

Abstract
EPAC1 and EPAC2, two isoforms of exchange proteins directly activated by cAMP (EPAC), respond to the second messenger cAMP and regulate a wide variety of intracellular processes under physiological and pathophysiological circumstances. Herein, we report the chemical design, synthesis, and pharmacological characterization of three different scaffolds (diaryl sulfones, N,N-diarylamines, and arylsulfonamides) as highly potent and selective antagonists of EPAC2. Several selective EPAC2 antagonists have been identified including 20i (HJC0350), which has an apparent IC50 value of 0.3 µM for competing with 8-NBD-cAMP binding of EPAC2, and is about 133-fold more potent than cAMP. Compounds 1(ESI-05), 14c (HJC0338) and 20i, selected from each series, have exhibited no inhibition of EPAC1-mediated Rap1-GDP exchange activity at 25 µM, indicating that they are EPAC2-specific antagonists. Moreover, live-cell imaging studies using EPAC1, EPAC2, or PKA FRET sensor also demonstrate that 20i functions as an EPAC2 specific antagonist
INTRODUCTION
Identification and development of small-molecule probes with high potency and specificity represents a major effort for chemical biologists to explore and validate the roles of the proteins in a broader biological context.1 The most common and versatile second messenger, cyclic adenosine monophosphate (cAMP), regulates a striking number of physiological and pathophysiological processes through the signaling networks which interfere with many disease states including inflammatory disorders, immune-deficiencies, and cancer.2 Initially, the effects of cAMP were believed to be transduced by protein kinase A (PKA) and cAMP-gated ion channels. However, the contribution of more recently discovered cAMP target exchange proteins directly activated by cAMP (EPAC) is becoming more and more appreciated.3–7 EPAC proteins are now assumed to be implicated in a number of cellular processes, which researchers had previously thought to be associated only with PKA, including insulin secretion, neurotransmitter release, integrin-mediated cell adhesion, cell survival and apoptosis, gene transcription and chromosomal integrity.8–10 Therefore, advances in the understanding the role of EPAC proteins as regulators of cAMP-dependent signaling are critical for further elucidating the mechanism of cAMP signaling. cAMP analogs that selectively activate EPAC have been developed to help reveal the ability of EPAC to modulate ongoing physiological signaling.11–13 Nevertheless, novel EPAC-specific antagonists are desperately needed as diverse approaches to help elucidation of the mechanisms by which cAMP fulfills its function via EPAC.14,15 To this end, we direct our efforts on identification novel small molecules as selective EPAC antagonists to probe the functions of EPAC in relevant signaling pathways and act as potential therapeutics with unique targets.16–18
EPAC is a cAMP-activated guanine nucleotide exchange factor (cAMP-GEF) for the small GTP-binding proteins Rap1 and Rap2.3,4 There are two isoforms of EPAC, EPAC1 and EPAC2, which are coded by two independent genes with distinct tissue distributions in mammals. EPAC1 is ubiquitously expressed in all tissues with high levels of expression in the kidney, while EPAC2 is detectable most notably in the central nervous system, adrenal gland, and pancreas. Given that EPAC1 and EPAC2 share extensive sequence homology, developing EPAC1- or EPAC2-specific antagonists with the capability of discriminating the functions of EPAC1 and EPAC2 is quite essential in this field.
To identify new chemical probes with high capability of specifically inhibiting EPAC, a Maybridge Hitfinder compound library of 14,400 structurally diverse small molecules has been screened using a fluorescence-based high-throughput screening (HTS) assay.16–18 This assay was based on a fluorescent cyclic nucleotide analog, 8-NBD-cAMP, binding of which to purified full-length EPAC2 protein could lead to a dose-dependent increase in fluorescent signal, while cAMP or EPAC2 antagonists could compete with their binding and decrease the fluorescent signal in a dose-dependent manner.16 After analyzing the EPAC2 inhibition activity, some of the compounds screened from the library were applied to detect their selectivity using a secondary functional assay that could evaluate their cAMP-mediated EPAC1 GEF activity in the purified recombinant full-length EPAC1 protein. Two HTS hits 1 (ESI-05) and 2 (ESI-10) have been identified (Figure 1), which inhibit cAMP-mediated EPAC2 GEF activity with IC50 values of 0.5 µM and 18 µM, respectively. The hit compound 1 exhibits as a selective antagonist of EPAC2 without apparent activity towards EPAC1, while 2 is not exclusively specific for EPAC2.18b
Figure 1.

Structures of cAMP, HTS hits: 1 (ESI-05) and 2 (ESI-10).
In continuation of our efforts to identify novel potent and EPAC-specific antagonists,17 we have designed, synthesized and characterized three different series of new molecules namely diaryl sulfones, N,N-diarylamines, and arylsulfonamides based on the two HTS hits 1 and 2 as our lead molecules. Herein, we describe our findings on their potency and specificity as well as the structure-activity relationship (SAR) results of the new analogs.
RESULTS AND DISCUSSION
Chemistry
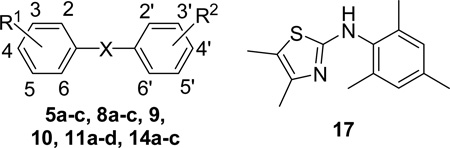
Our previous medicinal chemistry effort and molecular dockings have demonstrated that appropriate hydrophobic pharmacophores of the molecules play a crucial role for their potency and EPAC-isoform specificity.17 Therefore, in our initial drug design and SAR studies, we have attempted to retain 2,4,6-trimethyl phenyl ring of hit 1 as a privileged fragment to investigate the new molecules. As shown in Scheme 1, the diaryl sulfone derivatives 5a-c and 8a-b were prepared via Friedel–Crafts sulfonylation of 3a-c with 2,4,6-trimethylbenzene-1-sulfonyl chloride 4 or that of 7 with substituted benzenesulfonyl chlorides 6a-b in 69–96% yields. The target compound 9 was obtained by Suzuki coupling reaction of 8a with 2-fluoropyridine-5-boronic acid in the presence of Pd(dppf)Cl2 catalyst in 70% yield. Generation of 10 was achieved in 92% yield by demethylation of 8b using boron tribromide. Compounds 11a-d were produced in 77–90% overall yields by Mitsunobu reaction of 10, followed by subsequent Boc-deprotection with trifluoroacetic acid if necessary (e.g. 11c-d).
Scheme 1.

Synthesis of the Diaryl Sulfones Scaffolda
aReagents and conditions: (a) AlCl3, 25 °C, 69–96%; (b) Pd(dppf)Cl2, KOAc, ArB(OH)2, THF/EtOH/H2O, 80 °C, 70%; (c) BBr3, DCM, 0 °C to 25 °C, 92%; (d) for 11a and 11b: DIAD, PPh3, THF, 25 °C, 77–90%; for 11c and 11d: (i) DIAD, PPh3, THF, 25 °C, 87–90%; (ii) TFA, DCM, 0 °C, 98–99%.
The synthetic route to N,N-diarylamine derivatives is outlined in Scheme 2. N,N-diarylamine analogues 14a-c were prepared from substituted anilines 12a-c by palladium-catalyzed cross-coupling with 2,4,6-mesitylbromide 13 in 62–93% yields. The cyclization reaction of α-bromo ketone 15 with substituted thiourea 16 afforded N-phenylthiazoleamine 17 in 65% yield.
Scheme 2.

Synthesis of the N,N-Diarylamine Scaffolda
aReagents and conditions: (a) Pd2(dba)3, NaOtBu, (±)-BINAP, toluene, 120 °C, 62–93%; (b) EtOH, reflux, 65%.
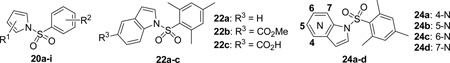
Scheme 3 illustrates the synthesis of diversely substituted arylsulfonamide analogues. Substituted pyrroles 18a-b were treated with various sulfonyl chlorides 19a-h, in the presence of 60% NaH as base, to give the corresponding arylsulfonamides 20a-i in 30–96% yields. The synthesis of indole and azaindole derivatives 22a-c and 24a-d was accomplished by the treatment of various indoles 21a-b or azaindoles 23a-d with 2,4,6-trimethylbenzene-1-sulfonyl chloride 4 in a similar fashion. The indole ester 22b was saponified to generate the carboxylic acid 22c in 66% yield.
Scheme 3.

Synthesis of the Arylsulfonamide Scaffolda
aReagents and conditions: (a) 60% NaH, THF, 0 °C to 25 °C, 30–96%; (b) 2 N LiOH/H2O, MeOH/H2O, 25 °C, 66%.
