

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) has been linked to the development of Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease (MCD). We have characterized the role of KSHV-encoded viral FLICE inhibitory protein (vFLIP) K13 in the modulation of anti-IgM-induced growth arrest and apoptosis in B cells. We demonstrate that K13 protects WEHI 231, an immature B-cell line, against anti-IgM-induced growth arrest and apoptosis. The protective effect of K13 was associated with the activation of the NF-κB pathway and was deficient in a mutant K13 with three alanine substitutions at positions 58 to 60 (K13-58AAA) and a structural homolog, vFLIP E8, both of which lack NF-κB activity. K13 upregulated the expression of NF-κB subunit RelB and blocked the anti-IgM-induced decline in c-Myc and rise in p27Kip1 that have been associated with growth arrest and apoptosis. K13 also upregulated the expression of Mcl-1, an antiapoptotic member of the Bcl2 family. Finally, K13 protected the mature B-cell line Ramos against anti-IgM-induced apoptosis through NF-κB activation. Inhibition of anti-IgM-induced apoptosis by K13 may contribute to the development of KSHV-associated lymphoproliferative disorders.
INTRODUCTION
B-cell receptor (BCR)-induced apoptosis of self-reactive immature B cells is one of the defining mechanisms underlying B-lymphocyte development and immune tolerance (1). Immature B cells in the bone marrow, upon successful rearrangement of their light chains, begin to express the BCR (IgM) on their surface (1, 2). Engagement of the BCR with self-antigens results in cells arresting, undergoing receptor editing and clonal deletion (2). Several rounds of this negative selection occur before the B cells leave the bone marrow to migrate to the spleen in an effort to prevent autoreactivity. Functional studies have highlighted a role for the NF-κB pathway in the survival of immature B cells during negative selection and an absolute critical role for this pathway during B-cell survival and development in the spleen (3).
Kaposi's sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV8), is a gammaherpesvirus 2 that was originally identified in Kaposi sarcoma (KS) lesions from HIV-1-infected individuals (4). Subsequently, it was shown that KSHV is also associated with two lymphoproliferative disorders, primary effusion lymphoma (PEL) and a plasmablastic variant of multicentric Castleman's disease (MCD) (5, 6). MCD is characterized by the presence of large abnormal polyclonal plasmablasts in the mantle zones of B-cell follicles that express high levels of cytoplasmic and surface immunoglobulin that is IgM(λ) restricted (7–9).
K13 protein is one of the few KSHV proteins that are expressed in latently infected cells. K13 contains two death effector domains and was initially classified as a viral FLICE inhibitory protein (vFLIP) based on its homology to the prodomain of caspase-8/FLICE (10). However, subsequent studies by this laboratory and others have shown that K13 does not act as an inhibitor of caspase-8 but, rather, acts as a potent activator of both the classic and the alternative nuclear factor-kappa B (NF-κB) pathways (11–14). K13 successfully utilizes the NF-κB pathway to initiate an extensive range of cellular processes that promote survival, proliferation, differentiation, cytokine secretion, and oncogenic transformation and protect cells against growth factor withdrawal-induced apoptosis (15–20).
Based on the ability of K13 to strongly activate the NF-κB pathway, we postulated that it may protect autoreactive B cells from BCR-induced growth arrest and apoptosis. In this report, we examined this hypothesis by employing two different cell line models of BCR-induced apoptosis. First, we used the phenotypically immature IgM-positive cell line WEHI 231, which represents a population that is characteristically similar to immature B lymphocytes both on the basis of surface markers and biological properties (21). Second, we used the phenotypically mature Burkitt lymphoma human B-cell line Ramos to examine whether the protective effective of K13 is sustained through B-cell maturation. We show that expression of K13 in both WEHI 231 and Ramos cell lines confers protection against anti-IgM-induced apoptosis through activation of the NF-κB pathway.
MATERIALS AND METHODS
Cell culture, plasmids, and reagents.
WEHI 231 cells were grown in Dulbecco's modified Eagle's medium (Invitrogen Life Technologies) supplemented with 10% (vol/vol) fetal bovine serum and 50 μM 2-β-mercaptoethanol. Ramos cells were grown in RPMI 1640 medium supplemented with 10% (vol/vol) fetal bovine serum (both from Life Technologies). Retrovirus constructs containing C-terminal FLAG epitope-tagged wild-type and mutant vFLIP K13 and E8 have been described previously (13, 20). K13-ERTAM cells stably expressing an NF-κB-driven luciferase reporter construct (K13-ERTAM-NF-κB-Luc) were generated in WEHI 231 cells as previously described (22). The wild-type and mutant relB luciferase reporter constructs were kind gifts of X. Dong (Mayo Clinic College of Medicine, Rochester, MN) (23). The MSCV-puro-WT-Mcl-1 (where MSCV is murine stem cell virus and WT is wild type) construct was a kind gift from J. Opferman (Department of Biochemistry, St Jude Children's Hospital, Memphis, TN). All infections were carried out in the presence of Polybrene (8 μg/ml; Sigma, St. Louis, MO). Goat anti-mouse IgM, μ chain specific, and goat anti-human IgM were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA); 4-hydroxytamoxifen (4OHT) and As2O3 were from Sigma (St. Louis, MO). JQ1 was generously provided by James Bradner (Dana-Farber Cancer Institute).
Cell viability and cell cycle assays.
Cells from exponentially growing cultures were plated in an untreated, flat-bottom 96-well plate at a density of 1 × 104 cells/well in the presence and absence of anti-IgM. Cell viability was measured after 48 h using MTS reagent [3-4,5-dimethylthiazol-2yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt] following the manufacturer's instructions (Promega, Madison, WI). K13-ERTAM cells were cultured with or without 4OHT (20 nM) and treated with anti-IgM for 24 h and 48 h. DNA content analysis was performed as previously described (24). Apoptotic analysis was conducted using an Annexin V-PE Apoptosis Detection Kit I according to the manufacturer's instructions (BD Biosciences San Jose, CA) and also analyzed on a BD Biosciences LSR II instrument.
Luciferase reporter assay.
293T cells were transfected in a 24-well plate with various test plasmids along with wild-type and mutant relB luciferase reporter constructs (75 ng/well) and a pRSV/LacZ (β-galactosidase) reporter construct (75 ng/well) as described previously (11). Cells were lysed 24 to 36 h later, and extracts were used for the measurement of firefly luciferase and β-galactosidase activities, respectively. Luciferase activity was normalized relative to the β-galactosidase activity to control for the difference in the transfection efficiencies.
Western blot analysis.
Western blot analysis was performed as previously described (25). Primary antibodies used in Western blotting experiments were FLAG-horseradish peroxidase (HRP; 1:25,000) (A8592; Sigma), A20 (1:1,000) (5630s; Cell Signaling), phospho-IκBα (1:1,000; Ser32) (2859s; Cell Signaling), Mcl-1 (1:1,000) (sc-19; Santa Cruz), RelB (1:1,000) (sc-226; Santa Cruz), IκB-α (1:1,000) (sc-864; Santa Cruz), p27(1:1,000) (sc-1641; Santa Cruz), c-Myc (1:10,000) (1472-1; Epitomics), and tubulin (1:10,000) (T9026; Sigma). A mouse monoclonal antibody against the full-length K13 protein (8F6) was generated in our laboratory. For Western blots developed by Odyssey Infrared Imaging System CLx (Li-Cor Biosciences), the secondary antibodies, IRDye 800CW conjugates of goat anti-rabbit IgG (Li-Cor) and goat anti-mouse IgG (Li-Cor), were used at a 1:10,000 dilution for detection of antibody targets in the 800-nm channel. The blots were scanned and analyzed using an Odyssey Infrared scanner using Odyssey imaging software, version 2.0.
Assays for nuclear NF-κB DNA binding activity and luciferase activity.
The DNA binding activity of the p65 subunit was measured in triplicate in the nuclear extracts of WEHI 231 and Ramos cells using an enzyme-linked immunosorbent assay (ELISA)-based assay as previously described (26). The NF-κB luciferase assay was performed essentially as previously described (27).
