

Abstract
Virus-like particles (VLPs) from hepatitis B and human papillomaviruses have been successfully used as preventative vaccines against these infectious agents. These VLPs consist of a self-associating capsid polymer formed from a single structure protein and are devoid of viral DNA. Since virions from herpesviruses consist of a large number of molecules of viral and cellular origin, generating VLPs from a subset of these would be a particularly arduous task. Therefore, we have adopted an alternative strategy that consists of producing DNA-free defective virus particles in a cell line infected by a herpesvirus mutant incapable of packaging DNA. We previously reported that an Epstein-Barr virus (EBV) mutant devoid of the terminal repeats (ΔTR) that act as packaging signals in herpesviruses produces substantial amounts of VLPs and of light particles (LPs). However, ΔTR virions retained some infectious genomes, and although these mutants had lost their transforming abilities, this poses potential concerns for clinical applications. Therefore, we have constructed a series of mutants that lack proteins involved in maturation and assessed their ability to produce viral DNA-free VLP/LPs. Some of the introduced mutations were deleterious for capsid maturation and virus production. However, deletion of BFLF1/BFRF1A or of BBRF1 resulted in the production of DNA-free VLPs/LPs. The ΔBFLF1/BFRF1A viruses elicited a potent CD4+ T-cell response that was indistinguishable from the one obtained with wild-type controls. In summary, the defective particles produced by the ΔBFLF1/BFRF1A mutant fulfill the criteria of efficacy and safety expected from a preventative vaccine.
INTRODUCTION
The Epstein-Barr virus (EBV) infects more than 90% of the human population worldwide (1). Primary infection usually occurs during early childhood and leads to asymptomatic lifelong persistence. However, if infection is delayed, infectious mononucleosis (IM), a self-limiting lymphoproliferative disease, can develop. IM has been reported to increase the risk of different types of lymphomas, including Hodgkin's lymphoma (2, 3).
Owing to a strong cellular immune response, EBV infection is usually rapidly controlled in healthy individuals (4). However, immunodeficiency caused by infectious agents such as human immunodeficiency virus or by iatrogenic treatment after organ transplantation can lead to the development of posttransplant lymphoproliferative disease (PTLD), an affliction that carries a high degree of morbidity and mortality (5). Moreover, EBV has been described as the etiological agent of several other malignancies such as nasopharyngeal carcinoma and gastrointestinal carcinoma (6, 7). Altogether, EBV infection probably causes as many cancer cases as hepatitis C virus infection (8). Therefore, generation of an EBV prophylactic or even of a therapeutic vaccine is of prime importance.
The recent years have witnessed different attempts at generating an EBV-specific vaccine (9). One approach focuses on the major EBV membrane glycoprotein gp350; other approaches use a recombinant vaccinia virus to deliver a restricted number of EBV peptide epitopes (10–12). All approaches led to the production of neutralizing antibodies against these virus proteins, and one of them reduced the frequency of IM but did not confer protection against wild-type virus infection (12). In recent decades, virus-like particles (VLPs) have emerged as attractive vaccine candidates. VLPs differ from wild-type viruses in that they lack the viral genome. Therefore, they cannot replicate and propagate but can elicit an immune response against their constituents (13).
VLPs derived from human papillomavirus (HPV) and hepatitis B virus (HBV) proved to efficiently prevent infections with these pathogens (14). These particles consist of a single protein that self-assembles to form capsomers and VLPs. Such an approach with EBV is probably inapplicable, as its virions comprise more than 40 viral proteins and include cellular membranes that form the virus envelope. Therefore, EBV-infected cells must be induced to produce VLPs following launching of EBV viral replication. We previously identified HEK293 cells infected with ΔTR, an EBV mutant that lacks the DNA packaging signals also known as the terminal repeats, as an abundant source of EBV VLPs and light particles (LPs) (15, 16). These defective virions were found to elicit a potent T-cell response in vitro (17). However, deletion of the terminal repeats did not completely block viral DNA incorporation, although it caused a complete loss of the wild-type EBV transforming properties (16). Thus, these VLPs did not fulfill the safety criteria for use in humans. VLPs could in principle also be obtained by eliminating the viral proteins required for viral DNA incorporation. Because very little is known about the EBV proteins that serve this function, we took advantage of the knowledge accumulated in alphaherpesviruses. BGRF1/BDRF1, BALF3, and BFRF1A are the EBV homologues of the alphaherpesvirus pUL15, pUL28, and pUL33, respectively. These proteins form the terminase complex that is responsible for cleavage of DNA concatemers during virus replication and DNA packaging into preassembled capsids (18, 19). The endonuclease pUL15 possesses a highly conserved ATP binding motif and is required for DNA cleavage, for incorporation into capsids (20), and for the translocation of pUL28, pUL33, and pUL6 into the nuclear replication centers (21, 22). The DNA binding protein pUL28 (23) interacts with pUL15 (24) and pUL33. pUL33 also stabilizes the terminase complex (25). pUL32, the EBV homologue of BFLF1, is another important protein that is involved not only in cleavage and DNA packaging but also in transport of viral capsids to nuclear DNA replication compartments (26, 27). In the present study, we generated a series of mutants that lack the EBV homologues of some of these proteins and report their phenotypes and ability to produce viral DNA-free VLPs.
MATERIALS AND METHODS
Primary cells and cell lines.
HEK293 is a neuroendocrine cell line obtained by transformation of embryonic epithelial kidney cells with adenovirus (28, 29). Raji is an EBV-positive Burkitt's lymphoma cell line (30). Elijah 5E5 is an EBV-negative Burkitt's lymphoma cell line (kindly provided by A. B. Rickinson). RK13 is a rabbit kidney cell line (ATCC; CCL-37). WI38 are primary human lung embryonic fibroblasts (31). Peripheral blood mononuclear cells were purified from fresh blood buffy coats by density gradient centrifugation. CD19-positive B cells were isolated using M-450 CD19 (PanB) magnetic beads (Invitrogen) followed by detachment of the B cells from Dynabeads using Detachabead (Invitrogen). All cell lines were routinely grown in RPMI 1640 medium supplemented with 10% fetal calf serum (Biochrom). Lymphoblastoid cell line (LCL) and T cells were prepared and maintained as previously described (17).
Recombinant plasmids.
A BZLF1 expression plasmid (p509) was used for initiation of virus lytic replication (32). The entire BBRF1 gene (B95.8 coordinates 114204 to 116045; GenBank accession number V01555) was PCR amplified and cloned into the pRK5 expression plasmid under the control of a human cytomegalovirus (HCMV) immediate-early promoter. The BFLF1 gene (B95.8 coordinates 56948 to 58525) was PCR amplified and placed under the control of the p38 parvovirus promoter. The BFRF1A gene (B95.8 coordinates 58524 to 58929) and the BGRF1 and BDRF1 exons (B95.8 coordinates 124938 to 125915 and 129188 to 130351, respectively) were PCR amplified and cloned behind the HCMV promoter in pcDNA3.1(+) expression plasmids. Sequencing confirmed the integrity of all gene sequences.
Recombinant EBV genomes.
EBV wild-type (EBV-WT) recombinant plasmid (p2089) carries the prokaryotic F factor origin of replication, the green fluorescent protein (GFP) gene, the chloramphenicol (CAM) resistance gene, and the hygromycin (HYG) resistance gene as described previously (33). The EBV BBRF1-negative mutant was obtained by exchanging a large part of the BBRF1 gene (B95.8 coordinates 114501 to 115441; GenBank accession number V01555) with the kanamycin (KAN) resistance gene using homologous recombination (34). To this end, forward and reverse primers labeled BBRF1-fwd and BBRF1-rev, whose internal parts (24 bp; underlined) are specific for the KAN resistance gene and whose external parts (40 bp) are specific for the BBRF1 gene, were synthesized (Table 1). The internal sequences of these primers were used to amplify the KAN resistance gene from plasmid pCP15 by PCR, whereas their external sequences drove homologous recombination of the amplified PCR product with the EBV wild-type genome. All PCR amplification products were treated with the restriction enzyme DpnI to remove traces of the parental plasmids and introduced by electroporation (1,000 V, 25 μF, 100 Ω) into Escherichia coli DH10B cells carrying the recombinant virus p2089 and the temperature-sensitive pKD46 helper plasmid encoding the phage lambda red recombinase to foster homologous recombination. Cells were grown in Luria broth (LB) with CAM (15 μg/ml) at 37°C for an hour and then plated onto LB agar plates containing CAM (15 μg/ml) and KAN (10 μg/ml). Incubation at 42°C led to a progressive loss of the helper plasmid. After CAM and KAN double selection, DNA of positive clones was purified and analyzed with the BamHI and HindIII restriction enzymes to confirm correct recombination.
