

To the Editor
Pancreatic ductal adenocarcinoma (PDA) accounts for the vast majority of all pancreatic neoplasms. Other frequently seen and well-described pancreatic neoplasms include pancreatic neuroendocrine tumors and cystic neoplasms of the pancreas. However, there is a subset of rare benign pancreatic pathologies which can mimic malignant processes. Differentiating between these processes and that of pancreatic carcinoma is of paramount importance in order to determine clinical treatment options and timely instigate therapy; associations between rare disease disorders may help accuracy of such determinations. Autoimmune lymphoproliferative syndrome (ALPS) is a rare condition with only 500 cases reported in the literature. To date, autoimmune pancreatitis (AIP) has not been described as one of the possible autoimmune manifestations of ALPS. We report a case of an ALPS patient with positron emission tomography (PET) avid pancreatic lesions referred to the surgical oncology service with concern for pancreatic carcinoma; physical, biochemical, radiologic and pathologic examination lead to the benign diagnosis of type I AIP. Relevant literature supporting an association of ALPS and AIP or IgG4-related disorders including its shared histopathology and immunologic manifestations is detailed.
A 65 year-old female with positron emission tomography (PET) avid pancreatic lesions (Figure 1.1) was referred to the surgical oncology service with the presumed diagnosis of pancreatic carcinoma. Ten years prior, the patient was diagnosed with ALPS through identification of a germline heterozygous FAS mutation (ALPS Type Ia; ALPS-FAS, c.1074delT; p. L278fs, Fig. 1.1). On physical examination she was found to have an enlarged right salivary gland and proptosis of her right eye. Magnetic resonance imaging (MRI) of the orbits revealed abnormal prominence of bilateral lacrimal glands (Fig. 1.2). Lacrimal gland biopsy was performed. Pathologic analysis of the lacrimal gland biopsy revealed a dense lymphocytic infiltrate and numerous IgG4 positive plasma cells (Fig. 1.3). No granuloma, lymphoma or other malignancies were identified. Following these findings, using the International Consensus Diagnostic Criteria for AIP the patient was given a diagnosis of type I AIP1. Non-operative management with steroids was implemented; current MRI of the abdomen revealed a normal appearing pancreas.
Figure 1.1.

Fas mutations. Black arrow depicts the patient presented, FAS mutation c.1074delT; p.L278fs (Figure adapted from Su, Helen C; and, Lenardo, Michael J (September 2009) Apoptosis: Inherited Disorders. In: Encyclopedia of Life Sciences (ELS). John Wiley & Sons, Ltd: Chichester.)
Figure 1.2.

(A) Computed Tomography imaging demonstrating a diffusely enlarged pancreas (white arrow). (B) Magnetic Resonance Imaging of enlarged and enhancing lacrimal glands (white arrow). (C) Positron Emission Tomography demonstrating pancreatic avidity, SUV 6 (black arrow)
Figure 1.3.
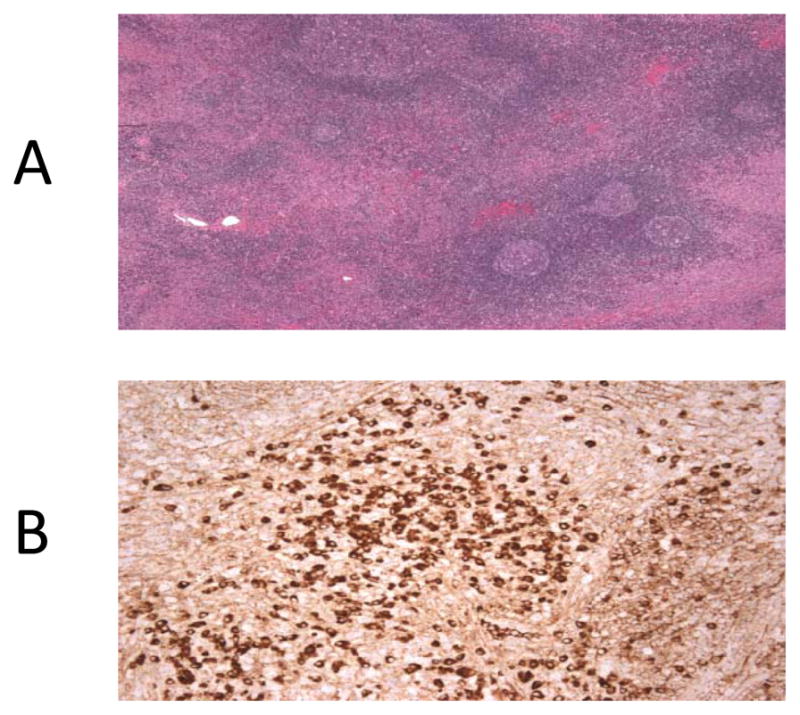
(A) H and E staining of lacrimal gland biopsy showing dense lymphocytic infiltration. (B) Immunohistochemistry staining of lacrimal gland biopsy showing IgG4 positive plasma cells
Autoimmune lymphoproliferative syndrome (ALPS) is a condition first described in the 1990’s after the analysis of a family with unexplained lymphadenopathy accompanied by autoimmune symptoms. An associated genetic mutation was found in the FAS gene, which codes for a death receptor that leads to programmed cell death or apoptosis 2. Mutations in the FAS gene result in decreased apoptosis and an increase of peripheral lymphocytes2. Consequent accumulation of lymphocytes leads to the expansion of lymphoid organs. The persistence of these cells results in expanded populations of lymphocytes and predisposes to autoimmune phenomena and lymphomas 2,3.
