

Abstract
Tumor necrosis factor related apoptosis-inducing ligand (TRAIL) has been shown to induce apoptosis in malignant cells while leaving normal cells unharmed, making it a desirable anticancer target. In the present study, metastatic melanoma cell lines were treated with lexatumumab (Human Genome Sciences, Inc.) a high-affinity monoclonal antibody agonistic to TRAIL receptor 2 (DR5). Binding of the antibody to the receptor led to activation of the extrinsic apoptosis pathway in approximately 20% of the treated cells. However, by combining subtoxic concentrations of the protein translation inhibitor anisomycin with lexatumumab, we obtained synergistic effects on cell viability compared with single agent treatment. Even the low doses of anisomycin could inhibit protein synthesis in melanoma cells with up to 30%, which might result in the shift in the levels of the proteins involved in apoptosis. Co-treatment with anisomycin increased activation of caspases and cleavage of the anti-apoptotic protein Livin, leading to formation of truncated p30-Livin α and p28-Livin β proteins with potential pro-apoptotic functions. Furthermore, ansiomcycin treatment decreased levels of antiapototic XIAP. In summary our results suggest that combinational treatment with anicomycin and lexatumumab represents a novel therapeutic strategy in the treatment of melanoma.
Keywords: melanoma, TRAIL, lexatumumab, anisomycin, livin, caspases, therapy
Introduction
Malignant melanoma arises from the transformation of melanocytes and is considered the most severe type of skin cancer that accounts for more than 80% of skin cancer related deaths.1 If diagnosed early, melanomas can be cured by excision of the primary lesion. However, treatment of melanoma patients with advanced disease represents a medical challenge due to low response rates to both chemotherapeutics and biotherapeutic drugs. Recently, highly promising therapeutic effects have been achieved using inhibitors targeting mutant BRAF protein which is found in up to 50% of melanomas.2 Unfortunately, most patients relapse and develop resistance to the drug after an initial period of response. Furthermore, efficient treatment options for patients with melanoma that do not have BRAF mutations are very poor. For this reason novel combinational and targeted therapies for metastatic disease are highly warranted.
In search for new therapeutic options, attention has been directed toward the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). In vitro studies have demonstrated that recombinant TRAIL induces apoptosis in a variety of human cancer cell lines, including melanoma, while having low toxicity toward normal cells.3-5 Furthermore, in mice TRAIL has been shown to suppress growth of human tumor xenografts.5 Due to this selectivity, TRAIL represents an attractive strategy for anti-cancer treatment and clinical evaluation of TRAIL and agonistic antibodies targeting TRAIL receptors is ongoing for several cancer types.6
Binding of TRAIL to its receptors 1 (death receptor 4) and 2 (death receptor 5) causes recruitment of Fas-Associated protein with Death Domain (FADD) and formation of the Death Inducing Stimulation Complex (DISC), ultimately leading to activation of initiator caspases-8 and -10. Activated caspase-8 or -10 then cleaves executioner caspases-3, -6 and -7 that in turn act on a number of substrates, many of which give rise to features of apoptosis. TRAIL may also activate the intrinsic apoptotic pathway by caspase-8 dependent cleavage of the pro-apoptotic protein Bid, which in its truncated form translocates to the mitochondria leading to release of cytochrome c and activation of the intracellular apoptotic cascade.7
Unfortunately, a major challenge associated with TRAIL-based therapy is decreased sensitivity of tumors to TRAIL-mediated apoptosis.8 Mechanisms underlying TRAIL resistance include absence or low expression of death receptors, increased expression of inhibitors of apoptosis proteins (IAPs) or overexpression of anti-apoptotic Bcl-2 family members. In order to overcome resistance, both chemotherapeutic and biological agents have been used with success to sensitize tumor cells to TRAIL-mediated apoptosis.9,10 Sensitization effects are suggested to occur by potentiation of the mitochiondrial apoptotic pathway, downregulation of IAP levels, inhibition of NFκB activation and upregulation of TRAIL receptors.11
Previous studies in mesothelioma, prostate and glioma cells have shown that treatment with the protein synthesis inhibitor anisomycin can increase the sensitivity to TRAIL induced apoptosis.12-14 Anisomycin binds the 60S ribosomal subunit and block peptide bond formation and DNA synthesis.15 In addition, anisomycin is commonly used as an activation agent of mitogen-activated protein kinases c-jun N-terminal kinase/stress-activated protein kinase (JNK) and p38 mitogen activated protein kinase (p38).16,17 Recently an in vivo study in mice showed that anisomycin has low toxicity and no significant side effects at effectively therapeutic doses.18
For this reason we have investigated if similar effects might be achieved when combining lexatumumab, an agonistic high-affinity monoclonal antibody (mAb) that binds to and activates TRAIL receptor 2/death receptor 5 (DR5) with subtoxic concentrations of anisomycin in metastatic melanoma cells.