RNA interference.
Short interfering RNA (siRNA) oligonucleotides against mouse RelB, mouse c-Myc, and mouse Mcl-1 and a control siRNA were purchased from Santa Cruz Biotechnology. WEHI 231 cells expressing K13 or K13-ERTAM (WEHI 231-K13 or WEHI 231-K13-ERTAM, respectively) were transfected with these siRNA oligonucleotides using a Neon transfection system (Invitrogen) as per the manufacturer's recommendations.
Real-time PCR.
Total RNA was isolated using an RNeasy Mini Kit (Qiagen), and cDNA was synthesized using the reverse transcriptase enzyme Superscript II (Invitrogen). Real-time quantitative reverse transcription-PCR (qRT-PCR) was performed with SYBR green, using c-Myc-specific PCR primers. Samples were run in triplicate, and PCR was performed by an ABI Step One Plus thermocycler (Applied Biosystems). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as housekeeping gene, and qRT-PCR data (threshold cycle [CT] values) were analyzed using the 2−ΔΔ C method as described earlier (26). The qRT-PCR data were presented as fold change in target gene expression ± standard deviation.
Statistical analysis.
Statistical analyses were performed using unpaired t tests. Values are expressed as means ± standard deviations (SD), with all experiments repeated a minimum of three times with triplicate samples. A P value of ≤0.05 was considered statistically significant.
RESULTS
K13 protects WEHI 231 cells from anti-IgM-induced growth inhibition.
Treatment of WEHI 231 cells with anti-IgM leads to growth inhibition and ultimately cell death (21). We generated polyclonal populations of WEHI 231 cells expressing FLAG-tagged K13 and an empty vector using retroviral gene transfer and confirmed expression of K13 by immunoblotting with an antibody against the FLAG epitope tag (Fig. 1A). Furthermore, immunoblotting with a monoclonal antibody (8F6) against K13 confirmed that the level of ectopically expressed K13 in WEHI 231 cells was comparable to the physiological level of K13 found in the PEL cell line BC1 (Fig. 1B). We next examined the ability of K13 to protect against anti-IgM-induced growth inhibition by growing WEHI 231 cells expressing an empty vector (WEHI 231-vector) and WEHI 231-K13 with increasing concentrations of anti-IgM. WEHI 231-vector cells showed a dramatic and dose-dependent reduction in cell number upon treatment with anti-IgM (Fig. 1C). In contrast, the expression of K13 conferred significant protection against the growth inhibition caused by the anti-IgM treatment (Fig. 1C).
Fig 1.
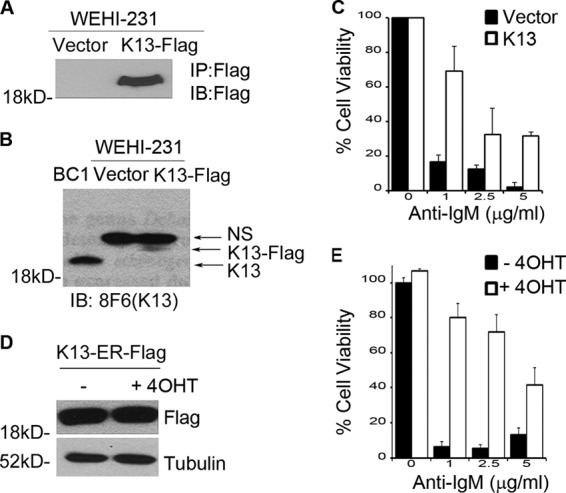
K13 protects WEHI 231 cells from anti-IgM induced growth inhibition. (A) Expression of FLAG-K13 in WEHI 231 cells. K13 was immunoprecipitated (IP) by FLAG antibody beads from WEHI 231 cells stably expressing an empty vector or FLAG-tagged K13 followed by immunoblot (IB) analysis with a FLAG antibody. (B) Level of ectopic K13 in WEHI 231 cells compared with its physiological level in the KSHV-infected PEL cell line BC1. Immunoblotting was performed using 8F6 monoclonal antibody against K13; a nonspecific band (NS) serves as a loading control. (C) WEHI 231 cells expressing an empty vector or K13 were grown in triplicate in a 96-well plate with the indicated concentrations of anti-IgM for 48 h. Cell viability was measured using the MTS assay (values are means ± SD; n = 3). (D) Expression of FLAG-K13-ERTAM in WEHI 231 cells, as measured by Western blotting. (E) WEHI 231 cells expressing K13-ERTAM grown in the presence and absence of 4OHT (20 nM) were treated in triplicate with the indicated concentrations of anti-IgM for 48 h. Cell viability was measured using the MTS assay (values are means ± SD; n = 3).
We also generated stable populations of WEHI 231 cells expressing a K13-ERTAM fusion construct (Fig. 1D). The K13-ERTAM fusion protein is expressed constitutively in WEHI 231 cells but becomes active only on the addition of 4-hydroxytamoxifen (4OHT), thus allowing posttranslational control of K13 activity (17). We grew WEHI 231-K13-ERTAM cells in the presence and absence of 4OHT (20 nM) for 16 h and then treated them with increasing concentrations of anti-IgM. While the untreated WEHI 231-K13-ERTAM cells showed dramatic growth inhibition upon anti-IgM treatment, 4OHT-treated cells were relatively resistant (Fig. 1E). Taken collectively, the above results demonstrate that K13 confers protection against anti-IgM-induced growth inhibition in WEHI 231 cells.
K13 inhibits anti-IgM-induced cell cycle arrest and anti-IgM-induced apoptosis in WEHI 231 cells.
To study the mechanism by which K13 protects against anti-IgM-induced growth arrest, we carried out cell cycle analysis. WEHI 231-K13-ERTAM cells were grown in the presence and absence of 20 nM 4OHT and subsequently treated with anti-IgM for 48 h. The cell cycle distribution profiles of propidium iodide-stained cells were measured by flow cytometry (Fig. 2A). Treatment of WEHI 231-K13-ERTAM cells with anti-IgM for 48 h resulted in an increase in cells in G0/G1 phase from 30% to 94% and a reduction of cells in S phase from 56% to 3%, reflecting cell cycle arrest in the G0/G1 phase. In contrast, anti-IgM treatment of K13-ERTAM cells that had been pretreated with 4OHT resulted in an increase in cells in G0/G1 phase from 27% to 50% and a reduction of cells in S phase from 57% to 37%. Thus, K13 was able to significantly block the G0/G1 cell cycle arrest induced by anti-IgM treatment.
Fig 2.
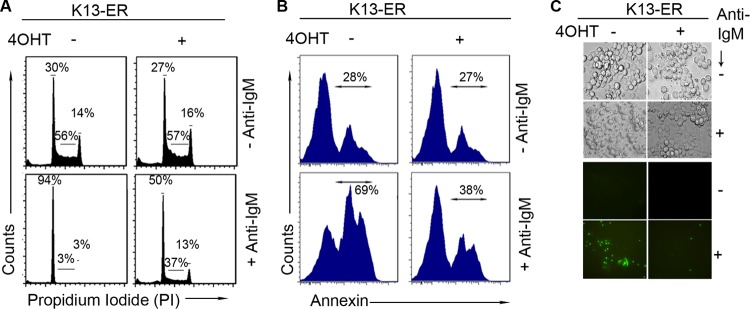
K13 protects WEHI 231 cells from anti-IgM-induced growth arrest and apoptosis. (A) WEHI 231-K13-ERTAM cells were grown in the presence and absence of 4OHT (20 nM) and then treated with and without anti-IgM (1 μg/ml) for 48 h. Cells were stained with propidium iodide, and cell cycle analysis was performed using flow cytometry. (B) WEHI 231-K13-ERTAM cells grown in the presence or absence of 4OHT (20 nM) were stained with phycoerythrin-labeled annexin V after treatment with anti-IgM (1 μg/ml) for 48 h and examined by flow cytometry. (C) WEHI 231-K13-ERTAM cells grown in the presence and absence of 4OHT were treated with 1 μg/ml of anti-IgM. Cells were then stained with Sytox Green, a cell-impermeable nuclear dye that stains the nuclei of dead cells; cells were then examined under a fluorescence microscope or under phase-contrast microscope and photographed.
