD5-analog peptide, from 0.3906 nM to 100 nM, with 2-fold-increasing concentrations in that order. Changes in mass, density, and thickness at the surface are shown, with increased tendencies of mass and thickness and a decrease in density to a different extent for each HD5 analog. The KD value of each HD5 analog and HSV-2 gD was calculated using the DPI explorer software, based on the mass changes absorbed on the chip surface (30). Asterisks indicate significant difference compared to HD5 (P < 0.05).
In vivo protection from HSV-2 infection.
Given these encouraging in vitro results, we chose a well-known murine genital herpes model to test the ability of the peptides to protect cells against HSV-2 in vivo. The E21R-HD5 mutant and natural HD5 were focused on in these experiments for their prominent anti-HSV activity in vitro. By intravaginal instillation, 0.2 mg of each peptide in 20 μl of PBS was administered as a single prophylactic dose 1 h prior to challenge or as a single therapeutic dose 24 h postinfection. Inspiringly, both E21R-HD5 and HD5 demonstrated a strong antiviral activity, no matter by prophylactic or therapeutic administration (Fig. 7). Compared to the controls (only 10% survival rate), significantly increased rates of survival were conferred by the peptides E21R-HD5 (70% and 60%) and HD5 (50% and 40%) given before and after HSV-2 infection, respectively (P < 0.05) (Fig. 7a). Treatment with the peptides reduced both the onset and the severity of the disease (Fig. 7b), which was associated with at least 60-fold reduction of viral load in the spinal cords (Fig. 7c). We did not find any significant difference in viral load or disease scores among the peptide-treated groups (Fig. 7b and c). Remarkably, there was a strong trend that mice treated with E21R-HD5 by prophylactic administration developed fewer clinical signs and light severity of the disease (Fig. 7b), with more reduction in viral DNA content (Fig. 7c). This supported the finding that more mice that received prophylactic treatment with E21R-HD5 survived in this experimental setting (Fig. 7a).
Fig 7.

Effects of local prophylactic or therapeutic administration of HD5 or E21R-HD5 peptides on protection of mice from lethal vaginal HSV infection. All groups of female BALB/c mice (n = 10 per group) were intravaginally inoculated with the lethal dose of 105 PFU/mouse of HSV-2. The mice were administered by a single-dose intravaginal instillation of 20 μl of PBS solution containing 0.2 mg of HD5 or E21R-HD5 peptide 1 h prior to challenge (indicated by dots) or 24 h postinfection. Control animals were treated with PBS alone at the same time point. The animals were observed for any signs of disease for 14 days postinfection. Panels a and b show survival percent and mean pathology score following prophylactic or therapeutic treatment of the peptides, respectively. Panel c shows HSV-2 genome copies in the spinal cord determined by quantitative PCR on day 7. Data are expressed in log scale as medians and the 25% and 75% percentiles (boxes) with the minimum and maximum responses (n = 5 per group).
Anti-HIV-1 activity.
To explore the potential activity of HD5 and its mutant against HIV-1, Jurkat T cell-derived JLTRG cells (1.0 × 105/well) (27) were grown in RPMI 1640 medium and preincubated with serial 2-fold-diluted HD5, E21R-HD5 (0.39063 to 50 μg/ml), or C34 (0.00064 to 3.2 μg/ml) peptide for 1 h, followed by addition of 1.0 × 104 H9/HIVHTLV-IIIB cells/well and further coculture for 3 days before flow cytometric analysis of the levels of EGFP-expressing cells that reflect the viral infectivity and, inversely, the antiviral activity of test peptides (28). Clearly, the test peptides showed noteworthy anti-HIV-1 activities without observable cell cytotoxicity. E21R-HD5 possessed a >2-fold-higher antiviral potency against laboratory-adaptive strain HIVHTLV-IIIB over its parental peptide, HD5, with IC50 of 3.53 μg/ml and 7.51 μg/ml, respectively (Fig. 8). The IC50 for the positive-control peptide C34 was determined to be 0.005 μg/ml in our experimental settings, which is consistent with the previously published value of 0.003 μg/ml (31).
Fig 8.