In Vitro Evaluation of EPAC2 Inhibition
All compounds have been evaluated for their inhibitory activity against the recombinant fusion protein EPAC2 using 8-NBD-cAMP as the artificial substrate to determine IC50 values, while cAMP competes with 8-NBD-cAMP in binding EPAC2 with an IC50 of 40 µM.16 Table 1 shows apparent IC50 values of the diaryl sulfones and N,N-diarylamine scaffolds for competing with 8-NBD-cAMP in binding EPAC2. Among these new diaryl sulfone analogues, compound 5a is the most potent antagonist with an IC50 value of 0.7 µM. When one aryl ring of the diaryl sulfones is substituted with too bulky groups including 4-pentyl, 4-cyclohexyl, and 4-O-piperidinyl, these compounds such as 5b-c, 9, and 11a-d display very low to no activity. When the methyl group in one ring of hit 1 is replaced by iodo, methoxy or hydroxyl, compounds 8a-b and 10 exhibit a slightly lower potency with IC50 of 4 µM, 1.9 µM and 13.5 µM, respectively. Among N,N-diarylamine derivatives, compound 14a is a new analogue of hit 2 with the 4-amino group of one phenyl ring replaced by 4-methyl substituent while 5-amino-benzenesulfonic acid moiety replaced by 2,4,6-trimethylphenyl fragment. 14a demonstrates a substantially enhanced activity with an IC50 of 3.8 µM, when compared with the hit lead 2. Interestingly, analogues 14b-c with the 3,5-dichloro or 2,5-dichloro exhibit a significantly better potency with IC50 values of 0.9 µM and 0.4 µM, respectively, than the 4-methyl derivative 14a. Compound 17 containing a thiazole ring as one of the aromatic moieties was also explored, and found no activity.
Table 1.
Apparent IC50 values of the diaryl sulfones and N,N-diarylamine scaffolds for competing with 8-NBD-cAMP in binding EPAC2.
 | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | X | IC50 (µM) |
Relative Potency (RA)* |
| cAMP | 40 | 1 | |||
| 1 | 4-methyl | 2’,4’,6’-trimethyl | SO2 | 0.5 | 80 |
| 2 | 4-NH2 | 2’-SO3H-4’-NH2 | NH | 18 | 2.2 |
| 5a | 2,4,5-trimethyl | 2’,4’,6’-trimethyl | SO2 | 0.7 | 57.1 |
| 5b | 4-pentyl | 2’,4’,6’-trimethyl | SO2 | >300 | <0.13 |
| 5c | 4-cyclohexyl | 2’,4’,6’-trimethyl | SO2 | >300 | <0.13 |
| 8a | 4-iodo | 2’,4’,6’-trimethyl | SO2 | 4 | 10 |
| 8b | 4-methoxy | 2’,4’,6’-trimethyl | SO2 | 1.9 | 21.1 |
| 9 | 4-(2-fluoro-5-pyridinyl) | 2’,4’,6’-trimethyl | SO2 | >300 | <0.13 |
| 10 | 4-hydroxyl | 2’,4’,6’-trimethyl | SO2 | 13.5 | 3.0 |
| 11a | 4-O-cyclohexyl | 2’,4’,6’-trimethyl | SO2 | >300 | <0.13 |
| 11b | 4-O-piperidinyl-1-Boc | 2’,4’,6’-trimethyl | SO2 | >300 | <0.13 |
| 11c | 4-O-piperidinyl | 2’,4’,6’-trimethyl | SO2 | >300 | <0.13 |
| 11d | 4-O-2-ethylamino | 2’,4’,6’-trimethyl | SO2 | >300 | <0.13 |
| 14a | 4-methyl | 2’,4’,6’-trimethyl | NH | 3.8 | 10.5 |
| 14b | 3,5-dichloro | 2’,4’,6’-trimethyl | NH | 0.9 | 44.4 |
| 14c | 2,5-dichloro | 2’,4’,6’-trimethyl | NH | 0.4 | 100 |
| 17 | NH | >300 | <0.13 | ||
RA = IC50, cAMP/ IC50, compound
Based upon the structural features of hits 1 and 2 as well as the preliminary SAR results from the above efforts, we have designed and investigated a series of arylsulfonamides as a new chemical entity of EPAC2 antagonists. The findings of the arylsulfonamide analogues are summarized in Table 2. Compound 20a with 2-ethylpyrrole moiety displays a high potency of EPAC2 inhibition with an IC50 of 0.5 µM. Analogues 20b-h with 2,4,6-trimethylphenyl moiety replaced by other substituted phenyl groups exhibit apparently dampened activities in comparison with 20a. Interestingly, 4-trifluoromethyl substituent on the phenyl ring of analogue 20e results in a dramatic loss of EPAC inhibitory activity. Compound 20i bearing the 2,4-dimethylpyrrole fragment displays a remarkable activity with an IC50 value of 0.3 µM, which is about 133-fold more potent than cAMP (Table 2). Analogues 22a-c and 24a-d have been investigated to replace the pyrrole scaffold with the indole or azaindole moieties. Among these new molecules, the indole analogue 22a and the 7-azaindole analogue 24d display an almost retained activity of EPAC2 inhibition in contrast with pyrrole derivatives. Analogues 22c and 24a-c are less active with a moderate potency. An ester group on indole ring of 22b results in a dramatic loss of activity, while the saponification of the ester 22b to carboxylic acid 22c can significantly regain the inhibitory activity of EPAC2. Taken together, the preliminary SAR results support that appropriate hydrophobic pharmacophores of the molecules play a crucial role for their activity targeting the EPAC proteins, and more potent ligands may be achieved through further systematic and extensive structural optimizations.
Table 2.
Apparent IC50 values of the arylsulfonamide scaffold for competing with 8-NBD-cAMP in binding EPAC2.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | IC50 (µM) |
Relative Potency (RA)* |
Entry | IC50 (µM) | Relative Potency (RA)* |
| 20a | 2-ethyl | 2,4,6-trimethyl | 0.5 | 80 | 22a | 1.2 | 33.3 |
| 20b | 2-ethyl | 4-methyl | 4 | 10 | 22b | >300 | <0.13 |
| 20c | 2-ethyl | 4-methoxy | 8 | 5 | 22c | 10.8 | 3.7 |
| 20d | 2-ethyl | 4-chloro | 8 | 5 | 24a | 3.8 | 10.5 |
| 20e | 2-ethyl | 4-trifluoromethyl | >300 | <0.13 | 24b | 8.9 | 4.5 |
| 20f | 2-ethyl | 2-methyl | 5.3 | 7.5 | 24c | 12.6 | 3.2 |
| 20g | 2-ethyl | 3,5-dimethyl | 4.7 | 8.5 | 24d | 2.4 | 16.7 |
| 20h | 2-ethyl | 2,4-dimethyl | 1.3 | 30.8 | cAMP | 40 | 1 |
| 20i | 2,4-dimethyl | 2,4,6-trimethyl | 0.3 | 133 | |||
RA = IC50, cAMP/ IC50, compound
Selectivity for EPAC2
To investigate the EPAC-isoform specificity, the inhibitory potencies of three most potent compounds 1, 14c and 20i selected from three different scaffolds have been evaluated in suppressing cAMP-mediated EPAC1 and EPAC2 GEF activities using purified recombinant full-length EPAC1 and EPAC2 proteins (Figure 3).16,19,20 These three analogues have shown to be able to inhibit EPAC2 GEF activity to basal levels at 25 µM concentration in the presence of equal concentration of cAMP. All of these compounds were found not to inhibit EPAC1-mediated Rap1-GDP exchange activity at 25 µM in the presence of equal concentration of cAMP, indicating that they are EPAC2-specific antagonists. To further investigate their specificity for EPAC over PKA, we performed counter-screening assays that measure type I and II PKA holoenzyme activities, respectively.16,21,22 All these analogues have demonstrated not to significantly alter cAMP-induced type I and II PKA holoenzymes activation at 25 µM concentration, while the selective PKA inhibitor H89, the positive control, blocked the type I or II PKA activities completely. These results suggest that compounds 1, 14c (HJC0338) and 20i (HJC0350) are EPAC2-specific antagonists that selectively block cAMP-induced EPAC2 activation but do not inhibit cAMP-mediated PKA activation. It should be emphasized that while these compounds exert no activity towards PKA, their pharmacological properties should be further evaluated to ensure that they do not disrupt other components of the cyclic nucleotide signaling pathways, such as the cyclic-nucleotide regulated ion channels (CNG and HCN channels), phosphodiesterases or adenylyl cyclases.
Figure 3. Specificity of EPAC antagonists 1, 14c and 20i.

