

Abstract
Five-coordinate boryl complexes relevant to Ir mediated C–H borylations have been synthesized, providing a glimpse of the most fundamental step in the catalytic cycle for the first time.
In many transition metal catalyzed processes, ligand dissociation or some other rearrangement of the metal “catalyst” is required to generate the metal species that reacts with substrate(s) in the catalytic cycle. Such is the case for Ir-catalyzed borylations of C–H bonds, where the five-coordinate boryl complexes believed to be responsible for C–H functionalization have not been observed.1,2 The closest to the five-coordinate complexes in phosphine and dipyridyl ligated systems are the six-coordinate complexes 1 and 2. While both of these compounds will borylate sp2-hybridized C–H bonds, ligand dissociation precedes the key C–H interaction between the hydrocarbon and the Ir centre (Scheme 1).
Scheme 1.
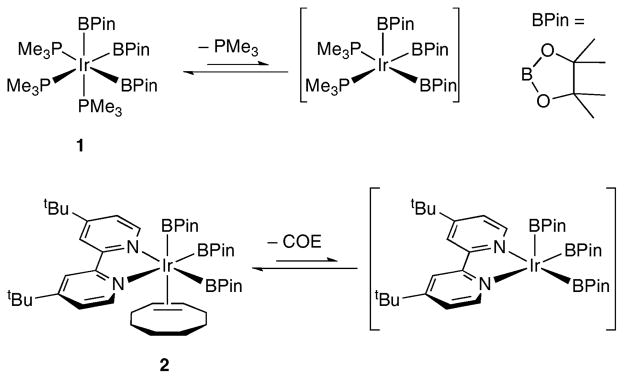
Generation of five-coordinate, reactive intermediates in C–H borylations.
Because Ir-catalyzed borylations exhibit unusual chemo-and regioselectivities, the reactivity of five-coordinate trisboryl complexes could offer important information regarding the most fundamental step in the catalytic cycle—C–H bond scission. Fully cognizant of the pitfalls embodied in Halpern’s well-known maxim that true reactive intermediates are rarely isolable,3 we set out to prepare five-coordinate complexes that could react directly with arenes and heterocycles. Our preliminary results are described herein.4
We expected that the choice of Ir starting material ligand would be critical for the successful preparation of five-coordinate complexes. Since compound 2 retains an η2-olefin from the starting material, we felt that displacement of an arene with a bidentate chelating ligand might yield the desired five-coordinate complexes since η2-coordination of the expelled arene would carry the penalty of disrupting its aromaticity (Scheme 2). The mesitylene trisboryl complex, 3, was selected as the starting material, since the mesitylene methyl groups block access to the arene sp2 C–H bonds.1
Scheme 2.
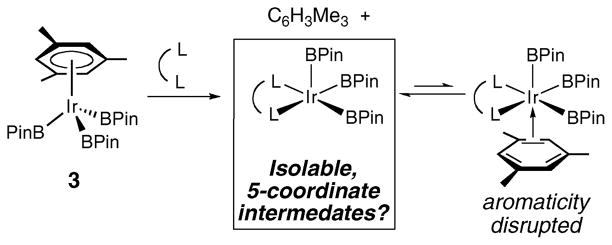
Arene displacement route to five-coordinate boryl complexes.
Complex 3 reacts cleanly with dmpe (1,2-bis(dimethylphosphino) ethane) to yield a new species, 4. 31P NMR spectra revealed two chemically inequivalent P environments present in a 2: 1 ratio. 1H NMR spectra of crude reaction mixtures showed unreacted 3 was present in addition to resonances for 4 with a 1: 2 ratio of 3: 4. At this point, it was clear that 4 was a dinuclear species, which could be obtained in good yield by adjusting the stoichiometry (eqn (1)). While equimolar solutions of 3 and dmpe catalyze C–H borylation, no conversion is observed when pure compound 4 is used. This is consistent with our previous observation that catalysis ceases abruptly when P: Ir ratios reach 3: 1. The formation of 4 suggests that a significant portion of the Ir may be sequestered in an inactive form. Consequently, the efficiency of the active forms of Ir phosphine catalysts may have been grossly underestimated.
