

Summary
Peptide immunotherapy using soluble peptides containing allergen‐derived immunodominant T‐cell epitopes holds therapeutic promise for allergic asthma. Previous studies in BALB/c mice using the immunodominant peptide epitope of chicken ovalbumin (p323–339) have been unable to demonstrate therapeutic effects in ovalbumin‐induced allergic airway inflammation. We have previously shown that intravenous application of p323–339 can effectively tolerise p323–339‐reactive T cells in a non‐allergic model in C57BL/6 mice. This study aimed to assess the effects of using p323–339 immunotherapy in a C57BL/6 model of ovalbumin‐induced allergic airway inflammation, identify any additional epitopes recognized by the ovalbumin‐responsive T‐cell repertoire in C57BL/6 mice and assess the effects of combination peptide immunotherapy in this model. Ovalbumin‐reactive T‐cell lines were generated from ovalbumin‐immunized C57BL/6 mice and proliferative responses to a panel of overlapping peptides covering the ovalbumin sequence were assessed. Soluble peptides (singly or combined) were administered intravenously to C57BL/6 mice before the induction of ovalbumin‐induced allergic airway inflammation. Peptide immunotherapy using the 323–339 peptide alone did not reduce the severity of allergic airway inflammation. An additional immunodominant T‐cell epitope in ovalbumin was identified within the 263–278 sequence. Combination peptide immunotherapy, using the 323–339 and 263–278 peptides together, reduced eosinophilia in the bronchoalveolar lavage and ovalbumin‐specific IgE, with apparent reductions in interleukin‐5 and interleukin‐13. Characterization of the T‐cell response to a model allergen has allowed the development of combination peptide immunotherapy with improved efficacy in allergic airway inflammation. This model holds important potential for future mechanistic studies using peptide immunotherapy in allergy.
Keywords: allergy, asthma, epitopes, ovalbumin, peptide immunotherapy
Introduction
Allergic asthma is the commonest form of asthma, causing significant morbidity (and sometimes mortality), particularly in the paediatric population.1–3 In the absence of disease‐modifying therapy, there remains substantial unmet clinical need for improved approaches to treat allergic asthma.4 Allergen‐reactive CD4+ T cells (particularly those of the T helper type 2 subset) play a key role in the pathogenesis of allergic asthma, via the provision of help for B cells and the generation of allergen‐specific IgE antibodies, and by producing cytokines that are integral to the recruitment of inflammatory cells such as eosinophils to the lungs and for goblet cell development.5,6 Allergen‐specific CD4+ T cells therefore remain a key therapeutic target.7
Specific immunotherapy, whereby whole protein allergen is applied to induce allergen‐specific tolerance,8,9 can improve allergic disease,10,11 and may also modify its progression.12,13 However, specific immunotherapy comes with the risk of inducing severe allergic reactions, including anaphylaxis, as a consequence of pre‐existing allergen‐specific IgE binding to conformation‐dependent epitopes within the protein allergen and prompting mast cell degranulation.10,14 Peptide immunotherapy (PIT), using short allergen‐derived peptides containing known immunodominant CD4+ T‐cell epitopes, but not containing IgE‐binding epitopes, can overcome this risk.15–17 In the experimental setting, PIT has most often been studied in the context of autoimmune disease, where varied different approaches have been shown to reduce and prevent disease.18–23
PIT has been used in some early clinical trials in patients with allergies, predominantly for cat allergy, but clinical outcomes have varied.17,24–26 There is therefore a pressing need for further understanding of the mechanisms of action of PIT to maximize effective clinical translation. Recently, in clinical studies, there has been a move towards administering multiple allergen‐derived peptides rather than a single peptide.24 This aims both to increase the number of allergen‐derived T‐cell epitopes that can be tolerised against, and confers an advantage when treating an MHC diverse patient population.27 The development of experimental models that are more akin to this type of approach would therefore be beneficial.
Chicken ovalbumin (OVA) has been widely used as a model allergen for the study of allergic airway inflammation (AAI),28 primarily because of the mechanistic insights that can be obtained through the use of transgenic mice with T‐cell receptors reactive to OVA.29 In both BALB/c and C57BL/6 mice, the immunodominant OVA T‐cell epitope is reported to be contained within the 323–339 peptide (p323–339).29,30 However, previous PIT studies using p323–339 in models of OVA‐driven allergic disease in BALB/c mice, have not shown therapeutic effects.31,32 In contrast, we have previously developed a protocol using high‐dose, intravenous p323–339 PIT to induce robust tolerance in a non‐allergic model in C57BL/6 mice.33,34 The adoptive transfer of traceable p323–339‐reactive T cells revealed T‐cell deletion as the major component of tolerance in those studies. Here, we developed an OVA‐driven AAI model in C57BL/6 mice and also found no therapeutic benefit of PIT using p323–339 alone. Detailed characterization of the T‐cell response to OVA revealed an additional T‐cell epitope and we show that a combination approach, using both peptides, provides improved modulation of immunological and histopathological parameters of AAI.
Materials and methods
Mice
Age‐matched, 6‐ to 10‐week‐old female C57BL/6J mice were sourced from Charles River (Kent, UK) and maintained in specific pathogen‐free conditions, on an OVA‐free diet, at the University of Edinburgh, UK. Mice were from mixed litters and randomly allocated to each experimental group. Experiments were conducted under a UK Home Office licence and were locally approved by the University of Edinburgh ethics review panel.
Antigens
Chicken OVA was purchased from Worthington Biochemical Corporation (Lakewood, NJ). The p323–339 peptide was synthesized by PepLogic (Essex, UK). A panel of 75 overlapping 15‐mer peptides (10 amino acid overlap, five amino acid shift) covering the entire sequence of OVA were synthesized by Peptide 2·0 (Chantilly, VA). The peptide panel is shown in Table 1. OVA peptide 263–278 (p263–278) with the sequence KLTEWTSSNVMEERKI was synthesized by Cambridge Research Biochemicals (Cleveland, UK).
Table 1.