(A) cAMP-mediated EPAC1 GEF activity measured in the presence or absence of EPAC antagonists: open squares, EPAC1 alone; closed squares: EPAC1 in the presence of 25 µM cAMP; closed triangles up, EPAC1 with 25 µM cAMP and 25 µM 1; open circles, EPAC1 with 25 µM cAMP and 25 µM 14c; closed circles, EPAC1 with 25 µM cAMP and 25 µM 20i. (B) cAMP-mediated EPAC2 GEF activity measured in the presence or absence of EPAC antagonists: open squares, EPAC2 alone; closed squares: EPAC2 in the presence of 25 µM cAMP; closed triangles up, EPAC2 with 25 µM cAMP and 25 µM 1; open circles, EPAC2 with 25 µM cAMP and 25 µM 14c; closed circles, EPAC2 with 25 µM cAMP and 25 µM 20i.
To test whether 20i is capable of modulating EPAC activation in living cells and to further validate that 20i is an EPAC2 isoform-specific antagonist, we tested the compound using HEK293 cells stably expressing an EPAC2- or EPAC1-based fluorescence resonance energy transfer (FRET) sensor, EPAC2-FL or EPAC1-FL.23 As expected, stimulation of HEK293/EPAC2-FL cells by 8-(4-chlorophenylthio)-2’-O-methyladenosine-3’,5’-cyclic monophosphate acetoxymethyl ester (007-AM), a membrane permeable EPAC selective cAMP analog, led to a decrease of FRET measured as an increase of the 485/535 nm FRET ratio (Figure 4A). Pretreatment of HEK293/EPAC2-FL cells with 10 µM 20i fully blocked the 007-AM induced decrease of FRET (Figure 4B). In contrast, 20i was found incapable of blocking the 007-AM induced decrease of FRET changes in HEK293/EPAC1-FL (Figure 4C & 4D). Using HEK293 cells stably expressing the PKA sensor AKAR3,24 it was also possible to demonstrate the stimulatory action of 8-Br-cAMP-AM at PKA, as measured as an increase FRET with a corresponding decrease of the 485/535 nm FRET ratio (Figure 5A). This action of 8-Br-cAMPAM to activate PKA was not inhibited by 20i (Figure 5B). These live-cell results are in complete agreement with our biochemical data and further validate that 20i is an effective EPAC2-isoform specific antagonist.
Figure 4. Effects of 20i on 007-AM mediated cellular activation of EPAC.

For HEK293 cells stably expression EPAC2-FL (A, B) or EPAC1-FL (C, D), it was demonstrated that 10 µM 20i blocked EPAC2 but not EPAC1 activation in response to 10 µM 007-AM.
Figure 5. Effects of 20i on 8-Br-cAMP-AM mediated cellular activation of PKA.

HEK293 cells stably expression AKAR3, it was demonstrated that 10 µM 20i did not affect PKA activation in response to 10 µM 8-Br-cAMP-AM.
CONCLUSION
In summary, this work presents three series of new molecules that exhibit outstanding selectivity and potency for the inhibition of EPAC2 through a preliminary hit-to-lead optimization process. Analogues 14c and 20i have been identified as the most potent EPAC2-specific antagonists with IC50 values of 0.4 and 0.3 µM, which are about 100- and 133-fold more potent than cAMP, respectively. Accumulating studies support that EPAC proteins may represent promising targets for the design of new therapies for various human disorders such as diabetes, heart disease and cancer.25–29 Therefore, these compounds may not only be valuable pharmacological tools to explore physiological and pathological processes related to signaling pathways that are regulated by EPAC proteins, but also have great potential to be developed as novel molecular therapeutics for relevant human diseases. Further structural optimization of these scaffolds based upon our SAR findings and in vivo pharmacological evaluation of selected EPAC antagonists in disease models are currently underway.
EXPERIMENTAL SECTION
General Chemistry Information
All commercially available starting materials and solvents were reagent grade, and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Brucker-600 (1H, 600 MHz; 13C, 150 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: Nano ESI spray voltage was 1.8 kV; Capillary temperature was 275 °C and the resolution was 60,000; Ionization was achieved by positive mode. Melting points were measured on a Thermo Scientific Electrothermal Digital Melting Point Apparatus and uncorrected. Purities of final compounds were established by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/VIS). HPLC analysis conditions: Waters µBondapak C18 (300 × 3.9 mm); flow rate 0.5 mL/min; UV detection at 270 and 254 nm; linear gradient from 10% acetonitrile in water to 100% acetonitrile in water in 20 min followed by 30 min of the last-named solvent (for 11c and 11d, the same mobile phase with 0.1% TFA). All biologically evaluated compounds are > 95% pure.
1,3,5-Trimethyl-2-(2,4,5-trimethylbenzenesulfonyl)benzene (5a)
A mixture of mesitylsulfonyl chloride (219 mg, 1.0 mmol), 1,2,4-trimethyl-benzene (125 mg, 1.05 mmol) and AlCl3 (266 mg, 2.0 mmol) in DCM (5 mL) was stirred for 2 h at room temperature. The mixture was then poured into 10 mL of 5% HCl (aq.), and extracted by DCM (30 mL). The organic phase was washed by aqueous KHCO3 solution, brine, and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was purified by silica gel column chromatography (Hexane/EtOAc = 10/1) to give the desired product as a white solid (290 mg, 96%); mp 147-148 °C; HPLC purity 98.4% (tR = 25.33 min). 1H NMR (600 MHz, DMSO-d6) δ 7.70 (s, 1H), 7.11 (s, 1H), 7.05 (s, 2H), 2.38 (s, 6H), 2.27 (s, 6H), 2.25 (s, 3H), 2.07 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 143.0, 142.3, 138.5, 138.4, 134.4, 134.3, 133.7, 133.3, 132.0, 128.2, 21.8, 20.5, 19.1, 18.9, 17.8. HRMS (ESI) calcd for C18H23O2S 303.1413 (M + H)+, found 303.1407.
1,3,5-Trimethyl-2-(4-pentylbenzenesulfonyl)benzene (5b)
Compound 5b was prepared in 90% yield by a procedure similar to that used to prepare compound 5a. The title compound was obtained as a pale yellow oil. HPLC purity 97.2% (tR = 26.16 min). 1H NMR (600 MHz, CDCl3) δ 7.68 (d, 2H, J = 7.2 Hz), 7.25 (d, 2H, J = 6.6 Hz), 6.93 (s, 2H), 2.63 (t, 2H, J = 7.2 Hz), 2.49 (s, 6H), 2.28 (s, 3H), 1.57-1.62 (m, 2H), 1.28-1.32 (m, 4H), 0.88 (t, 2H, J = 6.6 Hz). 13C NMR (150 MHz, CDCl3) δ 148.4, 143.3, 140.9, 140.1, 134.3, 132.3, 128.9, 126.4, 35.9, 31.5, 30.8, 22.9, 22.6, 21.1, 14.1. HRMS (ESI) calcd for C20H27O2S 331.1726 (M + H)+, found 331.1734.
2-(4-Cyclohexylbenzenesulfonyl)-1,3,5-trimethylbenzene (5c)
Compound 5c was prepared in 75% yield by a procedure similar to that used to prepare compound 5a. The title compound was obtained as a white solid (mp 102-103 °C). HPLC purity 99.8% (tR = 26.29 min). 1H NMR (600 MHz, CDCl3) δ 7.69 (d, 2H, J = 8.4 Hz), 7.27 (d, 2H, J = 8.4 Hz), 6.93 (s, 2H), 2.60 (s, 6H), 2.52-2.55 (m, 1H), 2.27 (s, 3H), 1.82-1.83 (m, 4H), 1.73-1.75 (m, 1H), 1.34-1.41 (m, 4H), 1.22-1.26 (m, 1H). 13C NMR (150 MHz, CDCl3) δ 153.3, 143.2, 141.0, 140.0, 134.3, 132.2, 127.4, 126.4, 44.6, 34.2, 26.7, 26.0, 22.9, 21.0. HRMS (ESI) calcd for C21H27O2S 343.1726 (M + H)+, found 343.1730.
2-(4-Iodobenzenesulfonyl)-1,3,5-trimethylbenzene (8a)
A mixture of 4-Iodo-benzenesulfonyl chloride (302 mg, 1.0 mmol), mesitylene (120 mg, 1.0 mmol) and AlCl3 (150 mg, 1.2 mmol) in DCM (5 mL) was stirred for 2 h at room temperature. The mixture was then poured into 10 mL of 5% HCl (aq.), and extracted by DCM (30 mL). The organic phase was washed by aqueous KHCO3, brine, and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was purified by silica gel column chromatography (Hexane/EtOAc = 10/1) to give the desired product (266 mg, 69%) as a white solid (mp 123-124 °C). HPLC purity 96.0% (tR = 23.82 min). 1H NMR (600 MHz, CDCl3) δ 7.82 (d, 2H, J = 8.4 Hz), 7.48 (d, 2H, J = 7.8 Hz), 6.95 (s, 2H), 2.58 (s, 6H), 2.30 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 143.9, 143.4, 140.2, 138.3, 133.5, 132.5, 127.8, 100.0, 23.0, 21.2. HRMS (ESI) calcd for C15H16IO2S 386.9910 (M + H)+, found 386.9915.