In addition to inducing cell cycle arrest, B-cell receptor stimulation of WEHI 231 cells is known to result in apoptosis. To determine if K13 also protects against anti-IgM-induced apoptosis, cells were analyzed using flow cytometry following staining with annexin V, a marker of apoptosis. Treatment of WEHI 231-K13-ERTAM cells with anti-IgM resulted in an almost doubling of annexin V-positive cells, which was significantly blocked by pretreatment with 4OHT (Fig. 2B). The protective effect of K13 against anti-IgM-induced apoptosis was confirmed by staining with Sytox Green, a membrane-impermeable nuclear dye that stains the nuclei of only dead and dying cells that have lost cell membrane integrity. K13-ERTAM cells were treated with 4OHT for 16 h or left untreated and were subsequently grown for 48 h in the presence or absence of 1 μg/ml anti-IgM. Apoptotic cells were evaluated under phase-contrast and fluorescence microscopy. Anti-IgM treatment of K13-ERTAM cells resulted in the appearance of cells with brightly stained, condensed and fragmented nuclei, suggestive of loss of membrane integrity and induction of apoptosis (Fig. 2C). The Sytox Green-positive cells were markedly reduced among anti-IgM-treated K13-ERTAM cells that had been grown in the presence of 4OHT (Fig. 2C).
Protective effect of K13 against anti-IgM-induced apoptosis is associated with upregulation of NF-κB activity.
The NF-κB pathway has been reported to be constitutively active in the WEHI 231 cells, and anti-IgM-induced apoptosis of these cells has been shown to be associated with a drop in NF-κB activity (28, 29). Since K13 is known to activate the NF-κB pathway, we next examined whether it protects against anti-IgM-induced apoptosis by blocking the drop in NF-κB activity observed following anti-IgM treatment. For this purpose, WEHI 231-vector and -K13-ERTAM cells were grown in the presence and absence of 4OHT and then stimulated with anti-IgM for 24 h. The status of the NF-κB pathway was examined using a nuclear p65/RelA DNA binding assay. Consistent with published reports (28, 29), nuclear extracts from WEHI 231-vector cells that had been grown in the absence or presence of 4OHT showed modest basal p65/RelA DNA binding activity, which was significantly reduced following anti-IgM treatment (Fig. 3A). Essentially similar results were obtained in WEHI 231-K13-ERTAM cells grown in the absence of 4OHT. In contrast, the p65/RelA DNA binding activity was approximately 3-fold higher in the WEHI 231-K13-ERTAM cells that had been exposed to 4OHT, suggesting activation of the NF-κB pathway by K13 (Fig. 3A). More importantly, treatment with anti-IgM resulted in only a minor reduction in the nuclear p65/RelA DNA binding activity in the 4OHT-treated WEHI 231-K13-ERTAM cells (Fig. 3A). In fact, the 4OHT-treated WEHI 231-K13-ERTAM cells maintained nearly 2-fold higher p65/RelA DNA binding activity even after anti-IgM treatment than the basal p65/RelA DNA binding activity observed in the cells that had not been treated with 4OHT or anti-IgM (Fig. 3A).
Fig 3.

K13 activates the NF-κB pathway in WEHI 231 cells. (A) An ELISA-based NF-κB binding assay showing increased binding of p65/RelA DNA binding activity in the nuclear extracts of WEHI 231-vector and WEHI 231-K13-ERTAM cells grown in the presence or absence of 4OHT and treated with 1 μg/ml of anti-IgM for 24 h. Values shown are the mean ± SD from one representative experiment out of three performed in duplicate. Asterisks indicate significance at a P level of ≤0.05. OD 655nm, optical density at 655 nm. (B) A luciferase-based reporter assay showing induction of NF-κB transcriptional activity by K13. WEHI 231 cells stably expressing the K13-ERTAM-NF-κB-Luc construct were grown in the presence and absence of 4OHT and treated with 1 μg/ml of anti-IgM for 24 h, and cell lysates were used for a luciferase reporter assay. Values shown are the mean ± SD from one representative experiment out of three performed in duplicate. Asterisks indicate significance at a P level of ≤0.05. (C) Western blots showing upregulation of A20 and RelB expression in 4OHT-treated WEHI 231 K13-ERTAM cells and no significant decline following treatment with anti-IgM (1 μg/ml). WEHI 231-vector cells treated with 4OHT did not show any increase in A20 and RelB. Tubulin served as a loading control. (D) Wild-type K13, but not vFLIP E8 or K13-58AAA, induces RelB promoter activity. 293T cells were transfected with a control vector and a vector encoding wild-type K13 or the K13 mutant K13-58AAA or vFLIP E8 (250 ng/ml) along with an RelB-Luc reporter construct (75 ng/well) and a pRSV/LacZ (β-galactosidase) reporter construct (75 ng/well), and the reporter assay was performed as described in Materials and Methods. The values shown are mean ± SD of one representative experiment out of three in which each transfection was performed in duplicate. (E) Expression levels of wild-type FLAG-tagged K13, the K13 mutant K13-58AAA, and vFLIP E8 in 293T cells. (F) K13 activates RelB promoter through NF-κB the pathway. 293T cells were transfected with a control vector or a vector encoding K13 along with either a WT-RelB-Luc or RelB-Luc construct containing mutations in NF-κB RE I (Mut I), NF-κB RE II (Mut II), or both (Mut 1+II). The experiment was performed as described in for panel D. (G) Dominant negative mutants of IκBα lacking the N-terminal 36 amino acids (IκBαΔN) and a superrepressor form of IκBα (IκBα SS32/36AA) block K13-induced RelB promoter activity. 293T cells were transfected either with an empty vector or K13, along with an RelB luciferase reporter construct and a pRSV/LacZ reporter construct, as described for panel F. The amount of inhibitor plasmids (500 ng/well) was 5 times the amount of vector or K13 (100 ng/well) plasmid, and the total amount of transfected DNA was kept constant by adding an empty vector. The values shown are the mean ± SD of one representative experiment out of three in which each transfection was performed in duplicate. (H) As2O3, an inhibitor of NF-κB, blocks K13-induced RelB promoter activation. 293T cells were transfected with an empty vector or a vector encoding K13 along with RelB-Luc and pRSV/LacZ reporter constructs. Approximately 3 h after transfection, cells were treated with control vehicle or 2.5 μM As2O3 for 18 h before cell lysis and measurement of reporter activities.
To demonstrate that the increase in p65/RelA DNA binding activity in the 4OHT-treated WEHI 231-K13-ERTAM cells is associated with a corresponding increase in NF-κB transcriptional activity, we used lentivirus-mediated gene transfer to generate a polyclonal population of WEHI 231-K13-ERTAM cells stably expressing an NF-κB-driven luciferase reporter construct. We grew the WEHI 231-K13-ERTAM-NF-κB-Luc cells in the presence and absence of 4OHT and subsequently treated them with anti-IgM for 24 h before cell lysis and measurement of luciferase activity. As shown in Fig. 3B, WEHI 231-K13-ERTAM-NF-κB-Luc cells showed low basal NF-κB-Luc activity, which was further reduced by anti-IgM treatment. In contrast, not only did 4OHT treatment result in a significant increase in the basal NF-κB-Luc activity, but also this activity remained significantly higher than the activity observed in the uninduced cells even after treatment with anti-IgM (Fig. 3B). Thus, K13 is able to sustain NF-κB signaling even in the presence of anti-IgM.