Overlapping peptide panel encompassing the sequence of ovalbumin
| Peptide number | Positional sequence | Amino acid sequence |
|---|---|---|
| 1 | 1–15 | GSIGAASMEFCFDVF |
| 2 | 6–20 | ASMEFCFDVFKELKV |
| 3 | 11–25 | CFDVFKELKVHHANE |
| 4 | 16–30 | KELKVHHANENIFYC |
| 5 | 21–35 | HHANENIFYCPIAIM |
| 6 | 26–40 | NIFYCPIAIMSALAM |
| 7 | 31–45 | PIAIMSALAMVYLGA |
| 8 | 36–50 | SALAMVYLGAKDSTR |
| 9 | 41–55 | VYLGAKDSTRTQINK |
| 10 | 46–60 | KDSTRTQINKVVRFD |
| 11 | 51–65 | TQINKVVRFDKLPGF |
| 12 | 56–70 | VVRFDKLPGFGDSIE |
| 13 | 61–75 | KLPGFGDSIEAQCGT |
| 14 | 66–80 | GDSIEAQCGTSVNVH |
| 15 | 71–85 | AQCGTSVNVHSSLRD |
| 16 | 76–90 | SVNVHSSLRDILNQI |
| 17 | 81–95 | SSLRDILNQITKPND |
| 18 | 86–100 | ILNQITKPNDVYSFS |
| 19 | 91–105 | TKPNDVYSFSLASRL |
| 20 | 96–110 | VYSFSLASRLYAEER |
| 21 | 101–115 | LASRLYAEERYPILP |
| 22 | 106–120 | YAEERYPILPEYLQC |
| 23 | 111–125 | YPILPEYLQCVKELY |
| 24 | 116–130 | EYLQCVKELYRGGLE |
| 25 | 121–135 | VKELYRGGLEPINFQ |
| 26 | 126–140 | RGGLEPINFQTAADQ |
| 27 | 131–145 | PINFQTAADQARELI |
| 28 | 136–150 | TAADQARELINSWVE |
| 29 | 141–155 | ARELINSWVESQTNG |
| 30 | 146–160 | NSWVESQTNGIIRNV |
| 31 | 151–165 | SQTNGIIRNVLQPSS |
| 32 | 156–170 | IIRNVLQPSSVDSQT |
| 33 | 161–175 | LQPSSVDSQTAMVLV |
| 34 | 166–180 | VDSQTAMVLVNAIVF |
| 35 | 171–185 | AMVLVNAIVFKGLWE |
| 36 | 176–190 | NAIVFKGLWEKTFKD |
| 37 | 181–195 | KGLWEKTFKDEDTQA |
| 38 | 186–200 | KTFKDEDTQAMPFRV |
| 39 | 191–205 | EDTQAMPFRVTEQES |
| 40 | 196–210 | MPFRVTEQESKPVQM |
| 41 | 201–215 | TEQESKPVQMMYQIG |
| 42 | 206–220 | KPVQMMYQIGLFRVA |
| 43 | 211–225 | MYQIGFRVASMASE |
| 44 | 216–230 | LFRVASMASEKMKIL |
| 45 | 221–235 | SMASEKMKILELPFA |
| 46 | 226–240 | KMKILELPFASGTMS |
| 47 | 231–245 | ELPFASGTMSMLVLL |
| 48 | 236–250 | SGTMSMLVLLPDEVS |
| 49 | 241–255 | MLVLLPDEVSGLEQL |
| 50 | 246–260 | PDEVSGLEQLESIIN |
| 51 | 251–265 | GLEQLESIINFEKLT |
| 52 | 256–270 | ESIINFEKLTEWTSS |
| 53 | 261–275 | FEKLTEWTSSNVMEE |
| 54 | 266–280 | EWTSSNVMEERKIKV |
| 55 | 271–285 | NVMEERKIKVYLPRM |
| 56 | 276–290 | RKIKVYLPRMKMEEK |
| 57 | 281–295 | YLPRMKMEEKYNLTS |
| 58 | 286–300 | KMEEKYNLTSVLMAM |
| 59 | 291–305 | YNLTSVLMAMGITDV |
| 60 | 296–310 | VLMAMGITDVFSSSA |
| 61 | 301–315 | GITDVFSSSANLSGI |
| 62 | 306–320 | FSSSANLSGISSAES |
| 63 | 311–325 | NLSGISSAESLKISQ |
| 64 | 316–330 | SSAESLKISQAVHAA |
| 65 | 321–335 | LKISQAVHAAHAEIN |
| 66 | 326–340 | AVHAAHAEINEAGRE |
| 67 | 331–345 | HAEINEAGREVVGSA |
| 68 | 336–350 | EAGREVVGSAEAGVD |
| 69 | 341–355 | VVGSAEAGVDAASVS |
| 70 | 346–360 | EAGVDAASVSEEFRA |
| 71 | 351–365 | AASVSEEFRADHPFL |
| 72 | 356–370 | EEFRADHPFLFCIKH |
| 73 | 361–375 | DHPFLFCIKHIATNA |
| 74 | 366–380 | FCIKHIATNAVLFFG |
| 75 | 371–385 | IATNAVLFFGRCVSP |
Immunizations and PIT
For the induction of AAI, mice were sensitized on days 0 and 14 by two intraperitoneal injections of 100 μg OVA (Worthington Biochemical Corporation), or PBS (Gibco, Paisley, UK) as a control, adsorbed to 2 mg aluminium hydroxide‐containing adjuvant (alum; Alum Imject Pierce Biotechnology, Rockford, IL).35 Commencing 14 days after the second sensitization, mice were challenged with three 50‐μg doses of OVA by direct airway instillation under anaesthesia. Mice were culled 2 days after the last airway challenge and AAI disease parameters were assessed as previously described.35
For short‐term immunization experiments and T‐cell line generation, mice were immunized subcutaneously into both hind limbs (50 μl per injection) with a total of 200 μg OVA emulsified in complete Freund's adjuvant (CFA) containing 4 mg/ml heat‐killed Mycobacterium tuberculosis H37a (Sigma‐Aldrich, Dorset, UK).
Peptides were diluted in sterile PBS. Seven days before immunization or sensitization soluble peptides (500 μg p323–339, 500 μg p263–278, 500 μg of each peptide in combination, or PBS as a control) were given intravenously via the tail vein.
Assessment of lymphoid cell recall responses
Mediastinal lymph nodes were isolated from mice used in AAI experiments. Inguinal and para‐aortic lymph nodes were obtained 10 days after OVA/CFA immunization. Single cell suspensions were generated and cells were cultured using RPMI‐1640 (Gibco) supplemented with 2 mm l‐glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (all from PAA, Pasching, Austria), 50 μm 2β‐mercaptoethanol and 5% heat‐inactivated fetal calf serum (Gibco). x‐Vivo 15™ serum‐free medium (BioWhittaker, Maidenhead, UK) supplemented with l‐glutamine and 2β‐mercaptoethanol as above, was used for experiments that involved CFA immunization. Cells were cultured in 96‐well flat‐bottomed plates (Costar UK Ltd, Buckinghamshire, UK) at concentrations of 6 × 105/well in the presence of a dose‐range of OVA, p323–339 or p263–278. To determine proliferation, wells were pulsed after 48 hr with [3H]thymidine (0·5 μCi/well; Amersham Biosciences, Buckinghamshire, UK). [3H]Thymidine incorporation was assessed using a liquid scintillation β‐counter (Wallac, Turku, Finland) 16 hr later. Cytokines were measured in culture supernatants after 72 hr by ELISA, as described previously,33 or using a FlowCytomix multiple analyte detection system (eBioscience, Vienna, Austria) as per the manufacturer's instructions.
Short‐term OVA‐reactive T‐cell lines
Lymph node cells were harvested 10 days after OVA/CFA immunization. CD4+ T‐cell lines were generated using in vitro stimulation with OVA, as previously described.37,38 For proliferation assays 2 × 104/well T cells were cultured with 5 × 105/well of irradiated syngeneic splenocytes and antigen. Proliferation and interferon‐γ production were assessed as above.
Bronchoalveolar lavage
Lungs were lavaged using 1 ml sterile PBS by cannulating the trachea. Cytospins were prepared and stained with Quick‐Diff red and Quick‐Diff blue stains (Gamidor Technical Services, Didcot, UK). Under blinded conditions, differential cell counts were determined via light microscopy, 300 cells were counted per slide.
Histological analysis
Sterile PBS (Gibco) was used to perfuse the lungs via the heart. Lungs were inflated and fixed in Methacarn fixative, before embedding in paraffin and processing for haematoxylin & eosin and periodic acid‐Schiff staining. The percentage of goblet cells was determined by counting the number of goblet cells and the number of non‐goblet cells in each airway. Ten consecutive small airways at × 200 magnification were scored for each mouse, and the average percentage of goblet cells in the airways was then calculated, as previously described.36 Scoring was carried out blinded to experimental conditions.