2-(4-Methoxybenzenesulfonyl)-1,3,5-trimethylbenzene (8b)
Compound 8b was prepared in 86% yield by a procedure similar to that used to prepare compound 8a. The title compound was obtained as a white solid (mp 132-133 °C). HPLC purity 98.9% (tR = 21.80 min). 1H NMR (600 MHz, CDCl3) δ 7.72 (d, 2H, J = 8.4 Hz), 6.93 (d, 2H, J = 7.2 Hz), 6.92 (s, 2H), 3.84 (s, 3H), 2.60 (s, 6H), 2.28 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 162.9, 143.2, 140.0, 135.4, 134.7, 132.3, 128.6, 114.2, 55.7, 23.0, 21.1. HRMS (ESI) calcd for C16H19O3S 291.1049 (M + H)+, found 291.1056.
2-Fluoro-5-[4-(2,4,6-trimethylbenzenesulfonyl)phenyl]pyridine (9)
To a solution of 8a (77 mg, 0.2 mmol) and 2-Fluoropyridine-5-boronic acid (28 mg, 0.2 mmol) in THF/EtOH/H2O (1 mL/1 mL/1 mL) was added KOAc (59 mg, 0.6 mmol) and then Pd(dppf)Cl2 (16 mg, 0.02 mmol). The resulting mixture was deoxygenated via five vacuum/N2-refill cycles. The mixture was stirred at 80 °C for 18 h, and was then concentrated under vacuum. The residue was partitioned between EtOAc (50 mL) and H2O (20 mL). The organic layer was separated and washed with brine (10 mL), dried over anhydrous Na2SO4, filtrated and concentrated to give an oily residue. This residue was purified with silica gel column (Hexane/EtOAc = 3/1) to obtain 9 (50 mg, 70%) as a red solid (mp 150-151 °C). HPLC purity 95.1% (tR = 22.50 min). 1H NMR (600 MHz, CDCl3) δ 8.39-8.41 (m, 1H), 7.95-7.98 (m, 1H), 7.88 (d, 2H, J = 8.4 Hz), 7.62-7.65 (m, 2H), 7.03 (d, 1H, J = 8.4 Hz), 6.96 (s, 2H), 2.61 (s, 6H), 2.30 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 164.5, 162.9, 146.3, 143.8, 143.3, 141.0, 140.2, 140.1, 133.6, 133.2, 132.4, 127.6, 127.2, 110.1, 109.9, 23.0, 21.1. HRMS (ESI) calcd for C20H19FNO2S 356.1115 (M + H)+, found 356.1120.
4-(2,4,6-Trimethylbenzenesulfonyl)phenol (10)
To a solution of 8b (350 mg, 1.2 mmol) in 10 mL of DCM was added 1 N BBr3/DCM (1.45 mL, 1.45 mmol) at 0 °C. The resulting mixture was stirred at r.t. for 16 h. The solution was diluted with EtOAc (50 mL), washed with H2O (10 mL) and brine (10 mL). The organic layer was dried over anhydrous Na2SO4 and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (Hexane/EtOAc = 3/1) to give the desired product (306 mg, 92%) as a white solid (mp 180-181 °C). HPLC purity 99.4% (tR = 19.46 min). 1H NMR (600 MHz, CDCl3) δ 7.64 (d, 2H, J = 8.4 Hz), 6.93 (s, 2H), 6.86 (d, 2H, J = 8.4 Hz), 6.23 (bs, 1H), 2.58 (s, 6H), 2.29 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 160.0, 143.4, 140.0, 135.1, 134.4, 132.4, 128.8, 115.8, 23.0, 21.1. HRMS (ESI) calcd for C15H17O3S 277.0893 (M + H)+, found 277.0898.
2-(4-Cyclohexyloxybenzenesulfonyl)-1,3,5-trimethylbenzene (11a)
To a solution of 10 (50 mg, 0.18 mmol) and Ph3P (58 mg, 0.22 mmol) in THF (5 mL) was added cyclohexanol (36 mg, 0.36 mmol) and DIAD (44 mg, 0.22 mmol). The reaction mixture was stirred at r.t. for 16 h, and then it was partitioned between EtOAc (50 mL) and H2O (20 mL). The organic layer was washed with brine (10 mL), dried with anhydrous Na2SO4, and concentrated to give the crude product. This residue was purified with silica gel column (hexane/EtOAc = 7/1) to afford the desired product as a colorless oil (50 mg, 77%). HPLC purity 99.7% (tR = 25.64 min). 1H NMR (600 MHz, CDCl3) δ 7.69 (d, 2H, J = 7.2 Hz), 6.91 (s, 2H), 6.91 (d, 2H, J = 6.6 Hz), 4.30 (s, 1H), 2.60 (s, 6H), 2.27 (s, 3H), 1.93-195 (m, 2H), 1.75-1.77 (m, 2H), 1.47-1.57 (m, 3H), 1.29-1.39 (m, 3H). 13C NMR (150 MHz, CDCl3) δ 161.4, 143.1, 139.9, 134.8, 134.8, 132.3, 128.6, 115.6, 75.8, 31.6, 25.6, 23.7, 23.0, 21.1. HRMS (ESI) calcd for C21H27O3S 359.1675 (M + H)+, found 359.1680.
4-[4-(2,4,6-Trimethylbenzenesulfonyl)phenoxy]-piperidine-1-carboxylic acid tert-butyl ester (11b)
Compound 11b was prepared in 90% yield by a procedure similar to that used to prepare compound 11a. The title compound was obtained as a colorless oil. HPLC purity 99.9% (tR = 24.54 min). 1H NMR (600 MHz, CDCl3) δ 7.71 (d, 2H, J = 9.0 Hz), 6.93 (s, 2H), 6.91 (d, 2H, J = 8.4 Hz), 4.52–4.54 (m, 1H), 3.65-3.69 (m, 2H), 3.33-3.37 (m, 2H), 2.60 (s, 6H), 2.29 (s, 3H), 1.90-1.93 (m, 2H), 1.73-1.77 (m, 2H), 1.46 (s, 9H).
4-[4-(2,4,6-Trimethylbenzenesulfonyl)phenoxy]piperidine (11c)
To a solution of 11b (128 mg, 0.28 mmol) in DCM (5 mL) was added TFA (1 mL) at 0 °C. The mixture was stirred at 0 °C for 1 h. The reaction mixture was concentrated, and the residue was partitioned between EtOAc (50 mL) and 1 N NaHCO3 (10 mL). The organic layer was washed with brine (10 mL), dried with anhydrous Na2SO4, and concentrated to give the crude product. This residue was purified with silica gel column (DCM/MeOH = 10/1) to provide 11c (100 mg, 99%) as a white solid (mp 95-97 °C). HPLC purity 99.5% (tR = 17.63 min). 1H NMR (600 MHz, CDCl3) δ 8.55 (bs, 1H), 7.70 (d, 2H, J = 7.2 Hz), 6.92 (d, 2H, J = 9.0 Hz), 6.91 (s, 2H), 4.67 (s, 1H), 3.28 (t, 2H, J = 8.4 Hz), 3.12-3.14 (m, 2H), 2.57 (s, 6H), 2.27 (s, 3H), 2.17 (t, 2H, J = 7.8 Hz), 2.02-2.04 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 159.8, 143.4, 139.8, 136.1, 134.2, 132.3, 128.7, 115.6, 68.9, 40.2, 27.2, 22.9, 21.0. HRMS (ESI) calcd for C20H26NO3S 360.1628 (M + H)+, found 360.1631.