Results
Anisomycin enhances inhibitory effects of TRAIL-R2 agonist lexatumumab in metastatic melanoma cell lines
To examine if low dose anisomycin could sensitize melanoma cells to TRAIL-R2 agonist induced apoptosis, the melanoma cell lines FEMX-1 and WM239 were treated with anisomycin and lexatumumab as single agents or in combination for up to 48 h and analyzed for cell viability using the MTS assay. As single agent 40 nM anisomycin reduced the cell viability in both cell lines by 25% while 0.75 μg/ml lexatumumab led to a 30% reduction as compared with the untreated controls (Fig. 1A). When co-administered, a reduction in cell viability by 45% for FEMX-1 and 55% for WM239 cells was observed. The istotype-matched antibody control did not affect cell viability alone or in combination with anisomycin for the given concentrations (data not shown).

Figure 1. (A) Cell viability of FEMX-1 (black bars) and WM239 cells (white bars) treated with 0.75 ng/ml lexatumumab and 40 nM anisomycin for 48 h assessed by the MTS assay (*p = 0.0042 and **p = 0.0041). (B) Assessment of cytoplasmic histone-associated DNA fragments indicative of apoptosis in FEMX-1 and WM239 cells after treatment as in (A). Results from three biological replicate experiments.
Since co-treatment led to a significant decrease in cell viability compared with the individual treatments, we evaluated whether a synergistic effect was achieved. The cell viability data obtained following treatment with increasing amounts of anisomycin (40, 60 and 80 nM) and lexatumumab (0.75, 1.5 and 3.0 μg/ml) alone or in combination was analyzed by the Combination Index Method.21 Moderate to strong synergism was observed for all concentrations used in both cell lines CI < 0.9 (Table 1).
Table 1. Assessment of synergistic interactions between anisomycin and lexatumumab in FEMX-1 and WM239 cells.
| |
Combination index* |
||
|---|---|---|---|
| ANIS |
LEXA |
FEMX-1 | WM239 |
| (nM) | (μg/ml) | ||
| 40 |
0.75 |
0.57 |
0.6 |
| 40 |
1.5 |
0.52 |
0.55 |
| 40 |
3 |
0.59 |
0.62 |
| 60 |
0.75 |
0.55 |
0.44 |
| 60 |
1.5 |
0.54 |
0.45 |
| 60 |
3 |
0.55 |
0.48 |
| 80 |
0.75 |
0.53 |
0.34 |
| 80 |
1.5 |
0.5 |
0.36 |
| 80 | 3 | 0.5 | 0.42 |
The combination index (CI) values are defined as synergy < 0.9, additive effect 0.9–1.1 and antagonistic interaction > 1.1.
Anisomycin increases lexatumumab-induced apoptosis
In order to evaluate whether the decrease in cell viability was due to inhibition of proliferation or increased susceptibility to apoptosis, the cells were treated with drugs for 48 h and analyzed using The Cell Death ELISA Plus Kit allowing specific detection and quantitation of mono- and oligonucleosomes that are released into the cytoplasm of cells dying from apoptosis. As shown in Figure 1B, treatment with 40 nM anisomycin as single agent did not influence the percentage of apoptotic cells beyond the basal levels in either of the cell lines, indicating that the decrease in cell viability observed using the MTS assay is due to inhibition of proliferation rather than apoptosis. Lexatumumab (0.75 μg/ml) treatment increased the degree of apoptosis by 2 and 5 fold in FEMX-1 and WM239 cells, respectively. Furthermore, when 40 nM anisomycin was administered together with 0.75 μg/ml lexatumumab, approximately a 2-fold increase in number of apoptotic cells was observed in both cell lines as compared with lexatumumab as a single treatment.
As shown in Figure 2 apoptotic cell death was also confirmed when expression levels and activation of pro-caspases-8, -9 and -3 was examined by immunoblotting. Cleavage of the pro-caspases was observed in both cell lines 3 h post-lexatumumab treatment and was sustained for up to 24 h. Treatment with anisomycin as single agent did not lead to cleavage of the pro-caspases. When lexatumumab was combined with anisomycin an additional increase in cleavage of the pro-caspase 3, producing fragments p17, p15 and p12 was observed after 8 h in both cell lines as well as the cleavage of the nuclear enzyme poly ADP-ribose polymerase (PARP).

Figure 2. Immuno-blot analysis of caspase-8, caspase-9 and caspase-3 cleavage and activation and cleavage of PARP after treatment with 0.75 ng/ml lexatumumab and/or 40 nM anisomycin for the indicated time in FEMX-1 and WM239 cells. α-tubulin is included as a loading control.
In order to evaluate if the observed synergistic effects were caspase-dependent, the cells were treated with 40 nM anisomycin and/or 0.75 μg/ml lexatumumab alone or in combination with 10 μM of either the broad spectrum caspase inhibitor z-VAD-fmk or the caspase-8 specific inhibitor z-IEDT-fmk for 48 h. As shown in Figure 3. cell viability was almost restored to basal level in both cell lines when the caspase inhibitors were added together with lexatumumab alone or in combination with anisomycin. Inhibition of caspase activity could not restore the 25% reduction in cell viability caused by 40 nM anisomycin treatment alone, supporting the previous observation that anisomycin inhibits proliferation rather than inducing apoptosis.