OVA‐specific IgE
Ovalbumin‐specific IgE was detected as described previously.35 High binding EIA/RIA 96‐well plates (Costar UK Ltd) were coated overnight with 5 μg/ml OVA in 0·05 m carbonate buffer (0·795 g Na2CO3, 1·465 g NaHCO3 plus 500 ml dH2O, pH 9·6; all from Sigma‐Aldrich). Serum was IgG depleted using fast flow protein G–Sepharose beads (Sigma‐Aldrich), as described previously.35 Following IgG depletion, serial dilutions of sera were transferred to OVA‐coated plates and incubated at 4° overnight. Bound antibody was detected using biotin‐conjugated rat anti‐mouse IgE antibody (2 μg/ml, Clone R35‐118; BD Bioscience, Oxford, UK), plates were incubated for 1 hour at 37°. For detection, horseradish peroxidase‐labelled streptavidin (R+D Systems Ltd, Oxon, UK) (1 hr at room temperature) followed by 3,3′,5,5′‐tetramethylbenzidine (TMB; Invitrogen Ltd, Paisley, UK) were used. The reaction was stopped using 2 m H2SO4 (Sigma‐Aldrich). All plates were developed for the same length of time (20 min) to enable comparisons between samples. Plates were read at 450 nm (with a wavelength correction of 630 nm) using a Biotek Synergy HT plate reader (Biotek, Bedfordshire, UK) and gen5 software (Biotek).
Statistical analysis
Unpaired t‐tests were used when comparing two groups. To compare three or more groups a one‐way analysis of variance (anova) was used, the data being log10 transformed to correct for non‐normality when necessary. Post hoc tests were performed with Bonferroni correction for multiple comparisons and IgE measurements were analysed using non‐parametric two‐way anova because the data were non‐normal and heteroscedastic. Within each dilution the IgE data were ranked and the effect of treatment was tested using two‐way anova. P < 0·05 was considered significant for all tests. Statistical analyses were performed using prism 4 (Microsoft UK, Reading, UK) and minitab 15 software (Minitab Ltd., Coventry, UK).
Results
High‐dose intravenous PIT with p323–339 alone does not reduce the severity of OVA‐induced AAI in C57BL/6 mice
Previous studies delivering p323–339 via the subcutaneous31 or intranasal32 routes have been unable to modulate OVA‐driven AAI or OVA sensitization in BALB/c mice. Given this background of uncertainty regarding the tolerogenic potential of p323–339, we chose to use systemic, intravenous administration of high‐dose p323–339, which induces tolerance robustly in C57BL/6 mice in a non‐allergic model, as demonstrated previously.33,34,39
To study PIT using p323–339 in the context of OVA‐induced AAI, we established this model in C57BL/6 mice. A standard regimen of OVA sensitization and airway challenge reliably provoked features of AAI including pulmonary cellular infiltration, goblet cell metaplasia, eosinophilia in bronchoalveolar lavage (BAL) and the generation of OVA‐specific IgE (Fig. 1a–h). To assess the effects of p323–339 PIT in this model, 500 μg p323–339 (or PBS as a control) was given intravenously 7 days before OVA sensitization. No significant reductions in the number of total cells or eosinophils in BAL or the percentage of goblet cells in the airways were found following administration of p323–339 (Fig. 2a,b,d). A small but significant increase in OVA‐specific IgE was noted in the PIT group compared with PBS‐treated controls (Fig. 2e), confirming findings reported by others.31 Hence, despite its known capacity to induce tolerance of p323–339‐reactive T cells, PIT using p323–339 did not reduce the severity of any of the parameters of AAI tested.
Figure 1.
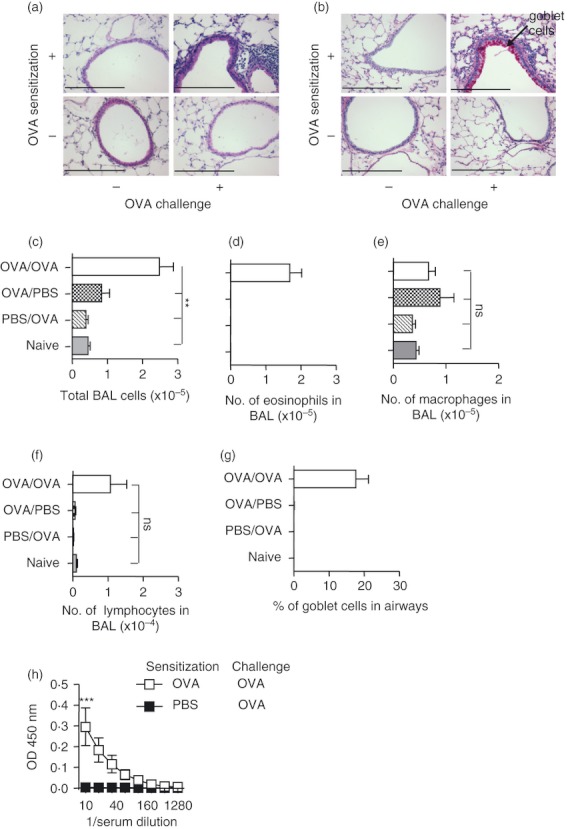
Ovalbumin (OVA) sensitization and challenge induces allergic airway inflammation (AAI). Mice were sensitized and challenged with OVA, sensitized and mock challenged, mock sensitized and OVA challenged or untreated (naive). Mice were killed 2 days after the last challenge and lungs were fixed and stained with (a) haematoxylin & eosin or (b) periodic acid–Schiff stains. Numbers of total cells (c), eosinophils (d), macrophages (e) and lymphocytes (f) in bronchoalveolar lavage, the percentage of goblet cells in the airways (g) and levels of OVA‐specific IgE (h) were determined. (c–g) represent cumulative data from three experiments, n = 3 for naive group, n ≥ 6 for all other groups; (h) represents one experiment n ≥ 4. Data are shown as mean ± SEM. **P < 0·01, ***P < 0·001, determined by analysis of variance with Bonferroni post tests (c–g) or two‐way analysis of variance (h).
Figure 2.
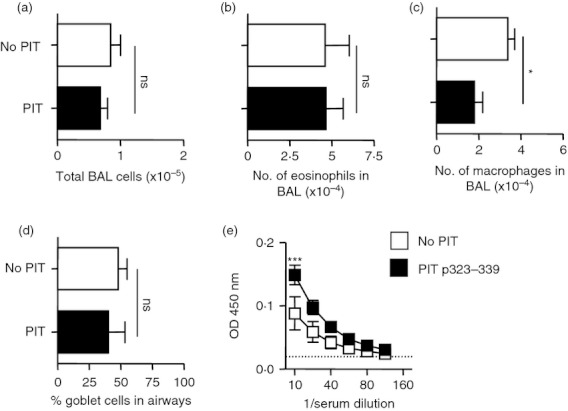
Peptide immunotherapy (PIT) using p323–339 before induction of ovalbumin (OVA) ‐induced allergic airway inflammation (AAI) does not reduce disease severity. PIT using 500 μg of p323–339 or PBS as a control was administered to mice intravenously 7 days before the induction of AAI. Mice were culled 2 days after the last challenge, lungs were fixed and and sections were stained with periodic acid–Schiff. Numbers of total cells (a), eosinophils (b) and macrophages (c) in bronchoalveolar lavage (BAL) were determined. The percentage of goblet cells in the airways was quantified (d) and levels of OVA‐specific IgE (e) were determined. Figure shows representative data from one of two repeat experiments, n = 4. Data are shown as mean ± SEM. ns, not significant, *P < 0·05, ***P < 0·001 as determined by an unpaired t‐test (a–d) and two‐way analysis of variance (e).