2-[4-(2,4,6-Trimethylbenzenesulfonyl)phenoxy]ethylamine (11d)
Compound 11d was prepared in 85% yield (two steps) by a procedure similar to that used to prepare compounds 11a and 11c. The title compound was obtained a pale red oil. HPLC purity 97.7% (tR = 16.68 min). 1H NMR (600 MHz, CDCl3) δ7.64 (d, 2H, J = 7.2 Hz), 6.89 (s, 2H), 6.88 (d, 2H, J = 7.2 Hz), 5.40-5.48 (bs, 2H), 4.02-4.03 (m, 2H), 3.10-3.12 (m, 2H), 2.52 (s, 6H), 2.23 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 161.6, 143.4, 139.8, 135.7, 134.2, 132.3, 128.5, 114.7, 67.1, 40.0, 22.8, 21.1. HRMS (ESI) calcd for C17H22NO3S 320.1315 (M + H)+, found 320.1319.
p-Tolyl-(2,4,6-trimethylphenyl)amine (14a)
NaOtBu (115 mg, 1.2 mmol), Pd2(dba)3 (92 mg, 0.1 mmol) and (±)-BINAP (124 mg, 0.2 mmol) were placed into a flask and dissolved into distilled toluene (5 mL). To this solution was added mesityl bromide (995 mg, 5.0 mmol) and p-tolylamine (107 mg, 1.0 mmol) dropwise with stirring at room temperature and the mixture was refluxed at 120 °C for 24 h. After the mixture was cooled, 10 mL of 5% HCl (aq.) was added and extracted with EtOAc (50 mL). The combined organic layer was washed with NaHCO3 and dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/EtOAc = 10/1) to give the desired product as a pale yellow oil (210 mg, 93%). HPLC purity 99.4% (tR = 24.58 min). 1H NMR (600 MHz, CDCl3) δ 7.11 (d, 2H, J = 7.8 Hz), 7.09 (s, 2H), 6.57 (d, 2H, J = 7.8 Hz), 5.12 (s, 1H), 2.47 (s, 3H), 2.40 (s, 3H), 2.33 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 144.4, 136.1, 135.7, 135.1, 129.8, 129.3, 127.1, 113.5, 21.0, 20.5, 18.3. HRMS (ESI) calcd for C16H20N 226.1590 (M + H)+, found 226.1593.
(3,5-Dichlorophenyl)-(2,4,6-trimethylphenyl)amine (14b)
Compound 14b was prepared in 68% yield by a procedure similar to that used to prepare compound 14a. The title compound was obtained as a pale yellow solid (mp 99-100 °C). HPLC purity 98.8% (tR = 27.53 min). 1H NMR (600 MHz, CDCl3) δ 7.00 (s, 2H), 6.73 (s, 1H), 6.37 (s, 2H), 5.21 (s, 1H), 2.36 (s, 3H), 2.20 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 148.8, 136.7, 136.4, 135.7, 133.9, 129.6, 117.7, 111.3, 21.0, 18.2. HRMS (ESI) calcd for C15H16Cl2N 280.0654 (M + H)+, found 280.0653.
(2,5-Dichlorophenyl)-(2,4,6-trimethylphenyl)amine (14c)
Compound 14c was prepared in 62% yield by a procedure similar to that used to prepare compound 14a. The title compound was obtained as a pale yellow solid (mp 91-93 °C). HPLC purity 99.5% (tR = 27.32 min). 1H NMR (600 MHz, CDCl3) δ 7.22 (d, 1H, J = 8.4 Hz), 6.98 (s, 2H), 6.62 (dd, 1H, J1 = 7.8 Hz, J2 = 1.8 Hz), 6.14 (d, 1H, J = 2.4 Hz), 5.67 (s, 1H), 2.33 (s, 3H), 2.16 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 143.8, 136.9, 136.5, 133.9, 133.8, 130.1, 129.6, 117.8, 117.2, 112.1, 21.1, 18.1. HRMS (ESI) calcd for C15H16Cl2N 280.0654 (M + H)+, found 280.0656.
(4,5-Dimethylthiazol-2-yl)-(2,4,6-trimethylphenyl)amine (17)
To a solution of (2,4,6-Trimethyl-phenyl)-thiourea (49 mg, 0.25 mmol) in EtOH (5 mL) was added 3-bromo-butan-2-one (38 mg, 0.25 mmol). The mixture was stirred at 90 °C for 1 h. The solution was diluted with EtOAc (30 mL), and washed with H2O (10 mL) and brine (10 mL). The organic layer was dried over Na2SO4, and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/EtOAc = 3/1) to give the desired product (40 mg, 65%) as a pale yellow solid (mp 180-182 °C). HPLC purity 97.5% (tR = 22.67 min). 1H NMR (600 MHz, CDCl3) δ 7.70-8.00 (bs, 1H), 6.94 (s, 2H), 2.30 (s, 3H), 2.27 (s, 6H), 2.09 (s, 3H), 2.02 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 167.4, 143.1, 137.4, 136.9, 135.5, 129.5, 113.2, 21.1, 18.2, 14.5, 11.1. HRMS (ESI) calcd for C14H19N2S 247.1263 (M + H)+, found 247.1267.
2-Ethyl-1-(2,4,6-trimethylbenzenesulfonyl)-1H-pyrrole (20a)
To a solution of 2-ethyl-1H-pyrrole (~80% purity) (24 mg, 0.25 mmol) and mesitylsulfonyl chloride (110 mg, 0.5 mmol) in 5 mL of THF was added 60% NaH (24 mg, 0.6 mmol) at 0 °C. The resulting mixture was stirred at r.t. for 16 h. The solution was diluted with EtOAc (50 mL), washed with 1 N HCl (aq.) (10 mL) and brine (10 mL). The organic layer was dried over anhydrous Na2SO4 and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (Hexane/EtOAc = 10/1) to give the desired product as a pale yellow solid (20 mg, 36%); mp 74-75 °C; HPLC purity 95.2% (tR = 25.07 min). 1H NMR (600 MHz, CDCl3) δ 7.29-7.30 (m, 1H), 6.95 (s, 2H), 6.13-6.14 (m, 1H), 5.96-5.98 (m, 1H), 2.46 (s, 6H), 2.34-2.38 (m, 2H), 2.31 (s, 3H), 1.07 (t, 3H, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 144.0, 140.2, 136.5, 133.5, 132.3, 122.3, 110.0, 109.1, 22.4, 21.2, 19.3, 12.4. HRMS (ESI) calcd for C15H20NO2S 278.1209 (M + H)+, found 278.1205.
2-Ethyl-1-(toluene-4-sulfonyl)-1H-pyrrole (20b)
Compound 20b was prepared in 30% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a colorless oil. HPLC purity 98.0% (tR = 22.25 min). 1H NMR (600 MHz, CDCl3) δ 7.64 (d, 2H, J = 8.4 Hz), 7.28 (d, 2H, J = 8.4 Hz), 7.27 (d, 1H, J = 8.4 Hz), 6.20 (t, 1H, J = 3.0 Hz), 5.99 (d, 1H, J = 3.0 Hz), 2.68 (q, 2H, J = 7.2 Hz), 2.40 (s, 3H), 1.16 (t, 3H, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 144.8, 137.5, 136.7, 130.1, 126.9, 122.4, 111.3, 111.1, 21.7, 20.6, 12.8. HRMS (ESI) calcd for C13H16NO2S 250.0896 (M + H)+, found 250.0900.
2-Ethyl-1-(4-methoxybenzenesulfonyl)-1H-pyrrole (20c)
Compound 20c was prepared in 38% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a pale red solid (mp 73-74 °C). HPLC purity 99.5% (tR = 21.63 min). 1H NMR (600 MHz, CDCl3) δ 7.72 (d, 2H, J = 10.2 Hz), 7.28 (t, 1H, J = 1.2 Hz), 6.95 (d, 2H, J = 11.4 Hz), 6.19 (t, 1H, J = 3.0 Hz), 5.99 (d, 1H, J = 1.8 Hz), 3.85 (s, 3H), 2.69 (q, 2H, J = 7.8 Hz), 1.16 (t, 3H, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 163.7, 137.4, 131.1, 129.2, 122.2, 114.7, 111.2, 111.0, 55.8, 20.6, 12.8. HRMS (ESI) calcd for C13H16NO3S 266.0845 (M + H)+, found 266.0849.
1-(4-Chlorobenzenesulfonyl)-2-ethyl-1H-pyrrole (20d)
Compound 20d was prepared in 76% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a pale red solid (mp 61-62 °C). HPLC purity 95.6% (tR = 22.68 min). 1H NMR (600 MHz, CDCl3) δ 7.68 (d, 2H, J = 7.2 Hz), 7.47 (d, 2H, J = 7.2 Hz), 7.27 (d, 1H, J = 3.6 Hz), 6.23 (t, 1H, J = 3.6 Hz), 6.02 (d, 1H, J = 3.6 Hz), 2.68 (q, 2H, J = 7.2 Hz), 1.18 (t, 3H, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 140.4, 138.1, 137.6, 129.8, 128.3, 122.4, 111.9, 111.6, 20.7, 12.8. HRMS (ESI) calcd for C12H13ClNO2S 271.0350 (M + H)+, found 270.0355.