To examine if an increase in NF-κB signaling by K13 translates into an increase in the expression of downstream targets of the NF-κB pathway, we examined the status of A20 and RelB, two proteins that are known to be upregulated by NF-κB activation. Western blot analyses revealed low basal expression of A20 and RelB in uninduced WEHI 231-K13-ERTAM cells that was further reduced by IgM treatment. In contrast, the basal levels of both A20 and RelB were significantly higher in 4OHT-treated WEHI 231-K13-ERTAM cells and remained so following IgM treatment. As a control, we also examined the levels of A20 and RelB in WEHI 231-vector cells and observed no significant difference with 4OHT treatment (Fig. 3C).
K13 upregulates RelB expression in WEHI 231 cells through NF-κB activation.
RelB is not only a member of the NF-κB family but also an NF-κB target gene (30). Previous studies have shown that CD40 signaling protects WEHI 231 cells against anti-IgM-induced apoptosis by upregulating RelB expression (31). To determine the role of RelB in K13-induced protection against anti-IgM-induced apoptosis, we examined the level of RelB expression in WEHI 231-K13-ERTAM cells. As shown in Fig. 3C, expression of RelB was significantly increased in WEHI 231-K13-ERTAM cells upon treatment with 4OHT, and this elevated level was sustained even following treatment with anti-IgM. In contrast, 4OHT had no effect on RelB level in WEHI 231-vector cells (Fig. 3C). To determine the mechanism by which K13 upregulates RelB expression, we cotransfected a relB promoter-driven luciferase reporter construct with an empty vector or a K13 expression plasmid. K13 strongly induced relB promoter activity compared to vector-transfected cells (Fig. 3D). In contrast, no induction of relB promoter activity was observed upon expression of an NF-κB-defective mutant of K13 with three alanine substitutions at positions 58 to 60 (K13-58AAA) (Fig. 3D). The equine herpesvirus 2-encoded vFLIP E8 resembles K13 in structure but lacks the ability to activate the NF-κB pathway (11). As shown in Fig. 3D and E, vFLIP E8 failed to activate the relB promoter. Collectively, the above results support the argument that K13 activates the relB promoter via NF-κB activation.
The human relB promoter contains two NF-κB response elements located at positions −247 to −238 and −175 to −166 from the translation start site and designated NF-κB RE I and NF-κB RE II, respectively (23). To delineate the contribution of these κB sites in K13-induced relB promoter activation, we tested the ability of K13 to activate luciferase reporter constructs driven by either the wild-type relB promoter or relB promoters containing mutations in one or both of the κB response elements. As shown in Fig. 3F, while K13 strongly activated the wild-type relB promoter and the promoter containing a mutation in the NF-κB RE I, it failed to activate relB promoters containing mutations in NF-κB RE II or both NF-κB RE I and II. Thus, K13 upregulates relB transcription through the NF-κB RE II. Finally, to confirm the role of the classical NF-κB pathway in K13-induced relB promoter activity, we used genetic and pharmacological inhibitors of this pathway. K13-induced relB promoter activity was blocked by a superrepressor form of IκBα in which the two critical serine residues have been mutated to alanine (IκBα SS32/36AA) and a deletion mutant of IκBα lacking the N-terminal 36 amino acids (IκBαΔN) (Fig. 3G). Finally, As2O3, an agent known to block the K13-induced classical NF-κB pathway, effectively blocked relB promoter activity induced by K13 expression (Fig. 3H). Taken collectively, the above results confirm that K13 upregulates RelB expression through classical NF-κB activation.
Protective effect of K13 against anti-IgM-induced growth arrest and apoptosis is specific to its ability to activate the NF-κB pathway and upregulate RelB.
The studies in the preceding sections demonstrated that the protective effect of K13 against anti-IgM-induced growth arrest and apoptosis is associated with the activation of the NF-κB pathway. To determine if NF-κB activation is functionally involved in the above processes, we generated stable polyclonal populations of WEHI 231 cells expressing the K13-58AAA mutant and vFLIP E8, which lack NF-κB activity (11, 20). The expression of K13-58AAA and the vFLIP E8 in WEHI 231 cells was confirmed by immunoblotting (Fig. 4A). Consistent with their inability to activate the NF-κB pathway, K13-58AAA and the vFLIP E8 failed to induce the expression of A20 and RelB, while robust activation of these proteins was seen in the wild-type K13-expressing cells (Fig. 4B). We next compared the ability of wild-type K13, K13-58AAA, and vFLIP E8 to protect against IgM-induced apoptosis. In contrast to K13, both E8 and K13-58AAA conferred no protective effect against anti-IgM-induced apoptosis in WEHI 231 cells (Fig. 4C). Finally, to confirm the functional involvement of RelB in the protective effect conferred by K13 expression, we studied the effect of its downregulation on anti-IgM-induced cell death. Downregulation of RelB in the WEHI 231-K13 cells by siRNA resulted in a significant reduction in cell viability following anti-IgM treatment compared to the control siRNA-transfected cells (Fig. 4D and E). Essentially similar results were obtained upon silencing of RelB in K13-ERTAM cells (Fig. 4F). Thus, the protective effect of K13 against anti-IgM-induced growth arrest and apoptosis is conferred through upregulation of RelB via increased classical NF-κB activity.
Fig 4.

The protective effect of K13 against anti-IgM-induced apoptosis is dependent on NF-κB signaling. (A) Expression of K13, K13-58AAA, and vFLIP E8 in WEHI 231 cells as determined by immunoprecipitation with FLAG beads followed by immunoblot analysis with a FLAG antibody. The asterisk denotes a nonspecific band. (B) Western blot analysis showing expression of A20 and RelB in WEHI 231 cells expressing wild-type K13, K13-58AAA, and vFLIP E8. GAPDH served as a loading control. The blot was imaged using an Odyssey Infrared Imaging System. (C) WEHI 231 cells expressing wild-type K13, K13-58AAA, and the vFLIP E8 were subjected to anti-IgM treatment at the indicated concentrations. Cell viability was measured using an MTS assay (values are means ± SD; n = 3). (D) Western blot analysis showing siRNA-mediated knockdown of RelB expression in WEHI 231-K13 cells. The blot was reprobed with an antibody against GAPDH (bottom panel) to show equal loading and specificity of gene silencing. The blot was imaged using an Odyssey Infrared Imaging System. G3PDH, glyceraldehyde-3-phosphate dehydrogenase. (E and F) Downregulation of RelB in the WEHI 231-K13 and WEHI 231-K13-ERTAM cells by siRNA results in a significant reduction in cell viability following anti-IgM (1 μg/ml) treatment compared to the control siRNA-transfected cells. The experiment was performed as described in the legend of Fig. 3C. Values shown are the means ± SD from one representative experiment out of two performed in triplicate. Asterisks indicate statistical significance at a P level of ≤0.05.
Protective effect of K13 against anti-IgM-induced growth arrest in WEHI 231 cells is associated with modulation of c-Myc and p27Kip1 expression.