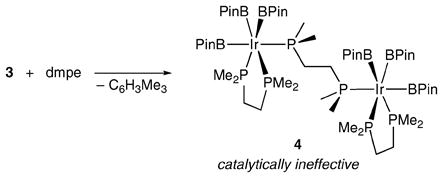 |
(1) |
The most obvious tact for preparing a five-coordinate complex is to increase the steric demands of the phosphine ligand. Indeed, 3 reacts cleanly with 1,2-bis(di-tert-butylphosphino) ethane (dtbpe) in cyclohexane or THF to afford a new complex, 5, whose 31P NMR and 1H NMR spectra were consistent with a five-coordinate structure (eqn (2)). This was confirmed by X-ray crystallography and the structure of 5 is shown in Fig. 1.‡ The geometry about Ir is a distorted square pyramid. The Ir atom lies only 0.14 Å above the least-squares plane defined by the phosphorus and basal boron atoms. The apical boron atom shows the most pronounced distortion as the Ir–B1 vector is ~15° away from being normal to the basal plane, canting towards the boryl groups and away from the phosphine ligand. Unique boryl resonances cannot be discerned in low temperature 1H NMR spectra, indicating either isochronous chemical shifts or rapidly exchanging boryl environments. Likewise, a single resonance is observed for the tert-butyl methyl groups down to −80 °C.
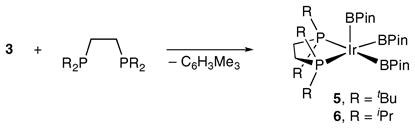
Fig. 1.
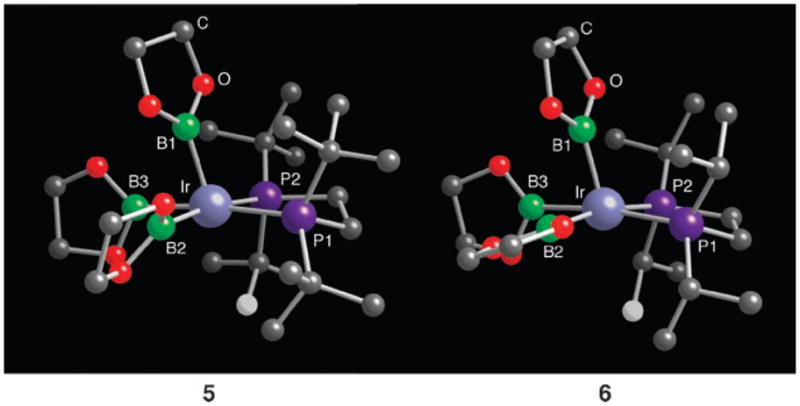
X-Ray structures of compounds 5 and 6 with BPin methyl groups omitted. The white carbons are those with the closest Ir contacts (Ir···C = 3.169 Å in 5 and 3.419 Å in 6). In both cases, the Ir–C distances are significantly longer than those in compounds with Ir C–H agostic interactions.
 |
(2) |
The structure of 5 differs substantially from the most closely related boron compound, trans-IrCl(BCat)2(PEt3)2 (Cat = catecholate), which is a distorted trigonal bipyramid with trans axial phosphines.5 Given that many nominally five-coordinate d6 transition metal structures are stabilized by agostic interactions from C–H bonds in their ligand periphery,6 we have examined this possibility for 5. NMR and IR spectra lack the signatures associated with agostic C–H interactions, which is consistent with the solid state structure where the closest Ir–C distance of 3.169(9) Å falls outside the range of 2.65–2.94 Å for Ir compounds with bona fide agostic interactions,7–12 and is ~0.5 Å longer than the agostic C–H distances for five-coordinate IrIII examples.7,11
Complex 5 reacts only slowly with arenes at room temperature. This is not surprising considering the crowded environment at the metal centre (Fig. 1). This is also consistent with the low catalytic activity of in situ generated catalysts using dtbpe as the chelating ligand. To ease the steric crowding at the Ir centre, compound 6, the isopropyl analogue of 5, was prepared in analogous fashion. The spectroscopic data for 6 are similar to those for 5 (Fig. 1). Refinement of the X-ray crystal structure was less straightforward as the boryl group containing B2 exhibited rotational disorder about the Ir–B2 vector. The conformer population was optimized by least-squares analysis in the usual way and the structure of the major conformer (73% of the total population) is the one shown in Fig. 1. The closest Ir–C methyl contact (3.419 Å) is even longer than that in 5, reflecting a bona fide five-coordinate structure for 6.
Relative to 5, 6 is significantly more reactive at room temperature as the borylation with 1,3-C6H4(CF3)2 in Scheme 3 attests, and borylation of 2-methylthiophene by 6 is even more facile, which is consistent with the greater reactivities of aromatic heterocycles in C–H borylations.1,13 While the reactions are very clean with respect to the organic substrates and products, it is evident from 1H and 31P NMR spectra that a number of Ir hydride species form.14 The differences in relative reactivities also translate to catalytic reactions. Specifically, borylations of 2-methylthiophene were examined in cyclohexane-d12 solutions at 100 °C. When catalysts were generated in situ by adding 10 mol% of the diphosphine ligand to 10 mol% (η5-indenyl)Ir(η4-1,5-cyclooctadiene), 93% conversion was observed for the dippe catalyst after 2 h, whereas the analogous reaction with the dtbpe catalysts yielded only 12% conversion. Similar relative rates were seen using isolated compounds 5 and 6, although the absolute rates approximately doubled. This is not surprising since in situ generation will have an induction period and the conversion of the precatalyst to its active form may not be quantitative.