2-Ethyl-1-(4-trifluoromethyl-benzenesulfonyl)-1H-pyrrole (20e)
Compound 20e was prepared in 58% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a pale red solid (mp 55-57 °C). HPLC purity 96.0% (tR = 22.82 min). 1H NMR (600 MHz, CDCl3) δ 7.86 (d, 2H, J = 7.8 Hz), 7.76 (d, 2H, J = 7.8 Hz), 7.28-2.30 (m, 1H), 6.25 (d, 1H, J = 1.8 Hz), 6.03-6.05 (m, 1H), 2.68 (q, 2H, J = 7.2 Hz), 1.18 (t, 3H, J = 7.8 Hz). HRMS (ESI) calcd for C13H13F3NO2S 304.0614 (M + H)+, found 304.0602.
2-Ethyl-1-(toluene-2-sulfonyl)-1H-pyrrole (20f)
Compound 20f was prepared in 73% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a pale red oil (mp 55-57 °C). HPLC purity 98.3% (tR = 22.42 min). 1H NMR (600 MHz, CDCl3) δ 7.50 (d, 2H, J = 7.2 Hz), 7.31-7.36 (m, 3H), 6.24-6.26 (m, 1H), 6.07-6.09 (m, 1H), 2.55 (q, 2H, J = 7.2 Hz), 2.54 (s, 3H), 1.13 (t, 3H, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 138.6, 138.0, 137.6, 133.7, 132.9, 128.3, 126.5, 123.0, 110.7, 110.3, 20.3, 20.0, 12.6. HRMS (ESI) calcd for C13H16NO2S 250.0896 (M + H)+, found 250.0900.
1-(3,5-Dimethylbenzenesulfonyl)-2-ethyl-1H-pyrrole (20g)
Compound 20g was prepared in 67% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a pale red solid (mp 67-70 °C). HPLC purity 98.2% (tR = 23.09 min). 1H NMR (600 MHz, CDCl3) δ 7.36 (s, 2H), 7.29 (s, 1H), 7.19 (s, 1H), 6.21 (t, 1H, J = 3.0 Hz), 6.00 (s, 1H), 2.70 (q, 2H, J = 7.2 Hz), 2.35 (s, 6H), 1.17 (t, 3H, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 139.6, 139.4, 137.5, 135.5, 124.3, 122.4, 111.2, 111.0, 21.4, 20.6, 12.9. HRMS (ESI) calcd for C14H18NO2S 264.1053 (M + H)+, found 264.1056.
1-(2,4-Dimethylbenzenesulfonyl)-2-ethyl-1H-pyrrole (20h)
Compound 20h was prepared in 76% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a white solid (mp 75-77 °C). HPLC purity 98.3% (tR = 24.40 min). 1H NMR (600 MHz, CDCl3) δ 7.46 (d, 1H, J = 7.2 Hz), 7.33-7.35 (m, 1H), 7.10-7.12 (m, 2H), 6.22-6.24 (m, 1H), 6.04-6.06 (m, 1H), 2.55 (q, 2H, J = 7.2 Hz), 2.47 (s, 3H), 2.39 (s, 3H), 1.13 (t, 3H, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 144.7, 137.9, 137.4, 135.5, 133.6, 128.7, 127.1, 122.9, 110.6, 110.1, 21.4, 20.3, 19.8, 12.6. HRMS (ESI) calcd for C14H18NO2S 264.1053 (M + H)+, found 264.1058.
2,4-Dimethyl-1-(2,4,6-trimethylbenzenesulfonyl)-1H-pyrrole (20i)
Compound 20i was prepared in 58% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a pale red solid (mp 98-100 °C). HPLC purity 95.7% (tR = 25.20 min). 1H NMR (600 MHz, CDCl3) δ 7.01 (s, 1H), 6.95 (s, 2H), 5.77 (s, 1H), 2.49 (s, 6H), 2.31 (s, 3H), 2.00 (s, 3H), 1.99 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 143.8, 140.2, 133.8, 132.2, 130.2, 119.7, 119.2, 114.5, 23.4, 21.1, 12.6, 11.8. HRMS (ESI) calcd for C15H20NO2S 278.1209 (M + H)+, found 278.1205.
1-(2,4,6-Trimethylbenzenesulfonyl)-1H-indole (22a)
Compound 22a was prepared in 84% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a white solid (mp 110-112 °C). HPLC purity 98.9% (tR = 24.73 min). 1H NMR (600 MHz, CDCl3) δ 7.59-7.61 (m, 1H), 7.54-7.55 (m, 1H), 7.34-7.35 (m, 1H), 7.17 (s, 2H), 6.93 (s, 2H), 6.61-6.62 (m, 1H), 2.52 (s, 6H), 2.26 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 144.1, 140.3, 134.7, 133.1, 132.5, 130.3, 126.8, 124.2, 122.8, 121.5, 112.5, 106.5, 22.7, 21.1. HRMS (ESI) calcd for C17H18NO2S 300.1053 (M + H)+, found 300.1057.
1-(2,4,6-Trimethylbenzenesulfonyl)-1H-indole-5-carboxylic acid methyl ester (22b)
Compound 22b was prepared in 84% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a white solid (mp 131-133 °C). HPLC purity 99.4% (tR = 23.80 min). 1H NMR (600 MHz, CDCl3) δ 8.30 (s, 1H), 7.88 (d, 1H, J = 9.0 Hz), 7.64 (d, 1H, J = 3.6 Hz), 7.40 (d, 1H, J = 8.4 Hz), 6.95 (s, 2H), 6.70 (d, 1H, J = 3.6 Hz), 3.90 (s, 3H), 2.51 (s, 6H), 2.28 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 167.3, 144.6, 140.4, 137.3, 132.8, 132.6, 130.0, 128.0, 125.6, 125.1, 123.9, 112.3, 107.1, 52.2, 22.7, 21.2. HRMS (ESI) calcd for C19H20NO4S 358.1108 (M + H)+, found 358.1114.
1-(2,4,6-Trimethylbenzenesulfonyl)-1H-indole-5-carboxylic acid (22c)
To a solution of 22b (72 mg, 0.2 mmol) in MeOH/H2O (4 mL/1 mL) was added 2N LiOH (0.4 mL, 0.8 mmol). The mixture was stirred at r.t. for 16 h. The solution was diluted with EtOAc (30 mL), washed with 1 N HCl (aq.) (10 mL) and brine (10 mL). The organic layer was dried over Na2SO4, and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/EtOAc = 1/1) to give the desired product (45 mg, 66%) as a pale yellow solid (mp 223-224 °C). HPLC purity 98.4% (tR = 14.89 min). 1H NMR (600 MHz, DMSO-d6) δ 12.80 (bs, 1H), 8.28 (s, 1H), 7.83-7.86 (m, 2H), 7.40 (d, 1H, J = 8.4 Hz), 7.15 (m, 2H), 6.94 (d, 1H, J = 3.6 Hz), 2.44 (s, 6H), 2.27 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 167.3, 144.7, 139.6, 136.3, 132.5, 131.9, 129.7, 128.5, 125.6, 125.4, 123.7, 111.8, 107.6, 21.9, 20.5. HRMS (ESI) calcd for C18H18NO4S 344.0951 (M + H)+, found 344.0954.
1-(2,4,6-Trimethylbenzenesulfonyl)-1H-pyrrolo[3,2-b]pyridine (24a)
Compound 24a was prepared in 96% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a white solid (mp 105-107 °C). HPLC purity 99.8% (tR = 21.84 min). 1H NMR (600 MHz, CDCl3) δ 8.48 (d, 1H, J = 3.6 Hz), 7.74-7.75 (m, 1H), 7.71 (d, 1H, J = 8.4 Hz), 7.09-7.11 (m, 1H), 6.94 (s, 2H), 6.81 (d, 1H, J = 3.0 Hz), 2.50 (s, 6H), 2.25 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 148.1, 145.8, 144.7, 140.4, 132.6, 132.5, 129.6, 128.3, 119.8, 118.7, 107.8, 22.7, 21.1. HRMS (ESI) calcd for C16H17N2O2S 301.1005 (M + H)+, found 301.1010.
1-(2,4,6-Trimethylbenzenesulfonyl)-1H-pyrrolo[3,2-c]pyridine (24b)
Compound 24b was prepared in 84% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a white solid (mp 119-120 °C). HPLC purity 99.7% (tR = 22.77 min). 1H NMR (600 MHz, CDCl3) δ 8.88 (s, 1H), 8.33 (d, 1H, J = 6.0 Hz), 7.54 (d, 1H, J = 3.6 Hz), 7.29 (d, 1H, J = 5.4 Hz), 6.95 (s, 2H), 6.68 (d, 1H, J = 3.6 Hz), 2.50 (s, 6H), 2.26 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 144.8, 144.3, 143.6, 140.4, 139.0, 132.6, 132.3, 127.2, 126.4, 107.6, 105.1, 22.6, 21.1. HRMS (ESI) calcd for C16H17N2O2S 301.1005 (M + H)+, found 301.1009.
