

Abstract
Abnormally expanded polyglutamine domains in proteins are associated with several neurodegenerative diseases, including Huntington's disease. Expansion of the polyglutamine (polyQ) domain facilitates aggregation of the affected protein, and several studies directly link aggregation to neurotoxicity. Studies of synthetic polyQ peptides have contributed substantially to our understanding of the mechanism of aggregation. In this report, polyQ fibrils were immobilized onto a sensor, and their elongation by polyQ peptides of various length and conformation was examined using quartz crystal microbalance with dissipation monitoring (QCM-D). The rate of elongation increased as the peptide length increased from 8 to 24 glutamines (Q8, Q20, and Q24). Monomer conformation affected elongation rates: insertion of a β-turn template d-Pro-Gly in the center of the peptide increased elongation rates several-fold, while insertion of Pro-Pro dramatically slowed elongation. Dissipation measurements of the QCM-D provided qualitative information about mechanical properties of the elongating fibrils. These data showed clear differences in the characteristics of the elongating aggregates, depending on the specific identity of the associating polyQ peptide. Elongation rates were sensitive to the pH and ionic strength of the buffer. Comparison of QCM-D data with those obtained by optical waveguide lightmode spectroscopy revealed that very little water was associated with the elongation of fibrils by the peptide containing d-Pro-Gly, but a significant amount of water was associated when the fibrils were elongated by Q20. Together, the data indicate that elongation of polyQ fibrils can occur without full consolidation to the fibril structure, resulting in variations to the aggregate structure during elongation.
Keywords: aggregation, fibril elongation, polyglutamine, quartz crystal microbalance with dissipation monitoring (QCM-D), optical waveguide lightmode spectroscopy (OWLS)
Introduction
Nine distinct neurodegenerative diseases including Huntington's disease1 share in common the expansion of CAG codons in disease-specific genes. Gene expression results in the production of proteins with expanded polyglutamine (polyQ) domains that are prone to aggregation, invariably leading to the formation of nuclear inclusions.2,3 The proteins share no other sequence, structural, or compositional homologies.4 The disease phenotype requires expansion of the polyQ domain beyond a disease-specific critical threshold length;5 further increases in the number of glutamine residues result in earlier ages of onset and more severe symptoms.5 The polyQ domain varies in both threshold length and location within the affected protein.6
Aggregation of proteins containing expanded polyQ regions has been causally linked to cellular toxicity. As one example, expression of an expanded polyQ region in hypoxanthine phosphoribosyl transferase, a protein unrelated to any known polyQ diseases, produced polyQ-like aggregates and neurodegeneration.7 Protein misfolding is the central molecular event of polyQ toxicity: several studies have shown that overexpression of chaperones ameliorates aggregation and toxicity in both animal and cellular models.8–10 Toxicity likely results from a gain of toxic function in the impacted protein rather than a loss of normal function.11 The identity of the toxic species remains unresolved; misfolded monomers,12 soluble oligomers,8,13 and insoluble aggregates14,15 all have been posited.
Synthetic polyQ peptides have been profitably employed as models to study polyQ conformation and aggregation.14,16–22 PolyQ peptides display many of the important aggregation-related behaviors of disease-implicated polyQ proteins. For example, polyQ peptides display more rapid aggregation kinetics in vitro as length increases,14 polyQ peptide aggregates delivered to the nuclei of mammalian cells were toxic,15 and polyQ peptides cleaved from a green fluorescent protein-ubiquitin-polyQ construct in a cellular model aggregated and caused cell death.23 These results indicate that the polyQ region in the absence of a full-length protein can still produce toxicity.
Two mechanisms for polyQ peptide aggregation have been proposed. In an adaptation of the nucleation–elongation model, a thermodynamically unfavorable β-sheet nucleus, consisting of one to four monomers in equilibrium with the bulk disordered monomer, is posited to serve as the nucleus.14,24,25 Fibrils grow from the nucleus by sequential addition of monomers. Data on monomer loss from solutions of synthetic peptides containing from 28 to 47 glutamines are well described by a kinetic model based on this mechanism.14,24,25 However, a number of alternative kinetic mechanisms fit these data equally well, and so the nucleation–elongation model cannot be construed as a unique solution to explain the collected data.26,27 In an alternative mechanism, which we refer to as the association–conformational conversion model, disordered monomers self-associate into soluble, unstructured oligomers.19,20,28–32 Within the “liquid-like” interior of the oligomers,27 conformational rearrangement leads to formation of β-sheet “nodes,” which then propagate through the oligomer, ultimately leading to formation of insoluble fibrillar aggregates with high β-sheet structural content and little water.20
Once fibrils form, further elongation can occur through addition of monomers to the fibril tip. Two possible mechanisms of monomer addition are illustrated in Fig. 1. In one mechanism (Fig. 1a), disordered monomer binds to (“docks”) and consolidates (“locks”) onto the fibril tip. Further rounds of binding happen only subsequent to consolidation—in other words, locking of one monomer must occur before docking of another. In an alternative mechanism (Fig. 1b), disordered monomers continuously add to the growing aggregate without requiring full consolidation to the β-sheet structure of the fibril. Eventually, conformational rearrangement to β-sheet is expected to propagate through the elongating aggregate. The differences in these mechanisms give rise to expected differences in the characteristics of the elongating aggregates (Fig. 1). In the first mechanism, most of the added material is irreversibly associated with the fibril, and the β-sheet-rich fibril structure remains unchanged as it elongates. In the second mechanism, a higher degree of reversibility is anticipated since the added material does not immediately fully consolidate to the fibril, and the structure of the elongating section differs from the base fibril prior to propagation of the β-sheet. Our previous work20,32 hinted that the second scenario may be correct. Specifically, when polyQ peptides were allowed to aggregate in solution, the aggregates displayed a high degree of lateral alignment, which we attributed to interactions between growing oligomers before they fully mature into fibrils. However, if the polyQ peptides were synthesized with the β-turn template d-Pro-Gly, the resulting fibrillar aggregates mostly lacked lateral alignment. We hypothesized that peptides containing a β-turn template will mature rapidly into fibrils, before they can laterally associate.32
Fig. 1.

Proposed schema of polyQ fibril elongation. Shaded regions represent the original fibril. (a) Monomer must fully consolidate (“lock”) to the fibrillar structure before an additional monomer can add (“dock”). (b) Full consolidation of newly added monomer is not required to support further elongation. This results in changes to the structure of the fibril as it elongates. Ultimately, the β-sheet-rich structure of the monomer propagates through the fibril, but this is not prerequisite for further elongation. As sketched, the first three steps are addition only and the latter steps show slow consolidation. Under this mechanism, it would be possible to have simultaneous addition and consolidation, but with consolidation slower than addition.
The work reported here has two objectives: to measure fibril elongation rates as a function of polyQ length and monomer conformation and to ascertain which, if either, of the proposed mechanisms best describes elongation. We employed quartz crystal microbalance with dissipation monitoring (QCM-D) for the majority of our fibril elongation experiments. The QCM-D allows sensitive (~1 ng cm−2 sensitivity in liquid), label-free, real-time investigation of biomolecule deposition. In QCM-D, a piezoelectric quartz sensor crystal is oscillated at its resonant frequency for a short time period. Attachment of mass to the sensor produces a decrease in frequency (Δf).33 For rigid adsorbed layers (adlayers), Δf can be related to the change in mass (Δm) using the Sauerbrey relation.34 The sensed mass Δm reflects the mass of the deposited biomolecule and that of solvent molecules bound to the protein or hydrodynamically coupled to the adsorbed layer. Adlayers of biomolecules (e.g., protein) are often nonrigid and do not fully couple to the crystal's oscillation, resulting in energy dissipation.35 By simultaneously measuring Δf and the change in the energy dissipation factor (ΔD), QCM-D allows quantitative investigation of attachment kinetics and provides qualitative information on the mechanical properties of the deposited mass (e.g., viscoelasticity of laterally homogeneous adlayers).35–37 Change in deposited mass with time is measured by oscillating the crystal for a short period of time, switching off the driving current, and sampling the decay curve as the oscillation dampens. Fitting a damped sinusoidal function to the decay curve allows one to obtain Δf and ΔD as a function of time.36,37 Rapid sampling can be achieved because oscillations are dampened within a few milliseconds. QCM-D has been successfully employed in a number of protein aggregation studies but has not yet been applied to polyQ.38–42
Normalization of ΔD by Δm allows qualitative characterization of the added material. Higher values of ΔD/Δm generally indicate a softer, looser, or more flexible adlayer, while lower values correspond to a denser or more rigid adlayer. ΔD/Δm can provide some insight into the shape and length of molecules associated with the surface.43 In a study of biotinylated, double-stranded DNA adsorbing to NeutrAvidin, Tsortos et al. noted that ΔD/Δm increased with increasing length of DNA. Additionally, for DNA with the same number of base pairs, straight or extended structures produced larger ΔD/Δm than bent or “triangular” DNA.44 With adsorbed polymers, Zhang and Wu showed that ΔD/Δm increases when the polymer swells and decreases when the polymer collapses due to changes in solvent.45 This feature has been applied to characterize aggregates of glucagon41 and beta-amyloid (Aβ),40 and we use it here to probe differences in the mechanical properties of elongating aggregates as a function of the specific polyQ peptide.
In a subset of experiments, we complemented our QCM-D measurements with those made by optical waveguide lightmode spectroscopy (OWLS) to examine the role of added water on polyQ fibril elongation. As noted above, the mass sensed by QCM-D includes that of both the depositing protein and water bound to or trapped in the proteinaceous adlayer. In contrast, OWLS is a “solvent-blind” technique and measures the “dry” mass of the deposited protein.46 OWLS employs an optical grating to incouple laser light into a planar waveguide. The incoupling angles for the transverse electric and magnetic modes are sensitive to the refractive index (nR) at the waveguide surface.47 Changes in nR caused by the presence of molecules of known refractive index increment (dnR/dc, where c is concentration) allow determination of the mass density of these molecules near the surface.47 The change in effective nR is measured relative to an initial, analyte-free state, allowing only non-solvent molecules to be detected. The OWLS technique allows non-solvent molecules to be sensed within 100–200 nm of the waveguide surface with a sensitivity on the order of 1 ng cm−2.46,47
Peptides were chosen for this study with the goal of gaining insight into the role played by polyQ length and structure in polyQ fibril elongation. The peptides are of the type K2WQxAK2, where x=8, 20, or 24. In addition, two Q20 peptides were synthesized containing either a Pro-Pro insert or the β-turn template d-Pro-Gly in the center of the glutamine repeat domain (K2WQ10XXQ10AK2, with XX=PP or DPG). Peptides are subsequently referred to by the length of the glutamine region (Q8, Q20, and Q24) or by their interrupting residues (PP and PG). In previous work, we found that Q20 and Q24, but not Q8, assembled rapidly into soluble oligomers that then slowly precipitated as fibrillar aggregates. Aggregation was significantly faster for Q24 than for Q20.20 PP and PG interrupting residues significantly affected both monomer conformation and aggregation properties.32 Specifically, PP increased poly-Pro II-like secondary structure of the monomer and prevented fibril formation, while PG increased β-turn content in the monomer and accelerated fibril formation. The peptides under study, and their aggregation properties, are summarized in Table 1. While we examined overall aggregation rates in our previous work, in this study, we focused specifically on fibril elongation.