Table 1.
Sequences of primers used for generation of the different virus mutants and for qPCR detection of EBV DNAa
| Primer | Sequence |
|---|---|
| BBRF1-fwd | 5′-CTGCATCCAGAAGCGATGGCCGAGCGATGACTCGTGTGCGAACAGCTATGACCATGATTACGCC-3′ |
| BBRF1-rev | 5′-ACTGCGGCGGGGGCTGACGCCGCTGGGTTGTTGCCGCCGGCCAGTCACGACGTTGTAAAACGAC–3′ |
| BFLF1/BFRF1A-fwd | 5′-GCGTAATTAACGAGTCACAGACCCCCTGTTCCAGATTCTGAACAGCTATGACCATGATTACGCC-3′ |
| BFLF1/BFRF1A-rev | 5′-GTCCTTGTGGCGGTGTCCCAGAAACTGGTGCCGACCCCGGCCAGTCACGACGTTGTAAAACGAC-3′ |
| BGRF1-fwd | 5′-CAATGCTTCGAGCATACTGCCGCCACTATGGCCCCAGGCCCAACAGCTATGACCATGATTACGCC-3′ |
| BGRF1-rev | 5′-TTGTTTCTGGAATGCTTCTAGGCGGGCGGGCTTGTGCTTGCCAGTCACGACGTTGTAAAACGAC-3′ |
| BDRF1-fwd | 5′-AGGATGCTCAGGAGCGGCTGCTGAACGTGGTAAGTTATGTAACAGCTATGACCATGATTACGCC-3′ |
| BDRF1-rev | 5′-TTCTTGAACGTGTGCTTCTGTGTTGTTGCGAGAAAATGAGCCAGTCACGACGTTGTAAAACGAC-3′ |
| Pol-fwd | 5′-CTTTGGCGCGGATCCTC-3′ |
| Pol-rev | 5′-AGTCCTTCTTGGCTAGTCTGTTGAC-3′ |
| Pol-probe | 5′-FAM-CATCAAGAAGCTGCTGGCGGCC-TAMRA-3′ |
fwd, forward; rev, reverse. pCP15-specific sequences are underlined.
The BFLF1/BFRF1A double mutant was constructed using the same strategy. Here again, the BFLF1/BFRF1A genes (B95.8 coordinates 57091 to 58525) were replaced by the KAN resistance gene using specific primers (Table 1). The generation of the BGRF1/BDRF1 double-deletion mutant required two successive steps. First, a mutant devoid of BGRF1 was constructed by exchanging part of BGRF1 (B95.8 coordinates 125159 to 125423) against the kanamycin resistance gene flanked by FLP recombination sites as described previously (34). We excised this cassette using the FLP recombinase, and this genome served as a template for the construction of the double mutant that was obtained by deleting BDRF1 (B95.8 coordinates 129423 to 130302) through homologous recombination.
Stable clone selection.
HEK293 cells were transfected with recombinant EBV plasmid DNA using Metafectene (Biontex) as described previously (35). One day posttransfection, the cells were transferred to a cell culture dish (150 mm in diameter) and HYG (100 μg/ml) was added to the culture medium for selection of stable HEK293 clones carrying EBV recombinant plasmids. Outgrowing GFP-positive colonies were expanded for further investigation. The cell clones used in this study are referred to as 293/ΔBBRF1, 293/ΔBFLF1/BFRF1A, and 293/ΔBGRF1/BDRF1 to denote producer cells carrying the respective knockout mutants.
Plasmid rescue in E. coli.
A denaturation-renaturation method (36) was used for extraction of circular plasmid DNA from 293/ΔBBRF1, 293/ΔBFLF1/BFRF1A, and 293/ΔBGRF1/BDRF1 cells. E. coli strain DH10B was transformed with the viral recombinant DNA by electroporation as described before (34), and clones were selected on LB plates containing CAM (15 μg/ml). Single bacterial colonies were expanded, and the plasmid DNA preparation was subjected to digestion with restriction enzymes BamHI and HindIII.
Virus production and infection of B cells.
Virus lytic replication in the producer cells was induced by transfection of the various producer cell lines with a BZLF1 expression plasmid (0.5 μg per 4 × 105 cells) using lipid micelles (Metafectene; Biontex). In cases where the virus progeny was required for cell infection experiments, the BALF4 gene expression plasmid (0.5 μg) was cotransfected. In trans-complementation assays, 293/ΔBBRF1 cells were cotransfected with a BBRF1 expression plasmid (0.5 μg), 293/ΔBFLF1/BFRF1A cells with BFLF1 (0.5 μg) and BFRF1A (1 μg) expression plasmids, and 293/ΔBGRF1/BDRF1 cells with the BGRF1/BDRF1 expression plasmid (1 μg). Empty expression plasmids were used as negative controls. Virus supernatants were harvested 4 days posttransfection, filtered through a 0.45-μm-pore-size filter, and frozen at −80°C. Viral titers were determined by quantitative real-time PCR (qPCR). To study virus infectivity, 104 Raji cells were incubated with increasing dilutions of virus supernatants. Three days after infection, GFP-positive Raji cells were counted using a fluorescence microscope. To analyze virus/VLP immortalization ability, 105 primary B cells were exposed to 1.4 × 106 EBV-WT particles, i.e., a multiplicity of infection (MOI) of 10 mature particles, or to supernatant from lytically induced mutant producer cells that contain 1.4 × 107 ΔBFLF1/BFRF1A, 6 × 106 ΔBBRF1, and 1.4 × 107 ΔTR defective particles, respectively, as determined by electron microscopic (EM) analysis of pelleted virus particles (see below). B cell infections with supernatants from lytically induced and complemented producer cells were performed at an MOI of 10 as determined by qPCR (see below). Sixteen hours postinfection, 103 B cells were plated per well of a U-bottomed 96-well plate coated with gamma-irradiated WI38 feeder cells (50 Gy; 103 cells/well) in RPMI 1640 medium supplemented with 20% fetal calf serum. The number of wells containing outgrowing LCL clones was recorded 4 weeks postinfection.
Electron microscopy.
Three days after induction of the lytic cycle, cells were harvested, washed in phosphate-buffered saline (PBS), and pelleted. Alternatively, 5-ml samples of virus supernatants were centrifuged for 2 h at 30 000 × g to obtain virus pellets. Both preparations were fixed with 2.5% glutaraldehyde in PBS for 20 min at 4°C. Samples were further processed as previously described (37). Ultrathin sections were examined with a Zeiss electron microscope. The number of viral particles present in the studied supernatants was estimated from the observation of pellet sections. Pseudorabies mutant viruses were propagated in RK13 cells at an MOI of 1 as previously described (26), followed by fixation and embedding (38), and ultrathin sections were analyzed in an electron microscope (Tecnai 12; Philips).
EBV binding assay.
Virus supernatant (1 ml) containing EBV-WT, ΔBLLF1 (39), or mutant viruses lacking a packaging protein was incubated for 3 h on ice with 5 × 105 EBV-negative Elijah B cells. Subsequently, the cells were washed three times with ice-cold PBS and fixed on glass slides with pure acetone for 20 min. Immunostaining with a monoclonal antibody directed against EBV glycoprotein gp350/220 (ATCC 72A1 hybridoma) was performed as previously described (16), and slides were examined with an inverted fluorescence microscope (Leica DM IRB).