1-(2,4,6-Trimethylbenzenesulfonyl)-1H-pyrrolo[2,3-c]pyridine (24c)
Compound 24c was prepared in 87% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a white solid (mp 132-134 °C). HPLC purity 99.8% (tR = 21.77 min). 1H NMR (600 MHz, CDCl3) δ 8.69 (s, 1H), 8.35 (d, 1H, J = 5.4 Hz), 7.73 (d, 1H, J = 3.6 Hz), 7.48 (d, 1H, J = 5.4 Hz), 6.96 (s, 2H), 6.63 (d, 1H, J = 3.0 Hz), 2.53 (s, 6H), 2.28 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 144.9, 142.1, 140.6, 135.8, 135.1, 132.8, 132.4, 131.8, 130.0, 115.9, 105.5, 22.7, 21.2. HRMS (ESI) calcd for C16H17N2O2S 301.1005 (M + H)+, found 301.1011.
1-(2,4,6-Trimethylbenzenesulfonyl)-1H-pyrrolo[2,3-b]pyridine (24d)
Compound 24d was prepared in 87% yield by a procedure similar to that used to prepare compound 20a. The title compound was obtained as a white solid (mp 139-141 °C). HPLC purity 99.6% (tR = 22.81 min). 1H NMR (600 MHz, CDCl3) δ 8.22 (d, 1H, J = 4.2 Hz), 7.85 (d, 1H, J = 3.0 Hz), 7.83 (d, 1H, J = 7.8 Hz), 7.08-7.11 (m, 1H), 6.93 (s, 2H), 6.57 (d, 1H, J = 3.0 Hz), 2.71 (s, 6H), 2.27 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 147.6, 144.7, 144.0, 141.3, 132.8, 132.1, 129.3, 126.9, 122.4, 118.6, 103.8, 23.0, 21.2. HRMS (ESI) calcd for C16H17N2O2S 301.1005 (M + H)+, found 301.0981.
Biological Evaluation Methods
Primary Screen Assay
Fluorescence intensity of 8-NBD-cAMP in complex with EPAC2 has been used as the readout in the primary screen assay. Briefly, 50 nM EPAC2 solution was prepared in 20 mM Tris buffer, pH 7.5, containing 150 mM NaCl, 1 mM EDTA and 1 mM DDT. 8-NBD-cAMP was added to EPAC2 solution up to 60 nM from stock solution in water. Sample was dispensed into 96-well plate (100 µL/well) and test compounds were added (1 µL/well) from 96-well mother plates. Test compounds were added from 10 mM stock solutions in DMSO. Samples with cAMP addition (1 µL/well from 30 mM stock solution in water) and no additions were used as a positive and a negative control. Fluorescence intensity signal from 8-NBD probe was recorded at room temperature before and after tested compounds were added using SpectaMaxM2 microplate reader (Molecular Devices, Silicon Valley, CA, USA) with excitation/emission wavelengths set at 470/540 nm.
Secondary Confirmation Assay
Measurement of in vitro guanine nucleotide exchange factor (GEF) activity of EPAC was adapted from a well known fluorescence-based assay using a fluorescent guanine nucleotide analog,30 and used as a functional confirmation assay for the compounds identified from primary screen. Briefly, 0.2 µM of Rap1B (1-167) loaded with the fluorescent GDP analog (Mant-GDP), was incubated with EPAC in 50 mM Tris buffer pH 7.5, containing 50 mM NaCl, 5 mM MgCl2, 1 mM DTT and a 100-fold molar excess of unlabeled GDP (20 µM) in the presence of 25 µM tested compound and 25 µM cAMP. Exchange of Mant-GDP by GDP was measured as a decrease in fluorescence intensity over time using a FluoroMax-3 spectrofluorometer with excitation/ emission wavelengths set at 366/450 nm. Typically, decay in the fluorescence intensity was recorded over a time course of 6000 s with data points taken every 60 s.
Counter Screening Assay
Kinase activities of the type I and II PKA holoenzymes were measured spectrophotometrically in a 96-well plate with a coupled enzyme assay as described previously.31 In this assay, the formation of the ADP is coupled to the oxidation of NADH by the pyruvate kinase/lactate dehydrogenase reactions so the reaction rate can be determined by following the oxidation of NADH, reflected by a decrease in absorbance at 340 nm. The kinase reaction mixture (100 µL) contained 50 mM Mops (pH 7.0), 10 mM MgCl2, 1 mM ATP, 1 mM PEP, 0.1 mM NADH, 8 U of pyruvate kinase, 15 U of lactate dehydrogenase, fixed amount of type I or type II PKA holoenzyme and 0.1 mM cAMP, with or without 25 µM of test compound. Reactions were pre-equilibrated at room temperature and initiated by adding the Kemptide substrate (final concentration 0.26 mM). PKA activities measured in the presence of 25 µM H89, a selective PKA inhibitor, were used as positive controls of PKA inhibition.
FRET-Based Assay for EPAC1, EPAC2, or AKAR3 Activation
HEK293 cells stably transfected with genetically-encoded FRET biosensors for EPAC1 (EPAC1 FL), EPAC2 (EPAC2 FL) or PKA (AKAR3) were used in this study. EPAC1 FL and EPAC2 FL were generated by sandwiching the full-length human EPAC1 or EPAC2 proteins with a Cerulean variant of cyan fluorescent protein (CFP) at their N-termini and a Venus variant of yellow fluorescent protein (YFP) at their C-termini.23 Activation of EPAC1 or EPAC2 in response to cAMP was measured as a decrease of FRET, and it was plotted as an increase of the CFP/YFP emission ratio (i.e., 485/535 nm emission ratio). The function core of AKAR3 contains a PKA substrate motif and a phospho-amino acid-binding domain connected by a flexible linker, which is flanked by a variant of CFP at its N-terminus and a variant of YFP at its C-terminus.24 Activation of AKAR3 in response to cAMP was measured as an increase of FRET, and it was plotted as a decrease of the 485/535 nm emission ratio. To test the effect of 20i on EPAC1, EPAC2 or PKA activation in live cells, single-cell suspensions of EPAC1-C9, EPAC2-C1, and AKAR3-C12 clones were plated onto 96-well clear bottom assay plates (Costar 3904) coated with rat tail collagen (Invitrogen). The clones were maintained in DMEM (25 mM glucose, 10% FBS) for 24 hr in order to allow the cell cultures to become 80-90% confluent. Cells were pretreated with various concentration of 20i or vehicle control before activated by membrane permeable cAMP analogs. Fluorescence spectra were recorded as described previously32 using a FlexStation 3 microplate reader controlled using SoftMax Pro software (Molecular Devices) with the excitation wavelength set at 435/9 nm (455 nm cut-off), and the emission wavelength set at 485/15 nm (CFP) or 535/15 nm (YFP). The time course of the change of FRET ratio was plotted after exporting data to Origin v.7.5 (OriginLab).
Supplementary Material
Figure 2. Relative potency of EPAC2 antagonist 20i(HJC0350).

Dose-dependent competition of EPAC2 antagonists with 8-NBD-cAMP in binding to EPAC2: closed circles, 20i; closed squares, cAMP.
ACKNOWLEDGMENTS
This work was supported by grants P30DA028821, R21MH093844 (JZ), R01GM066170, R21NS066510 (XC), and R01GM106218 (XC & JZ) from the National Institute of Health, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from Gulf Coast Consortia (GCC) for Chemical Genomics, and John Sealy Memorial Endowment Fund. We would like to thank Dr. Jin Zhang of Johns Hopkins School of Medicine for providing the EPAC1, EPAC2, and AKAR3 FRET reporters. We also want to thank Drs. Lawrence C. Sowers, Carol L. Nilsson, Jacob A. Theruvathu and Huiling Liu for mass spectrometry assistance and Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance.