Table 1.
Panel of polyQ peptides
| Abbreviation | Sequence | Monomer conformation | Aggregation properties |
|---|---|---|---|
| Q8 | KKWQ8AKK | Disordered | None |
| Q20 | KKWQ20AKK | Disordered | Soluble oligomers initially, slow maturation into insoluble fibrillar aggregates |
| Q24 | KKWQ24AKK | Disordered | Soluble oligomers initially, moderate rate of maturation into insoluble fibrillar aggregates |
| PG | KKWQ10DPGQ10AKK | β-Turn+disordered | Soluble oligomers initially, very rapid maturation into insoluble fibrillar aggregates |
| PP | KKWQ10PPQ10AKK | polyProII+disordered | Soluble oligomers initially, growth in size of aggregates but aggregates are not fibrillar |
Results
Immobilization of fibrils to surface
To use QCM-D for polyQ fibril elongation studies, we first developed a procedure to stably immobilize polyQ on the sensor surface and a method to block unoccupied sites. The blocking step is critical because we wanted to ensure that any adsorption of polyQ from solution was specifically to the immobilized polyQ and not nonspecifically to the surface. Initial efforts focused on using gold surfaces and thiol chemistry to covalently attach a modified dextran layer to the surface, a procedure commonly used in surface plasmon resonance studies.48 The modified dextran layer contains carboxymethyl groups, which form covalent bonds with amine groups when activated and allow immobilization of polyQ peptides via the flanking lysine residues.48 Using this approach, we covalently coupled polyQ fibrils to dextran-coated surfaces (data not shown). However, when active sites in the dextran layer were blocked by ethanolamine or polyethylene glycol modified with a primary amine group in control experiments, polyQ fibrils stably associated with the surface in significant amounts (data not shown), indicating that noncovalent association to this surface also occurred. Thus, we were unable to establish a good negative control for dextran-coated surfaces. We therefore investigated alternative surfaces and polyQ immobilization strategies to produce stable associations of polyQ on the surface that would prevent nonspecific association of polyQ monomer and aggregates when an appropriate blocking agent was used. PolyQ aggregates were found to stably adsorb to many surfaces, including dextran, gold, aluminum oxide, and a fluoropolymer, but successful blocking of adsorption proved difficult. Ultimately, a SiO2-coated sensor crystal, with poly-l-lysine (PLL) as a blocking agent, was found to be suitable. PLL stably associated with bare SiO2-coated sensors under the experimental conditions. No increase in mass was detected when Q8 monomers, or PG monomers and aggregates, were exposed to the PLL-coated sensor (data not shown), indicating lack of nonspecific adsorption and thus a suitable negative control surface.
We decided to immobilize fibrils rather than monomers for two reasons. First and foremost, our objective was to examine mechanisms of fibril elongation, in the absence of the confounding process of fibril initiation. Second, we were unable to stably immobilize monomers: when adsorbed monomers were exposed to PLL during the blocking step, PLL displaced all or nearly all of the monomer from the SiO2 surface (data not shown). We chose to immobilize polyQ fibrils made from PG because PG forms mature fibrillar aggregates much more readily than do uninterrupted polyQ peptides of similar length, and the fibrils have a more homogenous appearance, with less lateral alignment.32 PG fibrils were prepared by a freeze–thaw method (monomer equivalent concentration of 10 μM) in water adjusted to pH 3 with trifluoroacetic acid (TFA). The fibrillar nature of the aggregates was confirmed by transmission electron microscopy (Fig. 2).
Fig. 2.
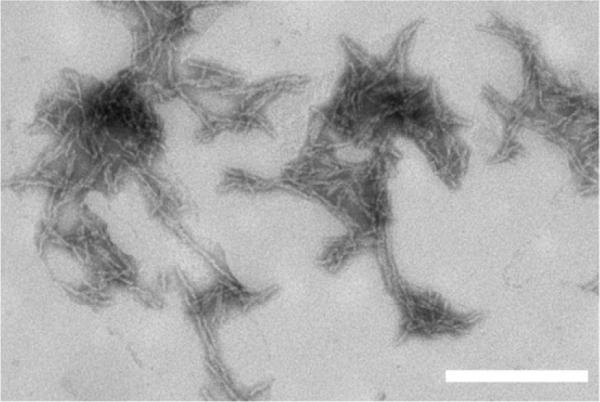
Transmission electron microscopy image of PG aggregates. Aggregates were prepared via freeze–thaw cycles of PG peptide in water adjusted to pH 3 with TFA. Image is representative of a large number of fields examined. The scale bar represents 200 nm.
To immobilize fibrils, we mounted the SiO2-coated sensor crystal in the QCM-D flow cell chamber and equilibrated it in water until a stable baseline was attained. A suspension of fibrils in water adjusted to pH 3 with TFA was then pumped across the sensor surface for 5 min. The low pH was chosen to prevent further growth of aggregates during the immobilization procedure. The positively charged fibrils readily associated with the negatively charged SiO2 surface, as detected by a decrease in the frequency measurement. The rate of attachment was initially rapid and declined as surface coverage increased (Fig. 3), indicating that the fibrils attach to the surface but do not continue to grow under the chosen solvent conditions, which was the desired response during immobilization. The Sauerbrey equation was used to convert Δf/n measurements to Δm values for all QCM-D experiments. The change in dissipation with the change in frequency, ΔD/(Δf/n), was small, less than 4×10−7 Hz−1; thus, use of the Sauerbrey equation to calculate adsorbed mass is justified.35 The adsorbed mass density approached 220 ng cm−2 after a 5-min exposure. (This compares to a plateau at 60 ng cm−2 with adsorption of PG monomer to the surface. As pointed out earlier, PG monomer was not stably adsorbed, in contrast to PG fibrils.) Interestingly, dissipation (ΔD) was relatively invariant with adsorbed mass until the density reached 50–100 ng cm−2, at which point ΔD rose linearly with mass (Fig. 3b). This pattern may indicate that at low surface coverage, fibrils attach flat and close to the surface, but when more of the sensor is occupied, fibrils extend further into the solution. During the linear phase, the change in dissipation per change in mass, ΔD/Δm, was small, ~0.007×10−6 cm2 ng−1. Rinsing the surface with Hepes buffer caused a slight disturbance in the baseline of the adsorbed mass, likely due to the change in the density of the solution. The surface was then blocked by pumping PLL in Hepes across the fibril-coated surface. A small decrease in mass was observed (Fig. 3a), which we interpret to indicate that PLL competes with more weakly associated PG aggregates for binding and removes aggregates that are not stably attached. The lack of an increase in mass upon exposure to PLL provides further evidence that most of the binding sites on the SiO2 surface are occupied by fibrils. Phosphate-buffered saline with azide (PBSA) was then pumped over the sensor for 30 min and no further change in mass density was detected (data not shown), indicating that the fibrils remain stably immobilized over the course of the experiment. We also observed that, once fibrils were immobilized, increasing pH to 12 did not cause dissociation of the fibrils or PLL (data not shown).
Fig. 3.

Immobilization of PG aggregates on SiO2 surface and blocking of unoccupied sites with PLL. Vertical lines indicate changes in solvent conditions. Initially, water at pH 3 was pumped across the surface, followed by PG aggregates (in pH 3 water). The solution was then switched to Hepes buffer (pH 7); the slight shift in signal is likely due to the solvent switch. Finally, the sensor was exposed to PLL to block any remaining surface sites. (a) Mass associated. (b) Mass associated–dissipation plot for the period of peptide exposure.
In all subsequent experiments, except where otherwise specified, freshly prepared (monomeric) solutions of polyQ peptide were pumped over the PG fibril-coated sensors for 5 min, and changes in resonant frequency and dissipation were measured over time. The sensors were then rinsed with buffer for 10 min to examine reversibility. Experiments were conducted with the peptides listed in Table 1. In these experiments, ΔD/(Δf/n) was <4×10−7 Hz−1, and the Sauerbrey equation was used to calculate adsorbed mass. The dissipation values were small and reflected subtle differences in adlayer mechanical properties or fibril/surface contact or both.35 For reference, all experimental results are summarized in Tables 2 and 3 as the total increase in associated mass over the 5-min exposure time (Δmmax), mass removed during dissociation (Δmdis), change in dissipation (at its largest extent) (ΔDmax), and ΔDmax/Δmmax.
Table 2.
PG addition to immobilized PG fibrils
| Concentration (μM) | pH | Ionic strength (M) | Δmmax(ng cm−2) | Δmdis(ng cm−2) | ΔDmax(×10−6) | ΔDmax/Δmmax(×10−9 cm2 ng−1) |
|---|---|---|---|---|---|---|
| 10 | 7.4 | 0.17 | 42±7 | 6±2 | 0.3±0.0 | 7.6±2 |
| 20 | 83±7 | 24±2 | 0.7±0.1 | 8.8±0.3 | ||
| 40 | 155±4 | 32±5 | 1.3±0.1 | 8.2±0.1 | ||
| 5 | 12 | 0.17 | 41±7 | 0±0 | 0.1±0.0 | 2.6±0.4 |
| 20 | 320±20 | 0±0 | 0.5±0.0 | 1.6±0.3 | ||
| 20 | 7.4 | 0.03 | 66±4 | 13±4 | 0.4±0.0 | 6.1±0.5 |
| 0.17 | 83±7 | 24±2 | 0.7±0.1 | 8.8±0.3 | ||
| 0.32 | 154±9 | 4±0 | 2.2±0.1 | 15±1 | ||
| 0.52 | 54±4 | 0±0 | 0.1±0.0 | 1.9±0.2 | ||
| 20 (aggregated) | 7.4 | 0.17 | 780±40 | 0±0 | 9.6±0.1 | 12±1 |
| 20 (OWLS) | 7.4 | 0.17 | 81±1 | 16±1 | N/A | N/A |
In all cases, solutions containing PG were contacted with immobilized fibrils for 5 min before rinsing with buffer. The maximum increase in associated mass (Δmmax), mass dissociated upon rinsing with buffer (Δmdis), maximum increase in dissipation (ΔDmax), and the ratio of the maximum dissipation increase to the maximum mass increase (ΔDmax/Δmmax) are reported as the mean and range of two independent experiments.