Quantification of inhibiting activity of HD5 and E21R-HD5 against strain HIVHTLV-IIIB. JLTRG cells (1 × 105) grown in 24-well plates were preincubated with 2-fold serially diluted HD5 or E21R-HD5 peptide (0.3906 to 50 μg/ml) or positive-control peptide C34 (0.00064 to 3.2 μg/ml) for 1 h. H9/HIVHTLV-IIIB cells (1 × 104) were then added to initiate viral infection, and the coculture was maintained for 3 days. Thereafter, the cell samples were analyzed for EGFP expression using flow cytometry. Data presented are means of two independent experiments, with triplicate samples for each point (C34 [a], HD5, and E21R-HD5 [c]). Percent inhibition was calculated by using the formula 100 × [1 − (net percentage of cells EGFP+ × mean fluorescence intensity value of cells EGFP+ in test group)/that in mock group].
DISCUSSION
Recurrence and prevalent transmission of HSV infection remain a major global health problem. Furthermore, the increase in HSV strains resistant to acyclovir-like nucleoside analogs urgently demands development of novel antiviral agents (4). In this study, four synthesized novel HD5 mutants were examined for their activities against HSV-2 subtype viruses (in vitro and in vivo). Their efficacy was demonstrated by the high binding affinity for HSV-2 capsid protein gD and broad neutralization of virus particles. The lack of toxicity in CaSki cells confirmed that their action was due to their efficacy instead of cell toxicity. Noticeably, the E21R-HD5 mutant exhibited enhanced inhibition of HSV-2 adhesion to and entry into target cells in vitro and was more efficient at preventing or treating HSV-2 infection in vivo, with a favorable 50% inhibition concentration. Here, we showed a facile method to endow E21R-HD5 with enhanced antiviral activity by a single Arg substitution.
Up to now, more than 1,500 antibacterial and 100 antiviral peptides from plants, insects, mammals, and humans have been identified (http://aps.unmc.edu/AP/main.php), which are often classified into amphipathic α-helical and β-sheet peptides. Especially, defensins with a β-sheet structure have been gaining much interest because of their antiviral property against HSV (13). Studies of the relationship between structure and function suggested that sequence determinant and secondary structure may be two factors crucial to antiviral activity (32, 33). Interestingly, alpha-defensins derived from different mammals had only approximately 30% sequence identity (Fig. 1). Apart from the conserved residues of Cys at positions 3, 5, and 10, Arg at position 6, Glu at position 14, and Gly at positions 18 and 24, which stabilize the peculiar secondary structure, the other residues vary freely. This may explain why different defensins show different antiviral efficacies and strain specificities. In addition, these sequence features are just the results of natural evolution occurring in struggles with fast-evolving microbes. Thus, we speculate that the mutation of amino acids at specific sites may alter the antiviral potency of defensins. A previous study indicated that HD5 mutants with Ala or charge-neutral Lys substituted for the reserved Arg residue were at least partly deprived of antibacterial potential (17), which suggested that this Arg residue in HD5 may play an important role in its antibacterial function (14). More importantly, replacing lysines with arginines in a series of model α-helical peptides resulted in significantly higher heparan sulfate affinity and improved antiviral efficacy against HSV-2 (32). Inspiringly, through sequence analysis of antiviral defensins, we found that Arg was strongly selected at multiple sites of the evolutionary positions corresponding to HD5 (16) (shown in Fig. 1). Therefore, we designed and synthesized four mutants with Arg substitution of Thr7, Leu16, Glu21, or Ser23 at the adaptive evolution sites of HD5 to screen for anti-HSV-2 peptides with improved bioactivity.
Our results demonstrated that HD5 derivatives with a single Arg replacement possess different antiviral efficacies. This may be explained by the alteration of the antiviral determinant of the peptides, which is likely caused by the difference in side chain chemistry of amino acids (34). Our assays indicate that T7R-HD5 and S23R-HD5 have similar antiviral potencies equivalent to that of natural HD5. This may be due to the following two factors: (i) the hydroxyl provided by Thr/Ser is active in terms of formation of hydrogen bonds, equivalent to amido of Arg (5); (ii) single-site mutation, such as the replacement of Thr7 and Ser23 by Arg, which are at the first and the second β-hairpin regions of HD5, respectively, does not introduce changes in the overall structure or topological features (17). The studies related to crystal structures suggest that residues which lie in the loop region and are also exposed on the molecular surfaces should be responsible for the biological activities of alpha-defensins (5). This assumption is validated by the observation in this study that E21R-HD5 has significantly enhanced antiviral activity against HSV-2 compared to natural HD5 (Fig. 2 to 5 and Fig. 7). On the other hand, the replacement of Leu16 by Arg in HD5 resulted in weakened antiviral potency. This may be ascribed to the breakage of conserved secondary structure, because hydrophobic Leu16 instead of positively charged Arg takes part in the formation of the second β-sheet strand (5). As can be expected, the structural alignment of these mutants and wild-type HD5 can reveal detailed structural changes caused by Arg substitution and possibly further explain the relationship between structure and antiviral activity in the future.