The murine oncogene c-myc has been extensively studied with respect to its role in growth arrest and induction of apoptosis in WEHI 231 cells upon engagement of IgM (32–34). Cross-linking of IgM results in a transient increase followed by a rapid decrease in c-myc transcript levels (32, 33). The c-myc gene contains κB binding sites, and the NF-κB pathway is known to modulate c-myc activity (35). To ascertain if the protective effect of K13 against anti-IgM-induced growth arrest and cell death is due in part to maintenance of c-myc level, we utilized quantitative real-time PCR analysis to determine changes in c-myc transcript levels upon anti-IgM treatment. While anti-IgM led to a significant reduction in c-myc mRNA levels in the untreated WEHI 231-K13-ERTAM cells, it had no significant effect on c-myc mRNA in cells that had been pretreated with 4OHT to induce K13 activity (Fig. 5A). Furthermore, the basal level of c-Myc protein was significantly higher in the WEHI 231-K13 cells than in cells expressing an empty vector, K13-58AAA, or vFLIP E8, supporting the involvement of K13-induced NF-κB activity in the upregulation of c-Myc (Fig. 5B). More importantly, while anti-IgM treatment resulted in a 2-fold reduction in the level of c-Myc protein in the WEHI 231-vector cells, it had only a marginal effect on the c-Myc protein level in the WEHI 231-K13 cells (Fig. 5C). In fact, the level of c-Myc protein in WEHI 231-K13 cells even following anti-IgM treatment was higher than its basal level in the untreated WEHI 231-vector cells (Fig. 5C). To determine the functional involvement of c-Myc in the protective effect conferred by K13, we used siRNA-mediated gene silencing to downregulate its expression (Fig. 5D). siRNA-mediated silencing of c-Myc expression in WEHI 231-K13 cells resulted in reduced cell viability following anti-IgM treatment compared to the control siRNA-transfected cells (Fig. 5E). Essentially similar results were obtained upon c-Myc silencing in WEHI 231-K13-ER cells (Fig. 5F). We also used a pharmacological approach to downregulate c-Myc expression. JQ1 is a recently described small-molecule inhibitor of BET bromodomains that downregulate c-myc mRNA and expression of its downstream target genes (36). Treatment of WEHI 231-K13 cells with 50 nM and 100 nM JQ1 resulted in a dose-dependent suppression of c-Myc expression, which was accompanied by reduction in cell viability following IgM treatment (Fig. 5G and H). Taken collectively, the above results support the argument that K13 protects WEHI 231 cells against anti-IgM-induced cell death via upregulation of c-Myc.
Fig 5.

K13 exerts its protective effect against anti-IgM-induced apoptosis through modulation of c-Myc and p27Kip1 levels. (A) Real-time RT-PCR analysis showing decline in c-myc levels in WEHI 231-K13 cells following treatment with anti-IgM for 24 h that was blocked by induction of K13 activity by treatment with 4OHT. Results shown are the means ± SD (n = 2). (B) Western blot analysis showing expression of c-Myc in the WEHI 231 cells expressing wild-type K13, K13-58AAA, and vFLIP E8. G3PDH served as a loading control. Blots were imaged using an Odyssey Infrared Imaging system. (C) Immunoblot analysis of WEHI 231-K13 cells grown in the presence and absence of 4OHT (20 nM) when treated with anti-IgM (1 μg/ml) for 24 h. K13 blocks anti-IgM-induced downregulation of c-Myc. Tubulin served as a loading control. (D) Immunoblot analysis with Odyssey Infrared Imaging System showing siRNA-mediated knockdown of c-Myc expression in WEHI 231-K13 cells. (E and F) Silencing of c-Myc in the WEHI 231-K13 and WEHI 231-K13-ERTAM cells results in a significant reduction in cell viability following anti-IgM treatment compared to the control siRNA-transfected cells. Values shown are the means ± SD from one representative experiment out of two performed in triplicate. (G) Western blot analysis showing suppression of c-Myc expression by a 24-h treatment with the indicated doses of JQ1. The G3PDH blot shows equal loading. (H) WEHI 231-K13 cells treated with JQ1 show a reduction in cell viability following anti-IgM (1 μg/ml) treatment. (I) Immunoblot analysis of WEHI 231-K13 cells grown in the presence and absence of 4OHT (20 nM) when treated with anti-IgM (1 μg/ml) for 24 h. K13 blocks anti-IgM-induced upregulation of p27Kip1. Tubulin served as a loading control.
The cyclin-dependent kinase inhibitor p27Kip1 has been shown to promote cell cycle arrest and apoptosis in WEHI 231 cells upon engagement of the BCR (33, 34). p27Kip1 is repressed by c-Myc, and a reduction in c-Myc expression has been shown to be sufficient to induce p27Kip1 expression (33, 34). As shown in Fig. 5I, treatment with anti-IgM resulted in upregulation of p27Kip1 levels in WEHI 231-vector cells, which was blocked in WEHI 231-K13 cells. Collectively, the above results suggest that K13 protects against anti-IgM-induced growth arrest by blocking the fall in c-Myc level and resulting upregulation of p27Kip1.
Protective effect of K13 against anti-IgM-induced apoptosis in WEHI 231 cells is associated with modulation of Mcl-1 expression.
We have previously shown that K13 protects against growth factor withdrawal-induced apoptosis by upregulating the expression of Mcl-1, an antiapoptotic member of the Bcl2 family (18, 24). To determine if upregulation of Mcl-1 is also involved in the protective effect of K13 against BCR-induced apoptosis, we examined its status by Western blotting. As shown in Fig. 6A, the expression of Mcl-1 was increased in WEHI 231-K13-ERTAM cells upon 4OHT treatment, and this elevated expression was maintained following treatment with anti-IgM. To investigate the functional role of Mcl-1 upregulation in the K13-mediated protection against anti-IgM-induced apoptosis, we generated a stable population of WEHI 231 cells with ectopic Mcl-1 expression by retrovirus-mediated gene transfer (Fig. 6B). WEHI 231 cells expressing Mcl-1 or an empty vector were treated with anti-IgM for 48 h. As shown in Fig. 6C, WEHI 231-Mcl-1 cells were significantly protected from anti-IgM-induced cell death compared to the WEHI 231-vector cells. To determine the functional role of Mcl-1 in the protection conferred by K13, we used siRNA-mediated gene silencing to downregulate its expression (Fig. 6D). WEHI 231-K13 cells transfected with Mcl-1 siRNA showed reduced cell viability following anti-IgM treatment compared to the control siRNA-transfected cells (Fig. 6E). Essentially similar results were obtained upon Mcl-1 silencing in the WEHI 231-K13-ERTAM cells (Fig. 6F). Taken collectively, the above results suggest that K13 protects against anti-IgM-induced apoptosis by upregulating Mcl-1 expression.
Fig 6.
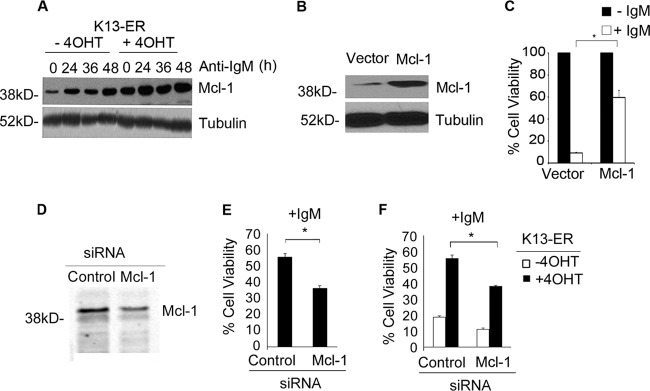
K13 exerts its protective effect against anti-IgM-induced apoptosis through upregulation of Mcl-1. (A) Immunoblot showing expression of Mcl-1 in K13-ERTAM cells that had been left untreated or treated with 4OHT and then exposed to anti-IgM (1 μg/ml) over a 48-h time period. Tubulin served as a loading control. (B) Immunoblot analysis showing ectopic expression of Mcl-1 in WEHI 231 cells. Tubulin served as a loading control. (C) WEHI 231 cells expressing an empty vector or Mcl-1 were grown in triplicate in a 96-well plate in the presence or absence of anti-IgM (1 μg/ml). Cell viability was measured 48 h later using an MTS assay (values are means ± SD; n = 3). (D) Western blot analysis showing siRNA-mediated knockdown of Mcl-1 expression in WEHI 231-K13 cells. (E and F) Downregulation of Mcl-1 in the WEHI 231-K13 and WEHI 231-K13-ERTAM cells by siRNA results in a significant reduction in cell viability following anti-IgM treatment compared to the control siRNA-transfected cells. Values shown are the means ± SD from one representative experiment out of two performed in triplicate. Asterisks indicate statistical significance at a P level of ≤0.05.
K13 is able to protect the mature human B-cell line Ramos from anti-IgM induced apoptosis.