Laboratory findings include elevation of double negative T cells (CD4−CD8−), decreased apoptosis by in vivo culture of lymphocytes and elevated interleukins such as interleukin-2 and 10 4. Also, these patients embody a unique histopathology, demonstrating paracortical expansion of T cells in lymph nodes with many CD4−CD8− T cells and follicular hyperplasia with prominent vascularity and marked plasmacytosis 3,5. While germline heterozygous FAS mutations comprise the majority of ALPS mutations, recent reports have introduced variants in other apoptosis-related genes like FAS-LG (Fas ligand), CASP8, and CASP10 (caspase 8 and 10). The 2009 revised classification of ALPS-related disorders now follows a genetic classification of this syndrome acknowledging the unique genotype-phenotype expressions of this syndrome (Fig. 1.4)6. Currently there are approximately 500 cases reported worldwide, however cases of AIP or IgG4-related disorders have not been described in this patient population 5.
Figure 1.4.
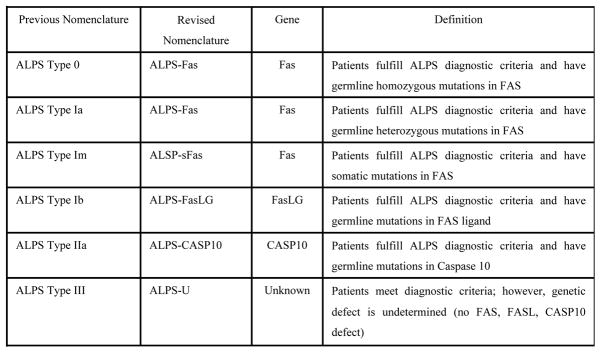
Revised ALPS Classification (ALPS; Autoimmune lymphoproliferative syndrome, FasLG; Fas-ligand, CASP10; Caspase 10) 5
Four histological findings diagnose type I AIP: A) marked IgG4+ plasma cell infiltration B) obliterative phlebitis C) prominent lymphoid follicles D) Prominent storiform fibrosis 1. The inflammatory infiltrate may extend to involve the peripancreatic soft tissues and consists predominantly of T lymphocytes with scattered B cells 1,7.
What are possible shared pathophysiological mechanisms of AIP and IgG4-related disorders, and how can AIP be connected to ALPS, an immunogenetic disease caused by germline mutations in one of the apoptosis regulating genes?
Shared genetic variants of AIP and ALPS
Several non-HLA genetic variants have been found to be associated with both AIP and IgG4-related disorders, among the best studied CTLA-4, Fc receptor like 3, and tumor necrosis factor α (TNFα). In a sentinel study of 70 patients with non-hereditary, histopathologically proven chronic pancreatitis and 286 control subjects, Chang MC et al found a strong association of TNFα promoter polymorphisms and haplotypes and the occurrence of pancreatitis 8. While no functional data of these polymorphisms on the expression levels of TNFα are presented it is conceivable that reduced, or altered, expression levels of TNFα, a strong apoptosis mediator in immune cells, may shift immune cell homeostasis favoring a survival of immune cell populations which are typically seen in ALPS. Reduced TNFα expression would lead to the same phenotype as loss-of-function Fas receptor (FasR) mutations, the gene typically afflicted by germline mutations in ALPS (also known as as apoptosis antigen 1 (APO-1 or APT) or tumor necrosis factor receptor superfamily member 6; TNFRSF6) as TNFα is one of the ligands of FasR hence providing a possible genetic link between ALPS and one of the genomic abnormalities described in IgG4-related disorders.