Table 3.
PolyQ peptide association to immobilized PG fibrils
| Peptide | pH | Δmmax (ng cm−2) | Δmdis (ng cm−2) | ΔDmax (×10−6) | ΔDmax/Δmmax (×10−9 cm2 ng−1) |
|---|---|---|---|---|---|
| Q8 | 7.4 | 7±2 | 4±1 | −0.1±0.0 | −11±2 |
| Q20 | 33±3 | 5±1 | 0.1±0.0 | 1.4±0.3a | |
| Q24 | 70±4 | 16±2 | 0.2±0.1 | 3.8±0.4 | |
| PG | 83±7 | 24±2 | 0.7±0.1 | 8.8±0.3 | |
| PP | 23±7 | 16±2 | 0.1±0.0 | 5.8±0.9b | |
| Q8 | 12 | 13±1 | 7±1 | 0.0±0.0 | −2.6±0.2 |
| Q20 | 100±10 | 13±4 | 0.2±0.0 | 2.3±0.1 | |
| Q24 | 250±3 | 11±1 | 0.3±0.0 | 1.2±0.1 | |
| PG | 320±20 | 0±0 | 0.5±0.0 | 1.6±0.3 | |
| PP | 36±4 | 21±2 | 0.3±0.0 | 9.4±0.9 | |
| Q20 (OWLS) | 7.4 | 20±1 | 6±0 | N/A | N/A |
| PG (OWLS) | 81±1 | 16±1 | N/A | N/A |
Solutions containing the indicated peptide (20 μM) were contacted with immobilized fibrils for 5 min before rinsing with buffer. The maximum increase in associated mass (Δmmax), mass dissociated upon rinsing (Δmdis), maximum increase in dissipation (ΔDmax), and ΔDmax/Δmmax are reported as the mean and range of two independent experiments.
ΔD/Δm changes over the course of the 5-min exposure because after an initial rise in dissipation, D levels off. Initially, ΔD/Δm ~4×10−9 cm2 ng−1.
ΔD/Δm changes over the course of the 5-min exposure because after an initial rise in dissipation, D levels off. Initially, ΔD/Δm ~9×10−9 cm2 ng−1.
PG monomer addition to immobilized PG fibrils
The dPG insert was designed as a template for a β-turn in the center of this polyQ peptide; adoption of β-turn was confirmed by circular dichroism, which further showed that the PG monomer remains partially disordered.32 We prepared solutions of PG in phosphate-buffered saline (PBS) and then immediately pumped the solution across the PG fibril-coated sensor. Previous experiments indicate that immediately after preparation, PG solutions are largely monomeric but contain a small (likely <1%) oligomer population.32 Upon exposure to the PG monomer, the mass associated with the fibril-coated sensor increased continuously over the 5-min exposure period (Fig. 4a). The rate of association increased nearly linearly with increasing PG concentration. Association was faster early in the exposure, then slowed somewhat with time. This is not due to depletion of monomer in solution, as the peptide concentration was in far excess and was continually delivered to the flow cell during peptide exposure. Peptide continued to adsorb to the immobilized fibrils throughout the 5-min exposure. About 15–30% of the newly adsorbed material dissociated upon rinsing, indicating that PG addition to the immobilized fibrils is partly reversible. In another experiment, even after a 10-h batch exposure to PG, we observed continued association (data not shown), indicating that aggregate growth can be maintained indefinitely provided peptide remains in solution.
Fig. 4.

Effect of PG monomer concentration on elongation kinetics. Concentrations used were 10 μM (◯), 20 μM (■), and 40 μM (▲) in PBSA. Vertical lines indicate changes in solvent conditions. Results are representative of two independent experiments. (a) Mass associated. (b) Dissipation measurements. (c) Mass associated–dissipation plot for the period of peptide exposure.
Dissipation also increased upon exposure of the surface to the PG monomer (Fig. 4b). The increase in dissipation was fastest initially and slowed with time, similar to the observed increase in mass. When plotted as the change in dissipation versus change in mass (ΔDversus Δm, Fig. 4c), all of the data fall on the same line, regardless of peptide concentration or time of exposure. Furthermore, ΔD/Δm≈0.008 × 10−6 cm2 ng−1, very close to the value obtained during the second phase of immobilization of PG fibrils. Together, these analyses indicate that the dissipative properties of the elongating aggregates are independent of monomer concentration, remain constant at all stages of growth, and are the same whether the fibrils are preformed or grown from monomer addition.
Effect of ionic strength
We next tested whether changes in ionic strength (I) affected elongation. PG monomer addition to immobilized PG fibrils was measured at ionic strengths ranging from 0.026 M to 0.52 M, using NaCl as the electrolyte. From I=0.026 to 0.32 M, Δmmax, ΔDmax, and ΔDmax/Δmmax all increased with increasing I (Table 2). Surprisingly, with a further increase to I=0.52 M, all three properties decreased markedly. At low and near-physiological salt concentrations (up to 0.15 M), 20–25% of the adsorbed material dissociated with a buffer rinse. In contrast, at higher ionic strength, association was irreversible.
Effect of pH
Flanking lysines, which are positively charged at neutral pH, were incorporated into the polyQ peptides to facilitate synthesis and initial solubilization. Both simulation and experiment have demonstrated that flanking charges affect conformation and oligomerization.20,49 To examine the effect of lysine charge on fibril elongation, we conducted experiments at pH 12, well above the pKa of lysine (10.5 for the free amino acid). Upon raising the pH to 12, some drift in the baseline caused by ionization of the SiO2 surface was noted, but there was no indication that immobilized fibrils dissociated from the sensor (data not shown). (Control experiments also showed that immobilized PLL did not dissociate from the sensor at pH 12, data not shown.) Dissipation measurements for the immobilized PG fibrils decreased (after accounting for the shift in baseline, data not shown). Thus, it is likely that the immobilized fibrils are oriented differently on the surface at pH 12 than at pH 7.4, forming a somewhat more rigid or denser layer, presumably because of reduced repulsion between fibrils. PG monomer solution was prepared at pH 12 and immediately pumped over the immobilized fibrils. (Some aggregation likely occurs in solution at this pH; however, no turbidity or precipitates were observed.) The total mass of PG adsorbed to the immobilized fibrils during the 5-min exposure period was much larger at pH 12 compared to pH 7.4 (Table 2). In contrast to pH 7.4, at pH 12, the rate of association was constant over the 5-min exposure time (Fig. 5). The initial rate of association (first 15 s) at pH 7.4 was 57±2 ng cm−2 min−1, nearly equal to the constant rate of 64±4 ng cm−2 min−1 observed throughout the exposure at pH 12. The rate of association was strongly concentration dependent: Δmmax at 5 μM was approximately one-eighth that at 20 μM (Fig. 5). Dissociation of newly associated monomer was undetectable when the solution was rinsed with pH 12 buffer, similar to the lack of reversibility observed at high ionic strength. A plot of ΔDversus Δm again results in a straight line (Fig. 5c) with ΔD/ Δm=0.0016×10−6 cm2 ng−1 at pH 12. This is about fivefold lower than the value obtained at pH 7.4 but very similar to the value obtained at the highest salt concentration. These results indicate that neutralization (pH 12) or screening (I=0.52 M) of lysine charges has similar effects on the irreversibility of monomer addition and on the dissipative properties of the fibrils.
Fig. 5.
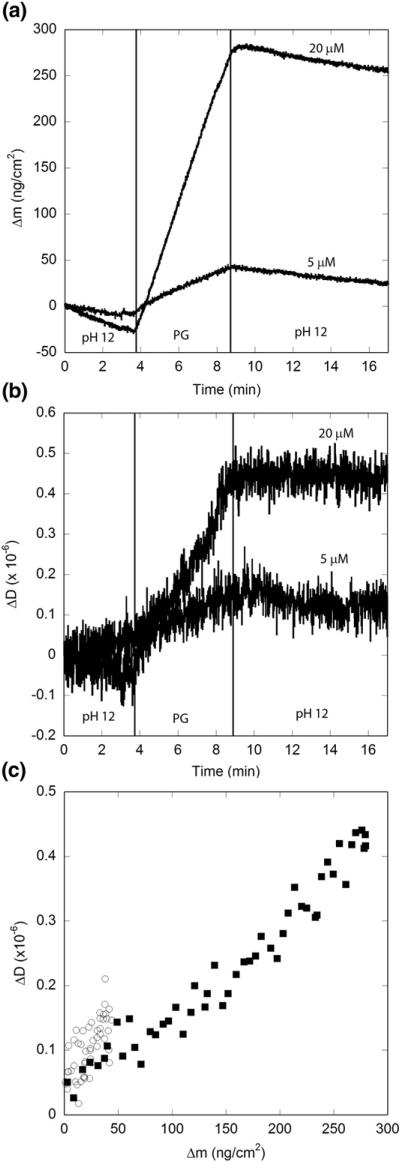
Effect of pH on PG elongation kinetics. Solutions of PG were prepared in pH 12 phosphate buffer at 20 μM (■) and 5 μM (◯). Vertical lines indicate times at which solutions were changed. Results are representative of two independent experiments. (a) Mass associated. Drift in the baseline is caused by ionization of the SiO2 surface at pH 12. (b) Dissipation measurements. (c) Mass associated–dissipation plot for the period of peptide exposure.