Virus purification using Dynabeads.
Dynal magnetic beads (1 mg) (M-270 Epoxy; Invitrogen) were coated with 15 μg anti-gp350 monoclonal antibody (clone 72A1 [40]) as recommended by the manufacturer. Antibody-coated beads (1 mg) were incubated with 1 ml of supernatant from lytically induced producer cell lines for 1 h at room temperature. Attached virus particles were collected with a magnet, washed three times with RPMI medium, and diluted in the initial volume of supernatant. Purified virus particle preparations were used for quantitative real-time PCR (qPCR) and Western blot analysis.
qPCR analysis.
DNA content in virus supernatants of lytically induced virus producer cells or of particles purified with magnetic beads, coupled to gp350-specific antibody, was determined by quantitative real-time PCR. Virus titers were calculated as genome equivalents per milliliter virus supernatant. Cell supernatants or purified virus particles were digested with DNase I (1 IU/ml supernatant) at 37°C for 1 h, followed by heat inactivation for 10 min at 70°C. Virus particles were lysed by adding proteinase K (1 mg/ml) 1:1 (vol/vol) and incubation for 60 min at 50°C, followed by proteinase K inactivation for 20 min at 75°C. Next, the DNA preparations were subjected to qPCR using primers and probe specific for BALF5 (referred to as Pol-fwd, Pol-rev, and Pol-probe in Table 1). Amplification reactions were performed in a total volume of 25 μl, including 12.5 μl of TaqMan universal PCR master mix (Applied Biosystems), 2.5 μl each of Pol-fwd and Pol-rev primers (2 μM), 1 μl of 5 μM FAM-labeled Pol-probe, 1.5 μl of water, and 5 μl of virus supernatant or purified particles. After initial activation of the DNA polymerase for 10 min at 95°C, samples were amplified for 40 cycles (15 s at 95°C and 60 s at 60°C). The ABI 7300 real-time PCR system (Applied Biosystems) was used for detection of the fluorescent signals. A serial dilution of an EBV bacterial artificial chromosome (BAC) preparation (p2089) was included to calculate the viral DNA content of the different supernatants.
Immunofluorescence.
Lytically induced cells were pelleted 3 days posttransfection, washed three times in PBS, and fixed on glass slides for 20 min using pure acetone at room temperature. Next, slides were incubated for 30 min with an antibody directed against EBV glycoprotein gp350/220 (ATCC 72A1 hybridoma) or early antigen EA-D (Chemicon/Millipore; MAB818). Slides were washed three times in PBS, incubated for 30 min with a secondary Cy3-conjugated goat-anti-mouse antibody (Dianova), washed several times in PBS, and embedded with 90% glycerol. Immunofluorescence was recorded using a fluorescence microscope coupled to a charge-coupled-device (CCD) camera (Leica).
Western blot analysis.
Purified virus particles were resuspended in PBS and denatured in Laemmli buffer for 5 min at 95°C. Denatured proteins were separated on a 10% SDS-polyacrylamide gel and electroblotted onto a nitrocellulose membrane (Hybond ECL; GE Healthcare). After blocking in 3% milk in PBS, the blot was incubated with a polyclonal serum against the EBV tegument protein BNRF1 (1:20,000 dilution) (41) for 1 h, followed by several washings in 0.1% Tween in PBS and incubation with horseradish peroxidase-coupled protein A (Promega) for 1 h. Bound antibody was detected using an ECL reagent (PerkinElmer).
T-cell activation assay.
T-cell activation assays were performed as described previously (17). Briefly, 5 × 104 LCLs were pulsed with 1 μg, 100 ng, or 10 ng BNRF1 or gp350 peptide, with 150 μl, 15 μl, and 1.5 μl supernatants, or with defective particles present in 150 μl of supernatant that were purified with a gp350-specific antibody coupled to magnetic Dynal beads. Tested virions and defective particles were obtained from lytically induced 293/EBV-WT, 293/ΔBBRF1, and 293/ΔBFLF1/BFRF1A cells. A 150-μl portion of EBV-WT supernatant corresponds to an MOI of 30 and contains approximately 2 × 106 virus particles. Stimulated LCLs were cocultured with 1 × 105 cells from a CD4+ T-cell clone specific for BNRF1 (1H7) (42) or for gp350 (ID6) in T-cell medium in 96-well flat-bottomed plates. Gamma interferon (IFN-γ) released by the stimulated T cells was measured by an enzyme-linked immunosorbent assay (ELISA).
RESULTS
Construction of a set of mutants lacking proteins involved in EBV maturation.
We generated a BBRF1-negative mutant and two double-deletion mutants that lack BFLF1/BFRF1A and BGRF1/BDRF1, respectively (Fig. 1). The BFLF1/BFRF1A double knockout was constructed by deletion of 1,395 bp of the BFLF1 open reading frame as a result of an exchange against the KAN resistance gene by homologous recombination in E. coli. The first 179 nucleotides of the gene open reading frame were not modified since they entail the promoter of the BFLF2 gene. The deletion also impaired expression of the BFRF1A open reading frame, which is encoded on the opposite strand. Restriction enzyme analysis with BamHI evidenced a 7.4-kb fragment in the EBV wild-type genome that compares with a 7.5-kb fragment generated by digestion of the ΔBFLF1/BFRF1A genomes. Digestion of the wild-type DNA with HindIII gave rise to a 29.3-kb fragment that contrasted with the two 9.1-kb and 20.2-kb fragments that resulted from the deletion (Fig. 1A).
Fig 1.

Construction of the recombinant viruses. Left panel, schematic maps of the EBV genome fragments that encompass four EBV genes presumably involved in virus maturation, before and after recombination with targeting vectors. Recombination results in an exchange between the targeted genes and the kanamycin (kana) resistance cassette. Schematic maps of targeted EBV gene regions around BFLF1/BFRF1A (A), around BBRF1 (B), and around BGRF1/BDRF1 (C). Promoters (arrows), enzyme restriction sites, and the predicted sizes of the resulting fragments after cleavage with BamHI or HindIII are indicated. Right panel, results of the restriction fragment analysis of the various mutant genomes after construction in E. coli or after rescue from the producer cell lines are presented. The results of this analysis concur with the predicted restriction pattern. EBV-WT, Epstein-Barr virus wild type.
The BBRF1 gene knockout was obtained by deletion of 940 bp of the BBRF1 gene open reading frame. The first 297 and the last 601 bp of this gene were kept intact since they encompass the promoters for BBLF4 and BBRF2 genes, respectively. The DNA of the resulting recombinant EBV genome was subjected to a BamHI restriction enzyme analysis. The wild-type and mutant genomes differed in that the BBRF1 mutant carries a larger BamHI fragment (10.3 kb compared to 9.7 kb in wild-type viruses) (Fig. 1B). Furthermore, the 25.9-kb HindIII fragment that spans the BBRF1 gene in the wild-type virus was split into a 3.6-kb and a 23-kb fragment in the BBRF1 mutant as expected (Fig. 1B).
BGRF1 and BDRF1 are two exons separated from each other by a 3.28-kb intron and are spliced together to form the BGRF1/BDRF1 mature mRNA. Therefore, the mutant construction included two independent and sequential rounds of deletion. BGRF1 and BDRF1 are located in a complex viral locus where most genes partly overlap and share promoters and polyadenylation signals. The first 221 nucleotides of BGRF1, which include the BGLF3 gene promoter, and the last 489 nucleotides, including part of the BGLF2 gene, were spared, but all remaining sequences were deleted. This mutant was used for construction of the double-deletion mutant. This recombination deleted 879 bp from the BDRF1 gene but left the BDLF4 promoter and the BDLF3 gene untouched. These alterations were identified by a BamHI cleavage that produced a 6.5-kb and an 8-kb fragment that, respectively, contain BGRF1 and BDRF1 in the wild-type genome fragment but shift to a 6.4-kb and an 8.7-kb fragment in the mutant DNA (Fig. 1C). Sequencing of all mutants confirmed the results of the restriction analyses.