Figure 3. Cell viability of FEMX-1 (black bars) and WM239 cells (white bars) treated with 0.75 μg/ml lexatumumab and/or 40 nM anisomycin alone or in combination with 10 μM of the caspase inhibitors Z-Vad-FMK and Z-IEDT-FMK for 48 h assessed by the MTS assay. Results from three biological replicate experiments.
Anisomycin decreases levels of antiapoptotic proteins
Since our data suggest that anisomycin can enhance apoptosis induced by binding of the lexatumumab to DR5, we further examined possible mechanisms of its action. As a protein synthesis inhibitor, anisomycin might promote cell death by causing generalized cellular stress or by shifting the balance between anti- and pro-apoptotic proteins The ability of anisomycin to inhibit protein synthesis was evaluated by a [3H]-leucine incorporation assay. As shown in Figure 4 treatment with low anisomycin doses (10−100 nM) inhibited de novo protein synthesis in FEMX1 and WM239 with up to 20% and 30%, respectively, while higher inhibition was achieved with doses from 1−10 μM.
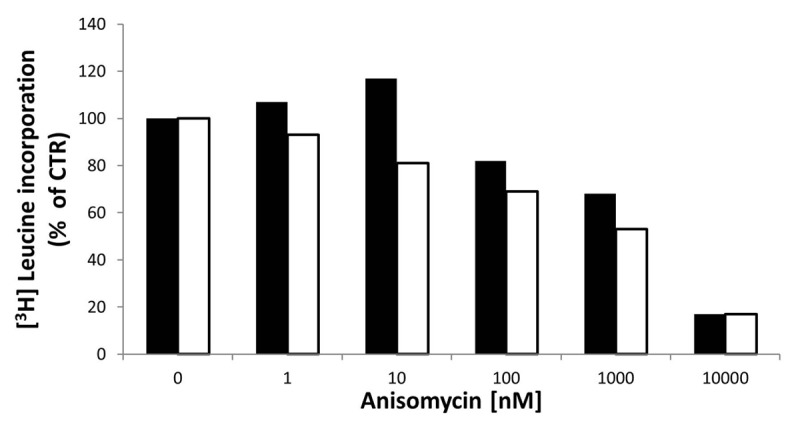
Figure 4. [3H]-leucine incorporation assay in FEMX-1(black bars) and WM239 cells (white bars) following 3 h of treatment with increasing doses of anisomycin. The results are reported as percentage leucine incorporation related to untreated cells. Results from two biological replicate experiments.
In order to evaluate if anisomycin treatment affects the levels of proteins involved in the extrinsic and intrinsic regulation of apoptosis we extended the analysis of apoptosis pathways using immunoblotting and flow cytometry. As shown in Figure 5A lexatumumab induced cleavage of Bid, a caspase-8 substrate that functions as an intermediate between the extrinsic and intrinsic apoptotic pathways, which was was further enhanced in the presence of anisomycin. However, we did not observe any changes in the levels or phosphorylation status of other Bcl-2 family members including Bcl-2, Bad, Bim, Bax or Mcl-1 after treatment with lexatumumab, anisomycin as single agents or in combination (data not shown). Analysis of the anti-apoptotic proteins in the IAP family, including Xiap and Livin, showed that co-treatment with anisomycin led to a slight additional decrease in the XIAP protein levels after 24 h (Figure 5A). Three hours after lexatumumab treatment alone, cleavage of α and β isoforms of Livin, producing truncated forms p30-Livin α and p28-Livin β was detected. A marked increase in α and β Livin cleavage was observed 8 h after co-treatment with anisomycin, andsustained for up to 24 h. Anisomycin treatment alone did not lead to cleavage of Livin.

Figure 5. (A) Immuno-blot analysis of Bid, XIAP and Livin in FEMX-1 and WM239 cells after treatment as described in Figure 2. Densiometric analysis of the blots was performed using ImageJ software where all lanes were normalized against their respective α-tubulin loading controls and related to 1 h untreated control. Ratio between Livin 30/39 and 28/30 is calculated for each time point between lexatumumab and co-treatment. (B) Flow cytometry analysis showing DR5 surface expression on FEMX-1 (black bars) and WM239 cells (white bars) one hour after treatment with 0.75 μg/ml lexatumumab and 40 nM anisomycin alone or in combination assessed by flow cytometry. Results from 3 biological replicate experiments.
Previously it has been reported that chemotherapeutic agents can enhance the sensitivity to TRAIL induced cell death by increasing the surface expression of TRAIL receptors.22 To investigate if sensitivity of FEMX-1 and WM239 cells to the combinational treatment could be attributed to upregulation of death receptors, cells were exposed to the respective treatments for one hour and analyzed for DR5 surface expression by flow cytometry. As shown in Figure 5B 80% of WM239 and FEMX-1 cells expressed DR5 which is in agreement with previous reports.4 Treatment with 0.75 μg/ml lexatumumab led to a marginal decrease of DR5 level while 40 nM anisomycin alone or in combination with lexatumumab had no effect in either cell line, suggesting that the reduced cell viability, and enhanced activation of the proteolytic caspase cascade is not due to altered receptor levels.