Elongation with uninterrupted polyQ peptides of varying length
In the next set of experiments, we examined the effect of the length of the polyQ sequence on elongation. Freshly prepared solutions of Q8, Q20, and Q24 in PBS were exposed to immobilized PG aggregates, and the changes in mass and dissipation were measured (Fig. 6). Association of Q8 was nearly undetectable above baseline levels, and most dissociated upon rinsing with PBS (Table 3). Additionally, no increase in dissipation was observed for Q8. These results show that Q8 weakly associates with preformed PG fibrils and does not elongate fibrils. In contrast, both Q20 and Q24 associated with the immobilized PG fibrils in a length-dependent manner: Δmmax for Q20 was approximately half that of Q24 (Table 3). The rate of mass increase was rapid initially but slowed over time. The fraction of peptide dissociating upon rinsing was ~20–25% for both Q20 and Q24 (Table 3). These data are consistent with our prior work in which we observed fibril formation in solutions of Q20 and Q24, but not Q8, with a faster rate of aggregation for Q24.20
Fig. 6.
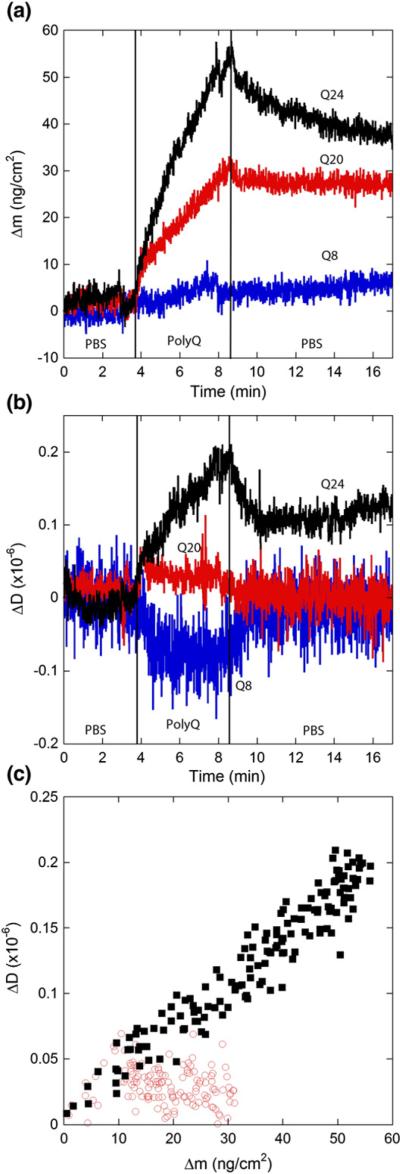
Effect of polyQ length on elongation kinetics. Solutions (20 μM) of monomeric Q8, Q20 (◯), or Q24 (■) in PBSA was exposed to a PG fibril-coated SiO2 sensor. Vertical lines indicate times at which solutions were changed. Results are representative of two independent experiments. (a) Mass associated. (b) Dissipation measurements. (c) Mass associated–dissipation plot for the period of peptide exposure (Q8 not shown for clarity).
With Q24, dissipation increased with time, and a plot of ΔDversus Δm was nearly linear (Fig. 6). A more complex situation was observed with Q20: initially, dissipation increased with mass, but after about 1 min, dissipation was flat, although mass continued to increase (Fig. 6). Taking just the initial data, ΔD/Δm ~0.004×10−6 cm2 ng−1 for both Q20 and Q24, roughly half the value observed for PG. These results indicate that the dissipative properties of the adsorbed peptide for Q20 and Q24 differ from those of PG and also that these properties change for Q20 over the course of exposure. All the dissipation values are small and reflect subtle differences in adlayer mechanical properties or fibril/surface contact or both.35
We also examined the impact of polyQ length at pH 12 (Fig. 7). Association of Q8 was minimal, the small amount of freshly associated material dissociated upon rinsing with buffer, and dissipation did not change. This indicates that eight glutamines in the repeat domain are insufficient for stable binding to preexisting PG fibrils, even if the lysine charge is neutralized. For Q20 and Q24, Δmmax at pH 12 was three- to fourfold higher than at pH 7.4 (Table 3), similar to PG (Table 2), and the rate of increase was constant with time (Fig. 7a). Unlike PG, association of Q20 and Q24 was partially reversible with rinsing, although the fraction of peptide dissociated was reduced at pH 12 compared to pH 7.4. Interestingly, for Q20, dissipation increased throughout the exposure period, in contrast to the pattern observed at pH 7.4. When plotted as ΔDversus Δm (Fig. 7c), the data for Q20 and Q24 collapse to a single curve. For Δm <100 ng cm−2, ΔD/Δm ~0.002×10−6 cm2 ng−1 for both Q20 and Q24, similar to the value obtained for PG, but decreased slightly to ~0.001×10−6 cm2 ng−1 for Q24 later in the elongation process (Fig. 7c). These are all very small values, indicating a rigid adlayer.
Fig. 7.
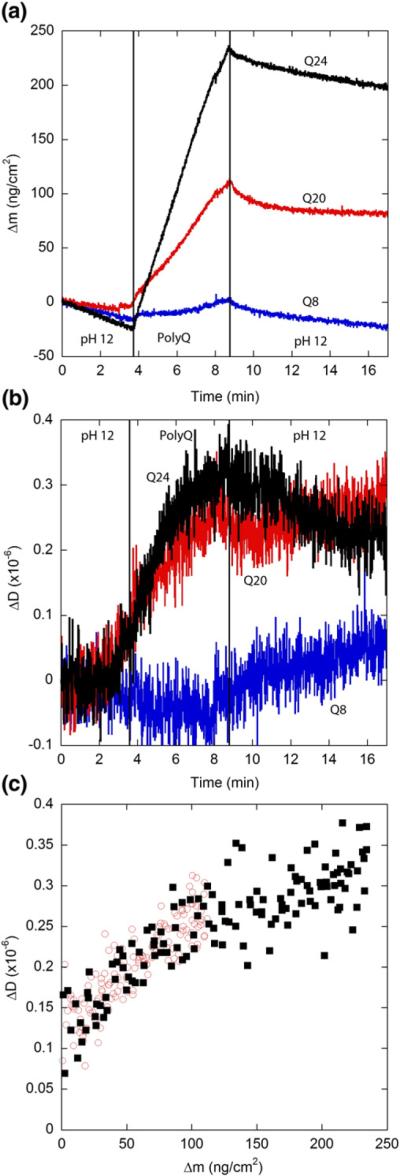
Elongation kinetics of polyQ peptides at pH 12. Solutions (20 μM) of monomeric Q8, Q20 (◯), or Q24 (■) in a phosphate buffer adjusted to pH 12 was exposed to a PG fibril-coated SiO2 sensor. Vertical lines indicate times at which solutions were changed. Results are representative of two independent experiments. (a) Mass associated. Drift in the baseline is caused by ionization of the SiO2 surface at pH 12. (b) Dissipation measurements. (c) Mass associated–dissipation plot for the period of peptide exposure (Q8 not shown for clarity).
Effect of monomer conformation
We previously showed that insertion of two prolines in the center of a string of 20 glutamines (PP) completely inhibits fibril formation and that even mixing PG with PP does not initiate fibril growth.32 We also showed that PP self-associates into large soluble, predominantly non-fibrillar aggregates.32 This was postulated to occur because the rigid PP sequence prevented adoption of a β-turn but could not thwart polyQ-mediated self-association. Immobilized PG fibrils were exposed to PP monomer in both PBS and pH 12 buffer to determine whether PP would interact with preexisting fibrils (Fig. 8). Δmmax for PP over the 5-min exposure time was 23 ng cm−2 at pH 7.4 and 36 ng cm−2 at pH 12, about two-thirds and one-third that observed for Q20, respectively, indicating that the PP interruption considerably slows but does not totally prevent attachment to the surface. More significantly, about 70% of the freshly associated PP dissociated from the fibrils upon rinsing. This indicates that PP does not effectively consolidate to the PG fibril. At both pH values, dissipation increased initially but then leveled off despite the continued increase in mass (Fig. 8b). Curiously, ΔD/Δm for PP was much larger for PP than for Q20 or Q24. Rinsing with buffer rapidly returned ΔD to a level near its baseline before exposure to PP. This again suggests that association of PP with the immobilized fibrils is not stable.
Fig. 8.
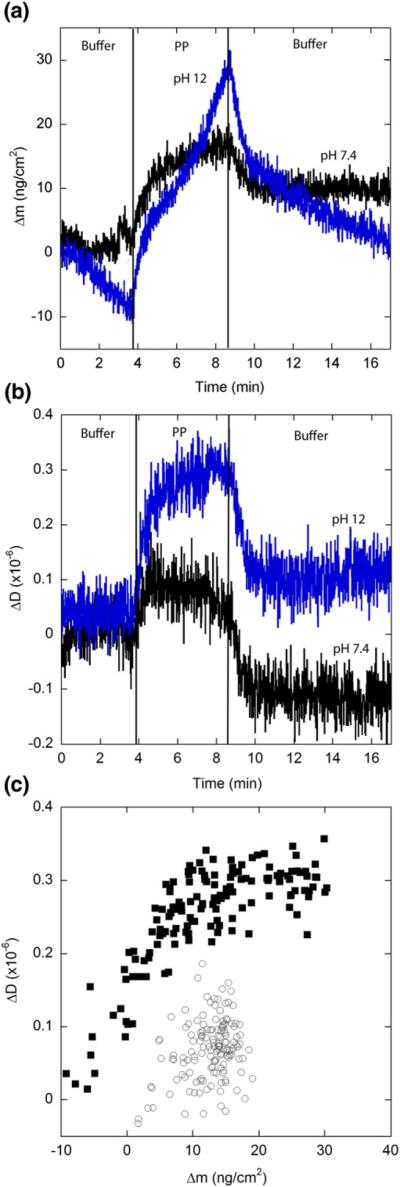
Elongation kinetics of PP. Solutions (20 μM) of monomeric PP in PBS (◯) or pH 12 buffer (■) was exposed to a fibril-coated SiO2 sensor. Results are representative of two independent experiments. Vertical lines indicate times at which solutions were changed. (a) Mass associated. Drift in the baseline is caused by ionization of the SiO2 surface at pH 12. (b) Dissipation measurements. (c) Mass associated–dissipation plot for the period of peptide exposure.