Purified DNAs from each virus mutant were then transfected into HEK293 cells and subjected to HYG selection. Ten HYG-resistant and GFP-positive outgrowing cell clones were expanded and tested for their ability to support the viral lytic replication. Induction of lytic replication was achieved through transient expression of the immediate-early protein BZLF1. The 293/EBV-WT cell line, which contains the wild-type virus, was used as a positive control. Three days after transfection, cells were stained for BMRF1 and glycoprotein gp350, early and late markers of lytic replication, respectively, and clones showing a high percentage of positive cells were selected for further experiments. The mutant viral genomes present in these clones were extracted and transformed in E. coli cells, and their restriction profile was determined. The results of this analysis, presented in Fig. 1, confirm that the mutant genomes have retained an intact structure upon transfer into HEK293 cells. With the exception of the expected size shifts of the relevant BamHI or HindIII restriction fragments, no alterations of the mutant genomes were visible.
Phenotypic traits of the defective mutants.
The various producer cell lines were lytically induced, and electron micrographs of cells fixed 3 days later were examined (Fig. 2A, left panel). We found that all tested mutants produced capsids in replicating cells and that all the capsids were exclusively of type A or B, i.e., were devoid of electron-dense cores that indicate the presence of nucleic acids. This confirmed that the proteins lacking in the tested mutants are involved in viral DNA packaging. However, aside from this common trait, differences between the mutants were also readily visible.
Fig 2.

Electron micrographs of replicating producer cell lines carrying wild-type EBV or various mutants. (A) Induced producer cells with or without gene complementation. (B) Pelleted supernatants from induced cells. Insets show viral progenies at various stages of maturation. Bars, 200 nm; N, nucleus; C, cytoplasm.
The average number of capsids in replicating cells was reduced in induced 293/ΔBGRF1/BDRF1 cells relative to wild-type cells (Fig. 3A). In contrast, replicating 293/ΔBFLF1/BFRF1A, 293/ΔBBRF1, or 293/ΔTR cells contained approximately twice as many capsids as the wild-type cells. The total numbers and the percentages of viral particles observed in the cytoplasm of replicating cells were higher when they contained wild-type genomes than when they contained any of the mutant viruses (Fig. 3B). We conclude from these observations that BGRF1/BDRF1 is important for capsid assembly and/or stability as well as for nuclear egress, that all proteins studied are essential for viral DNA packaging, and that A and B capsids are preferentially though not exclusively retained in the nucleus.
Fig 3.
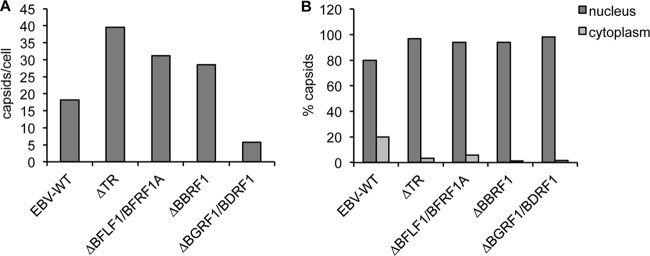
Results of the electron microscopic analysis of producer cells. (A) Absolute numbers of EBV capsids per replicating producer cells. A total of 350 cells per mutant were analyzed. (B) The percentages of capsids observed in the nucleus or cytoplasm of the different producer cell lines studied are indicated.
All producer cell lines, with the exception of 293/ΔTR, were complemented by cotransfection of expression plasmids that carried the genes missing in the respective recombinant viruses. The levels of gp350 protein expression in complemented and noncomplemented cells were similar (data not shown). In each case, induced and complemented cells were examined by electron microscopy and found to contain both mature and immature capsids in their nuclei (Fig. 2A, right panel). Therefore, we conclude that the genetic defects present in the studied viral knockouts are limited to the mutations intentionally introduced. The 293/ΔTR mutant obviously cannot be complemented by proteins, but we previously reported that a 293 cell line carrying this mutant can efficiently package plasmids that contain the EBV terminal repeats. This mutant can therefore be cis complemented (15).
To calculate the approximate amount of virus particles (virions/VLPs/LPs) present in 1 ml virus supernatant from lytically induced EBV-WT or mutant producer cell lines, we pelleted 1 ml of each supernatant and examined ultrathin slices by EM. The bulk of virus particles is concentrated in certain areas of the virus pellet. We counted both LPs and VLPs and calculated the number of EBV-WT and mutant virus particles per micrometer of slice area. Knowledge of the total area containing virus particles allowed calculation of the total number of viruses present in 1 ml of supernatant. Since qPCR analysis of EBV-WT revealed that 1 ml of supernatant contains 107 genomes, and considering that only 70% of the particles carry DNA, 1 ml EBV-WT supernatant contains 1.4 × 107 virus particles. Taking into account the number of particles present per slice area in EM, we could estimate the number of mutant particles as being approximately 1.4 × 107 ΔTR, 1.4 × 107 ΔBFLF1/BFRF1A, and 6 × 106 ΔBBRF1 particles per ml supernatant, respectively.
293/ΔBFLF1/BFRF1A cells produce large amounts of DNA-free VLPs/LPs.
We then conducted a series of experiments that aimed at defining the characteristics of the VLPs/LPs present in the supernatants from induced 293/ΔBFLF1/BFRF1A and 293/ΔBBRF1 cells. To this aim, we performed infection studies both on primary B cells and on the B cell lymphoma cell line Raji. The results of these experiments are given in Table 2 and Fig. 4. A total of 105 primary B cells were exposed to approximately 1.4 × 106 EBV-WT, 1.4 × 107 ΔTR, 1.4 × 107 ΔBFLF1/BFRF1A, and 6 × 106 ΔBBRF1 particles. None of the mutants was able to transform primary B cells. The amount of EBV-WT particles used in the transformation assay corresponds to an MOI of 10. The number of mutant particles was found to be at least one order of magnitude higher than that of EBV-WT. Complementation of these mutants restored their ability to produce infectious viruses, and complemented ΔBFLF1/BFRF1A, ΔBBRF1, and ΔBGRF1/BDRF1 viruses transformed B cells as efficiently as wild-type viruses (data not shown). Therefore, these proteins do not contribute to B cell transformation, although the performed experiments cannot formally exclude a role for these proteins in the initial stages of infection, e.g., in case they become incorporated into the mature virion and serve a function immediately after infection and before a new round of viral transcription can be initiated.
Table 2.
Phenotypic traits of the various EBV mutants
| EBV virus | Statusg of: |
|||||
|---|---|---|---|---|---|---|
| Lytic replicationa | Raji B-cell infectionb | B-cell transformationc | VLP productiond | Packaged DNAe | T-cell stimulationf | |
| WT | + | + (2.5 × 105) | + | + | + (1 × 107) | + |
| ΔTR | + | + (1.4 × 102) | − | + | + (1 × 104) | + |
| ΔBFLF1/BFRF1A | + | − | − | + | − | + |
| ΔBBRF1 | + | − | − | + | − | + |
| ΔBGRF1/BDRF1 | + | − | − | − | NA | NA |
Lytically induced virus producer cells were immunostained for EA-D and gp350.
Presented as green Raji units per ml supernatant.
Outgrowth of lymphoblastoid cells was recorded 4 weeks postinfection.
Measured by Western blot analysis of purified particles using a BNRF1-specific antibody, by electron microscopy of pelleted supernatants from induced producer cell lines, and by binding to Elijah B cells, followed by immunostaining with a gp350-specific antibody.
Measured by qPCR analysis and presented as genomes per ml supernatant.
IFN-gamma production of stimulated T cells as measured by ELISA.