To ascertain if the protective effect of K13 is not developmentally restricted, we used the mature human B-cell line Ramos. Polyclonal populations of Ramos cells expressing K13 (Ramos-K13) or an empty vector (Ramos-vector) were generated using retroviral gene transfer, and the expression of K13 was confirmed by immunoblotting (Fig. 7A). The level of retrovirally expressed K13 in the Ramos-K13 cells was comparable to the physiological level of K13 found in the PEL cell line BC1 (Fig. 7A). Next, the ability of K13 to protect against anti-IgM-induced growth inhibition was examined by growing Ramos-K13 and Ramos-vector cells with anti-IgM. Expression of K13 conferred a protective effect against anti-IgM-induced growth arrest as measured by the MTS-based assay (Fig. 7B). Anti-IgM treatment of Ramos-vector cells resulted in a decline in cells in S and G2 phases from 41% to 32% and from 18% to 10%, respectively. However, this decline in cells in the S and G2 phases was not mirrored by a corresponding increase in cells in the G0/G1 phase due to an increase in apoptosis, as reflected by an increase in cells in the sub-G0/G1 fraction from 7% to 25% (Fig. 7C). All of the above responses to anti-IgM were attenuated in K13-expressing Ramos cells. As shown in Fig. 7C, anti-IgM treatment of Ramos-K13 cells resulted in a smaller decline in cells in the S (from 40% to 37%) and G2 (from 22% to 17%) phases and a corresponding smaller increase in cells in the sub-G0/G1 fraction (from 7% to 11%). Thus, in contrast to WEHI 231 cells, Ramos cells primarily undergo apoptosis in response to anti-IgM treatment, and this response is blocked upon K13 expression.
Fig 7.

K13 protects against anti-IgM-induced apoptosis in the mature B-cell line Ramos through activation of the NF-κB pathway. (A) Expression of retrovirally expressed K13-FLAG in Ramos cells and endogenous K13 in BC1 cells as determined by immunoblotting with 8F6 monoclonal antibody. Tubulin serves as a loading control. (B) Ramos cells expressing an empty vector or K13 were grown in triplicate in a 96-well plate with anti-IgM (30 μg/ml). Cell viability was measured using the MTS assay (values are means ± SD; n = 3). Asterisks indicate significance at a P level of ≤0.05. (C) Ramos cells expressing an empty vector or K13 were treated with or without anti-IgM (30 μg/ml) for 24 h. Cells were stained with propidium iodide, and cell cycle analysis was performed using flow cytometry. Values shown are the percentage of cells in the different phases of the cell cycle. (D) An ELISA-based NF-κB binding assay showing p65/RelA DNA binding activity in the nuclear extracts of Ramos cells expressing an empty vector or K13 treated with or without 30 μg/ml of anti-IgM for 24 h. The values shown are means ± SD of one representative experiment out of three in which p65 DNA binding was measured in duplicate. Asterisks indicate significance at a P level of ≤0.05. (E) Immunoblotting showing expression of RelB, Mcl-1, and Bcl2 in Ramos cells expressing an empty vector or K13 when treated with anti-IgM (30 μg/ml) over a 48-h time period.
To determine the mechanism by which K13 protects Ramos cells against IgM-induced arrest and apoptosis, we examined the status of NF-κB and its downstream target genes. Treatment of Ramos-vector cells with anti-IgM resulted in downregulation of NF-κB as measured by a nuclear p65/RelA DNA binding assay (Fig. 7D). Consistent with the ability of K13 to activate the NF-κB pathway, the basal level of nuclear p65/RelA was significantly higher in Ramos-K13 cells than in Ramos-vector cells (Fig. 7D). Although anti-IgM treatment resulted in a decline in the nuclear p65/RelA in the Ramos-K13 cells as well, its level was significantly higher than in the anti-IgM-treated Ramos-vector cells (Fig. 7D). We next compared the status of several downstream targets of the NF-κB pathway in the Ramos-vector and -K13 cells by Western blotting. The activation of the NF-κB pathway in the Ramos-K13 cells was associated with a significant increase in the expression of RelB, and its elevated level was maintained following anti-IgM treatment (Fig. 7E). Additionally, while anti-IgM treatment of Ramos-vector cells resulted in a 2-fold reduction in the expression of Mcl-1 protein by 48 h, the expression of Mcl-1 was maintained in the Ramos-K13 cells. Finally, Bcl-2 expression was detected only in the Ramos-K13 cells, and its level was maintained during anti-IgM exposure (Fig. 7E). Taken collectively, these results provide further evidence that K13 is able to protect B cells from anti-IgM-induced apoptosis through activation of the NF-κB pathway.
DISCUSSION
The signaling mechanisms involved in BCR-induced apoptosis are complex and have been extensively studied in both mature and immature B cells (2, 37). These studies have revealed that BCR-mediated apoptosis is primarily mediated via the intrinsic apoptosis pathway and is associated with enhanced permeability of the mitochondrial outer membrane and the activation of caspase-9 and -3 (38). On the other hand, BCR-mediated apoptosis is believed to be independent of FADD (Fas-associated death domain) and caspase-8, which are components of the extrinsic apoptosis pathway (38). In addition to the intrinsic apoptosis pathway, the NF-κB pathway has also been shown to play a key role in BCR-induced apoptosis, and it has been shown that BCR-induced apoptosis in WEHI 231 cells is preceded by a drop in the basal NF-κB activity (28, 29, 31).
K13 was originally believed to act as an inhibitor of caspase-8/FLICE that protected virally infected cells against death receptor-induced apoptosis (10). However, subsequent studies revealed that K13 is not an inhibitor of caspase-8/FLICE but, instead, is a strong activator of the NF-κB pathway (11, 12, 15, 20). In this study, we demonstrate that K13 protects against BCR-induced apoptosis via NF-κB activation, independent of its activity as a caspase-8 inhibitor. This conclusion is supported by our results showing that the protective effect of K13 against BCR-induced apoptosis was associated with not only an increase in basal NF-κB activity in these cells but also maintenance of this activity following treatment with anti-IgM. It has previously been reported that BCR-induced apoptosis in WEHI 231 cells can be reversed by treatment with CD40 ligand (CD40L) (28). Interestingly, similar to K13, CD40L rescue was shown to prevent the drop in NF-κB/Rel binding induced by anti-IgM treatment (31). Thus, K13 mimics CD40 signaling to protect against BCR-induced apoptosis. The involvement of the NF-κB pathway in the K13 rescue is further supported by our results with the NF-κB defective mutant of K13, K13-58AAA, which failed to confer protection against BCR-induced apoptosis. Finally, the vFLIP E8, which has been shown to protect against Fas-induced apoptosis by blocking caspase-8/FLICE but lacks the ability to activate NF-κB, failed to protect against BCR-induced apoptosis. Taken collectively, the above results demonstrate that K13 protects against BCR-induced apoptosis through NF-κB activation and not via inhibition of caspase-8. These results are also consistent with the previous studies demonstrating a lack of involvement of FADD and caspase-8 in BCR-induced apoptosis (1, 2).
The CD40L rescue of WEHI 231 cells against BCR-induced apoptosis was also associated with increased expression of the NF-κB subunit RelB (31). More importantly, siRNA-mediated knockdown of RelB in WEHI 231 cells was shown to substantially increase their susceptibility to anti-IgM-induced apoptosis while ectopic expression of RelB was shown to confer protection (31). Based on these reports, we examined the status of RelB and observed significant upregulation of RelB expression in the K13-expressing WEHI 231 and Ramos cells. The relB promoter contains two κB binding sites, and previous studies have reported that the NF-κB RE II is involved in transcriptional activation of relB promoter by agents known to activate the classical NF-κB pathway, such as tumor necrosis factor (TNF) and lipopolysaccharide (LPS) (30). Consistent with these studies, we observed that K13 strongly activated a luciferase reporter construct driven by the wild-type relB promoter but failed to activate a construct containing a mutation in the NF-κB RE II. Furthermore, K13-induced relB promoter activation was effectively blocked by genetic and pharmacological inhibitors of the classical NF-κB pathway. Taken collectively, these results demonstrate that K13 upregulates relB gene expression via activation of the classical NF-κB pathway. Although RelB expression was strongly upregulated in K13-expressing WEHI 231 and Ramos cells, it is important to clarify that we are not contending that RelB is the sole NF-κB subunit responsible for the protective effect of K13 against BCR-induced apoptosis. Instead, we favor the hypothesis that RelB works in concert with other NF-κB subunits to regulate the expression of genes responsible for protection against BCR-induced apoptosis.