Scheme 3.

Relative reactivities of 5 and 6 with 1,3-bis(trifluoromethyl)- benzene and 6 with 2-methylthiophene at 25 °C.
The enhanced reactivity of 6 relative to 5 arises from steric relief in the metal coordination sphere that is incumbent with removing four methyl groups from the diphosphine ligand. Fig. 2 depicts a putative transition state 7 for the C–H cleavage step. It is similar to that found by Sakaki in his computational studies of related complexes with chelating nitrogen ligands where the hydrogen is transferred from the arene to one of the boryl ligands.15,16 For compound 6, the isopropyl substituents make the metal centre more accessible for arene approach relative to 5. Fig. 2 points out a subtler feature that may contribute to the lowered barrier—namely, the orientation of the boryl ligands. In Sakaki’s transition state, the boryl ligand that facilitates transfer must be oriented such that the boron and its pinacolate oxygen atoms lie in the basal plane of the square pyramid. As seen in Fig. 2, the dihedral angle φ between the plane defined by B2 and its oxygen atoms and the basal plane in 5 is significantly larger (φ = 48°) than the corresponding angle in 6 (φ = 21°). Thus, transition state 7 should be more readily accessible from structure 6 relative to 5. The disorder of the BPin ligand containing B2 in compound 6 is also consistent with greater flexibility afforded by the isopropyl-substituted diphosphinoethane as no similar disorder was evident in the tert-butyl analogue, 5. Despite these observations, the fact remains that compounds 5 and 6 are fluxional, indicating that barriers for boryl reorientation are small, and we hesitate to ascribe too much significance to the boryl orientations in these ground states on the relative transition state energies.
Fig. 2.
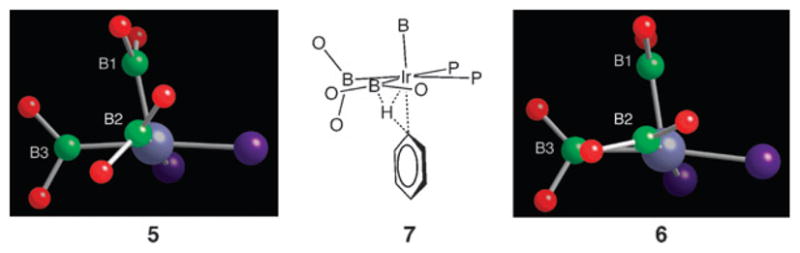
A comparison of the orientation for the B2 boryl ligands in structures 5 and 6 with the qualitative transition state 7. For compound 5, rotation about the Ir–B bond is required to reach the transition state, whereas the boryl orientation in 6 is nearly ideal for cleaving the arene C–H bond.
In summary, the steric influence of the chelating diphosphinoethane ligands has a dramatic effect on the structures and reactivities of the trisboryl complexes obtained in their reactions with arene complex 3. It is interesting that even though equimolar solutions of 3 and dmpe are catalytically active for borylation, the major product 4 obtained by reaction of 3 and dmpe is ineffective. Consequently, it is conceivable that the active catalytic species for certain phosphine-supported systems are present in very low concentration. Perhaps most importantly, the synthesis of five-coordinate boryl complexes that react directly with arenes makes it possible to probe the rate-limiting and most fundamental step in the catalytic cycle, while avoiding the potential for obfuscation by ligand dissociation that is incumbent to most precatalysts. We are actively pursuing further reactivity of compounds 5 and 6, with other arenes and heterocycles, as well as boranes. In addition, we are actively developing routes to related five-coordinate structures with other bidentate ligands.
Supplementary Material
Acknowledgments
We thank BASF, Inc. for a gift of HBPin, Professor Gregory Hillhouse for a gift of dtbpe, and the Michigan Economic Development Corp., the NIH (GM63188 to M.R.S.), the Pharmaceutical Roundtable of the ACS Green Chemistry Institute, Pfizer, Inc., and the Astellas USA Foundation (to R.E.M.) for generous financial support.