In this study, the peptides preincubated with HSV-2 virions efficiently protected CaSki cells from infection but had little effect when only added to cells prior to inoculation. This phenomenon suggests that HD5 analogs exhibit anti-HSV activity by direct interaction with viral particles but not with target cells. Temporal antiviral assays further revealed that the peptides mainly interfere with early events of the viral infectious cycle, including adhesion and entry processes. Earlier studies indicated that HD5 binds O-linked and N-linked carbohydrate moieties on HSV-2 glycoprotein B (gB) with high affinity (13), which plays a key role in viral binding (35). This can reasonably explain the definite action of HD5 mutants in blocking HSV-2 attachment to CaSki cells in our experiments. Interestingly, to interpret how the peptides block virus entry to target cells, we conducted DPI assays, which first disclosed that HD5 analogs interacted with viral capsid protein gD at different affinities. HSV entry is a complex process that requires the concerted activities of gB, gD, and hetero-oligomers of glycoproteins H and L (gH-gL) (1). Because all of the capsid proteins of HSV are glycosylated and HD5 is a lectin that binds O-linked and N-linked glycans (36), it seems plausible that HD5 analogs may also bind other essential glycoproteins. Hereby, these interactions between peptides and viral capsid proteins may cause aggregation of virions to prevent viruses from normal binding to target cells (12). Another possibility is that the binding of HD5 derivatives to the virion envelope blocks the epitopes of interaction between viruses and cell receptors, which are necessary for viral adsorption or entry into host cells (37). Therefore, the tightened binding between E21R-HD5 and gD with higher affinity can, at least in part, explain why it has enhanced activity against HSV-2 compared to HD5.
Human peptides 1 to 4 from neutrophils and human beta-defensins 2 and 3 from epithelial cells have been reported to have anti-HIV-1 activity by acting on HIV virions and/or target cells (38–41). In this study, we found that HD5 and its variant E21R-HD5 also possess specific activity of inhibiting HIV-1 infection. Our finding is in contrast to that reported by Rapista et al. (42), who demonstrated that HD5 enhanced HIV-1 infectivity through promoting HIV attachment. This discrepancy is most likely due to the choice of different experimental systems, including virus model and target cells, as opposite results with cellulose sulfate against HIV-1 have also been reported (43, 44). Published work showing enhancement of HIV infectivity by HD5 was primarily based on assays of periodical interaction of HD5 and pseudotyped HIVJR-FL luciferase reporter virus, which is replication-defective pseudovirus deprived of the complete infection characteristics of the wild-type virus strain (42, 45, 46). In such assays, only a single round of viral replication can happen and the effect of antiviral agents on late-stage of HIV-1 life cycle cannot be meaningfully determined. In contrast, we employed an assay system in which CD4+ T cells (JLTRG) are constantly exposed to a fully replication-competent wild-type HIVHTLV-IIIB strain which may better mimic the conditions of viral infection and replication in vivo. Under our conditions, both E21R-HD5 and HD5 exhibited definitive anti-HIV activities, albeit relatively weaker than that of the C34 peptide. These seemingly controversial results highlight the complexity of the interaction among defensins, HIV, and host cells and indicate that HD5 and its derivatives may act on multiple virus or host targets and/or in multiple stages of the HIV-1 life cycle such as viral entry, reverse transcription, integration, maturation, and so on (39, 47). Since the JLTRG-H9 (HIVHTLV-IIIB infected) cell coculture system partially mimics the in vivo niche of mucosal surfaces where dendritic cells capture HIV-1 particles and present them to CD4+ T cells via cell-cell contact, our results suggest the possibility that HD5 may play a role in inhibiting HIV transmission via cell-cell contact. Nevertheless, further elucidation of its mechanism is warranted.