−, negative; +, positive; NA, not applicable.
Fig 4.
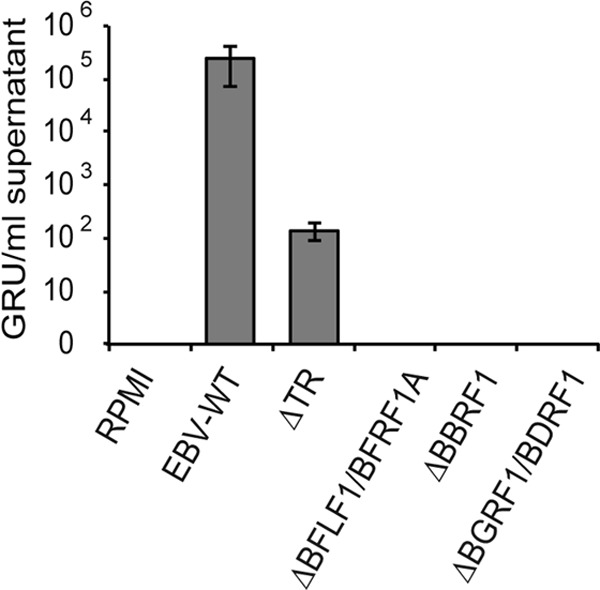
Deletion of BFLF1/BFRF1A, BBRF1, or BGRF1/BDRF1 curbs the virus ability to infect B cells. Raji cells were exposed to increasing dilutions of supernatants from induced producer cell lines. Three days postinfection, cells were examined under UV light and the number of GFP-positive cells was counted. Titers are given as green Raji units (GRU) per ml of supernatant.
Raji cell infections were largely confirmative of the transformation assays, in that none of the mutants could infect these cells with the notable exception of supernatants from induced 293/ΔTR cells that displayed infectious titers on the order of 1 × 102 Raji cells per ml of supernatants (Fig. 4), as previously reported (16). Therefore, the VLPs/LPs produced by 293/ΔBFLF1/BFRF1A and 293/ΔBBRF1 are devoid of infectious DNA.
VLPs/LPs used in vivo must fulfill multiple safety criteria, among which the absence of viral DNA from a transforming virus is of prime importance. Therefore, we investigated the presence of contaminating EBV DNA in VLPs/LPs produced by some of the mutants using qPCR, as this method might be more sensitive than the infection experiments previously described (293/ΔBGRF1/BDRF1 were excluded from this analysis as they generate no or only rare LP/VLPs). However, we deemed it important to restrict our analysis to purified virions so as to exclude potential DNA contaminants from nonviral structures. To that aim, we coupled antibodies directed against the viral envelope protein gp350 to magnetic beads to purify viral structures. Incubation of these coupled antibodies with supernatants from induced 293/ΔBFLF1/BFRF1A, 293/ΔBBRF1, 293/EBV-WT, or 293/ΔTR cells followed by collection with a magnet allowed multiple washings and purification of viral structures. These were submitted in parallel to a qPCR analysis using primers and probe specific for the BALF5 gene (EBV-Pol) and to Western blot analysis with a BNRF1-specific antibody. The latter assay confirmed the presence of BNRF1-positive viral structures in supernatants from induced 293/ΔBFLF1/BFRF1A, 293/ΔBBRF1, 293/EBV-WT, and 293/ΔTR cells (Fig. 5A and Table 2). It also confirmed the relative paucity of VLPs/LPs in supernatants from induced 293/ΔBBRF1 cells. The detection of BNRF1, an EBV tegument protein, within the material purified with the gp350-specific antibodies fits with the previous results and strongly suggests that, with the exception of 293/ΔBBRF1, the mutants produce VLPs/LPs at approximately the same rate as 293/EBV-WT produces infectious viruses.
Fig 5.

Purification and analysis of DNA-free defective virus particles. (A) Virus particles from 1 ml of the indicated supernatants were pulled down using gp350-specific antibodies and protein extract thereof were separated through an acrylamide gel. Blotted proteins were incubated with a rabbit antiserum specific for BNRF1 tegument protein. Wild-type viruses were used as a positive control. (B) Elijah B cells were incubated with 1 ml of the various virus supernatants, and bound particles were visualized by immunostaining using a gp350-specific antibody. Wild-type virus was used as a positive control and gp350 knockout mutant virus (ΔBLLF1) as a negative control. (C) The viral DNA content per ml of purified supernatant was determined by qPCR analysis using an EBV-specific probe. This assay used RPMI and wild-type viruses as negative and positive controls, respectively. The lower limit of quantitative measurement is 5 × 103 DNA molecules per ml of supernatant. The mean results and standard deviations from eight independent experiments are given.
These findings were confirmed by an EBV particle binding assay. We incubated EBV-negative Elijah B cells with supernatants from lytically induced wild-type or mutant producer cell lines and performed an immunostaining with a gp350-specific antibody to visualize bound particles (Fig. 5B and Table 2). We found that induced 293/ΔBFLF1/BFRF1A and 293/ΔTR mutants produced at least as many viral particles as 293/EBV-WT. In contrast, 293/ΔBBRF1 generated around 10 times less particles than the wild type and the 293/ΔBGRF1/BDRF1 mutant did not produce any particles. Gp350 deletion mutant (ΔBLLF1) particles were used as a negative control and, as expected, did not bind to B cells.
The qPCR assay performed with supernatants from induced 293/ΔBBRF1 or 293/ΔBFLF1/BFRF1A cells delivered signals indistinguishable from those obtained with virus-free medium control; i.e., these supernatants contained fewer than 103 DNA molecules per ml. This corresponds to fewer than 0.00036 DNA molecules per ΔBFLF1/BFRF1A VLP/LP or fewer than 0.0008 DNA molecules per ΔBBRF1 VLP/LP (Fig. 5C and Table 2). In contrast, this assay allowed the detection of viral DNA within purified ΔTR virions, although it was a thousand times less abundant than in wild-type viruses.
ΔBBRF1 and ΔBFLF1/BFRF1A elicit a CD4-positive T-cell response.
Having ascertained that producer cell lines that lack BBRF1 or BFLF1/BFRF1A produce DNA-free VLPs/LPs, we wished to determine whether these maintain their ability to induce a T-cell response. To this aim, we incubated virus supernatants or purified defective particles from these producer cell lines with LCLs that were subsequently cocultivated with BNRF1- or gp350-specific T-cell clones. Evidence for T-cell activation was recorded by measuring IFN-γ production using ELISA. We found that the VLPs/LPs elicited a dose-dependent IFN-γ release response that was similar in amplitude to the one observed in the wild-type positive control (Fig. 6A and B). The intensity of T-cell stimulation with antibody-purified particle preparations was reduced compared to that in nonpurified virus supernatants. This may result from impaired T-cell recognition due to the presence of magnetic beads (Fig. 6A and B, gray bars). However, the efficiency of T-cell stimulation with purified wild-type or ΔBFLF1/BFRF1A defective particles was within the same range. As expected, ΔBBRF1 VLPs/LPs delivered a weak signal.
Fig 6.
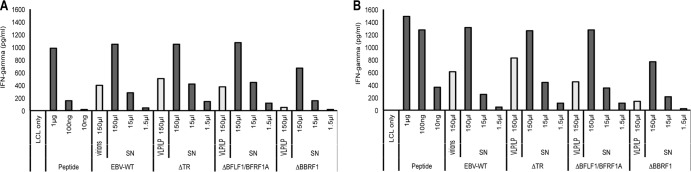
B cells exposed to defective or wild-type EBV particles are recognized by BNRF1- and gp350-specific CD4+ T cells. B cells were pulsed with antibody-purified particles (VLP/LP or virions) (gray bars) or supernatants (SN) from induced producer cells (dark gray bars) that carry wild-type virus or defective virus mutants as indicated and were incubated with syngeneic T cells that are specific for BNRF1 (A) or gp350 (B). Incubation of T cells with B cells that were pulsed with dilutions of the respective peptides served as positive controls. IFN-γ released into the medium by stimulated T cells was determined by ELISA.