Attachment of PG aggregates in solution to immobilized PG
We next examined the ability of immobilized aggregates to recruit peptide from a solution containing PG aggregates. Unfiltered aggregated PG stock solution was diluted to 20 μM in PBS and exposed to the sensor in two consecutive exposures (Fig. 9). Flow was returned to PBS between each exposure. Much larger increases in associated mass (Δmmax=750–800 ng cm−2) were observed when preaggregated solutions were exposed to the fibril-coated surface compared to monomer under the same conditions (pH 7.4 PBS). The size of the increase was similar for both exposures, again supporting the notion that elongation is nonsaturable under the experimental conditions. No dissociation was observed upon rinsing with PBS for either exposure, indicating that attachment was irreversible. Both exposures also caused noticeable increases in dissipation (Fig. 9). The ΔD versus Δm plot (Fig. 9b) was nearly linear, with a slope (0.012×10−6 cm2 ng−1) about 50% larger than that observed for PG monomer addition.
Fig. 9.

Extension of immobilized PG fibrils by aggregates in solution. Vertical lines indicate times at which solutions were changed. Unfiltered PG aggregates were diluted into PBS to a concentration of 20 μM and exposed to a PG fibril-coated SiO2 sensor. Results are representative of two independent experiments. (a) Mass associated and dissipation measurements (secondary y-axis). (b) Mass associated–dissipation plot.
Comparison of mass addition by QCM and OWLS
The mass sensed by QCM-D includes not only that of the peptide but also the water molecules bound to the peptide or trapped in the adsorbed layer. To determine the amount of water associated with polyQ fibrils during elongation, we examined elongation of PG fibrils by Q20 and PG by OWLS.46,50 We use ΔmOWLS to distinguish mass change measured by OWLS from those measured by QCM-D. Fibrils were immobilized to the SiO2-coated OWLS waveguide and blocked with PLL as described for the QCM-D experiments. Immobilization of PG fibrils caused an increase in areal mass density, which asymptotically approached 130 ng cm−2 (data not shown), suggesting that 90 ng cm−2 of bound and/or coupled water is included in the QCM-D measurement of 220 ng cm−2 (Fig. 3a; i.e., the mass sensed by QCM-D was ~40% water). The immobilized PG fibrils were then exposed to PG or Q20 (20 μM, pH 7.4) as before. For comparison, QCM-D data are reproduced in Fig. 10a. The mass of PG attached, ΔmOWLS, increased to 81 ng cm−2; rinsing with buffer reduced the value to 65 ng cm−2′ (Fig. 10b). These results are similar to Δm obtained using QCM-D, indicating that little water was associated with the PG monomer as it elongates the fibrils. For Q20, an initial attachment of around 15 ng cm−2 occurred in the first minute of exposure. This initial attachment is similar for both OWLS and QCM-D. Subsequently, the rate of mass increase as measured by OWLS slowed significantly, with an areal mass density of only 20 ng cm−2 reached at the end of the exposure. Rinsing with buffer caused a decrease to 14 ng cm−2. These values are markedly lower than those obtained using QCM-D, indicating that a substantial amount of water is associated with the Q20 monomer during fibril elongation. The lower rate of mass addition sensed by OWLS corresponds closely to the decrease in the slope of the dissipation trace obtained by QCM-D (Fig. 6b). This strongly suggests a correlation between the change in the rate of elongation and in dissipation.
Fig. 10.
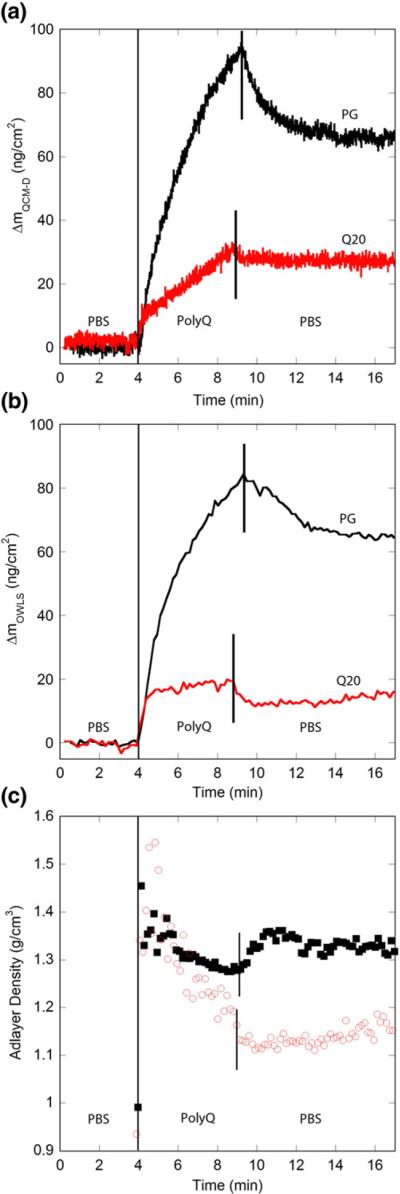
Comparison of elongation kinetics as measured by OWLS and QCM-D. Solutions (20 μM) of monomeric Q20 or PG in PBS were exposed to a PG fibril-coated SiO2 sensor. Vertical lines indicate times at which solutions were changed. (a) QCM-D data reproduced from Figs. 4 and 6. (b) OWLS data from PG and Q20. (c) Calculated density of the adlayer for PG (■) and Q20 (◯). Data from before the exposure of polyQ peptide have been removed for clarity. Results are representative of two independent experiments.
We calculated the average density of the adsorbed layer from the combined OWLS and QCM-D measurements using the method of Vörös46 (Fig.10c). After passing through small maxima, the adlayer densities for Q20 and PG diverge, with that for Q20 dropping while that for PG remaining high. This result reinforces the conclusion that elongation of Q20 is accompanied by a significant incorporation of water whereas PG elongation is relatively water free.
Discussion
In this study, we focused specifically on polyQ fibril elongation and considered two possible pathways by which monomer could add to a growing fibril (Fig. 1). We chose QCM-D because the technique allows simultaneous measurement of the rate of elongation and the dissipative properties of the elongating aggregate. Because we measured growth from immobilized fibrils, fibril elongation is measured in isolation from fibril initiation. The mass detected in QCM-D includes water coupled to the oscillations of the sensor (both water bound to the peptide and that present between fibrils in the layer). As a complementary measure of growth rate, we used OWLS, a solvent-blind technique allowing measurement of dry mass adsorbed. The combination of both techniques allows determination of the mass of water associated with the adlayer. To our knowledge, this is the first application of QCM-D and/or OWLS to the study of polyQ-mediated aggregation.
In interpreting the mass change measurements, we attributed all of the added mass to elongation. Mass could accumulate nonspecifically on the sides rather than on the tip of the growing fibril. We think this is unlikely because (a) in solution, growth is predominantly at the tip, and (b) the elongation data collected in this study closely match our previous data on the effect of polyQ length and conformation on fibril growth.20,32
Comparison of rates and reversibility of elongation
Although Q8 associates weakly with immobilized PG fibrils, much of the associated material readily dissociates upon rinsing (Fig. 6a). We conclude that short (Q<9) polyQ peptides are essentially incapable of elongating fibrils (Fig. 6a). Our results are consistent with an atomic force microscopy study, where Legleiter et al. detected some oligomerization but only minor fibrillization of a peptide containing seven glutamines.51 In contrast, longer polyQ peptides (Q20 and Q24) readily added to the immobilized PG fibrils. These experiments confirm previous studies showing that interactions between peptides with different glutamine repeat domains are possible.18,32 Only about 20% of the newly associated Q20 or Q24 dissociated during rinsing, indicating that elongation is partially, but not completely, irreversible with the longer polyQ. Taken together, these data suggest that all three peptides associate with preformed fibrils, but only the longer ones (Q20 and Q24) are able to partially consolidate to the fibrillar template. Partial reversibility is taken as evidence that full consolidation (“locking”) has not taken place in the time frame of the experiment.
Both PG and PP peptides contain 20 glutamines, like Q20, but the glutamines are arranged in two Q10 blocks interrupted by two amino acid residues that influence the conformation of the monomer: PG contains a β-turn template while the two prolines of PP enforce local stiffness. Elongation rates for PG were two to three times faster than for Q20 and slightly faster than for Q24 (Table 3). Previously, we observed that the kinetics of formation of insoluble fibrils in solution was ~500-fold faster for PG than for Q20.20,32 Why does the β-turn template impose a much larger increase in rate of sedimentation than in rate of elongation? (We define sedimentation rate here as the rate of formation of insoluble aggregated materials that can be centrifuged to form a precipitate.) We explain this observation by noting that elongation requires simply repeated association with the fibril tip, whereas sedimentation requires conformational rearrangement to an insoluble structure. We hypothesize that PG, with its “prewired” propensity to adopt a β-turn, enjoys a modest enhancement in the rate of association with a fibrillar template but a substantial enhancement in the rate of adoption of the fully fibrillar (and insoluble) structure. This explanation is consistent with our previous observations of large soluble oligomers that grew continuously in size in solutions of Q20 or Q24 for several days before sedimentation was detected.20 In contrast, small soluble oligomers of PG were present for only a short window of time prior to the onset of insolubility.32 It is interesting to note that association of PG with immobilized fibrils was partially reversible (Table 2 and Fig. 4a), indicating that full consolidation to the fibrillar structure is not immediate despite the β-turn template at neutral pH.
The rate of PP association with immobilized fibrils was slower than that of Q20, and, as with Q8, much of the PP dissociated with rinsing. These data demonstrate that PP can associate with PG, presumably via glutamine–glutamine interactions, but its local rigidity prevents adoption of a β-turn/β-sheet conformation. This conclusion is consistent with our previous observations that PP self-associates into large soluble non-fibrillar aggregates and that even in the presence of PG, PP does not form sedimentable aggregates.32 Thus, the ability to adopt or conform to the β-sheet fibrillar template is critical for elongation.
Bhattacharyya et al. used biotin-labeled peptide to estimate a solution-phase elongation rate constant for polyQ of ~11,400 L mol−1 s−1,25 which, based on their estimates of fibril size, we can convert to a deposition rate of 5 ng cm−2 min−1, a similar order of magnitude as our measurements. They report that the elongation rate is largely independent of polyQ length and that therefore most length dependence of aggregation is attributable to increased favorability of forming the nucleus with increasing polyQ length.14,25 This differs from our finding of a strong dependence of elongation rate on polyQ length. This could be attributed to the fact that they used longer peptides or to the difference in measurement method.