Epitope mapping the T‐cell response to OVA in C57BL/6 mice
Given that high‐dose intravenous p323–339 PIT induces a predominantly deletional, rather than regulatory, form of tolerance,33,34 p323–339 PIT would be unlikely to modulate the AAI provoked by OVA if the allergen contained additional T‐cell epitopes. To explore this as a potential underlying reason for the failure of p323–339 PIT to modulate OVA‐induced AAI, we formally characterized the T‐cell response induced in response to OVA in C57BL/6 mice. Short‐term CD4+ T‐cell lines were generated from OVA/CFA immunized C57BL/6 mice using previously described protocols.37,38 CFA was chosen over alum in these experiments to generate a strong immune response. T‐cell line proliferative responses were measured against a panel of 75 peptides constituting the entire sequence of OVA. These 15‐mer peptides incorporated a five amino acid shift, hence a 10 amino acid overlap, to facilitate epitope mapping of the OVA‐induced T‐cell response (individual sequences for this peptide panel are shown in Table 1).
Proliferative responses were observed towards peptides 321–335 and 326–340, both containing the 326–335 sequence, which lies at the core of the p323–339 peptide (Fig. 3a). Additional responses were evident against peptides 261–275 and 266–280 (Fig. 3a). Although responses were also found to peptide 26–40, it was notable that this peptide was found to be poorly soluble during reconstitution.
Figure 3.
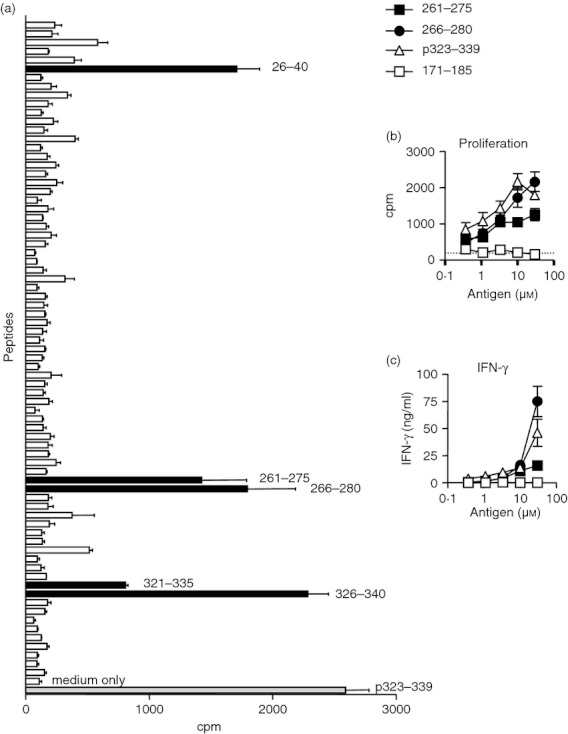
Mapping the T‐cell response of a C57BL/6‐derived ovalbumin (OVA)‐reactive short‐term T‐cell line to a panel of overlapping OVA‐derived peptides. OVA‐reactive short‐term T‐cell lines were generated from C57BL/6 mice immunized with OVA in complete Freund's adjuvant and recall assays conducted to a panel of overlapping OVA peptides at a concentration of 10 μm. Proliferative responses to overlapping peptides were determined using [3H]thymidine incorporation (a). Black bars indicate peptides encompassing the known T‐cell epitope in p323–339 (peptides 321–335 and 326–340) and potential additional OVA T‐cell epitopes (peptides 26–40, 261–275 and 266–280). Consistent results were found in a repeat experiment. Dose‐responsive proliferation (b) and interferon‐γ production (c) were determined for peptides identified from the panel. Peptide 171–185 was included as a negative control. Representative data from one of two experiments are shown as mean ± SEM. In (b) the dotted line shows the response to medium alone.
Both peptides 261–275 and 266–280 induced dose‐dependent proliferative and interferon‐γ responses, as did p323–339 (Fig. 3b,c). Peptide 26–40 did not induce either dose‐dependent T‐cell proliferation, or an interferon‐γ response (data not shown) and was therefore excluded from further study. We conclude that an additional T‐cell epitope is contained within the OVA 266–275 sequence (shared by peptides 261–275 and 266–280). To pursue T‐cell responses to this epitope in the context of PIT and AAI, we chose the peptide p263–278, containing 266–275 with three flanking residues at both the N‐terminus and the C‐terminus.
PIT using either p323–339 or p263–278 induces epitope‐specific tolerance
To assess the capacity of p263–278 to induce tolerance, mice received 500 μg of p323–339, p263–278, or a combination of the two intravenously (a control group received PBS), 7 days before immunization with OVA/CFA. Ten days after immunization, draining lymph node cells were harvested and proliferative responses to in vitro challenge with the individual epitopes were compared. In vivo administration of either peptide before immunization diminished the in vitro recall response to itself, but not to the other peptide (Fig. 4). Abrogation of responses to both peptides was only achieved when both peptides had first been given together in tolerogenic form (Fig. 4). The proliferative response to p323–339 was consistently boosted following PIT with p263–278 (Fig. 4a). We conclude that PIT can potently inhibit the generation of a T‐cell response to the peptide used, but it does not provoke a regulatory function capable of suppressing the response to an alternative epitope within the same immunizing antigen.
Figure 4.
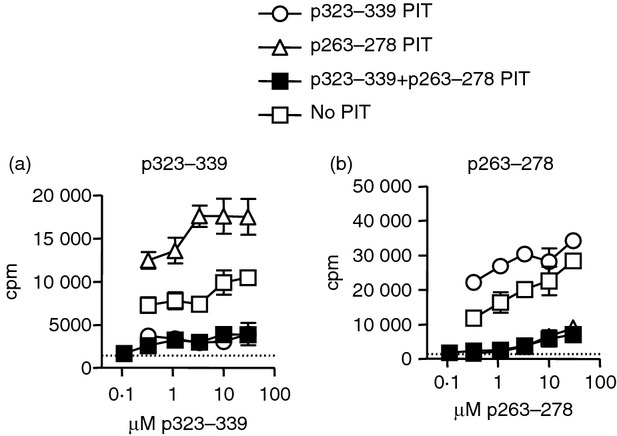
p263–278 is capable of inducing tolerance in vivo. C57BL/6 mice intravenously received p323–339, p263–278 or a combination of the two, or PBS as a control, 7 days before immunization with ovalbumin/complete Freund's adjuvant (OVA/CFA). Ten days after immunization, in vitro proliferative responses of inguinal and para‐aortic lymph node cells to (a) p323–339 and (b) p263–278, were assessed. Lymph node cells from three or four mice per group were pooled. Representative data from one of two experiments are shown as mean ± SEM of three technical replicates. Dotted line represents mean proliferative response to medium alone.
Combination PIT using both p323–339 and p263–278 is required to reduce both histopathological and immunological features of OVA‐induced AAI
Having confirmed that our PIT protocol was capable of preventing efficient T‐cell priming, we put this information to the test in the C57BL/6 OVA‐induced AAI model as characterized in Fig. 1. As we had already shown that PIT with p323–339 was unsuccessful (Fig. 2), for these experiments we concentrated on the tolerogenic effects of either p263–278, alone, or a combination of p263–278 + p323–339, given 7 days before the initial OVA sensitization for the induction of AAI.