Low dose anisomycin activates p38 signaling in melanoma cells
Anisomycin has previously been shown to activate the MAPK signaling pathways JNK and p38.16 For this reason we examined if these pathways are affected by anisomycin treatment also in melanoma cells. In addition, we examined the activation status of the MAPK/ERK1/2 and PI3K/Akt signaling pathways that are essential for melanoma cell survival.23 As shown in Figure 6 anisomycin treatment led to an accumulation of phosphorylated, activated p38 in both cell lines. Total p38 protein level, however, remained unchanged while pMAPKAPK2, the downstream target of active p38, was found to be activated with kinetics similar to that of p38. No changes in the phosphorylation and activation levels of JNK, MAPK/ERK1/2 or Akt were observed.

Figure 6. Representative immuno-blot analysis of p38, MAPK/ERK1/2, Akt and JNK pathways activation in FEMX-1 and WM239 cells after treatment as described in Figure 2.
In order to evaluate if activation of the p38 pathway contributes to the increase in apoptosis observed after combination treatment, the cells were pre-incubated with the p38-inhibitor SB203580 for 1 h followed by 40 nM anisomycin and/or 0.75 μg/ml lexatumumab treatment and assessed for cell viability after 24 h. As shown in Figure 7A and B, SB203580 inhibited the activity of p38 but did not affect the viability for any of the cell lines suggesting that activation of p38 pathway has a marginal role in the pro-apoptotic signaling pathway mediated by anisomycin.

Figure 7. (A) Cell viability assessed by the MTS assay FEMX-1 (black bars) and WM239 cells (white bars) treated with 0.75 μg/ml lexatumumab and 40 nM anisomycin alone or in combination with 2.5 μM of the p38 MAPK inhibitor SB203580 for 24 h. (B) Immuno-blot evaluation of p38 pathway inhibition. FEMX-1 and WM239 cells were treated with 2.5 and 5 μM of the p38 inhibitor SB203580, 0.75 μg/ml lexatumumab and 40 nM anisomycin alone or in combination for 24 h.
Discussion
Malignant melanoma is considered as a highly aggressive cancer form due to its ability to metastasize and develop resistance to all available therapy. As a therapeutic approach, TRAIL-induced apoptosis represents an attractive option due to its selective nature for eliminating cancer cells while leaving normal cells unharmed.3 Unfortunately many melanoma cell lines exhibit profound resistance to TRAIL treatment due to decreased expression of death receptors or increased levels of anti-apoptotic proteins.24,25 Several previous studies have suggested that TRAIL therapies will be most effective when used in combination with agents that can sensitize tumor cells to TRAIL.26,27 In accordance with these studies, we showed that melanoma cells are sensitized to lexatumumab induced apoptosis using subtoxic levels of the protein translation inhibitor anisomycin. Our results suggest that the increased cell death observed after treatment with lexatumumab and anisomycin is due to enhanced caspase activity and shifted balance between pro- and anti-apototic proteins.
Members of the inhibitor of apoptosis protein (IAP) family including XIAP, Survivin and Livin have been reported to be overexpressed in melanomas.28,29 Their expression is also correlated with reduced survival and drug resistance in other cancer types.30-32 Previously ours and other studies have suggested that downregulation of XIAP and Survivin increases the sensitivity of melanoma cells to TRAIL treatment in vitro and in vivo.33,34 Here we show that anisomycin can sensitize melanoma cells to lexatumumab by reducing the levels of XIAP and Livin. Previously it has been shown that cleavage of Livin generate truncated forms of Livin (tLivin) with marked proapoptotic activity in melanomas.35 Interestingly, our results show that combinational treatment shifts the balance between the long α and β anti-apoptotic forms of Livin in favor of the pro-apoptotic truncated p30-Livin α and p28-Livin β proteins which could account for the observed increase in apoptosis.
In mesothelioma cells anisomycin was shown to increase TRAIL mediated apoptosis by posttranslational potentiation of Bim, which primes cells for apoptosis via the death receptor pathway.12 We did however not, detect any changes in the level or phosphorylation of Bim. However, combined anisomycin-lexatumumab treatment led to additional decrease of Bid protein levels, which might further enhance activation of intrinsic apoptotic pathway.
Previously it has been shown that the sensitivity of cells to TRAIL induced apoptosis might be modulated by chemical agents that affect expression of TRAIL receptors DR4 and DR5.36,37 However, anisomycin is unlikely to act by this mechanism in melanoma cells since our results showed that the expression of the receptors were unchanged after anisomycin treatment.
Our results suggest that anisomycin can acts as a protein synthesis inhibitor in melanoma cells even at the low doses. For this reason it is possible that the observed decrease in the protein levels of the antiapototic Xiap, Livin and Bid might be the result of the protein synthesis inhibition. However, this decrease alone is not sufficient to initiate an apoptotic response but may rather enhance the apoptotic response initiated by lexatumumab.
Inhibition of protein synthesis by anisomycin has been shown to influence the levels of the caspase 8 inhibitor FLIP38 and thereby sensitizes prostate cancer cells to anoikis. In this study we tried to assess the effect of anisomycin on the levels of FLIP using several different antibodies. Unfortunately, none of the antibodies showed satisfactory specificity required to make reliable conclusions. Examination of mRNA levels of FLIP after 24 h of the respective treatments did not however reveal any differences between lexatumumab and co-treatment (data not shown). As a protein synthesis inhibitor, anisomycin can influence the synthesis of many proteins and we therefore cannot exclude the possibility that it might sensitize melanoma cells to lexatumumab by decreasing additional proteins not examined in our study.