Links between VLP production and capsid maturation in pseudorabies virus.
Although herpesvirus capsid maturation and egress were mainly studied in alphaherpesviruses, little has been reported on the nuclear egress of empty capsids and the production of VLPs in these infectious agents. Therefore, we reexamined the phenotypic traits of pseudorabies (PrV) mutants that lack pUL6 (homologue of BBRF1), pUL15 (homologue of BGRF1/BDRF1), pUL32 (homologue of BFLF1), or pUL33 (homologue of BFRF1A), which have already been demonstrated to produce large amounts of light particles (LPs) (26). We now confirm these results and additionally find that, with the exception of the pUL15 (BGRF1/BDRF1)-deleted virus, all PrV mutants also produce a minority of VLPs. As an example, viral particles released from RK13 cells infected with PrVΔUL32 are shown in Fig. 7. After analysis of multiple electron micrographs, we estimate that VLPs represent approximately 10% of the defective particles released by pUL6-, pUL32-, or pUL33-deleted PrV.
Fig 7.

Electron microscopy of cells infected with a PrV UL32 deletion mutant. (A) VLPs and LPs are released at the cell membrane of infected cells. The inset represents an example of a VLP (bar, 250 nm). (B) Multiple LPs and VLPs are present in the extracellular space. The inset on the left shows an LP and that on the right a VLP (bar, 500 nm).
DISCUSSION
The purpose of the present study was to identify proteins that are essential for EBV maturation and whose absence triggers abundant VLP/LP production. This search was guided by knowledge accumulated on alphaherpesviruses.
Herpesvirus maturation has been extensively studied in the alphaherpesviruses HSV1 and PrV. In HSV1, pUL15, pUL28, and pUL33, the EBV homologues of BGRF1/BDRF1, BALF3, and BFRF1A, respectively, form the terminase complex that cleaves monomeric genomes from the concatemers built during virus replication but also facilitates their translocation into preformed capsids (18, 19). pUL28 binds to the viral DNA (23) whereas pUL33 interacts with pUL28 to stabilize the terminase complex (25). pUL15 is an endonuclease that is also able to recruit ATP (20), which can be hydrolyzed to fuel viral DNA incorporation into capsids. This complex protein is involved in the transport of pUL28 and pUL33 and also of pUL6 to the nuclear replication centers (21, 22). In addition, pUL32, the EBV BFLF1 homologue, has been found to be important for cleavage and packaging of the HSV1 and PrV genomes. pUL32 is required for the transport of viral capsids to nuclear DNA replication compartments (26, 27).
The analysis of the EBV/ΔBFLF1/BFRF1A mutant revealed a defect in viral DNA packaging, but primary egress of empty capsids and the release of DNA-free VLPs were not impaired (Fig. 2 and 3). We could demonstrate that these DNA-free VLPs can induce a potent CD4+ T-cell response (Fig. 6). Because our mutant lacks both BFLF1 (pUL32 homologue) and BFRF1A (pUL33 homologue), it is difficult to delineate the function of both proteins. However, a PrV mutant that lacks pUL32 was found to generate exclusively immature capsids (26). Deletion of pUL33 from the PrV or HSV1 genome had similar effects (26, 43).
A more complex picture emerged from the study of the ΔBGRF1/BDRF1 mutant in the present paper and of previously published data on knockouts of its homologues in alphaherpesviruses (pUL15). A common feature of ΔBGRF1/BDRF1 and ΔUL15 in HSV1 and PrV is the absence of C-type capsids in cells lytically infected by these mutants (Fig. 2) (26, 44, 45). However, deletion of BGRF1/BDRF1, but not of UL15, also drastically reduced capsid synthesis (Fig. 3A). Furthermore, successful nuclear egress was among the phenotypic traits visible in cells infected with HSV1 ΔUL15 (45) but not in those infected with PrV ΔUL15 (26). This suggests that the three homologues diverge, at least to a certain extent, in their functions. As already mentioned, pUL15 has been implicated in protein transport of other lytic proteins, including HSV pUL28 and pUL33, but also in ATP binding (45, 46). This protein also contributes to DNA translocation into the capsid and possesses an endonuclease activity (20, 22). Thus, it is not unexpected that such a multifunctional protein should serve overlapping though not identical functions in different viruses.
The BBRF1 homologue pUL6, also known as the portal protein, forms homo-multimers that arrange into a circular structure that is incorporated into procapsids. The pUL6 complexes form a docking site for the terminase complex with which it interacts and builds the entry site for the viral genome into the capsids (47–49). HSV1 and PrV mutants that lack pUL6 are unable to incorporate their DNA into preformed capsids (50–52). However, rare immature capsids can undergo primary envelopment and gain access to the cytoplasm, although no information was available on VLP production in the extracellular milieu. Analysis of the EBV/ΔBBRF1 mutant confirms that in EBV this protein is also required for viral genome incorporation and is therefore very likely to be the functional homologue of pUL6. Lytically induced ΔBBRF1 cells produced capsids in large numbers, all of which were found to be of A or B type (Fig. 2). Most capsids located to the nucleus, but some could also be found in the cytoplasm (Fig. 3B). The cells also released DNA-free VLPs/LPs, although less efficiently than ΔBFLF1/BFRF1A mutant cells (Fig. 5). Therefore, EBV-derived viral structures that contain immature capsids can undergo primary and secondary egress.
This observation to some extent contradicts the current view on capsid maturation that proposes that only C-type capsids can undergo primary and, a fortiori, secondary envelopment and release (53). This prompted us to reexamine the morphology of viral progeny in PrV mutants that are defective for the genes involved in DNA packaging pUL6, pUL15, pUL32, or pUL33 and we found that, with the exception of the mutant devoid of pUL15, all mutants produce VLPs, although these viral structures were much less frequently seen than LPs (Fig. 7 and data not shown). Therefore, the absence of viral DNA within capsids does not fully inhibit egress, although we agree that it substantially impairs it. Similar observations were made in HSV-1-infected neurons (54). Our data confirm the high degree of functional homology of the proteins involved in DNA incorporation and capsid maturation across alpha- and gammaherpesvirus subfamilies.
The search for EBV mutants that produce VLPs/LPs was motivated by the observation that ΔTR mutants imperfectly block the viral genome incorporation into the capsid. In contrast, B cell infection studies (Fig. 4 and Table 2) and qPCR (Fig. 5B) could not detect any EBV DNA in VLPs/LPs obtained from induction of 293/ΔBFLF1/BFRF1A and 293/ΔBBRF1. The purified VLPs/LPs elicited a CD4+ T-cell response after incubation of B cells that acted as antigen-presenting cells as strongly as wild-type virus particles. Therefore, the ΔBFLF1/BFRF1A VLPs/LPs differ from those obtained with 293/ΔTR cells in that they are devoid of viral DNA contaminants. As such, the ΔBFLF1/BFRF1A VLPs/LPs represent promising candidates for a safe and efficient preventative vaccine.