While administration of p263–278 alone did not reduce the severity of BAL or goblet cell readouts (Fig. 5a–e), administration of p263–278 in combination with p323–339 resulted in a significant reduction in the percentage of eosinophils in the BAL (Fig. 5d). However, changes in numbers of total cells and eosinophils in the BAL and in the percentage of airway goblet cells did not reach statistical significance (Fig. 5a,b,c,e). OVA‐specific IgE levels were reduced significantly following p263–278 PIT and were reduced further following p323–339 + p263–278 PIT (Fig. 5f). Consistent with these findings, proliferative responses of mediastinal lymph node cells to OVA were only lowered by PIT if the mice received both p323–339 and p263–278 (Fig. 6a). PIT using p323–339 and p263–278 in combination resulted in an apparent reduction in interleukin‐5 (IL‐5) and IL‐13 production from mediastinal lymph node cells in response to OVA in two independent experiments (Fig. 6b,c). However, obtaining sufficient lymph node cells to perform these assays demanded the pooling of samples from multiple mice, preventing statistical analyses of these data. IL‐10, IL‐17, interferon‐γ, tumour necrosis factor‐α, IL‐27 and IL‐6 were below the limits of reliable detection (data not shown).
Figure 5.
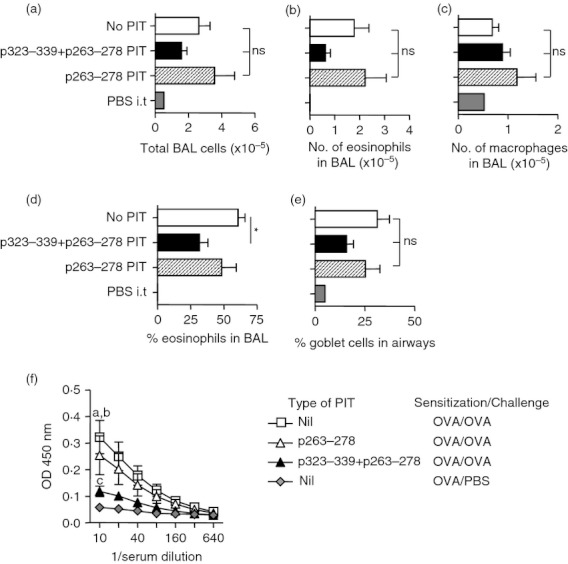
Peptide immunotherapy (PIT) using p263–278 and p323–339 in combination reduces bronchoalveolar lavage (BAL) eosinophilia and ovalbumin (OVA)‐specific IgE. C57BL/6 mice intravenously received 500 μg of either p323–339, p263–278 or a combination of the two, or PBS as a control, 7 days before the induction of allergic airway inflammation (AAI). Two control mice were sensitized to OVA, but challenged with PBS. Two days after the last airway challenge the number of total cells (a), eosinophils (b), and macrophages (c), and the percentage of eosinophils (d) in BAL were determined. The percentage of goblet cells in the airways (e), and levels of OVA‐specific IgE (f), were assessed. Figures represent pooled data from two separate experiments, n = 2 OVA‐sensitized PBS‐challenged group, n ≥ 8 all other groups. Data are shown as mean ± SEM. *P < 0·05, ns = not significant as determined by analysis of variance with Bonferroni post tests (a–e). In (f) a = P < 0·01 no PIT versus p263–278 PIT, b = P < 0·001 no PIT versus p323–339 + p263–278 PIT, c = P < 0·01 p263–278 PIT versus p323–339 + p263–278 PIT, all determined using a two‐way analysis of variance.
Figure 6.
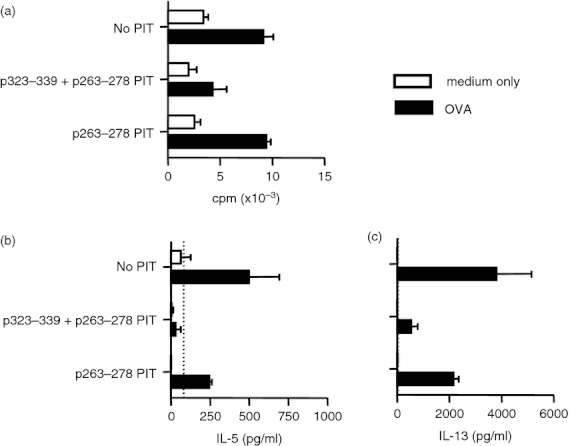
Peptide immunotherapy (PIT) using p263–278 and p323–339 in combination reduces mediastinal lymph node cell responses to ovalbumin (OVA). C57BL/6 mice intravenously received 500 μg of either p323–339, p263–278 or a combination of the two, or PBS as a control, 7 days before the induction of allergic airway inflammation (AAI). Two days after the last airway challenge, lymph nodes from mice within each group were pooled (four mice per group, n = 2) and (a) proliferative responses to 10 μm OVA or medium alone were assessed. After 72 hr of culture supernatants were tested using FlowCytomix multiple analyte detection system for (b) interleukin‐5 (IL‐5) and (c) IL‐13. Figures show representative data from one of two experiments. Data are shown as mean ± SEM of technical replicates. Dotted lines represent lower limits of detection.
Discussion
We formally identify p323–339 and p263–278 as immunodominant T‐cell epitopes that are important for immune responses to OVA in C57BL/6 mice and we demonstrate that their combined use in PIT facilitates the down‐modulation of BAL eosinophilia and OVA‐specific IgE in a model of OVA‐induced AAI.
It has previously been reported that immunization of mice with a vector encoding the 265–280 sequence of OVA resulted in efficient cytotoxic T‐cell responses against OVA.40 The choice of that sequence was directed by prediction algorithms for peptide binding to the Ab MHC class II molecule (Maecker HT and Levy S, personal communication). A limited number of studies have since used the 265–280 sequence as a component of immunogenic antigen preparations in C57BL/6 mice.41,42 The results of our more comprehensive approach (using peptides spanning the entire OVA sequence) (i) confirm the existence of this second OVA T‐cell epitope (our data in fact indicate that this is contained within the 266–275 sequence) and (ii) suggest that the T‐cell response following exposure to OVA is focused, if not entirely, then very particularly on 323–339 and 266–275. No other T‐cell epitopes could be definitively identified (we believe that the proliferation seen against peptide 26–40 in Fig. 3 was the result of the poor solubility of this peptide, rather than a genuine T‐cell‐receptor‐mediated response). CFA was chosen as adjuvant for the epitope mapping studies because of impracticalities associated with the weaker adjuvant alum for these types of studies. A caveat to this approach is that differences in immunodominant epitopes could exist when using different adjuvants. However, the successful reduction of components of T helper type 2‐dependent immunity (IgE and eosinophilia) seen here, support our conclusions that p263–278 is indeed an important epitope in OVA/alum‐driven disease.
Previous studies reported that intranasal administration of p323–339 before OVA/alum sensitization did not alter the generation of T‐cell or B‐cell (IgE and IgG1) immunity to OVA32 and that giving p323–339 subcutaneously after sensitization and before challenge did not have therapeutic value.31 In fact that study reported an augmentation of AAI,31 a later study concluding that this was because of a T helper type 2‐promoting effect of p323–339.43 Those reports in BALB/c mice, together with our data using C57BL/6 mice, show that PIT using p323–339 alone is unable to modulate OVA‐driven AAI. However, p323–339 is not, intrinsically, a ‘non‐tolerogenic’ peptide. We have previously reported its use in PIT models in which mice were subsequently immunized with p323–339/CFA.33,34,39 Our data presented here re‐iterate this; when immunizing with OVA/CFA, prior PIT with p323–339 strongly abrogates the T‐cell response to itself. Nevertheless, this did not impact on AAI following OVA/alum sensitization, because the T‐cell response to p263–278, and therefore to intact OVA protein, remained. Therefore, a sustained ability to provide T‐cell help to B cells presumably accounted for the sustained ability to generate anti‐OVA IgE (Fig. 2).