Dissipation rates, and an estimate of the binding sites on immobilized fibrils
In general, increases in dissipation paralleled increases in mass; this is apparent in the linearity of ΔD versus Δm plots (e.g., Figs. 4c and 5c). A few cases that do not conform to this pattern are worth further discussion. With Q20 in PBS, after the first minute, dissipation plateaus, whereas mass continues to increase (Fig. 6a and b). A kink in the mass addition rate is apparent at the point where dissipation levels off. This becomes even clearer when OWLS data (Fig. 10b) are compared to dissipation data (Fig. 6b). Comparison of QCM-D and OWLS data shows that a substantial amount of water is associated with Q20 during the second phase of addition. A very similar pattern is observed with PP (Fig. 8): again, a kink in the mass addition rate corresponds closely in time to the point at which dissipation stops changing. For both PP and Q20, as measured by QCM-D, Δm ~15 ng cm−2 at the kink. We hypothesize that this pattern of association indicates rapid initial binding of Q20 or PP to the exposed PG fibril tip, up to the point of saturation of the tips. In other words, the PG fibril tips accommodate 15 ng cm−2 of peptide in the first round of monomer addition. Although attached, Q20 and PP do not readily consolidate to the fibrillar structure, since Q20 is a weak fibril former and PP is essentially a non-fibril former. Once the first round of monomer addition to the PG fibril tips is complete, the nature of the exposed tip changes, and further association is much slower and less stable.
Aggregate characterization via dissipation measurements
In PBSA, we observed differences in ΔD/Δm when the immobilized fibrils were exposed to different peptides (Table 3). We hypothesize that these differences in ΔD/Δm, while small, indicate subtle differences in the structure of elongating aggregates. If so, this would provide support for the elongation mechanism depicted in Fig. 1b, because consolidation to a fibrillar structure is not required for continued addition of monomer. Specifically, ΔD/Δm increased in the order Q20<Q24<PG<PG aggregates (Table 3). We propose that the increasing ΔD/Δm in this series is generally indicative of longer aggregates that protrude further into the solution,44 due to increasing β-sheet character in the elongating aggregate. ΔD/Δm for PP is quite high compared to PG. Since PP is known not to form fibrils, one possible explanation is that PP attaches very loosely to the immobilized fibrils and the elongating aggregate is very swollen and flexible compared to other peptides.45
Other studies of fibril elongation have also benefited from insights into the mechanical properties of fibrils made possible by QCM-D dissipation measurements. Saraiva et al. determined that Aβ oligomers are softer than Aβ monomers: ΔD/Δm ~ 0.006 × 10− 6 cm2 ng−1 for Aβ(1–40) and 0.017×10− 6 cm2 ng−1 for the more aggregation-prone Aβ(1–42).40 These ΔD/Δm values are similar to those observed here. Hovgaard et al. found that large increases in dissipation coincided with the onset of glucagon fibril formation and the growth of fibrils,41 but differences in the experimental approach make direct numerical comparison to values observed here unfeasible.
Importance of flanking lysine residues
We previously showed that neutralizing the charge of flanking lysines affected aggregation in a length-dependent manner.20 Specifically, Q8 aggregation was not changed much by a shift from pH 7 to pH 12, Q20 aggregation rate increased modestly, but Q24 monomers became significantly more compact and associated into insoluble aggregates much faster.20 Consistent with these earlier results, neutralization of lysine charges did not stabilize Q8 association with immobilized PG fibrils. A similar result was obtained with PP in that charge neutralization modestly increased the amount of added mass, but association remained readily reversible. Thus, in cases where fibril formation is weak or nonexistent at neutral pH, either because the Q domain is too short or because the monomer conformation does not support fibrillogenesis, fibril elongation cannot be forced by neutralizing charges.
For Q20, Q24, and PG, in contrast, the increase in pH increased the elongation rate by three- to fourfold, and the fraction of peptide removed upon rinsing decreased, especially for Q24 and PG. Furthermore, the mass added increased linearly with time at pH 12, in contrast to the slowing rate of growth at pH 7.4. Thus, neutralization of flanking repulsive charges leads to more rapid and efficient consolidation of the monomer to the growing aggregate. It is interesting to note that with Q20, dissipation leveled off at pH 7.4 but not at pH 12. We interpret these data to indicate that Q20 is borderline for fibril growth and that neutralization of the lysine charges is needed to “push” Q20 into consolidating into fibrils.
Doubling the salt concentration doubled the elongation rate and reduced reversibility to near zero for PG, similar to the effects of increased pH and suggesting more efficient consolidation of monomer to the growing fibril, due to screening of repulsive lysine–lysine charges, increased contribution of hydrophobic interactions, or both. At moderately high ionic strength (0.32 M), ΔD/Δm increased, likely indicative of fibrils that are more extended from the SiO2 surface. A further increase in salt concentration led to a decrease in both elongation rate and ΔD/Δm; the latter may indicate a collapse of fibrils closer to the surface. Interestingly, during purification of huntingtin aggregates from brain homogenate, Diaz-Hernandez et al. noted that addition of 0.8 M NaCl resulted in the isolation of smaller aggregates than observed at a physiological NaCl concentration.52 This result suggests that high salt concentrations actually assist in disaggregating huntingtin plaques, and perhaps similar effects are evident here.
The role of water
Comparison of OWLS and QCM-D data reveals the different role of associated water in fibril elongation for weak (Q20) and strong (PG) fibril formers (Fig. 10). Upon initial exposure, Q20 binds rapidly until a coverage of ~15 ng cm−2 is reached, as measured by both OWLS and QCM-D. This step represents initial association of Q20 with the immobilized PG fibrils. Since both OWLS and QCM-D measure the same value for this initial binding period, these data indicate that either very little water is associated with the monomer initially attaching to the fibril or any water associated with Q20 displaces an equal mass of water as the peptide binds. The elongation rates for Q20 and PG during this stage were also very similar. This suggests that the initial binding event was dominated by the nature of the binding site (the fibril tip) and not the binding moiety (the PG or Q20 monomer). The subsequent large discrepancy between QCM and OWLS measurements for Q20 indicates that a substantial amount of water is bound to and/or trapped by Q20. The drop in adlayer density to ~1.15 g cm−3 (Fig. 10c) is a direct measure of the relatively high water content of the adsorbed Q20, and there is no further change once exposure of the surface to the peptide is stopped. With PG addition, there is a slight decrease in adlayer density over time that suggests a small amount of water is incorporated into the growing aggregate. The rate of water incorporation is much lower for PG than for Q20. Once the solution is switched to buffer, adlayer density rebounds. We speculate that the increase in adlayer density is due to expulsion of water from the remaining PG as the PG aggregate undergoes conformational conversion and maturation, as illustrated in Fig. 1b. In other words, the combination of QCM-D and OWLS may provide evidence of conformational conversion rates within the fibril. If so, this would represent an exciting result, although considerably more work would be required to confirm this provisional interpretation. PG reaches an equilibrium adlayer density of ~1.3 g cm−3, evidence that the mature PG aggregate is remarkably dry.
The predisposition for β-turn likely increases the number of intramolecular hydrogen bonds (both main chain and side chain) in PG, compared to Q20, which maintains a larger number of peptide–water hydrogen bonds. If PG, because of its β-turn template, partially or fully consolidates to the growing fibril structure whereas Q20 does not, the nature of the tip of the aggregate changes after the first round of addition. We hypothesize that the rate of addition slows only slightly if the tip is covered with PG but drops dramatically if the tip is covered with Q20. Consistent with our experimental results, recent simulations of the role of water in protein aggregation concluded that water associated with hydrophilic groups slows fibril growth rates, specifically for amide groups of polar side chains.53
Conclusions
We demonstrated the utility of QCM-D and OWLS for gaining insight into mechanisms of polyQ-containing peptide elongation. Differences in elongation rates as measured by these two techniques demonstrate the role of water and illustrate that mass measurements from QCM alone tend to overestimate the amount of bound peptide/protein. We proposed two alternative mechanisms for fibril elongation by monomer addition. In the “dock-lock” mechanism, one round of associated monomer must fully adopt a fibrillar conformation before a second round of addition (Fig. 1a). Alternatively, monomers could continue to add to the elongating aggregate without necessarily requiring conformational consolidation prior to further growth (Fig. 1b). The variation of ΔD/Δm with peptide structure indicates that the dissipative properties of the elongating aggregate depend on the conformation of the associating monomer; this result is consistent with the schematic of Fig. 1b. The mechanism of elongation may be biologically relevant, as evidence exists supporting the hypothesis that mature fibrils are more biologically inert than aggregates that are actively forming.54–56
Materials and Methods
Peptide synthesis and purification
All materials were from Fisher Scientific (Pittsburgh, PA) except where indicated. Peptides were synthesized using standard Fmoc solid-phase methods on a Protein Technologies Inc. Symphony synthesizer. Peptides of the type K2WQxAK2 were synthesized, where x equals 8, 20, or 24. Two additional Q20 peptides were synthesized, with either a Pro-Pro insert or the β-turn template D-Pro-Gly inserted in the center of the glutamine region. Alanine, glycine, proline, D-proline, glutamines with a trityl side-chain protecting group, and lysines and tryptophan with a Boc side-chain protecting group were purchased from Novabiochem (Gibbstown, NJ). Peptides were synthesized and purified as described previously.20 Flanking lysine residues were added to the polyQ core to increase solubility, consistent with previous work,14,20,32 and tryptophan was used for concentration determination.20,32 The N-terminus was acetylated and the C-terminus was amidated to eliminate charge interactions of the termini. Peptide identity was confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, which yielded molecular masses of 1854.0 Da (Q8, 1854.2 Da theoretical), 3390.7 Da (Q20, 3391.8 Da theoretical), 3903.9 Da (Q24, 3904.3 Da theoretical), 3586.9 Da (PP, 3585.9 Da theoretical), and 3545.8 Da (PG, 3545.8 Da theoretical).
Sample preparation
Lyophilized peptides were disaggregated using a protocol similar to that developed by others.57 Briefly, peptide was incubated overnight in a 1:1 solution of TFA and hexafluoroisopropanol. The solvent was evaporated under a gentle flow of N2, and the peptides were redissolved in water adjusted to pH 3 with TFA. Peptide stock solutions were filtered through a 0. 45-μm polyvinylidene fluoride filter, aliquoted, snap frozen in dry ice/ethanol, and stored at −80 °C. Concentrations of stock solutions (typically ~200 μM) were determined by tryptophan fluorescence with excitation at 295 nm and emission at 350 nm.20 Prior to each experiment, a vial was thawed and centrifuged at 19,500 rcf for 30 min and then the supernatant (top 75%) was removed and filtered through a 0. 22-μm polyvinylidene fluoride filter.