For in vivo experiments, dosing and timing of the peptide administration are obviously vital parameters that need to be optimized. However, based on our initial tests, we can claim that E21R-HD5 shows considerable promise as a prophylactic agent against HSV-2. The E21R-HD5 peptide efficiently prevented disease development when administered 1 h prior to HSV-2 inoculation, and it delayed both disease onset and severity when given 24 h after viral infection, consistent with the improved efficacy of blocking viral adhesion and entry into CaSki cells in vitro. Although the ability of topically administered HD5 to protect mice against an intravaginal challenge by HSV-2 has been demonstrated previously (13), ours is the first study that shows the improved antiviral characteristics of a novel human alpha-defensin 5 variant against HSV-2. In summary, our studies suggest that E21R-HD5 may be a promising microbicide against genital herpes and also provides an alternative way to search and design novel antiviral drugs based on natural antimicrobial peptide.
Supplementary Material
ACKNOWLEDGMENTS
A.W. and F.C. contributed equally to this article.
This project was supported in part by grants from the National Natural Science Foundation of China (no. 81172600, 81271898, and 30771892), Science Foundation for Distinguished Young Scholars of Zhejiang Province (no. R2100226), Academician Fund of Chongqing City (no. CSTC, 2007AB5022), Independent Research Grant of the State Key Laboratory for Diagnosis and Treatment of Infectious Diseases (no. A60903-4), and Natural Science Foundation of Chongqing City (no. CSTC, 2008BB5137).
Footnotes
Published ahead of print 26 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02209-12.
REFERENCES
- 1. Wilson SS, Fakioglu E, Herold BC. 2009. Novel approaches in fighting herpes simplex virus infections. Expert Rev. Anti Infect. Ther. 7:559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xu F, Sternberg MR, Kottiri BJ, McQuillan GM, Lee FK, Nahmias AJ, Berman SM, Markowitz LE. 2006. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 296:964–973 [DOI] [PubMed] [Google Scholar]
- 3. Glynn JR, Crampin AC, Ngwira BM, Ndhlovu R, Mwanyongo O, Fine PE. 2008. Herpes simplex virus type 2 trends in relation to the HIV epidemic in northern Malawi. Sex Transm. Infect. 84:356–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Piret J, Boivin G. 2011. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 55:459–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Szyk A, Wu Z, Tucker K, Yang D, Lu W, Lubkowski J. 2006. Crystal structures of human alpha-defensins HNP4, HD5, and HD6. Protein Sci. 15:2749–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bevins CL. 2006. Paneth cell defensins: key effector molecules of innate immunity. Biochem. Soc. Trans. 34:263–266 [DOI] [PubMed] [Google Scholar]
- 7. Com E, Bourgeon F, Evrard B, Ganz T, Colleu D, Jegou B, Pineau C. 2003. Expression of antimicrobial defensins in the male reproductive tract of rats, mice, and humans. Biol. Reprod. 68:95–104 [DOI] [PubMed] [Google Scholar]
- 8. Quayle AJ, Porter EM, Nussbaum AA, Wang YM, Brabec C, Yip KP, Mok SC. 1998. Gene expression, immunolocalization, and secretion of human defensin-5 in human female reproductive tract. Am. J. Pathol. 152:1247–1258 [PMC free article] [PubMed] [Google Scholar]
- 9. Spencer JD, Hains DS, Porter E, Bevins CL, DiRosario J, Becknell B, Wang H, Schwaderer AL. 2012. Human alpha defensin 5 expression in the human kidney and urinary tract. PLoS One 7:e31712 doi:10.1371/journal.pone.0031712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ericksen B, Wu Z, Lu W, Lehrer RI. 2005. Antibacterial activity and specificity of the six human alpha-defensins. Antimicrob. Agents Chemother. 49:269–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buck CB, Day PM, Thompson CD, Lubkowski J, Lu W, Lowy DR, Schiller JT. 2006. Human alpha-defensins block papillomavirus infection. Proc. Natl. Acad. Sci. U. S. A. 103:1516–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dugan AS, Maginnis MS, Jordan JA, Gasparovic ML, Manley K, Page R, Williams G, Porter E, O'Hara BA, Atwood WJ. 2008. Human alpha-defensins inhibit BK virus infection by aggregating virions and blocking binding to host cells. J. Biol. Chem. 283:31125–31132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hazrati E, Galen B, Lu W, Wang W, Ouyang Y, Keller MJ, Lehrer RI, Herold BC. 2006. Human alpha- and beta-defensins block multiple steps in herpes simplex virus infection. J. Immunol. 177:8658–8666 [DOI] [PubMed] [Google Scholar]