While several previous studies in other cancer types have shown that anisomycin can activate both the JNK and p38 pathways,16,39 the doses used in our study selectively activated only p38. However, activation of the JNK pathway could be achieved if doses higher that 500 nM were used (data not shown). In prostate cancer it has been shown that activation of JNK is required for sensitization of PC3 cells to TRAIL-induced apoptosis by translation inhibitors in cells that are otherwise TRAIL resistant.13 Our results suggest that the JNK pathway is not involved in sensitization of melanoma cells to lexatumumab by anisomycin. Furthermore, although anisomycin treatment led to activation of the p38 pathway, inhibition of p38 activity with SB203580 did not decrease apoptosis. This suggests that activation of p38 is not necessary for the sensitization of melanoma cells to lexatumumab by anisomycin.
Previously it has been shown that activation of the PI3k/Akt pathway can confer resistance to TRAIL induced apoptosis in breast and ovarian cancer cell lines,40 while in melanomas, activation of the ERK1/2 pathway is suggested to provide protection against apoptosis by inhibiting Smac/DIABLO release from mitochondria.41 We did, however, not observe any changes in phosphorylation of Akt or ERK1/2 after anisomycin treatment suggesting that the activation status of these pathways remained unchanged.
In conclusion, we have shown that combined treatment of lexatumumab and anisomycin compared with lexatumumab alone significantly enhanced apoptosis in melanoma cell lines. The synergistic effects is caused by an increased caspase cascade signaling and enhanced cleavage and decreased levels of anti-apoptotic proteins Bid, Livin and XIAP. Altogether these results suggest that anisomycin treatment might be a good strategy for enhancing the response of melanoma cells to TRAIL-R2 targeted therapies.
Material and Methods
Cell lines and culture conditions
The Wistar Melanoma (WM) cell lines were kindly provided by Dr. Meenhard Herlyn (Wistar Institute, Philadelphia, PA) and have been described in detail elsewhere.19 The cell line FEMX-I was established from a lymph node metastasis obtained from a melanoma patient treated at the Norwegian Radium Hospital, Oslo University Hospital.20 All cell lines were cultured in RPMI1640 medium (Bio Whittaker) supplemented with 5% fetal bovine serum (FBS, PAA Laboratories) and 2 mM L-glutamine (GibcoBRL). The cells were maintained at 37°C in a humidified 5% CO2 atmosphere and routinely checked for mycoplasma infections. Anisomycin was obtained from Sigma-Aldrich. The agonistic TRAIL-R2/DR5 human mAb, lexatumumab, and an isotype-matched control mAb of irrelevant specificity were provided by Human Genome Sciences. The general caspase inhibitor Z-VAD-FMK was obtained from Promega and the caspase-8 inhibitor Z-IEDT-FMK was obtained from R&D Systems.
Cell viability and CalcuSyn analysis
The growth inhibitory effect of mono and combined treatments was measured using the CellTiter 96 Aqueous One solution (MTS assay, Promega). Cells were seeded in 96-well plates at a density of 2 × 103 cells/well. The following day the cells were treated with mAb control, lexatumumab and anisomycin or the respective combinations. After 48 h incubation, 20 μl of the CellTiter 96 Aqueous One solution was added to each well and the absorbance at 490 nm was measured using a microplate reader (Wallac 1420 Multilabel counter, PerkinElmer). Viability of treated cells was normalized to the untreated control cells. Each experiment was performed with three parallel observations and repeated at least three times. The synergy calculations were performed using CalcuSyn (Biosoft). The combination index (CI) values were defined as: synergy < 0.9, additive effects 0.9−1.1 and antagonistic interactions > 1.1.
Apoptosis assay
The rate of apoptotic cell death was estimated using The Cell Death ELISA Plus Kit (Roche Diagnostics), which quantitates nucleosomes released into the cytoplasm of dying cells. Briefly, cells were collected by trypsinization, lysed, and incubated with a mixture of biotinylated antihistone antibody and peroxidase-conjugated anti-DNA antibody, both of which bind to histone DNA complexes and initiate a color reaction in the presence of the substrate 2,2′-azino-di(3-ethylbenzthiazolinesulfonate) (ABTS). The OD reading at 405 nm was expressed as fold increase in apoptosis as compared with the level in untreated control cells. Results presented are average of three biological replicate experiments.
Flow cytometry
Cells were seeded into T75 filter flasks at a density of 1 × 106 and left to adhere over night. The following day the cells were treated with 40 nM anisomycin and 0.75 μg/ml lexatumumab alone or in combination for 1 h. Detached cells were collected by centrifugation of cultured medium and pooled together with adherent cells harvested by EDTA treatment. The cells were then washed twice with PBS and 100 μl of cell suspension containing approximately 1x106 cells was divided into individual tubes for direct staining. The antibodies PE-anti-human-CD262 (DR5, TRAIL-R2), PE-anti-mouse-IgG1 and κ Isotype control (BioLegend) were applied as recommended by the producer and incubated in the dark at room temperature for 25 min. Cells were washed once in PBS and re-suspended in 400 μl PBS for further analysis. Staining of viable cells was done by adding 5 μl of Hoechst 33258 (Invitrogen/Life Technology) prior to analysis. Data acquisition and analysis were performed on Becton Dickinson LARII (Becton Dickinson Immunocytometry Systems) using Multifit software (FACSDiVa House Inc.). The median fluorescent intensity from the treated cells expressing DR5 was normalized to untreated control cells, and the percentage of DR5 expressing cells calculated. The experiment was repeated three times.