ABBREVIATIONS USED
- EPAC
exchange proteins directly activated by cAMP
- SAR
structure-activity relationship
- cAMP
cyclic adenosine monophosphate
- 8-NBD-cAMP
8-(2-[7-nitro-4-benzofurazanyl]aminoethylthio)adenosine-3’,5’-cyclic monophosphate
- GDP
guanosine diphosphate
- PKA
protein kinase A
- FRET
fluorescenceresonance energy transfer
- GEF
guanine nucleotide exchange factor
- GTP
guanosine triphosphate
- Rap
Ras-related protein
- HTS
high-throughput screening
- 007-AM
8-(4-chlorophenylthio)-2’-O-methyladenosine-3’,5’-cyclic monophosphate acetoxymethyl ester
- AKAR3
A-kinase activity reporter 3
- 8-Br-cAMPAM
8-bromoadenosine 3’,5’-cyclic monophosphate acetoxymethyl ester
- TLC
thin layer chromatography
- UV
ultraviolet
- TMS
tetramethylsilane
- HRMS
high-resolution mass spectrometry
- HPLC
high-performance liquid chromatography
- TFA
trifluoroacetic acid
- DCM
dichloromethane
- EtOAc
ethyl acetate
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetic acid
- DDT
dichlorodiphenyltrichloroethane
- ADP
adenosine diphosphate
- NADH
nicotinamide adenine dinucleotide
- PEP
phospho(enol)pyruvate
- H89
N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide dihydrochloride
- CFP
cyan fluorescent protein
- YFP
yellow fluorescent protein
- DMEM
Dulbecco’s Modified Eagle’s Medium
- FBS
fetal bovine serum
Footnotes
ASSOCIATED CONTENT
Supporting Information
1H and 13C NMR spectra of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Frye SV. The art of the chemical probe. Nature Chem. Biol. 2010;6:159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- 2.Beavo JA, Brunton LL. Cyclic nucleotide research - still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002;3:710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- 3.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. EPAC is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 4.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 5.Bos JL. EPAC: a new cAMP target and new avenues in cAMP research. Nat. Rev. Mol. Cell Biol. 2003;4:733–738. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- 6.Cheng X, Ji Z, Tsalkova T, Mei FC. EPAC and PKA: a tale of two intracellular cAMP receptors. Acta. Biochim. Biophys. Sin. (Shanghai) 2008;40:651–662. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bos JL. EPAC proteins: Multi-purpose cAMP targets. Trends Biochem. Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Gloerich M, Bos JL. EPAC: defining a new mechanism for cAMP action. Annu. Rev. Pharmacol. Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- 9.Grandoch M, Roscioni SS, Schmidt M. The role of EPAC proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br. J. Pharmacol. 2010;159:265–284. doi: 10.1111/j.1476-5381.2009.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breckler M, Berthouze M, Laurent AC, Crozatier B, Morel E, Lezoualc'h F. Rap-linked cAMP signaling EPAC proteins: compartmentation, functioning and disease implications. Cell. Signal. 2011;23:1257–1266. doi: 10.1016/j.cellsig.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 11.(a) Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of cAMP sensor EPAC. J. Physiol. 2006;577:5–15. doi: 10.1113/jphysiol.2006.119644. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Holz GG, Chepurny OG, Schwede F. EPAC-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell. Signal. 2008;20:10–20. doi: 10.1016/j.cellsig.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rehmann H, Schwede F, Døskeland SO, Wittinghofer A, Bos JL. Ligandmediated activation of the cAMP-responsive guanine nucleotide exchange factor EPAC. J. Biol. Chem. 2003;278:38548–38556. doi: 10.1074/jbc.M306292200. [DOI] [PubMed] [Google Scholar]
- 13.Dao KK, Teigen K, Kopperud R, Hodneland E, Schwede F, Christensen AE, Martinez A, Døskeland SO. EPAC1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition. J. Biol. Chem. 2006;281:21500–21511. doi: 10.1074/jbc.M603116200. [DOI] [PubMed] [Google Scholar]
- 14.Das R, Chowdhury S, Mazhab-Jafari MT, Sildas S, Selvaratnam R, Melacini G. Dynamically driven ligand selectivity in cyclic nucleotide binding domains. J. Biol. Chem. 2009;284:23682–23696. doi: 10.1074/jbc.M109.011700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selvaratnam R, Chowdhury S, VanSchouwen B, Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. U.S. A. 2011;108:6133–6138. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsalkova T, Mei FC, Cheng X. A Fluorescence-based High-Throughput assay for the discovery of exchange protein directly activated by cyclic AMP (EPAC) antagonists. PLos One. 2012;7:e30441. doi: 10.1371/journal.pone.0030441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H, Tsalkova T, Mei FC, Hu Y, Cheng X, Zhou J. 5-Cyano-6-oxo-1,6- dihydro-pyrimidines as potent antagonists targeting exchange proteins directly activated by cAMP. Bioorg. Med. Chem. Lett. 2012;22:4038–4043. doi: 10.1016/j.bmcl.2012.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, Sastry SK, Schwede F, Cheng X. A novel EPAC specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol. Pharmacol. 2013;83:1–8. doi: 10.1124/mol.112.080689. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) (b) Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL, Jr, Cheng X. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc. Natl. Acad. Sci. U.S. A. 2012;109:18613–18618. doi: 10.1073/pnas.1210209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsalkova T, Blumenthal DK, Mei FC, White MA, Cheng X. Mechanism of EPAC activation: structural and functional analyses of EPAC2 hinge mutants with constitutive and reduced activities. J. Biol. Chem. 2009;284:23644–23651. doi: 10.1074/jbc.M109.024950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li S, Tsalkova T, White MA, Mei FC, Liu T, Wang D, Woods VL, Jr, Cheng X. Mechanism of intracellular cAMP sensor EPAC2 activation: cAMP-induced conformational changes identified by amide hydrogen/deuterium exchange mass spectrometry (DXMS) J. Biol. Chem. 2011;286:17889–17897. doi: 10.1074/jbc.M111.224535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng X, Phelps C, Taylor SS. Differential binding of cAMP-dependent protein kinase regulatory subunit isoforms Ialpha and IIbeta to the catalytic subunit. J. Biol. Chem. 2001;276:4102–4108. doi: 10.1074/jbc.M006447200. [DOI] [PubMed] [Google Scholar]
- 22.Yu S, Mei FC, Lee JC, Cheng X. Probing cAMP-dependent protein kinase holoenzyme complexes I alpha and II beta by FT-IR and chemical protein footprinting. Biochemistry. 2004;43:1908–1920. doi: 10.1021/bi0354435. [DOI] [PubMed] [Google Scholar]
- 23.Herbst KJ, Coltharp C, Amzel LM, Zhang J. Direct activation of EPAC by sulfonylurea is isoform selective. Chem. Biol. 2011;18:243–251. doi: 10.1016/j.chembiol.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen MD, Zhang J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem. Biophys. Res. Commun. 2006;348:716–721. doi: 10.1016/j.bbrc.2006.07.136. [DOI] [PubMed] [Google Scholar]
- 25.Holz GG. EPAC: A new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes. 2004;53:5–13. doi: 10.2337/diabetes.53.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang CL, Katoh M, Shibasaki T, Minami K, Sunaga Y, Takahashi H, Yokoi N, Iwasaki M, Miki T, Seino S. The cAMP sensor EPAC2 is a direct target of antidiabetic sulfonylurea drugs. Science. 2009;325:607–610. doi: 10.1126/science.1172256. [DOI] [PubMed] [Google Scholar]
- 27.Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompre AM, Vandecasteele G, Lezoualc'h F. cAMP-binding protein EPAC induces cardiomyocyte hypertrophy. Circ. Res. 2005;97:1296–1304. doi: 10.1161/01.RES.0000194325.31359.86. [DOI] [PubMed] [Google Scholar]
- 28.Metrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, Lezoualc'h F. EPAC mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 2008;102:959–965. doi: 10.1161/CIRCRESAHA.107.164947. [DOI] [PubMed] [Google Scholar]
- 29.Lissitzky JC, Parriaux D, Ristorcelli E, Verine A, Lombardo D, Verrando P. Cyclic AMP signaling as a mediator of vasculogenic mimicry in aggressive human melanoma cells in vitro. Cancer Res. 2009;69:802–809. doi: 10.1158/0008-5472.CAN-08-2391. [DOI] [PubMed] [Google Scholar]
- 30.van den Berghe N, Cool RH, Horn G, Wittinghofer A. Biochemical characterization of C3G: an exchange factor that discriminates between Rap1 and Rap2 and is not inhibited by Rap1A(S17N) Oncogene. 1997;15:845–850. doi: 10.1038/sj.onc.1201407. [DOI] [PubMed] [Google Scholar]
- 31.Cook PF, Neville ME, Jr, Vrana KE, Hartl FT, Roskoski R., Jr Adenosine cyclic 3',5'-monophosphate dependent protein kinase: kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry. 1982;21:5794–5799. doi: 10.1021/bi00266a011. [DOI] [PubMed] [Google Scholar]
- 32.Chepurny OG, Leech CA, Kelley GG, Dzhura I, Dzhura E, Li X, Rindler MJ, Schwede F, Genieser HG, Holz GG. Enhanced Rap1 activation and insulin secretagogue properties of an acetoxymethyl ester of an EPAC-selective cyclic AMP analog in rat INS-1 cells: studies with 8-pCPT-2'-O-Me-cAMP-AM. J. Biol. Chem. 2009;284:10728–10736. doi: 10.1074/jbc.M900166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.