Dysregulation of Th2 cells and Tregs in AIP and IgG4-related disorders
Immunologically, AIP and IgG4-related disorders are characterized by a predominant Th2 cell blood phenotype with Th2-cell responses activated at affected sites as measured by elevated interleukin-4, -5, -10, and -13 levels which are particularly higher than in classic autoimmune conditions like autoimmune hepatitis, lupus erythematous, or rheumatoid arthritis 9. Another immunologic difference to autoimmune diseases is the activation of regulatory T (Treg) cells which are commonly depressed 9. In more detailed examination in patients with AIP, memory (CD45RA−) Tregs are significantly increased whereas circulatory naïve (CD45RA+) Tregs are depressed. Increased memory Tregs with increased interleukin-10 production have been observed in both pancreatic and extrapancreatic sites of manifestation suggesting inhibitory responses of memory Tregs to inflammation. This immunologic phenotype has parallels to ALPS which includes a Th2 cytokine profile as well as elevated interleukin-10 levels 9. Peripheral eosinophilia observed in 3 to 32 % of ALPS patients is seen in up to 40 % of AIP patients 9. It is tempting to speculate that variable degrees of imbalances of immune regulating cells due to disturbances in apoptosis regulation can induce an IgG4-related phenotype in some ALPS patients. Reduced production of TNFα and reduced apoptosis of select immune cell populations, either via reduced production due to depressed naïve T cells or because of the inhibition of CD4 CD8 effector cells, a common source of TNFα production, by an abnormally expanded memory Treg pool in IgG4-related disorders might be a common pathway explaining the observed association of these two rare disorders. However, there remains the possibility of a concomitant complication of IgG4-related disorders, including the recently described IgG4-positive multiorgan lymphoprolifetative syndrome (MOLPS), a more systemic variant of Sjogren’s syndrome, and ALPS with independent pathogenetic mechanisms. In this regard, it is interesting that AIP, and MOLPS, have also recently been reported in association with myelodysplastic syndrome (MDS) or antiphospholipid syndrome 10.
Although AIP and IgG4-related disorders have been linked to various autoimmune conditions, it has not previously been included as an entity of ALPS. We present the first case of AIP which mimicked pancreatic carcinoma in the ALPS population. Based on shared genetic and immunologic pathological features of both conditions we believe it is important to consider AIP in the myriad of findings in the ALPS population and maintain a high suspicion of this disease process when pancreatic abnormalities are identified.
Acknowledgments
This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute.
Footnotes
All authors declare no conflicts of interest.
References
- 1.Zhang L, Chari S, Smyrk TC, et al. Autoimmune pancreatitis (AIP) type 1 and type 2: an international consensus study on histopathologic diagnostic criteria. Pancreas. 2011;40:1172–9. doi: 10.1097/MPA.0b013e318233bec5. [DOI] [PubMed] [Google Scholar]
- 2.Turbyville JC, Rao VK. The autoimmune lymphoproliferative syndrome: A rare disorder providing clues about normal tolerance. Autoimmun Rev. 2010;9:488–93. doi: 10.1016/j.autrev.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Teachey DT, Seif AE, Grupp SA. Advances in the management and understanding of autoimmune lymphoproliferative syndrome (ALPS) Br J Haematol. 2010;148:205–16. doi: 10.1111/j.1365-2141.2009.07991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuss IJ, Strober W, Dale JK, et al. Characteristic T helper 2 T cell cytokine abnormalities in autoimmune lymphoproliferative syndrome, a syndrome marked by defective apoptosis and humoral autoimmunity. J Immunol. 1997;158:1912–8. [PubMed] [Google Scholar]
- 5.Oliveira JB, Bleesing JJ, Dianzani U, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116:e35–40. doi: 10.1182/blood-2010-04-280347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madkaikar M, Mhatre S, Gupta M, Ghosh K. Advances in autoimmune lymphoproliferative syndromes. European journal of haematology. 2011;87:1–9. doi: 10.1111/j.1600-0609.2011.01617.x. [DOI] [PubMed] [Google Scholar]
- 7.Mortenson MM, Katz MH, Tamm EP, et al. Current diagnosis and management of unusual pancreatic tumors. Am J Surg. 2008;196:100–13. doi: 10.1016/j.amjsurg.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Chang MC, Chang YT, Tien YW, Liang PC, Wei SC, Wong JM. Association of tumour necrosis factor alpha promoter haplotype with chronic pancreatitis. Gut. 2006;55:1674–6. doi: 10.1136/gut.2006.103085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stone JH, Zen Y, Deshpande V. IgG4-related disease. The New England journal of medicine. 2012;366:539–51. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 10.Tabata R, Tabata C, Okamoto T, Omori K, Terada M, Nagai T. Autoimmune pancreatitis associated with myelodysplastic syndrome. International archives of allergy and immunology. 2010;151:168–72. doi: 10.1159/000236007. [DOI] [PubMed] [Google Scholar]