BCR engagement in WEHI 231 cells, following anti-IgM treatment, is accompanied by a fall in c-Myc and a rise in p27Kip1 protein levels (32, 33). The changes in c-Myc and p27Kip1 levels have been shown to be functionally involved in anti-IgM-induced growth arrest and apoptosis in WEHI 231 cells (32, 33). Consistent with these prior studies, we also observed that a decline in NF-κB following anti-IgM treatment in WEHI 231 cells is accompanied by a decline in c-Myc and an increase in p27Kip1 protein levels. However, not only was the basal level of c-Myc significantly higher in the K13-expressing WEHI 231 cells than in the vector cells, but it also remained at elevated levels even following anti-IgM treatment. Similarly, the anti-IgM-induced rise in p27Kip1 was blocked in K13-expressing cells. Finally, the NF-κB-responsive genes that are known to protect against BCR-induced apoptosis also include members of the Bcl-2 family. Consistent with these studies, we observed significant upregulation of Mcl-1 expression in K13-expressing WEHI 231 and Ramos cells. Furthermore, ectopic expression of Mcl-1 in WEHI 231 cells resulted in protection against IgM-induced apoptosis. Taken collectively with previous studies, our results support the hypothesis that K13 protects against anti-IgM-induced growth arrest and apoptosis by preventing the fall in c-Myc levels, thereby blocking the rise in p27Kip1, and by upregulating Mcl-1.
The gammaherpesviruses that include KSHV, Epstein-Barr virus (EBV) and the murine gammaherpesvirus 68 (MHV-68), are able to establish life-long persistent infections in lymphocytes. The manipulation of BCR signaling events in order to successfully establish a latent infection in B lymphocytes is a common strategy among gammaherpesviruses. For example, EBV through the proteins LMP-1, which mimics CD40 signaling, and LMP-2A, which acts as a constitutively activated B-cell receptor, are able to deregulate BCR signaling and enhance cell survival and proliferation (39, 40). Similarly, the MHV-68 protein M2 has been shown to block BCR-induced arrest and apoptosis through upregulation of Vav activity (41). Here, we show that expression of K13 protects against BCR-mediated cell cycle arrest and apoptosis through NF-κB activation, providing yet another example of how gammaherpesviruses manipulate this signaling pathway to promote the survival and proliferation of virally infected cells.
Our results have significance for the pathogenesis of KSHV-associated lymphoproliferative disorders. The KSHV-positive plasmablasts in MCD express IgM (7–9), and K13-transgenic mice have been shown to develop an MCD-like disease (42). It is conceivable that inhibition of anti-IgM-induced apoptosis combined with the known ability of K13 to promote cellular proliferation (20) may contribute to the accumulation of IgM-expressing plasmablasts and autoimmune manifestations seen in MCD (43–45). Finally, our results may also have implications for the pathogenesis of KSHV-associated PEL. Although the PEL cells generally lack surface IgM expression, KSHV was recently shown to selectively establish infection in IgM(λ)-expressing B cells (46). This has led to the suggestion that PEL and MCD may share an IgM-expressing progenitor and that the lack of surface IgM expression in PEL cells is due to postinfection events that drive their differentiation into late-plasmablast stage, a stage known to be associated with loss of surface IgM (46). Therefore, K13 may also contribute to the pathogenesis of PEL by blocking anti-IgM-induced apoptosis of IgM(λ)-expressing PEL progenitor cells, which after accumulation of additional genetic and epigenetic alterations leads to the development of PEL. This hypothesis is consistent with the development of lymphoma with an immuno-phenotype resembling PEL in K13-transgenic mice (42, 47).
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (CA139119, DE019811, and CA124621) and the Leukemia and Lymphoma Society. Flow Cytometry was performed in the USC Flow Cytometry Core Facility that is supported in part by the National Cancer Institute Cancer Center Shared Grant award P30CA014089 and the USC Provost Office Dean's Development Funds.
Footnotes
Published ahead of print 12 December 2012
REFERENCES
- 1. Niiro H, Clark EA. 2002. Regulation of B-cell fate by antigen-receptor signals. Nat. Rev. Immunol. 2:945–956 [DOI] [PubMed] [Google Scholar]
- 2. Eeva J, Pelkonen J. 2004. Mechanisms of B cell receptor induced apoptosis. Apoptosis 9:525–531 [DOI] [PubMed] [Google Scholar]
- 3. Gerondakis S, Siebenlist U. 2010. Roles of the NF-κB pathway in lymphocyte development and function. Cold Spring Harbor Perspect. Biol. 2:a000182 doi:10.1101/cshperspect.a000182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 5. Nador RG, Cesarman E, Chadburn A, Dawson DB, Ansari MQ, Sald J, Knowles DM. 1996. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood 88:645–656 [PubMed] [Google Scholar]
- 6. Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, D'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 7. Du MQ, Liu H, Diss TC, Ye H, Hamoudi RA, Dupin N, Meignin V, Oksenhendler E, Boshoff C, Isaacson PG. 2001. Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in Castleman disease and associated lymphoproliferative disorders. Blood 97:2130–2136 [DOI] [PubMed] [Google Scholar]
- 8. Dupin N, Diss TL, Kellam P, Tulliez M, Du MQ, Sicard D, Weiss RA, Isaacson PG, Boshoff C. 2000. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 95:1406–1412 [PubMed] [Google Scholar]
- 9. Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U. S. A. 96:4546–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. 1997. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386:517–521 [DOI] [PubMed] [Google Scholar]
- 11. Chaudhary PM, Jasmin A, Eby MT, Hood L. 1999. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 18:5738–5746 [DOI] [PubMed] [Google Scholar]
- 12. Field N, Low W, Daniels M, Howell S, Daviet L, Boshoff C, Collins M. 2003. KSHV vFLIP binds to IKK-γ to activate IKK. J. Cell Sci. 116:3721–3728 [DOI] [PubMed] [Google Scholar]
- 13. Liu L, Eby MT, Rathore N, Sinha SK, Kumar A, Chaudhary PM. 2002. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Iκ B kinase complex. J. Biol. Chem. 277:13745–13751 [DOI] [PubMed] [Google Scholar]
- 14. Matta H, Chaudhary PM. 2004. Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP). Proc. Natl. Acad. Sci. U. S. A. 101:9399–9404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chugh P, Matta H, Schamus S, Zachariah S, Kumar A, Richardson JA, Smith AL, Chaudhary PM. 2005. Constitutive NF-κB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 102:12885–12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guasparri I, Keller SA, Cesarman E. 2004. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 199:993–1003 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17. Matta H, Surabhi RM, Zhao J, Punj V, Sun Q, Schamus S, Mazzacurati L, Chaudhary PM. 2007. Induction of spindle cell morphology in human vascular endothelial cells by human herpesvirus 8-encoded viral FLICE inhibitory protein K13. Oncogene 26:1656–1660 [DOI] [PubMed] [Google Scholar]