A recent paper by Ruiss et al. (55) has also suggested using VLPs from a similar though distinct producer cell line (293-VII+) for vaccination purposes. The EBV mutant contained in this cell line is based on the ΔTR mutant but also lacks multiple EBV latent genes (EBNA2, EBNA3, LMP1, LMP2) as well as the lytic BZLF1 gene. Since EBV latent genes are unlikely to modulate DNA incorporation into viral capsids, one would expect similar degrees of DNA contamination in the defective particles produced by the 293-VII+ cell line and in the 293/ΔTR clone (16). However, the authors report that they could not find evidence of contamination using an endpoint PCR analysis performed on VLPs/exosome preparations from induced producer cells. The reasons for this discrepancy are difficult to explain without parallel evaluation of the mutants. It would be important to estimate how many VLPs/LPs are produced by the 293-VII+ cell line, to purify them using gp350 antibodies, and to assess DNA content by sensitive PCR analysis. Clearly, the strategy pursued by Ruiss et al. aims at minimizing the potential adverse consequences of viral DNA contamination in a preventative EBV vaccine. Indeed, deletion of the latent genes and of BZLF1 massively reduces the risk of oncogenic transformation and of virus propagation, although the expression of BRLF1 is independent of BZLF1 and this transactivator can drive the expression of lytic proteins such as the bcl-2 homologue BHRF1 (56–60). In addition, other genetic elements such as EBNA1, BHRF1, and the BHRF1 micro RNAs (miRNAs) have been shown to be involved in EBV-mediated B cell transformation or in the induction of genetic abnormalities (59, 61–63). Moreover, very little is known about the persistence of EBV after primary infection but the consensus is that hardly any gene expression is required for persistence in B cells (64–66). Therefore, it is possible that the viral genome present in the 293-VII+ cell line might be able to persist in the bodies of vaccinated individuals along with the genes that encode GFP and the hygromycin resistance gene. A recent study has also provided persuasive evidence that attenuated herpesvirus vaccines can recombine in vivo and form virulent field viruses (67). Therefore, VLP/LPs EBV mutants that preclude viral DNA incorporation altogether might in the end be more suitable as preventative vaccines.
ACKNOWLEDGMENTS
We thank H. Bannert, H. Lips, P. Meyer, M. Jörn, and B. Hub for expert technical assistance, J. Mautner for providing the T-cell clones, and T. C. Mettenleiter for fruitful discussions.
This work was supported by the Association for International Cancer Research (AICR) (project grant 09-0517) and the German Centre for Infection Research (DZIF).
Footnotes
Published ahead of print 12 December 2012
REFERENCES
- 1. Rickinson AB, Kieff E. 2007. Epstein-Barr virus, p 2655–2700 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Goldacre MJ, Wotton CJ, Yeates DG. 2009. Associations between infectious mononucleosis and cancer: record-linkage studies. Epidemiol. Infect. 137:672–680 [DOI] [PubMed] [Google Scholar]
- 3. Hjalgrim H, Askling J, Sorensen P, Madsen M, Rosdahl N, Storm HH, Hamilton-Dutoit S, Eriksen LS, Frisch M, Ekbom A, Melbye M. 2000. Risk of Hodgkin's disease and other cancers after infectious mononucleosis. J. Natl. Cancer Inst. 92:1522–1528 [DOI] [PubMed] [Google Scholar]
- 4. Long HM, Taylor GS, Rickinson AB. 2011. Immune defence against EBV and EBV-associated disease. Curr. Opin. Immunol. 23:258–264 [DOI] [PubMed] [Google Scholar]
- 5. Knowles DM. 1999. Immunodeficiency-associated lymphoproliferative disorders. Mod. Pathol. 12:200–217 [PubMed] [Google Scholar]
- 6. Delecluse HJ, Feederle R, O'Sullivan B, Taniere P. 2007. Epstein Barr virus-associated tumours: an update for the attention of the working pathologist. J. Clin. Pathol. 60:1358–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kutok JL, Wang F. 2006. Spectrum of Epstein-Barr virus-associated diseases. Annu. Rev. Pathol. 1:375–404 [DOI] [PubMed] [Google Scholar]
- 8. zur Hausen H. 2006. Infections causing human cancer. Wiley-VCH Verlag GmbH & Co.KGaA, Weinheim, Germany [Google Scholar]
- 9. Cohen JI, Fauci AS, Varmus H, Nabel GJ. 2011. Epstein-Barr virus: an important vaccine target for cancer prevention. Sci. Transl. Med. 3:107fs107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duraiswamy J, Sherritt M, Thomson S, Tellam J, Cooper L, Connolly G, Bharadwaj M, Khanna R. 2003. Therapeutic LMP1 polyepitope vaccine for EBV-associated Hodgkin disease and nasopharyngeal carcinoma. Blood 101:3150–3156 [DOI] [PubMed] [Google Scholar]
- 11. Mackett M, Cox C, Pepper SD, Lees JF, Naylor BA, Wedderburn N, Arrand JR. 1996. Immunisation of common marmosets with vaccinia virus expressing Epstein-Barr virus (EBV) gp340 and challenge with EBV. J. Med. Virol. 50:263–271 [DOI] [PubMed] [Google Scholar]
- 12. Sokal EM, Hoppenbrouwers K, Vandermeulen C, Moutschen M, Leonard P, Moreels A, Haumont M, Bollen A, Smets F, Denis M. 2007. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J. Infect. Dis. 196:1749–1753 [DOI] [PubMed] [Google Scholar]
- 13. Crisci E, Barcena J, Montoya M. 2012. Virus-like particles: the new frontier of vaccines for animal viral infections. Vet. Immunol. Immunopathol. 148:211–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramqvist T, Andreasson K, Dalianis T. 2007. Vaccination, immune and gene therapy based on virus-like particles against viral infections and cancer. Expert Opin. Biol. Ther. 7:997–1007 [DOI] [PubMed] [Google Scholar]
- 15. Delecluse HJ, Pich D, Hilsendegen T, Baum C, Hammerschmidt W. 1999. A first-generation packaging cell line for Epstein-Barr virus-derived vectors. Proc. Natl. Acad. Sci. U. S. A. 96:5188–5193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feederle R, Shannon-Lowe C, Baldwin G, Delecluse HJ. 2005. Defective infectious particles and rare packaged genomes produced by cells carrying terminal-repeat-negative Epstein-Barr virus. J. Virol. 79:7641–7647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adhikary D, Behrends U, Feederle R, Delecluse HJ, Mautner J. 2008. Standardized and highly efficient expansion of Epstein-Barr virus-specific CD4+ T cells by using virus-like particles. J. Virol. 82:3903–3911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beard PM, Taus NS, Baines JD. 2002. DNA cleavage and packaging proteins encoded by genes UL28, UL15, and UL33 of herpes simplex virus type 1 form a complex in infected cells. J. Virol. 76:4785–4791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koslowski KM, Shaver PR, Casey JT, II, Wilson T, Yamanaka G, Sheaffer AK, Tenney DJ, Pederson NE. 1999. Physical and functional interactions between the herpes simplex virus UL15 and UL28 DNA cleavage and packaging proteins. J. Virol. 73:1704–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu D, Weller SK. 1998. Genetic analysis of the UL 15 gene locus for the putative terminase of herpes simplex virus type 1. Virology 243:32–44 [DOI] [PubMed] [Google Scholar]
- 21. Koslowski KM, Shaver PR, Wang XY, Tenney DJ, Pederson NE. 1997. The pseudorabies virus UL28 protein enters the nucleus after coexpression with the herpes simplex virus UL15 protein. J. Virol. 71:9118–9123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang K, Homa F, Baines JD. 2007. Putative terminase subunits of herpes simplex virus 1 form a complex in the cytoplasm and interact with portal protein in the nucleus. J. Virol. 81:6419–6433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adelman K, Salmon B, Baines JD. 2001. Herpes simplex virus DNA packaging sequences adopt novel structures that are specifically recognized by a component of the cleavage and packaging machinery. Proc. Natl. Acad. Sci. U. S. A. 98:3086–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abbotts AP, Preston VG, Hughes M, Patel AH, Stow ND. 2000. Interaction of the herpes simplex virus type 1 packaging protein UL15 with full-length and deleted forms of the UL28 protein. J. Gen. Virol. 81:2999–3009 [DOI] [PubMed] [Google Scholar]