Aggregate preparation
PolyQ aggregates were formed via freeze–thaw cycles of PG peptide stock solution. Frozen aggregate stocks were thawed and used for QCM-D experiments without further preparation. Images of aggregates were obtained using a Philips CM120 scanning transmission electron microscope. A drop of stock solution was applied to a pioloform-coated grid and stained with methylamine tungstate stain before imaging.
QCM-D flow cell cleaning
The QCM-D flow cell was cleaned before use each day using the following procedure: A gold-coated AT-cut quartz sensor crystal (QSX 301, Q-Sense, Gothenburg, Sweden) was mounted in the flow chamber and the chamber was rinsed with Milli-Q water for 5 min at a flow rate of 1 mL min−1. The chamber was then cleaned with a 2% solution of LpH (Steris, Mentor, OH) for 5 min (flow rate, 100 μL min−1), rinsed again with water for 10 min (flow rate, 1 mL/min), cleaned with a 2% solution of Hellmanex II for 5 min (flow rate, 100 μL min−1), a 2% solution of SDS (flow rate, 100 μL min−1), and rinsed with water for 15 min (flow rate, 1 mL min−1). The gold sensor crystal was removed and the chamber was dried under nitrogen before use.
QCM-D sensor preparation
SiO2-coated sensor crystals (50 nm) (QSX 303, Q-Sense) were cleaned before each use as follows: sensors were rinsed with water, then soaked in a 2% solution of LpH for 5 min, and rinsed with water again. Sensors were sonicated in a 2% SDS solution for 20 min, rinsed with water, ethanol, and water again before being dried under nitrogen. Immediately prior to use, the sensors were cleaned in a UV/Ozone ProCleaner (BioForce Nanosciences, Ames, IA) for 20 min.
QCM-D immobilization of aggregates
A flow rate of 100 μL min−1 was maintained for all experiments. The temperature was maintained at 37.0 °C (±0.05 °C) unless otherwise noted. All buffer solutions were degassed by sonication for 20 min prior to use. For each measurement, a clean SiO2-coated sensor crystal was mounted in the flow cell chamber of a Q-Sense E1 system (Q-Sense). Water was pumped over the sensor crystal surface until a stable baseline was established (|Δf/Δt|<0.1 Hz min−1), at which time resonant frequencies were found and data collection began. Frequency and dissipation data were collected for odd-numbered overtones 1 through 13. After 3 min of collecting baseline data in water, polyQ aggregates at a concentration of 10 μM in water adjusted to pH 3 with TFA were pumped through the flow cell for 5 min. The flow was then switched to a Hepes buffer (0.01 M, pH 7.0) for 3 min and then changed to a solution of PLL hydrobromide (molecular mass, 300 kDa; Sigma, St. Louis, MO) at a concentration of 10 μg mL−1 in Hepes buffer for 4 min to block any unoccupied sites on the SiO2 surface. Performing immobilization and blocking steps online allowed the process to be monitored and minimized variability between runs. All sensor surfaces were prepared in this manner before exposure to the buffer and peptide of interest.
Detection of aggregate elongation by QCM-D
After the aggregate immobilization and blocking steps, the flow was changed to the buffer of interest for 5 min during which time the PG fibril adlayer equilibrated with the buffer. Next, the flow was changed to the peptide solution in the same buffer for 5 min and then returned to buffer (without peptide) for 10 min. Buffers used included PBSA (10 mM Na2HPO4/NaH2PO4, 140 mM NaCl, and 3 mM sodium azide, pH 7.4), 10 mM phosphate buffers with varying amount of NaCl (0 M, 0.29 M, and 0.49 M NaCl, pH 7.4), and 10 mM sodium phosphate adjusted to pH 12 with NaOH. Peptides were diluted in buffer to a concentration of 20 μM unless otherwise noted, 1 min before use in QCM-D experiments. In all cases, the amount of monomer that associated with the fibril-coated surface was small enough that the concentration of monomer in bulk solution can be considered constant.
Changes in bulk solution density and viscosity impact the resonant frequency of the SiO2 sensor. Control surfaces consisting of bare SiO2 and PLL-coated surfaces were exposed to the solvent changes used in each experiment. Detectable PLL and polyQ fibrils did not dissociate from the surface under any of the experimental conditions employed. At pH 12, ionization of the SiO2 surface resulted in a steady, small drift in the baseline. This decrease was detectable on bare SiO2 surfaces as well as fibril-coated surfaces and was the same for both, indicating that ionization of SiO2 does not force dissociation of immobilized fibrils from the surface.
Analysis of QCM-D data
The Sauerbrey relationship34 applies to rigid thin films and was used to convert measured changes in the oscillation (or overtone) frequency (Δfn) to the change in mass associated with the sensor surface (Δm):
| (1) |
where n is the overtone number and C is the crystal constant (17.7 ng Hz−1 cm−2 for the 4.95-MHz quartz crystal). Use of the Sauerbrey equation is appropriate if ΔDn/(Δfn/n)<4×10−7 Hz−1.35 All reported masses and dissipation measurements reflect the fifth (~25 MHz) overtone.
Detection of aggregate elongation by OWLS
OWLS measurements were performed on an OWLS 210 system (MicroVacuum, Budapest, Hungary) containing a single sensor waveguide mounted in a laminar slit shear flow cell and employing a He–Ne laser (λ=632.8 nm). Prior to each measurement, the OWLS flow cell and SiO2-coated waveguides were cleaned following the manufacturer's recommended protocol. Aggregates of PG were immobilized and any unoccupied sites on the waveguide were blocked using PLL as described above for the QCM-D experiments. No increase in mass was detected upon exposure to PLL, suggesting that all of the polymer-accessible attachment sites were occupied by PG aggregates. Upon switching from Hepes buffer to PBS, significant drift in the baseline occurred, which was allowed to stabilize for 15 min instead of the 5 min used in the QCM-D experiments. Monomeric 20 μM PG or Q20 in PBS was exposed to the waveguide surface at a flow rate of 100 μL min−1 and a temperature of 37 °C (±0.1 °C). Measurements of the transverse electric and transverse magnetic modes were converted, using the BioSense software package (MicroVacuum), to surface mass density (ng cm−2) using de Feijter's equation:58–61
| (2) |
where ΔmOWLS is the surface mass density (g cm−2); dA is the thickness of the adlayer (cm); nR,A and nR,C are the refractive indices of the adlayer molecules and background solution, respectively; and dnR,A/dc is the refractive index increment of the adlayer molecules (g cm−3), taken to be 0.182 for all measurements.61
Calculation of adsorbed layer density
Under the flow conditions employed for both QCM-D and OWLS, with comparable wall shear rates and negligible depletion of bulk peptide, the density of the proteinaceous layer, ρlayer, can be calculated as:46
| (3) |
where the density of dry protein ρprotein was taken to be 1.33 g cm−3 and the density of the solvent ρsolvent=1.00 g cm−3. In Eq. (3), Δm is the mass change measured by QCM-D and ΔmOWLS is the mass change measured by OWLS.
Acknowledgements
This work was supported by grant CBET-085 2278 from the National Science Foundation and grant 5T32 GM-08349 (R.H.W. and K.H.J.) from the National Institutes of Health. We gratefully acknowledge the technical assistance of Dr. Gary Case and the UW Biotechnology Center mass spectrometry staff. Use of the OWLS instrument was graciously provided by Dr. Nita Sahai, Department of Geology, University of Wisconsin-Madison.