It seems that the only truly effective way to use a single peptide to reduce disease driven by immunity to multiple epitopes within an antigen is to induce a regulatory function in those T cells initially triggered by the tolerogenic peptide. There are various forms of suppressive CD4+ T cells, most notably Foxp3+‐induced T regulatory cells, which can have multiple modes of suppressive action, and Foxp3− cells that produce predominantly suppressive cytokines such as IL‐10 and transforming growth factor‐β.44,45 Some, but by no means all, studies of PIT have indicated a role for either of these populations,26,46,47 but the net effect can be the same – ‘linked‐suppression’ – in which therapeutic induction of tolerance to one peptide from a protein provides tolerance towards a different peptide in the same protein.21,46,48 We found no evidence supporting linked suppression in the AAI model used here; i.e. inducing tolerance using either p323–339 or p263–278 did not lead to the induction of tolerance towards the other. We have shown previously that the administration of p323–339 PIT using the dose and route as used here, primarily induces a deletional form of tolerance.33,34 It is reasonable to assume that the use of p263–278 here had a similar deletional effect (although this cannot easily be tested at present, in the absence of a p263–278‐responsive TCR transgenic mouse).
Clearly, deletion of antigen‐reactive T cells cannot provide a regulatory mechanism. It also explains the need to provide both p323–339 and p263–278 in combination to have any impact on OVA‐driven AAI. Our data are also consistent with a recent report using a model of OVA‐driven food allergy in BALB/c mice, in which subcutaneous administration of three OVA peptides was required to achieve a tolerogenic effect and reduce OVA‐specific IgE production.49,50 Interestingly, in those studies, p263–274 was described as containing a minor epitope. The combination of p329–343 with two additional peptides (p39–53 and p147–161), rather than p263–274, had therapeutic effects, highlighting the differences in the relative importance of different epitopes between the C57BL/6 and BALB/c strains.49,50 In that previous study, a modulation in responses towards T helper type 1 and T regulatory phenotypes was seen, perhaps as a consequence of the multi‐dose, subcutaneous delivery regimen used.50
In contrast to these series of findings using OVA peptide in allergic models, Campbell et al.46 found that administration of one cat allergen (Fel d I) ‐derived 17‐mer peptide to mice intradermally between airway challenge and rechallenge, reduced AAI and induced linked suppression. It may be that the choice of route (intradermal), dose (very low) and timing of administration (immediately after challenge and before rechallenge) in that study was responsible for driving a suppressive T‐cell phenotype. Exhaustive variations of each of these parameters, in search of the key feature(s) that drive regulatory versus deletional tolerance were beyond the scope of our study. Yet, the increased number of clinical studies using multiple peptides to treat allergic disease increases the requirement for greater understanding of the immunological effects of PIT using combinations of peptides. This is of particular importance because most allergic airways disease starts in children and young people, a population for which detailed understanding of modes of action of any novel therapeutics is required. This study therefore provides an important model of combination PIT in AAI.
Although tolerogenic administration of p323–339 and p263–278 in combination led to reduced levels of OVA‐specific IgE and BAL eosinophilia, goblet cell metaplasia was still evident. As discussed above, a pre‐emptive deletion of OVA‐responsive CD4+ T cells probably inhibited the generation of OVA‐specific IgE. However, OVA‐specific IgE is not a prerequisite for the development of AAI in mice51,52 and in humans, levels of allergen‐specific IgE do not necessarily correlate with disease severity.53 Furthermore, studies using the monoclonal IgE‐blocking antibody omalizumab in patients with allergic asthma vary in effectiveness, and are currently restricted to use in very severe cases.54,55 A deletion of OVA‐responsive CD4+ T cells also substantially reduced T helper type 2 cytokines produced in response to OVA from mediastinal lymph node cells. This probably limited airway eosinophilia, but a residual low level of OVA responsiveness may have been sufficient to trigger innate cytokine responses in the mucosa (e.g. IL‐33, IL‐25) and the induction of IL‐13 production by innate lymphoid cells, which has been shown to be sufficient for the development of goblet cell metaplasia.56,57 This could account for the lack of a reduction in the percentage of goblet cells seen here following PIT. It is also possible that innate lymphoid cell production of IL‐5 could contribute to the incomplete reduction of eosinophilia in the BAL seen following p323–339 and p263–278 combination PIT.56
It is interesting to speculate on which form of tolerance (regulatory or deletional) is most desirable, clinically. The expansion of T cells with regulatory function has benefits in that it may be possible to provide tolerance at the level of the tissue/organ, rather than the antigen; i.e. there would be no requirement to stringently identify all possible T‐cell epitopes and it is even conceivable that tolerant T cells could suppress allergic responses to non‐related allergens during co‐exposure. However, there are data from studies of experimental and natural tolerance to allergens to suggest that regulatory activity can be lost, with restoration of allergic function among T cells over time.58 Hence maintenance of a regulatory form of tolerance may well require long‐term treatment, consistent with the current consensus that 3 years of specific immunotherapy treatment is advisable to confer longer‐term tolerance.13,59 In contrast, a deletional mechanism should remove the pathogenic T cells, and with them concerns over the longevity of tolerance, but greater knowledge of the entirety of the T‐cell response to allergen, and the mechanisms implicated in deletional modes of tolerance, is required. The data presented here indicate that, once we are armed with this knowledge, efficacious application of PIT is within our grasp.
Acknowledgments
This work was funded by the UK Medical Research Council. KJM received an MRC Clinical Research Training Fellowship. We are grateful to Dr Margo Chase‐Topping at the Centre for Immunity, Infection and Evolution, The University of Edinburgh, for guidance on statistical analysis.
Disclosure
The authors do not have any conflict of interest pertaining to this study.