- 14. Tanabe H, Ouellette AJ, Cocco MJ, Robinson WE., Jr 2004. Differential effects on human immunodeficiency virus type 1 replication by alpha-defensins with comparable bactericidal activities. J. Virol. 78:11622–11631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Das S, Nikolaidis N, Goto H, McCallister C, Li J, Hirano M, Cooper MD. 2010. Comparative genomics and evolution of the alpha-defensin multigene family in primates. Mol. Biol. Evol. 27:2333–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lynn DJ, Lloyd AT, Fares MA, O'Farrelly C. 2004. Evidence of positively selected sites in mammalian alpha-defensins. Mol. Biol. Evol. 21:819–827 [DOI] [PubMed] [Google Scholar]
- 17. de Leeuw E, Rajabi M, Zou G, Pazgier M, Lu W. 2009. Selective arginines are important for the antibacterial activity and host cell interaction of human alpha-defensin 5. FEBS Lett. 583:2507–2512 [DOI] [PubMed] [Google Scholar]
- 18. Wu Z, Ericksen B, Tucker K, Lubkowski J, Lu W. 2004. Synthesis and characterization of human alpha-defensins 4-6. J. Pept. Res. 64:118–125 [DOI] [PubMed] [Google Scholar]
- 19. Schnölzer M, Alewood P, Jones A, Alewood D, Kent SB. 1992. In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int. J. Pept. Protein Res. 40:180–193 [DOI] [PubMed] [Google Scholar]
- 20. Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. 1995. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4:2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yasin B, Pang M, Turner JS, Cho Y, Dinh NN, Waring AJ, Lehrer RI, Wagar EA. 2000. Evaluation of the inactivation of infectious Herpes simplex virus by host-defense peptides. Eur. J. Clin. Microbiol. Infect. Dis. 19:187–194 [DOI] [PubMed] [Google Scholar]
- 22. Koch C, Reichling J, Schneele J, Schnitzler P. 2008. Inhibitory effect of essential oils against herpes simplex virus type 2. Phytomedicine 15:71–78 [DOI] [PubMed] [Google Scholar]
- 23. Parr MB, Kepple L, McDermott MR, Drew MD, Bozzola JJ, Parr EL. 1994. A mouse model for studies of mucosal immunity to vaginal infection by herpes simplex virus type 2. Lab. Invest. 70:369–380 [PubMed] [Google Scholar]
- 24. McCluskie MJ, Cartier JL, Patrick AJ, Sajic D, Weeratna RD, Rosenthal KL, Davis HL. 2006. Treatment of intravaginal HSV-2 infection in mice: a comparison of CpG oligodeoxynucleotides and resiquimod (R-848). Antiviral Res. 69:77–85 [DOI] [PubMed] [Google Scholar]
- 25. Namvar L, Olofsson S, Bergstrom T, Lindh M. 2005. Detection and typing of herpes simplex virus (HSV) in mucocutaneous samples by TaqMan PCR targeting a gB segment homologous for HSV types 1 and 2. J. Clin. Microbiol. 43:2058–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moore JD, Perez-Pardo MA, Popplewell JF, Spencer SJ, Ray S, Swann MJ, Shard AG, Jones W, Hills A, Bracewell DG. 2011. Chemical and biological characterisation of a sensor surface for bioprocess monitoring. Biosens. Bioelectron. 26:2940–2947 [DOI] [PubMed] [Google Scholar]
- 27. Jones J, Whitford W, Wagner F, Kutsch O. 2007. Optimization of HIV-1 infectivity assays. Biotechniques 43:589–590, 592,, 594 [DOI] [PubMed] [Google Scholar]
- 28. Ochsenbauer-Jambor C, Jones J, Heil M, Zammit KP, Kutsch O. 2006. T-cell line for HIV drug screening using EGFP as a quantitative marker of HIV-1 replication. Biotechniques 40:91–100 [DOI] [PubMed] [Google Scholar]
- 29. Lin S, Lee CK, Lin YH, Lee SY, Sheu BC, Tsai JC, Hsu SM. 2006. Homopolyvalent antibody-antigen interaction kinetic studies with use of a dual-polarization interferometric biosensor. Biosens. Bioelectron. 22:715–721 [DOI] [PubMed] [Google Scholar]