Immunoblotting
Cells were lysed in ice-cold NP-40 lysis buffer (1% NP-40, 10% glycerol, 20 mM Tris-HCl, pH 7.5, 137 mM NaCl, 100 mM sodium vanadate, 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.02 mg/ml each of aprotinin, leupeptin and pepstatin, and 10 μl/ml phosphatase inhibitor cocktail I and II (Sigma-Aldrich). Protein quantification was done using standard Bradford protein assay, and 25 μg protein/lane was resolved by SDS PAGE (PAGE). To ensure even loading, membranes were stained with naphthol-blue black (Sigma-Aldrich). All membranes were blocked for one hour in 5% non-fat milk in TBST buffers and probed overnight (4°C, gentle agitation) with the following antibodies; Caspase 3 (#AF-605-NA) and XIAP (#AF8221) from R&D System; Caspase 8 (#9746,), Caspase 9 (#9502), PARP (#9532), Bid (#2002,), Bim (#2819), BAD (#9292), pMAPKAPK2 (#3007), p38(#9212), p-p38 (#4631),JNK (#9258), p-JNK (#9255), AKT (#9272) and p-AKT (#9271) from Cell Signaling; BAX (#MS-711-P) Neo Markers; Bcl2 (#M0887) Dako; Livin (#IMG-347) BioSite; Erk2 (sc-154) and Mcl-1 (sc-819) Santa Cruz; pMAPK (V8031) Promega; and α-tubulin (#CP06) Calbiochem. The membranes were thereafter probed for one hour at room temperature with secondary HRP-conjugated anti-rabbit or anti-mouse IgG antibodies (Promega). The visualization was performed using ECL-plus chemifluorescent reagent (GE Healthcare). Densiometric analysis of the blots was performed using ImageJ software (Rasband WS, ImageJ, US National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2012.) where all lanes were normalized against their respective α-tubulin loading controls and presented as relative to untreated control or as an internal ratio between the bands
Protein synthesis inhibition assay
The ability of anisomycin to inhibit protein synthesis in FEMX1 and WM239 cells was evaluated by a [3H]-leucine incorporation assay. 5 × 104 cells were seeded in 48 well plates (Nunc) and left to adhere over night at 37°C. The following day the cells were washed twice in ice cold PBS w/0.1% HSA and treated for three hours with different doses of anisomycin (10, 1, 0.1, 0.01 and 0.001 μM) in leucine free medium containing 2 μCi/ml [3H]-leucine. Cells were then washed twice with 5% TCA for 10 and 5 min and dissolved in 0.1M KOH for 5 min. The cell solution was transferred to counting tubes and 2 ml of Liquid Scintillator Aquasafe 300 Plus (Zinsser Analytic) were added to each sample. Sample counts were determined in a liquid scintillation counter (LKB Wallac, Perkin Elmer). Counts (cpm) for treated cells were compared with cpm for untreated cells, and reported as percentage leucine incorporation. The control value was set to 100%.
Supplementary Material
Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
These studies were funded by the Norwegian Cancer Society and Astrid and Birger Torsteds legacy, the Functional Genomics Program at the Norwegian Research Council (#158954/S10) and the Gene Therapy Program at the Norwegian Radium Hospital. We would like to acknowledge Human Genome Sciences, Inc. for providing the human, lexatumumab mAb used in this study.