Abbreviations used
- Aβ
beta-amyloid
- PBS(A)
phosphate-buffered saline (with azide)
- QCM-D
quartz crystal microbalance with dissipation monitoring
- OWLS
optical waveguide lightmode spectroscopy
- PLL
poly-l-lysine
- polyQ
polyglutamine
- TFA
trifluoroacetic acid
References
- 1.Bates G. Huntingtin aggregation and toxicity in Huntington's disease. Lancet. 2003;361:1642–1644. doi: 10.1016/S0140-6736(03)13304-1. [DOI] [PubMed] [Google Scholar]
- 2.Orr HT. Beyond the Qs in the polyglutamine diseases. Genes Dev. 2001;15:925–932. doi: 10.1101/gad.888401. [DOI] [PubMed] [Google Scholar]
- 3.Wanker EE. Protein aggregation and pathogenesis of Huntington's disease: mechanisms and correlations. Biol. Chem. 2000;381:937–942. doi: 10.1515/BC.2000.114. [DOI] [PubMed] [Google Scholar]
- 4.Albrecht M, Golatta M, Wullner U, Lengauer T. Structural and functional analysis of ataxin-2 and ataxin-3. Eur. J. Biochem. 2004;271:3155–3170. doi: 10.1111/j.1432-1033.2004.04245.x. [DOI] [PubMed] [Google Scholar]
- 5.Gusella JF, MacDonald ME. Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 2000;1:109–115. doi: 10.1038/35039051. [DOI] [PubMed] [Google Scholar]
- 6.Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31:521–528. doi: 10.1016/j.tins.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ordway JM, Tallaksen-Greene S, Gutekunst CA, Bernstein EM, Cearley JA, Wiener HW, et al. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell. 1997;91:753–763. doi: 10.1016/s0092-8674(00)80464-x. [DOI] [PubMed] [Google Scholar]
- 8.Schiffer NW, Broadley SA, Hirschberger T, Tavan P, Kretzschmar HA, Giese A, et al. Identification of anti-prion compounds as efficient inhibitors of polyglutamine protein aggregation in a zebrafish model. J. Biol. Chem. 2007;282:9195–9203. doi: 10.1074/jbc.M607865200. [DOI] [PubMed] [Google Scholar]
- 9.Jana NR, Tanaka M, Wang GH, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- 10.Carmichael J, Chatellier J, Woolfson A, Milstein C, Fersht AR, Rubinsztein DC. Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington's disease. Proc. Natl Acad. Sci. USA. 2000;97:9701–9705. doi: 10.1073/pnas.170280697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 12.Nagai Y, Inui T, Popiel HA, Fujikake N, Hasegawa K, Urade Y, et al. A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol. 2007;14:332–340. doi: 10.1038/nsmb1215. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 2008;17:345–356. doi: 10.1093/hmg/ddm311. [DOI] [PubMed] [Google Scholar]
- 14.Chen SM, Ferrone FA, Wetzel R. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl Acad. Sci. USA. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum. Mol. Genet. 2002;11:2905–2917. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 16.Chen S, Berthelier V, Yang W, Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J. Mol. Biol. 2001;311:173–182. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 17.Chen SM, Berthelier V, Hamilton JB, O'Nuallain B, Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 18.Slepko N, Bhattacharyya AM, Jackson GR, Steffan JS, Marsh JL, Thompson LM, Wetzel R. Normal-repeat-length polyglutamine peptides accelerate aggregation nucleation and cytotoxicity of expanded polyglutamine proteins. Proc. Natl Acad. Sci. USA. 2006;103:14367–14372. doi: 10.1073/pnas.0602348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee CC, Walters RH, Murphy RM. Reconsidering the mechanism of polyglutamine peptide aggregation. Biochemistry. 2007;46:12810–12820. doi: 10.1021/bi700806c. [DOI] [PubMed] [Google Scholar]
- 20.Walters RH, Murphy RM. Examining polyglutamine peptide length: a connection between collapsed conformations and increased aggregation. J. Mol. Biol. 2009;393:978–992. doi: 10.1016/j.jmb.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers—their possible role in inherited neurodegenerative diseases. Proc. Natl Acad. Sci. USA. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc. Natl Acad. Sci. USA. 2002;99:5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raspe M, Gillis J, Krol H, Krom S, Bosch K, Veen H, Reits E. Mimicking proteasomal release of polyglutamine peptides initiates aggregation and toxicity. J. Cell Sci. 2009;122:3262–3271. doi: 10.1242/jcs.045567. [DOI] [PubMed] [Google Scholar]
- 24.Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat. Struct. Mol. Biol. 2011;18:328. doi: 10.1038/nsmb.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhattacharyya AM, Thakur AK, Wetzel R. Polyglutamine aggregation nucleation: thermodynamics of a highly unfavorable protein folding reaction. Proc. Natl Acad. Sci. USA. 2005;102:15400–15405. doi: 10.1073/pnas.0501651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernacki JP, Murphy RM. Model discrimination and mechanistic interpretation of kinetic data in protein aggregation studies. Biophys. J. 2009;96:2871–2887. doi: 10.1016/j.bpj.2008.12.3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vitalis A, Pappu RV. Assessing the contribution of heterogeneous distributions of oligomers to aggregation mechanisms of polyglutamine peptides. Biophys. Chem. 2011;159:14–23. doi: 10.1016/j.bpc.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vitalis A, Wang X, Pappu RV. Atomistic simulations of the effects of polyglutamine chain length and solvent quality on conformational equilibria and spontaneous homodimerization. J. Mol. Biol. 2008;384:279–297. doi: 10.1016/j.jmb.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vitalis A, Lyle N, Pappu RV. Thermodynamics of β-sheet formation in polyglutamine. Biophys. J. 2009;97:303–311. doi: 10.1016/j.bpj.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheibel T, Bloom J, Lindquist S. The elongation of yeast prion fibers involves separable steps of association and conversion. Proc. Natl Acad. Sci. USA. 2004;101:2287–2292. doi: 10.1073/pnas.0308754101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williamson TE, Vitalis A, Crick SL, Pappu RV. Modulation of polyglutamine conformations and dimer formation by the N-terminus of huntingtin. J. Mol. Biol. 2010;396:1295–1309. doi: 10.1016/j.jmb.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walters RH, Murphy RM. Aggregation kinetics of interrupted polyglutamine peptides. J. Mol. Biol. 2011;412:505–519. doi: 10.1016/j.jmb.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodahl M, Höök F, Krozer A, Brezezinski P, Kasemo B. Quartz-crystal microbalance setup for frequency and Q-factor measurements in gaseous and liquid environments. Rev. Sci. Instrum. 1995;66:3924–3930. [Google Scholar]
- 34.Sauerbrey G. Verwendung von Schwingquarzen zur Wägung dünner Schichten und der Mikrowägung. Z. Phys. 1959;155:206–222. [Google Scholar]
- 35.Reviakine I, Johannsmann D, Richter RP. Hearing what you can't see and visualizing what you hear: interpreting quartz crystal microbalance data from solvated interfaces. Anal. Chem. 2011;83:8838–8848. doi: 10.1021/ac201778h. [DOI] [PubMed] [Google Scholar]
- 36.Rodahl M, Höök F, Fredriksson C, Keller C, Krozer A, Brzezinski P, et al. Simultaneous frequency and dissipation factor QCM measurements of biomolecular adsorption and cell adhesion. Faraday Discuss. 1997;107:229–246. doi: 10.1039/a703137h. [DOI] [PubMed] [Google Scholar]
- 37.Rodahl M, Höök F, Kasemo B. QCM operation in liquids: an explanation of measured variations in frequency and Q factor with liquid conductivity. Anal. Chem. 1996;68:2219–2227. doi: 10.1021/ac951203m. [DOI] [PubMed] [Google Scholar]
- 38.Kotarek JA, Johnson KC, Moss MA. Quartz crystal microbalance analysis of growth kinetics for aggregation intermediates of the amyloid-beta protein. Anal. Biochem. 2008;378:15–24. doi: 10.1016/j.ab.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 39.Kotarek JA, Moss MA. Impact of phospholipid bilayer saturation on amyloid-beta protein aggregation intermediate growth: a quartz crystal microbalance analysis. Anal. Biochem. 2010;399:30–38. doi: 10.1016/j.ab.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 40.Saraiva AM, Pereira MC, Brezesinski G. Is the viscoelasticity of Alzheimer's A beta 42 peptide oligomers a general property of protein oligomers related to their toxicity? Langmuir. 2010;26:12060–12067. doi: 10.1021/la101203h. [DOI] [PubMed] [Google Scholar]
- 41.Hovgaard MB, Dong M, Otzen DE, Besenbacher F. Quartz crystal microbalance studies of multilayer glucagon fibrillation at the solid-liquid interface. Biophys. J. 2007;93:2162–2169. doi: 10.1529/biophysj.107.109686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White DA, Buell AK, Knowles TPJ, Welland ME, Dobson CM. Protein aggregation in crowded environments. J. Am. Chem. Soc. 2010;132:5170–5175. doi: 10.1021/ja909997e. [DOI] [PubMed] [Google Scholar]
- 43.Tellechea E, Johannsmann D, Steinmetz NF, Richter RP, Reviakine I. Model-independent analysis of QCM data on colloidal particle adsorption. Langmuir. 2009;25:5177–5184. doi: 10.1021/la803912p. [DOI] [PubMed] [Google Scholar]
- 44.Tsortos A, Papadakis G, Gizeli E. Shear acoustic wave biosensor for detecting DNA intrinsic viscosity and conformation: a study with QCM-D. Biosens. Bioelectron. 2008;24:836–841. doi: 10.1016/j.bios.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 45.Zhang G, Wu C. Quartz crystal microbalance studies on conformational change of polymer chains at interface. Macromol. Rapid Commun. 2009;30:328–335. doi: 10.1002/marc.200800611. [DOI] [PubMed] [Google Scholar]
- 46.Vörös J. The density and refractive index of adsorbing protein layers. Biophys. J. 2004;87:553–561. doi: 10.1529/biophysj.103.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vörös J, Ramsden JJ, Csucs G, Szendro I, De Paul SM, Textor M, Spencer ND. Optical grating coupler biosensors. Biomaterials. 2002;23:3699–3710. doi: 10.1016/s0142-9612(02)00103-5. [DOI] [PubMed] [Google Scholar]
- 48.Lofas S, Johnsson B. A novel hydrogel matrix on gold surfaces in surface-plasmon resonance sensors for fast and efficient covalent immobilization of ligands. J. Chem. Soc.: Chem. Commun. 1990:1526–1528. [Google Scholar]
- 49.Mao AH, Crick SL, Vitalis A, Chicoine CL, Pappu RV. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl Acad. Sci. USA. 2010;107:8183–8188. doi: 10.1073/pnas.0911107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Höök F, Vörös J, Rodahl M, Kurrat R, Boni P, Ramsden J, et al. A comparative study of protein adsorption on titanium oxide surfaces using in situ ellipsometry, optical waveguide lightmode spectroscopy, and quartz crystal microbalance/dissipation RID A-5843-2008. Colloids Surf., B. 2002;24:155–170. [Google Scholar]
- 51.Legleiter J, Mitchell E, Lotz GP, Sapp E, Ng C, DiFiglia M, et al. Mutant huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 2010;285:14777–14790. doi: 10.1074/jbc.M109.093708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diaz-Hernandez M, Moreno-Herrero F, Gomez-Ramos P, Moran MA, Ferrer I, Baro AM, et al. Biochemical, ultrastructural, and reversibility studies on huntingtin filaments isolated from mouse and human brain. J. Neurosci. 2004;24:9361–9371. doi: 10.1523/JNEUROSCI.2365-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thirumalai D, Govardhan R, Straub JE. Role of water in protein aggregation and amyloid polymorphism. Acc. Chem. Res. 2012;45:83–92. doi: 10.1021/ar2000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- 55.Diguet E, Petit F, Escartin C, Cambon K, Bizat N, Dufour N, et al. Normal aging modulates the neurotoxicity of mutant huntingtin. PLoS One. 2009;4:e4637. doi: 10.1371/journal.pone.0004637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 57.Chen SM, Wetzel R. Solubilization and disaggregation of polyglutamine peptides. Protein Sci. 2001;10:887–891. doi: 10.1110/ps.42301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurrat R, Ramsden JJ, Prenosil JE. Kinetic-model for serum-albumin adsorption—experimental verification. J. Chem. Soc., Faraday Trans. 1994;90:587–590. [Google Scholar]
- 59.Kurrat R, Textor M, Ramsden JJ, Boni P, Spencer ND. Instrumental improvements in optical waveguide light mode spectroscopy for the study of biomolecule adsorption. Rev. Sci. Instrum. 1997;68:2172–2176. [Google Scholar]
- 60.Ramsden JJ. Review of new experimental-techniques for investigating random sequential adsorption. J. Stat. Phys. 1993;73:853–877. [Google Scholar]
- 61.Defeijter JA, Benjamins J, Veer FA. Ellipsometry as a tool to study adsorption behavior of synthetic and biopolymers at air–water-interface. Biopolymers. 1978;17:1759–1772. [Google Scholar]