- 30. Tang Y, Mernaugh R, Zeng X. 2006. Nonregeneration protocol for surface plasmon resonance: study of high-affinity interaction with high-density biosensors. Anal. Chem. 78:1841–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stoddart CA, Nault G, Galkina SA, Thibaudeau K, Bakis P, Bousquet-Gagnon N, Robitaille M, Bellomo M, Paradis V, Liscourt P, Lobach A, Rivard ME, Ptak RG, Mankowski MK, Bridon D, Quraishi O. 2008. Albumin-conjugated C34 peptide HIV-1 fusion inhibitor: equipotent to C34 and T-20 in vitro with sustained activity in SCID-hu Thy/Liv mice. J. Biol. Chem. 283:34045–34052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jenssen H, Andersen JH, Mantzilas D, Gutteberg TJ. 2004. A wide range of medium-sized, highly cationic, alpha-helical peptides show antiviral activity against herpes simplex virus. Antiviral Res. 64:119–126 [DOI] [PubMed] [Google Scholar]
- 33. Trybala E, Olofsson S, Mardberg K, Svennerholm B, Umemoto K, Glorioso JC, Bergstrom T. 2004. Structural and functional features of the polycationic peptide required for inhibition of herpes simplex virus invasion of cells. Antiviral Res. 62:125–134 [DOI] [PubMed] [Google Scholar]
- 34. Xie C, Prahl A, Ericksen B, Wu Z, Zeng P, Li X, Lu WY, Lubkowski J, Lu W. 2005. Reconstruction of the conserved beta-bulge in mammalian defensins using D-amino acids. J. Biol. Chem. 280:32921–32929 [DOI] [PubMed] [Google Scholar]
- 35. Yasin B, Wang W, Pang M, Cheshenko N, Hong T, Waring AJ, Herold BC, Wagar EA, Lehrer RI. 2004. Theta defensins protect cells from infection by herpes simplex virus by inhibiting viral adhesion and entry. J. Virol. 78:5147–5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lehrer RI, Jung G, Ruchala P, Andre S, Gabius HJ, Lu W. 2009. Multivalent binding of carbohydrates by the human alpha-defensin, HD5. J. Immunol. 183:480–490 [DOI] [PubMed] [Google Scholar]
- 37. Akhtar J, Shukla D. 2009. Viral entry mechanisms: cellular and viral mediators of herpes simplex virus entry. FEBS J. 276:7228–7236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang TL, Klotman ME. 2004. Defensins: natural anti-HIV peptides. AIDS Rev. 6:161–168 [PubMed] [Google Scholar]
- 39. Chang TL, Vargas J, Jr, DelPortillo A, Klotman ME. 2005. Dual role of alpha-defensin-1 in anti-HIV-1 innate immunity. J. Clin. Invest. 115:765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eade CR, Wood MP, Cole AM. 2012. Mechanisms and modifications of naturally occurring host defense peptides for anti-HIV microbicide development. Curr. HIV Res. 10:61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Klotman ME, Chang TL. 2006. Defensins in innate antiviral immunity. Nat. Rev. Immunol. 6:447–456 [DOI] [PubMed] [Google Scholar]
- 42. Rapista A, Ding J, Benito B, Lo YT, Neiditch MB, Lu W, Chang TL. 2011. Human defensins 5 and 6 enhance HIV-1 infectivity through promoting HIV attachment. Retrovirology 8:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fletcher PS, Wallace GS, Mesquita PM, Shattock RJ. 2006. Candidate polyanion microbicides inhibit HIV-1 infection and dissemination pathways in human cervical explants. Retrovirology 3:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tao W, Richards C, Hamer D. 2008. Enhancement of HIV infection by cellulose sulfate. AIDS Res. Hum. Retroviruses 24:925–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ding J, Rapista A, Teleshova N, Lu W, Klotman ME, Chang TL. 2011. Mucosal human defensins 5 and 6 antagonize the anti-HIV activity of candidate polyanion microbicides. J. Innate Immun. 3:208–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Klotman ME, Rapista A, Teleshova N, Micsenyi A, Jarvis GA, Lu W, Porter E, Chang TL. 2008. Neisseria gonorrhoeae-induced human defensins 5 and 6 increase HIV infectivity: role in enhanced transmission. J. Immunol. 180:6176–6185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Quiñones-Mateu ME, Lederman MM, Feng Z, Chakraborty B, Weber J, Rangel HR, Marotta ML, Mirza M, Jiang B, Kiser P, Medvik K, Sieg SF, Weinberg A. 2003. Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. AIDS 17:F39–F48 doi:10.1097/00002030-200311070-00001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.