Supplemental Materials
Supplemental materials may be found here:
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/22953
References
- 1.Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004;351:998–1012. doi: 10.1056/NEJMra041245. [DOI] [PubMed] [Google Scholar]
- 2.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. BRIM-3 Study Group Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouralexis S, Findlay DM, Evdokiou A. Death to the bad guys: targeting cancer via Apo2L/TRAIL. Apoptosis. 2005;10:35–51. doi: 10.1007/s10495-005-6060-0. [DOI] [PubMed] [Google Scholar]
- 4.Lillehammer T, Engesaeter BO, Prasmickaite L, Maelandsmo GM, Fodstad O, Engebraaten O. Combined treatment with Ad-hTRAIL and DTIC or SAHA is associated with increased mitochondrial-mediated apoptosis in human melanoma cell lines. J Gene Med. 2007;9:440–51. doi: 10.1002/jgm.1036. [DOI] [PubMed] [Google Scholar]
- 5.Yang A, Wilson NS, Ashkenazi A. Proapoptotic DR4 and DR5 signaling in cancer cells: toward clinical translation. Curr Opin Cell Biol. 2010;22:837–44. doi: 10.1016/j.ceb.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Falschlehner C, Ganten TM, Koschny R, Schaefer U, Walczak H. TRAIL and other TRAIL receptor agonists as novel cancer therapeutics. Adv Exp Med Biol. 2009;647:195–206. doi: 10.1007/978-0-387-89520-8_14. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalvez F, Ashkenazi A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene. 2010;29:4752–65. doi: 10.1038/onc.2010.221. [DOI] [PubMed] [Google Scholar]
- 8.Koschny R, Walczak H, Ganten TM. The promise of TRAIL--potential and risks of a novel anticancer therapy. J Mol Med (Berl) 2007;85:923–35. doi: 10.1007/s00109-007-0194-1. [DOI] [PubMed] [Google Scholar]
- 9.Ashley DM, Riffkin CD, Lovric MM, Mikeska T, Dobrovic A, Maxwell JA, et al. In vitro sensitivity testing of minimally passaged and uncultured gliomas with TRAIL and/or chemotherapy drugs. Br J Cancer. 2008;99:294–304. doi: 10.1038/sj.bjc.6604459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun SY, Yue P, Hong WK, Lotan R. Augmentation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by the synthetic retinoid 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalene carboxylic acid (CD437) through up-regulation of TRAIL receptors in human lung cancer cells. Cancer Res. 2000;60:7149–55. [PubMed] [Google Scholar]
- 11.Amm HM, Oliver PG, Lee CH, Li Y, Buchsbaum DJ. Combined modality therapy with TRAIL or agonistic death receptor antibodies. Cancer Biol Ther. 2011;11:431–49. doi: 10.4161/cbt.11.5.14671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abayasiriwardana KS, Barbone D, Kim KU, Vivo C, Lee KK, Dansen TB, et al. Malignant mesothelioma cells are rapidly sensitized to TRAIL-induced apoptosis by low-dose anisomycin via Bim. Mol Cancer Ther. 2007;6:2766–76. doi: 10.1158/1535-7163.MCT-07-0278. [DOI] [PubMed] [Google Scholar]
- 13.Sah NK, Munshi A, Kurland JF, McDonnell TJ, Su B, Meyn RE. Translation inhibitors sensitize prostate cancer cells to apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) by activating c-Jun N-terminal kinase. J Biol Chem. 2003;278:20593–602. doi: 10.1074/jbc.M211010200. [DOI] [PubMed] [Google Scholar]
- 14.Xia S, Li Y, Rosen EM, Laterra J. Ribotoxic stress sensitizes glioblastoma cells to death receptor induced apoptosis: requirements for c-Jun NH2-terminal kinase and Bim. Mol Cancer Res. 2007;5:783–92. doi: 10.1158/1541-7786.MCR-06-0433. [DOI] [PubMed] [Google Scholar]
- 15.Grollman AP. Inhibitors of protein biosynthesis. II. Mode of action of anisomycin. J Biol Chem. 1967;242:3226–33. [PubMed] [Google Scholar]
- 16.Hori T, Kondo T, Tabuchi Y, Takasaki I, Zhao QL, Kanamori M, et al. Molecular mechanism of apoptosis and gene expressions in human lymphoma U937 cells treated with anisomycin. Chem Biol Interact. 2008;172:125–40. doi: 10.1016/j.cbi.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Croons V, Martinet W, Herman AG, Timmermans JP, De Meyer GR. The protein synthesis inhibitor anisomycin induces macrophage apoptosis in rabbit atherosclerotic plaques through p38 mitogen-activated protein kinase. J Pharmacol Exp Ther. 2009;329:856–64. doi: 10.1124/jpet.108.149948. [DOI] [PubMed] [Google Scholar]
- 18.Tang Z, Xing F, Chen D, Yu Y, Yu C, Di J, et al. In vivo toxicological evaluation of Anisomycin. Toxicol Lett. 2011 doi: 10.1016/j.toxlet.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Nesbit M, Nesbit HK, Bennett J, Andl T, Hsu MY, Dejesus E, et al. Basic fibroblast growth factor induces a transformed phenotype in normal human melanocytes. Oncogene. 1999;18:6469–76. doi: 10.1038/sj.onc.1203066. [DOI] [PubMed] [Google Scholar]
- 20.Fodstad O, Kjønniksen I, Aamdal S, Nesland JM, Boyd MR, Pihl A. Extrapulmonary, tissue-specific metastasis formation in nude mice injected with FEMX-I human melanoma cells. Cancer Res. 1988;48:4382–8. [PubMed] [Google Scholar]
- 21.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 22.Liu X, Yue P, Chen S, Hu L, Lonial S, Khuri FR, et al. The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res. 2007;67:4981–8. doi: 10.1158/0008-5472.CAN-06-4274. [DOI] [PubMed] [Google Scholar]