Footnotes
Electronic supplementary information (ESI) available: Spectral data for all new compounds, as well as general experimental procedures. CCDC 741048 and 741049. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b914736e
Crystal data for 5. C36H76O6B3P2Ir, M = 891.54, triclinic, P1̄, a = 11.651(4), b = 12.035(4), c = 17.151(4) Å, α = 73.82(3)°, β = 76.42(3)°, γ = 69.37(2)°, V = 2136.6(11) Å3, T = 293(2) K, Z = 2, 9677 reflections measured, 6106 unique (Rint = 0.0668), which were used in all calculations. The final R1 and wR2 were 0.0581 and 0.1428. CCDC 741048. Crystal data for 6. C32H68O6B3P2Ir, M = 835.43, monoclinic, P21/c, a = 17.821(4), b = 12.668(3), c = 18.491(4) Å, β = 104.80(3)°, V = 4035.9(14) Å3, T = 173(2) K, Z = 4, 34 198 reflections measured, 5816 unique (Rint = 0.0803), which were used in all calculations. The final R1 and wR2 were 0.0474 and 0.1035. CCDC 741049.
Contributor Information
Robert E. Maleczka, Jr, Email: maleczka@chemistry.msu.edu.
Milton R. Smith, III, Email: smithmil@msu.edu.
References
- 1.Cho JY, Tse MK, Holmes D, Maleczka RE, Jr, Smith MR., III Science. 2002;295:305–308. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]
- 2.Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J Am Chem Soc. 2002;124:390–391. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]
- 3.Halpern J. Science. 1982;217:401–407. doi: 10.1126/science.217.4558.401. [DOI] [PubMed] [Google Scholar]
- 4.We previously outlined this strategy for preparing 5-coordinate boryl complexes: Chotana GA, Maleczka RE, Jr, Smith MR., III Abstracts of Papers, 233rd ACS National Meeting; Chicago, IL, United States. March 25–29, 2007; 2007. p. INOR-188.
- 5.Clegg W, Lawlor FJ, Marder TB, Nguyen P, Norman NC, Orpen AG, Quayle MJ, Rice CR, Robins EG, Scott AJ, Souza FES, Stringer G, Whittell GR. J Chem Soc, Dalton Trans. 1998:301–309. [Google Scholar]
- 6.Kubas GJ. Metal Dihydrogen and Sigma-Bond Complexes: Structure, Theory, and Reactivity. Kluwer Academic/Plenum Publishers; New York: 2001. [Google Scholar]
- 7.Sau YK, Lee HK, Williams ID, Leung WH. Chem–Eur J. 2006;12:9323–9335. doi: 10.1002/chem.200600562. [DOI] [PubMed] [Google Scholar]
- 8.Scott NM, Pons V, Stevens ED, Heinekey DM, Nolan SP. Angew Chem, Int Ed. 2005;44:2512–2515. doi: 10.1002/anie.200463000. [DOI] [PubMed] [Google Scholar]
- 9.Clot E, Chen JY, Lee DH, Sung SY, Appelhans LN, Faller JW, Crabtree RH, Eisenstein O. J Am Chem Soc. 2004;126:8795–8804. doi: 10.1021/ja048473j. [DOI] [PubMed] [Google Scholar]
- 10.Dorta R, Goikhman R, Milstein D. Organometallics. 2003;22:2806–2809. [Google Scholar]
- 11.Clot E, Eisenstein O, Dube T, Faller JW, Crabtree RH. Organometallics. 2002;21:575–580. [Google Scholar]
- 12.Cooper AC, Streib WE, Eisenstein O, Caulton KG. J Am Chem Soc. 1997;119:9069–9070. [Google Scholar]
- 13.Takagi J, Sato K, Hartwig JF, Ishiyama T, Miyaura N. Tetrahedron Lett. 2002;43:5649–5651. [Google Scholar]
- 14.For example, the compounds (dippe)IrH3(BPin)2 and (dippe)- IrH4BPin can be seen in the milieu. These have been fully characterized and details will be published in due course. Some of these findings have been publicly disseminated. See: Vanchura BA, Ramanathan B, Chotana GA, Smith MR., III Abstracts of Papers, 237th ACS National Meeting; Salt Lake City, UT, United States. March 22–26, 2009; 2009. p. INOR-001.
- 15.Tamura H, Yamazaki H, Sato H, Sakaki S. J Am Chem Soc. 2003;125:16114–16126. doi: 10.1021/ja0302937. [DOI] [PubMed] [Google Scholar]
- 16.We find a transition state similar to 7 in computational studies of the phosphine ligated catalysts: D. A. Singleton and M. R. Smith, III, manuscript in preparation.
- 17.Hartwig and co-workers prepared a five-coordinate catecholate boryl analog of 6 using the route we previously disclosed in ref. 4. Please see: Liskey CW, Wei CS, Pahls DR, Hartwig JF. Chem Commun. 2009 doi: 10.1039/b913949d.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.