References
- 1.Kim HY, DeKruyff RH, Umetsu DT. The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat Immunol. 2010;11:577–84. doi: 10.1038/ni.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson HR, Gupta R, Strachan DP, Limb ES. 50 years of asthma: UK trends from 1955 to 2004. Thorax. 2007;62:85–90. doi: 10.1136/thx.2006.066407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Punekar YS, Sheikh A. Establishing the sequential progression of multiple allergic diagnoses in a UK birth cohort using the General Practice Research Database. Clin Exp Allergy. 2009;39:1889–95. doi: 10.1111/j.1365-2222.2009.03366.x. [DOI] [PubMed] [Google Scholar]
- 4.Holgate ST, Polosa R. Treatment strategies for allergy and asthma. Nat Rev Immunol. 2008;8:218–30. doi: 10.1038/nri2262. [DOI] [PubMed] [Google Scholar]
- 5.Larché M, Robinson DS, Kay AB. The role of T lymphocytes in the pathogenesis of asthma. J Allergy Clin Immunol. 2003;111:450–63. doi: 10.1067/mai.2003.169. [DOI] [PubMed] [Google Scholar]
- 6.Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just Th2 cells. Nat Rev Immunol. 2010;10:838–48. doi: 10.1038/nri2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akdis M, Akdis CA. Therapeutic manipulation of immune tolerance in allergic disease. Nat Rev Drug Discov. 2009;8:645–60. doi: 10.1038/nrd2653. [DOI] [PubMed] [Google Scholar]
- 8.Calderón MA, Casale TB, Togias A, Bousquet J, Durham SR, Demoly P. Allergen‐specific immunotherapy for respiratory allergies: from meta‐analysis to registration and beyond. J Allergy Clin Immunol. 2011;127:30–8. doi: 10.1016/j.jaci.2010.08.024. [DOI] [PubMed] [Google Scholar]
- 9.Frew AJ. Allergen immunotherapy. J Allergy Clin Immunol. 2010;125:S306–13. doi: 10.1016/j.jaci.2009.10.064. [DOI] [PubMed] [Google Scholar]
- 10.Abramson MJ, Puy RM, Weiner JM. Injection allergen immunotherapy for asthma. Cochrane Database Syst Rev (Online) 2010:CD001186. doi: 10.1002/14651858.CD001186.pub2. [DOI] [PubMed] [Google Scholar]
- 11.Calderon MA, Alves B, Jacobson M, Hurwitz B, Sheikh A, Durham S. Allergen injection immunotherapy for seasonal allergic rhinitis. Cochrane Database Syst Rev (Online) 2007:CD001936. doi: 10.1002/14651858.CD001936.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pajno GB, Barberio G, De Luca F, Morabito L, Parmiani S. Prevention of new sensitizations in asthmatic children monosensitized to house dust mite by specific immunotherapy. A six‐year follow‐up study. Clin Exp Allergy. 2001;31:1392–7. doi: 10.1046/j.1365-2222.2001.01161.x. [DOI] [PubMed] [Google Scholar]
- 13.Jacobsen L, Niggemann B, Dreborg S. Specific immunotherapy has long‐term preventive effect of seasonal and perennial asthma: 10‐year follow‐up on the PAT study. Allergy. 2007;62:943–8. doi: 10.1111/j.1398-9995.2007.01451.x. et al. [DOI] [PubMed] [Google Scholar]
- 14.Caubet JC, Eigenmann PA. Late side‐effects during systemic immunotherapy in children. Allergy. 2008;63:1561–2. doi: 10.1111/j.1398-9995.2008.01868.x. [DOI] [PubMed] [Google Scholar]
- 15.Larche M, Wraith DC. Peptide‐based therapeutic vaccines for allergic and autoimmune diseases. Nat Med. 2005;11:S69–76. doi: 10.1038/nm1226. [DOI] [PubMed] [Google Scholar]
- 16.Anderton SM. Peptide‐based immunotherapy of autoimmunity: a path of puzzles, paradoxes and possibilities. Immunology. 2001;104:367–76. doi: 10.1046/j.1365-2567.2001.01324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larché M. Peptide immunotherapy for allergic diseases. Allergy. 2007;62:325–31. doi: 10.1111/j.1398-9995.2006.01309.x. [DOI] [PubMed] [Google Scholar]
- 18.Metzler B, Wraith DC. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: influence of MHC binding affinity. Int Immunol. 1993;5:1159–65. doi: 10.1093/intimm/5.9.1159. [DOI] [PubMed] [Google Scholar]
- 19.Prakken BJ, van der Zee R, Anderton SM, van Kooten PJ, Kuis W, van Eden W. Peptide‐induced nasal tolerance for a mycobacterial heat shock protein 60 T cell epitope in rats suppresses both adjuvant arthritis and nonmicrobially induced experimental arthritis. Proc Natl Acad Sci USA. 1997;94:3284–9. doi: 10.1073/pnas.94.7.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daniel D, Wegmann DR. Protection of nonobese diabetic mice from diabetes by intranasal or subcutaneous administration of insulin peptide B‐(9–23) Proc Natl Acad Sci USA. 1996;93:956–60. doi: 10.1073/pnas.93.2.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderton SM, Wraith DC. Hierarchy in the ability of T cell epitopes to induce peripheral tolerance to antigens from myelin. Eur J Immunol. 1998;28:1251–61. doi: 10.1002/(SICI)1521-4141(199804)28:04<1251::AID-IMMU1251>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 22.Burkhart C, Liu GY, Anderton SM, Metzler B, Wraith DC. Peptide‐induced T cell regulation of experimental autoimmune encephalomyelitis: a role for IL‐10. Int Immunol. 1999;11:1625–34. doi: 10.1093/intimm/11.10.1625. [DOI] [PubMed] [Google Scholar]
- 23.Critchfield JM, Racke MK, Zuniga‐Pflucker JC, Cannella B, Raine CS, Goverman J, Lenardo MJ. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science. 1994;263:1139–43. doi: 10.1126/science.7509084. [DOI] [PubMed] [Google Scholar]
- 24.Alexander C, Tarzi M, Larche M, Kay AB. The effect of Fel d 1‐derived T‐cell peptides on upper and lower airway outcome measurements in cat‐allergic subjects. Allergy. 2005;60:1269–74. doi: 10.1111/j.1398-9995.2005.00885.x. [DOI] [PubMed] [Google Scholar]
- 25.Oldfield WLG, Larché M, Kay AB. Effect of T‐cell peptides derived from Fel d 1 on allergic reactions and cytokine production in patients sensitive to cats: a randomised controlled trial. Lancet. 2002;360:47–53. doi: 10.1016/s0140-6736(02)09332-7. [DOI] [PubMed] [Google Scholar]
- 26.Tarzi M, Klunker S, Texier C. Induction of interleukin‐10 and suppressor of cytokine signalling‐3 gene expression following peptide immunotherapy. Clin Exp Allergy. 2006;36:465–74. doi: 10.1111/j.1365-2222.2006.02469.x. et al. [DOI] [PubMed] [Google Scholar]
- 27.Larché M. Of cats and men: immunodominance and the role of HLA‐DP/DQ. Clin Exp Allergy. 2008;38:1709–11. doi: 10.1111/j.1365-2222.2008.03112.x. [DOI] [PubMed] [Google Scholar]
- 28.Kips JC, Anderson GP, Fredberg JJ. Murine models of asthma. Eur Respir J. 2003;22:374–82. doi: 10.1183/09031936.03.00026403. et al. [DOI] [PubMed] [Google Scholar]
- 29.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA‐based α‐ and β‐chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 30.Shimonkevitz R, Colon S, Kappler JW, Marrack P, Grey HM. Antigen recognition by H‐2‐restricted T cells. II. A tryptic ovalbumin peptide that substitutes for processed antigen. J Immunol. 1984;133:2067–74. [PubMed] [Google Scholar]
- 31.Janssen EM, Wauben MH, Jonker EH, Hofman G, Van Eden W, Nijkamp FP, Van Oosterhout AJ. Opposite effects of immunotherapy with ovalbumin and the immunodominant T‐cell epitope on airway eosinophilia and hyperresponsiveness in a murine model of allergic asthma. Am J Respir Cell Mol Biol. 1999;21:21–9. doi: 10.1165/ajrcmb.21.1.3519. [DOI] [PubMed] [Google Scholar]