- 18. Sun Q, Matta H, Chaudhary PM. 2003. The human herpes virus 8-encoded viral FLICE inhibitory protein protects against growth factor withdrawal-induced apoptosis via NF-κB activation. Blood 101:1956–1961 [DOI] [PubMed] [Google Scholar]
- 19. Sun Q, Matta H, Lu G, Chaudhary PM. 2006. Induction of IL-8 expression by human herpesvirus 8 encoded vFLIP K13 via NF-κB activation. Oncogene 25:2717–2726 [DOI] [PubMed] [Google Scholar]
- 20. Sun Q, Zachariah S, Chaudhary PM. 2003. The human herpes virus 8-encoded viral FLICE-inhibitory protein induces cellular transformation via NF-κB activation. J. Biol. Chem. 278:52437–52445 [DOI] [PubMed] [Google Scholar]
- 21. Benhamou LE, Cazenave PA, Sarthou P. 1990. Anti-immunoglobulins induce death by apoptosis in WEHI-231 B lymphoma cells. Eur. J. Immunol. 20:1405–1407 [DOI] [PubMed] [Google Scholar]
- 22. Matta H, Mazzacurati L, Schamus S, Yang T, Sun Q, Chaudhary PM. 2007. Kaposi's sarcoma-associated herpesvirus (KSHV) oncoprotein K13 bypasses TRAFs and directly interacts with the IκB kinase complex to selectively activate NF-κB without JNK activation. J. Biol. Chem. 282:24858–24865 [DOI] [PubMed] [Google Scholar]
- 23. Dong X, Craig T, Xing N, Bachman LA, Paya CV, Weih F, McKean DJ, Kumar R, Griffin MD. 2003. Direct transcriptional regulation of RelB by 1α,25-dihydroxyvitamin D3 and its analogs. J. Biol. Chem. 278:49378–49385 [DOI] [PubMed] [Google Scholar]
- 24. Yang Y, Groshong JS, Matta H, Gopalakrishnan R, Yi H, Chaudhary PM. 2011. Constitutive NF-κB activation confers interleukin 6 (IL6) independence and resistance to dexamethasone and Janus kinase inhibitor INCB018424 in murine plasmacytoma cells. J. Biol. Chem. 286:27988–27997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matta H, Punj V, Schamus S, Mazzacurati L, Chen AM, Song R, Yang T, Chaudhary PM. 2008. A nuclear role for Kaposi's sarcoma-associated herpesvirus-encoded K13 protein in gene regulation. Oncogene 27:5243–5253 [DOI] [PubMed] [Google Scholar]
- 26. Zhao J, Punj V, Matta H, Mazzacurati L, Schamus S, Yang Y, Yang T, Hong Y, Chaudhary PM. 2007. K13 blocks KSHV lytic replication and deregulates vIL6 and hIL6 Expression: a model of lytic replication induced clonal selection in viral oncogenesis. PLoS One 2:e1067 doi:10.1371/journal.pone.0001067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Punj V, Matta H, Schamus S, Yang T, Chang Y, Chaudhary PM. 2009. Induction of CCL20 production by Kaposi sarcoma-associated herpesvirus: role of viral FLICE inhibitory protein K13-induced NF-κB activation. Blood 113:5660–5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schauer SL, Bellas RE, Sonenshein GE. 1998. Dominant signals leading to inhibitor kappaB protein degradation mediate CD40 ligand rescue of WEHI 231 immature B cells from receptor-mediated apoptosis. J. Immunol. 160:4398–4405 [PubMed] [Google Scholar]
- 29. Wu M, Lee H, Bellas RE, Schauer SL, Arsura M, Katz D, FitzGerald MJ, Rothstein TL, Sherr DH, Sonenshein GE. 1996. Inhibition of NF-κB/Rel induces apoptosis of murine B cells. EMBO J. 15:4682–4690 [PMC free article] [PubMed] [Google Scholar]
- 30. Bren GD, Solan NJ, Miyoshi H, Pennington KN, Pobst LJ, Paya CV. 2001. Transcription of the RelB gene is regulated by NF-κB. Oncogene 20:7722–7733 [DOI] [PubMed] [Google Scholar]
- 31. Mineva ND, Rothstein TL, Meyers JA, Lerner A, Sonenshein GE. 2007. CD40 ligand-mediated activation of the de novo RelB NF-κB synthesis pathway in transformed B cells promotes rescue from apoptosis. J. Biol. Chem. 282:17475–17485 [DOI] [PubMed] [Google Scholar]
- 32. Wu M, Arsura M, Bellas RE, FitzGerald MJ, Lee H, Schauer SL, Sherr DH, Sonenshein GE. 1996. Inhibition of c-myc expression induces apoptosis of WEHI 231 murine B cells. Mol. Cell. Biol. 16:5015–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu M, Bellas RE, Shen J, Yang W, Sonenshein GE. 1999. Increased p27Kip1 cyclin-dependent kinase inhibitor gene expression following anti-IgM treatment promotes apoptosis of WEHI 231 B cells. J. Immunol. 163:6530–6535 [PubMed] [Google Scholar]
- 34. Yang W, Shen J, Wu M, Arsura M, FitzGerald M, Suldan Z, Kim DW, Hofmann CS, Pianetti S, Romieu-Mourez R, Freedman LP, Sonenshein GE. 2001. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene 20:1688–1702 [DOI] [PubMed] [Google Scholar]
- 35. Lee H, Arsura M, Wu M, Duyao M, Buckler AJ, Sonenshein GE. 1995. Role of Rel-related factors in control of c-myc gene transcription in receptor-mediated apoptosis of the murine B cell WEHI 231 line. J. Exp. Med. 181:1169–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. 2011. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146:904–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Herzog S, Reth M, Jumaa H. 2009. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat. Rev. Immunol. 9:195–205 [DOI] [PubMed] [Google Scholar]
- 38. Eeva J, Nuutinen U, Ropponen A, Mättö M, Eray M, Pellinen R, Wahlfors J, Pelkonen J. 2009. Feedback regulation of mitochondria by caspase-9 in the B cell receptor-mediated apoptosis. Scand. J. Immunol. 70:574–583 [DOI] [PubMed] [Google Scholar]
- 39. Vrzalikova K, Vockerodt M, Leonard S, Bell A, Wei W, Schrader A, Wright KL, Kube D, Rowe M, Woodman CB, Murray PG. 2011. Down-regulation of BLIMP1α by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood 117:5907–5917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Young LS, Dawson CW, Eliopoulos AG. 2000. The expression and function of Epstein-Barr virus encoded latent genes. Mol. Pathol. 53:238–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Madureira PA, Matos P, Soeiro I, Dixon LK, Simas JP, Lam EW-F. 2005. Murine γ-herpesvirus 68 latency protein M2 binds to Vav signaling proteins and inhibits B-cell receptor-induced cell cycle arrest and apoptosis in WEHI-231 B cells. J. Biol. Chem. 280:37310–37318 [DOI] [PubMed] [Google Scholar]
- 42. Ballon G, Chen K, Perez R, Tam W, Cesarman E. 2011. Kaposi sarcoma herpesvirus (KSHV) vFLIP oncoprotein induces B cell transdifferentiation and tumorigenesis in mice. J. Clin. Invest. 121:1141–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bower M, Newsom-Davis T, Naresh K, Merchant S, Lee B, Gazzard B, Stebbing J, Nelson M. 2011. Clinical features and outcome in HIV-associated multicentric Castleman's disease. J. Clin. Oncol. 29:2481–2486 [DOI] [PubMed] [Google Scholar]
- 44. Parravicini C, Corbellino M, Paulli M, Magrini U, Lazzarino M, Moore PS, Chang Y. 1997. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman's disease. Am. J. Pathol. 151:1517–1522 [PMC free article] [PubMed] [Google Scholar]
- 45. Waterston A, Bower M. 2004. Fifty years of multicentric Castleman's disease. Acta Oncol. 43:698–704 [DOI] [PubMed] [Google Scholar]
- 46. Hassman LM, Ellison TJ, Kedes DH. 2011. KSHV infects a subset of human tonsillar B cells, driving proliferation and plasmablast differentiation. J. Clin. Invest. 121:752–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ahmad A, Groshong JS, Matta H, Schamus S, Punj V, Robinson LJ, Gill PS, Chaudhary PM. 2010. Kaposi's sarcoma associated herpesvirus-encoded viral FLICE inhibitory protein (vFLIP) K13 cooperates with Myc to promote lymphoma in mice. Cancer Biol. Ther. 10:1033–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]