- 23.Nikolaou VA, Stratigos AJ, Flaherty KT, Tsao H. Melanoma: new insights and new therapies. J Invest Dermatol. 2012;132:854–63. doi: 10.1038/jid.2011.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhuang L, Lee CS, Scolyer RA, McCarthy SW, Zhang XD, Thompson JF, et al. Progression in melanoma is associated with decreased expression of death receptors for tumor necrosis factor-related apoptosis-inducing ligand. Hum Pathol. 2006;37:1286–94. doi: 10.1016/j.humpath.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 25.Hersey P, Zhang XD. How melanoma cells evade trail-induced apoptosis. Nat Rev Cancer. 2001;1:142–50. doi: 10.1038/35101078. [DOI] [PubMed] [Google Scholar]
- 26.Ivanov VN, Zhou H, Hei TK. Sequential treatment by ionizing radiation and sodium arsenite dramatically accelerates TRAIL-mediated apoptosis of human melanoma cells. Cancer Res. 2007;67:5397–407. doi: 10.1158/0008-5472.CAN-07-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phipps LE, Hino S, Muschel RJ. Targeting cell spreading: a method of sensitizing metastatic tumor cells to TRAIL-induced apoptosis. Mol Cancer Res. 2011;9:249–58. doi: 10.1158/1541-7786.MCR-11-0021. [DOI] [PubMed] [Google Scholar]
- 28.Kluger HM, McCarthy MM, Alvero AB, Sznol M, Ariyan S, Camp RL, et al. The X-linked inhibitor of apoptosis protein (XIAP) is up-regulated in metastatic melanoma, and XIAP cleavage by Phenoxodiol is associated with Carboplatin sensitization. J Transl Med. 2007;5:6. doi: 10.1186/1479-5876-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen N, Gong J, Chen X, Meng W, Huang Y, Zhao F, et al. Caspases and inhibitor of apoptosis proteins in cutaneous and mucosal melanoma: expression profile and clinicopathologic significance. Hum Pathol. 2009;40:950–6. doi: 10.1016/j.humpath.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Vogler M, Walczak H, Stadel D, Haas TL, Genze F, Jovanovic M, et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res. 2009;69:2425–34. doi: 10.1158/0008-5472.CAN-08-2436. [DOI] [PubMed] [Google Scholar]
- 31.Takeuchi H, Morton DL, Elashoff D, Hoon DS. Survivin expression by metastatic melanoma predicts poor disease outcome in patients receiving adjuvant polyvalent vaccine. Int J Cancer. 2005;117:1032–8. doi: 10.1002/ijc.21267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thayaparasingham B, Kunz A, Peters N, Kulms D. Sensitization of melanoma cells to TRAIL by UVB-induced and NF-kappaB-mediated downregulation of xIAP. Oncogene. 2009;28:345–62. doi: 10.1038/onc.2008.397. [DOI] [PubMed] [Google Scholar]
- 33.Engesæter BO, Sathermugathevan M, Hellenes T, Engebråten O, Holm R, Flørenes VA, et al. Targeting inhibitor of apoptosis proteins in combination with dacarbazine or TRAIL in melanoma cells. Cancer Biol Ther. 2011;12:47–58. doi: 10.4161/cbt.12.1.15714. [DOI] [PubMed] [Google Scholar]
- 34.Chawla-Sarkar M, Bae SI, Reu FJ, Jacobs BS, Lindner DJ, Borden EC. Downregulation of Bcl-2, FLIP or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2004;11:915–23. doi: 10.1038/sj.cdd.4401416. [DOI] [PubMed] [Google Scholar]
- 35.Nachmias B, Ashhab Y, Bucholtz V, Drize O, Kadouri L, Lotem M, et al. Caspase-mediated cleavage converts Livin from an antiapoptotic to a proapoptotic factor: implications for drug-resistant melanoma. Cancer Res. 2003;63:6340–9. [PubMed] [Google Scholar]
- 36.Sung B, Ravindran J, Prasad S, Pandey MK, Aggarwal BB. Gossypol induces death receptor-5 through activation of the ROS-ERK-CHOP pathway and sensitizes colon cancer cells to TRAIL. J Biol Chem. 2010;285:35418–27. doi: 10.1074/jbc.M110.172767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Prasad S, Ravindran J, Sung B, Pandey MK, Aggarwal BB. Garcinol potentiates TRAIL-induced apoptosis through modulation of death receptors and antiapoptotic proteins. Mol Cancer Ther. 2010;9:856–68. doi: 10.1158/1535-7163.MCT-09-1113. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Mawji IA, Simpson CD, Gronda M, Williams MA, Hurren R, Henderson CJ, et al. A chemical screen identifies anisomycin as an anoikis sensitizer that functions by decreasing FLIP protein synthesis. Cancer Res. 2007;67:8307–15. doi: 10.1158/0008-5472.CAN-07-1687. [DOI] [PubMed] [Google Scholar]
- 39.Mikhailov A, Patel D, McCance DJ, Rieder CL. The G2 p38-mediated stress-activated checkpoint pathway becomes attenuated in transformed cells. Curr Biol. 2007;17:2162–8. doi: 10.1016/j.cub.2007.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu J, Zhou JY, Wei WZ, Wu GS. Activation of the Akt survival pathway contributes to TRAIL resistance in cancer cells. PLoS One. 2010;5:e10226. doi: 10.1371/journal.pone.0010226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang XD, Borrow JM, Zhang XY, Nguyen T, Hersey P. Activation of ERK1/2 protects melanoma cells from TRAIL-induced apoptosis by inhibiting Smac/DIABLO release from mitochondria. Oncogene. 2003;22:2869–81. doi: 10.1038/sj.onc.1206427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.