- 32.Barbey C, Donatelli‐Dufour N, Batard P, Corradin G, Spertini F. Intranasal treatment with ovalbumin but not the major T cell epitope ovalbumin 323–339 generates interleukin‐10 secreting T cells and results in the induction of allergen systemic tolerance. Clin Exp Allergy. 2004;34:654–62. doi: 10.1111/j.1365-2222.2004.1929.x. [DOI] [PubMed] [Google Scholar]
- 33.Konkel JE, Frommer F, Leech MD, Yagita H, Waisman A, Anderton SM. PD‐1 signalling in CD4+ T cells restrains their clonal expansion to an immunogenic stimulus, but is not critically required for peptide‐induced tolerance. Immunology. 2010;130:92–102. doi: 10.1111/j.1365-2567.2009.03216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hochweller K, Anderton SM. Kinetics of costimulatory molecule expression by T cells and dendritic cells during the induction of tolerance versus immunity in vivo. Eur J Immunol. 2005;35:1086–96. doi: 10.1002/eji.200425891. [DOI] [PubMed] [Google Scholar]
- 35.de Vries A, Hazlewood L, Fitch PM, Seckl JR, Foster P, Howie SE. High‐fat feeding redirects cytokine responses and decreases allergic airway eosinophilia. Clin Exp Allergy. 2009;39:731–9. doi: 10.1111/j.1365-2222.2008.03179.x. [DOI] [PubMed] [Google Scholar]
- 36.Leech MD, Benson RA, deVries A, Fitch PM, Howie SEM. Resolution of Der p1‐induced allergic airway inflammation is dependent on CD4+ CD25+ Foxp3+ regulatory cells. J Immunol. 2007;179:7050–8. doi: 10.4049/jimmunol.179.10.7050. [DOI] [PubMed] [Google Scholar]
- 37.Anderton SM, Manickasingham SP, Burkhart C, Luckcuck TA, Holland SJ, Lamont AG, Wraith DC. Fine specificity of the myelin‐reactive T cell repertoire: implications for TCR antagonism in autoimmunity. J Immunol. 1998;161:3357–64. [PubMed] [Google Scholar]
- 38.Sweenie CH, Mackenzie KJ, Rone‐Orugboh A, Liu M, Anderton SM. Distinct T cell recognition of naturally processed and cryptic epitopes within the immunodominant 35–55 region of myelin oligodendrocyte glycoprotein. J Neuroimmunol. 2007;183:7–16. doi: 10.1016/j.jneuroim.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 39.Hochweller K, Sweenie CH, Anderton SM. Circumventing tolerance at the T cell or the antigen‐presenting cell surface: antibodies that ligate CD40 and OX40 have different effects. Eur J Immunol. 2006;36:389–96. doi: 10.1002/eji.200535506. [DOI] [PubMed] [Google Scholar]
- 40.Maecker HT, Umetsu DT, DeKruyff RH, Levy S. Cytotoxic T cell responses to DNA vaccination: dependence on antigen presentation via class II MHC. J Immunol. 1998;161:6532–6. [PubMed] [Google Scholar]
- 41.Mizukami S, Kajiwara C, Ishikawa H, Katayama I, Yui K, Udono H. Both CD4+ and CD8+ T cell epitopes fused to heat shock cognate protein 70 (hsc70) can function to eradicate tumors. Cancer Sci. 2008;99:1008–15. doi: 10.1111/j.1349-7006.2008.00788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gadola SD, Silk JD, Jeans A. Impaired selection of invariant natural killer T cells in diverse mouse models of glycosphingolipid lysosomal storage diseases. J Exp Med. 2006;203:2293–303. doi: 10.1084/jem.20060921. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janssen EM, van Oosterhout AJ, van Rensen AJ, van Eden W, Nijkamp FP, Wauben MH. Modulation of Th2 responses by peptide analogues in a murine model of allergic asthma: amelioration or deterioration of the disease process depends on the Th1 or Th2 skewing characteristics of the therapeutic peptide. J Immunol. 2000;164:580–8. doi: 10.4049/jimmunol.164.2.580. [DOI] [PubMed] [Google Scholar]
- 44.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Lloyd CM, Hawrylowicz CM. Regulatory T cells in asthma. Immunity. 2009;31:438–49. doi: 10.1016/j.immuni.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campbell JD, Buckland KF, McMillan SJ. Peptide immunotherapy in allergic asthma generates IL‐10‐dependent immunological tolerance associated with linked epitope suppression. J Exp Med. 2009;206:1535–47. doi: 10.1084/jem.20082901. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gabrysova L, Nicolson KS, Streeter HB, Verhagen J, Sabatos‐Peyton CA, Morgan DJ, Wraith DC. Negative feedback control of the autoimmune response through antigen‐induced differentiation of IL‐10‐secreting Th1 cells. J Exp Med. 2009;206:1755–67. doi: 10.1084/jem.20082118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Briner TJ, Kuo MC, Keating KM, Rogers BL, Greenstein JL. Peripheral T‐cell tolerance induced in naive and primed mice by subcutaneous injection of peptides from the major cat allergen Fel d 1. Proc Natl Acad Sci USA. 1993;90:7608–12. doi: 10.1073/pnas.90.16.7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang M, Mine Y. Novel T‐cell epitopes of ovalbumin in BALB/c mouse: potential for peptide‐immunotherapy. Biochem Biophys Res Commun. 2009;378:203–8. doi: 10.1016/j.bbrc.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 50.Yang M, Yang C, Mine Y. Multiple T cell epitope peptides suppress allergic responses in an egg allergy mouse model by the elicitation of forkhead box transcription factor 3‐ and transforming growth factor‐β‐associated mechanisms. Clin Exp Allergy. 2010;40:668–78. doi: 10.1111/j.1365-2222.2009.03442.x. [DOI] [PubMed] [Google Scholar]
- 51.Mehlhop PD, van de Rijn M, Goldberg AB, Brewer JP, Kurup VP, Martin TR, Oettgen HC. Allergen‐induced bronchial hyperreactivity and eosinophilic inflammation occur in the absence of IgE in a mouse model of asthma. Proc Natl Acad Sci USA. 1997;94:1344–9. doi: 10.1073/pnas.94.4.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korsgren M, Erjefalt JS, Korsgren O, Sundler F, Persson CG. Allergic eosinophil‐rich inflammation develops in lungs and airways of B cell‐deficient mice. J Exp Med. 1997;185:885–92. doi: 10.1084/jem.185.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Celik‐Bilgili S, Mehl A, Verstege A, Staden U, Nocon M, Beyer K, Niggemann B. The predictive value of specific immunoglobulin E levels in serum for the outcome of oral food challenges. Clin Exp Allergy. 2005;35:268–73. doi: 10.1111/j.1365-2222.2005.02150.x. [DOI] [PubMed] [Google Scholar]
- 54.Holgate ST. A look at the pathogenesis of asthma: the need for a change in direction. Discov med. 2010;9:439–47. [PubMed] [Google Scholar]
- 55.Rodrigo GJ, Neffen H, Castro‐Rodriguez JA. Efficacy and safety of subcutaneous omalizumab vs placebo as add‐on therapy to corticosteroids for children and adults with asthma: a systematic review. Chest. 2011;139:28–35. doi: 10.1378/chest.10-1194. [DOI] [PubMed] [Google Scholar]
- 56.Neill DR, Wong SH, Bellosi A. Nuocytes represent a new innate effector leukocyte that mediates type‐2 immunity. Nature. 2010;464:1367–70. doi: 10.1038/nature08900. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL‐33‐responsive lineage – CD25+ CD44hi lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188:1503–13. doi: 10.4049/jimmunol.1102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meiler F, Zumkehr J, Klunker S, Ruckert B, Akdis CA, Akdis M. In vivo switch to IL‐10‐secreting T regulatory cells in high dose allergen exposure. J Exp Med. 2008;205:2887–98. doi: 10.1084/jem.20080193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moldaver D, Larche M. Immunotherapy with peptides. Allergy. 2011;66:784–91. doi: 10.1111/j.1398-9995.2011.02610.x. [DOI] [PubMed] [Google Scholar]