- 25. Beilstein F, Higgs MR, Stow ND. 2009. Mutational analysis of the herpes simplex virus type 1 DNA packaging protein UL33. J. Virol. 83:8938–8945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fuchs W, Klupp BG, Granzow H, Leege T, Mettenleiter TC. 2009. Characterization of pseudorabies virus (PrV) cleavage-encapsidation proteins and functional complementation of PrV pUL32 by the homologous protein of herpes simplex virus type 1. J. Virol. 83:3930–3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamberti C, Weller SK. 1998. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J. Virol. 72:2463–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59–74 [DOI] [PubMed] [Google Scholar]
- 29. Shaw G, Morse S, Ararat M, Graham FL. 2002. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 16:869–871 [DOI] [PubMed] [Google Scholar]
- 30. Pulvertaft RJ. 1964. Phytohaemagglutinin in relation to Burkitt's tumour. (African lymphoma). Lancet ii:552–554 [DOI] [PubMed] [Google Scholar]
- 31. Hayflick L. 1965. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 37:614–636 [DOI] [PubMed] [Google Scholar]
- 32. Hammerschmidt W, Sugden B. 1988. Identification and characterization of oriLyt, a lytic origin of DNA replication of Epstein-Barr virus. Cell 55:427–433 [DOI] [PubMed] [Google Scholar]
- 33. Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. U. S. A. 95:8245–8250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Neuhierl B, Delecluse HJ. 2005. Molecular genetics of DNA viruses: recombinant virus technology. Methods Mol. Biol. 292:353–370 [DOI] [PubMed] [Google Scholar]
- 35. Janz A, Oezel M, Kurzeder C, Mautner J, Pich D, Kost M, Hammerschmidt W, Delecluse HJ. 2000. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of additional viral ligands. J. Virol. 74:10142–10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Griffin BE, Bjorck E, Bjursell G, Lindahl T. 1981. Sequence complexity of circular Epstein-Barr virus DNA in transformed cells. J. Virol. 40:11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Granato M, Feederle R, Farina A, Gonnella R, Santarelli R, Hub B, Faggioni A, Delecluse HJ. 2008. Deletion of Epstein-Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J. Virol. 82:4042–4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klupp BG, Granzow H, Mettenleiter TC. 2000. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 74:10063–10073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Busse C, Feederle R, Schnolzer M, Behrends U, Mautner J, Delecluse HJ. 2010. Epstein-Barr viruses that express a CD21 antibody provide evidence that gp350's functions extend beyond B-cell surface binding. J. Virol. 84:1139–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoffmann MK. 1980. Antigen-specific induction and regulation of antibody synthesis in cultures of human peripheral blood mononuclear cells. Proc. Natl. Acad. Sci. U. S. A. 77:1139–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feederle R, Neuhierl B, Baldwin G, Bannert H, Hub B, Mautner J, Behrends U, Delecluse HJ. 2006. Epstein-Barr virus BNRF1 protein allows efficient transfer from the endosomal compartment to the nucleus of primary B lymphocytes. J. Virol. 80:9435–9443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mautner J, Pich D, Nimmerjahn F, Milosevic S, Adhikary D, Christoph H, Witter K, Bornkamm GW, Hammerschmidt W, Behrends U. 2004. Epstein-Barr virus nuclear antigen 1 evades direct immune recognition by CD4+ T helper cells. Eur. J. Immunol. 34:2500–2509 [DOI] [PubMed] [Google Scholar]
- 43. al-Kobaisi MF, Rixon FJ, McDougall I, Preston VG. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180:380–388 [DOI] [PubMed] [Google Scholar]
- 44. Yang K, Poon AP, Roizman B, Baines JD. 2008. Temperature-sensitive mutations in the putative herpes simplex virus type 1 terminase subunits pUL15 and pUL33 preclude viral DNA cleavage/packaging and interaction with pUL28 at the nonpermissive temperature. J. Virol. 82:487–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang K, Wills EG, Baines JD. 2011. A mutation in UL15 of herpes simplex virus 1 that reduces packaging of cleaved genomes. J. Virol. 85:11972–11980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Higgs MR, Preston VG, Stow ND. 2008. The UL15 protein of herpes simplex virus type 1 is necessary for the localization of the UL28 and UL33 proteins to viral DNA replication centres. J. Gen. Virol. 89:1709–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Newcomb WW, Juhas RM, Thomsen DR, Homa FL, Burch AD, Weller SK, Brown JC. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 75:10923–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Patel AH, MacLean JB. 1995. The product of the UL6 gene of herpes simplex virus type 1 is associated with virus capsids. Virology 206:465–478 [DOI] [PubMed] [Google Scholar]
- 49. Trus BL, Cheng N, Newcomb WW, Homa FL, Brown JC, Steven AC. 2004. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J. Virol. 78:12668–12671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Patel AH, Rixon FJ, Cunningham C, Davison AJ. 1996. Isolation and characterization of herpes simplex virus type 1 mutants defective in the UL6 gene. Virology 217:111–123 [DOI] [PubMed] [Google Scholar]
- 51. Sherman G, Bachenheimer SL. 1988. Characterization of intranuclear capsids made by ts morphogenic mutants of HSV-1. Virology 163:471–480 [DOI] [PubMed] [Google Scholar]
- 52. Sherman G, Bachenheimer SL. 1987. DNA processing in temperature-sensitive morphogenic mutants of HSV-1. Virology 158:427–430 [DOI] [PubMed] [Google Scholar]
- 53. Yang K, Baines JD. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc. Natl. Acad. Sci. U. S. A. 108:14276–14281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ibiricu I, Huiskonen JT, Dohner K, Bradke F, Sodeik B, Grunewald K. 2011. Cryo electron tomography of herpes simplex virus during axonal transport and secondary envelopment in primary neurons. PLoS Pathog. 7:e1002406 doi:10.1371/journal.ppat.1002406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ruiss R, Jochum S, Wanner G, Reisbach G, Hammerschmidt W, Zeidler R. 2011. A virus-like particle-based Epstein-Barr virus vaccine. J. Virol. 85:13105–13113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Desbien AL, Kappler JW, Marrack P. 2009. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc. Natl. Acad. Sci. U. S. A. 106:5663–5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hardwick JM, Lieberman PM, Hayward SD. 1988. A new Epstein-Barr virus transactivator, R, induces expression of a cytoplasmic early antigen. J. Virol. 62:2274–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hong GK, Gulley ML, Feng WH, Delecluse HJ, Holley-Guthrie E, Kenney SC. 2005. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 79:13993–14003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kelly GL, Long HM, Stylianou J, Thomas WA, Leese A, Bell AI, Bornkamm GW, Mautner J, Rickinson AB, Rowe M. 2009. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog. 5:e1000341 doi:10.1371/journal.ppat.1000341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zalani S, Holley-Guthrie E, Kenney S. 1996. Epstein-Barr viral latency is disrupted by the immediate-early BRLF1 protein through a cell-specific mechanism. Proc. Natl. Acad. Sci. U. S. A. 93:9194–9199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Frappier L. 2012. Role of EBNA1 in NPC tumourigenesis. Semin. Cancer Biol. 22:154–161 [DOI] [PubMed] [Google Scholar]
- 62. Kamranvar SA, Masucci MG. 2011. The Epstein-Barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia 25:1017–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Seto E, Moosmann A, Gromminger S, Walz N, Grundhoff A, Hammerschmidt W. 2010. Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 6:e1001063 doi:10.1371/journal.ppat.1001063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA. 1998. EBV persistence in memory B cells in vivo. Immunity 9:395–404 [DOI] [PubMed] [Google Scholar]
- 65. Qu L, Rowe DT. 1992. Epstein-Barr virus latent gene expression in uncultured peripheral blood lymphocytes. J. Virol. 66:3715–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tierney RJ, Steven N, Young LS, Rickinson AB. 1994. Epstein-Barr virus latency in blood mononuclear cells: analysis of viral gene transcription during primary infection and in the carrier state. J. Virol. 68:7374–7385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee SW, Markham PF, Coppo MJ, Legione AR, Markham JF, Noormohammadi AH, Browning GF, Ficorilli N, Hartley CA, Devlin JM. 2012. Attenuated vaccines can recombine to form virulent field viruses. Science 337:188. [DOI] [PubMed] [Google Scholar]