

Abstract
The copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction has found broad application in myriad fields. For the most demanding applications requiring high yields at low substrate concentrations, highly active but air-sensitive copper complexes must be used. We describe here the use of an electrochemical potential to maintain catalysts in the active Cu(I) oxidation state in the presence of air. The simple procedure efficiently achieves excellent yields of CuAAC products involving both small molecule and protein substrates without the use of potentially damaging chemical reducing agents. A new water-soluble carboxylated version of the popular tris(benzyltriazolylmethyl)amine (TBTA) ligand is described. Cyclic voltammetry revealed reversible or quasi-reversible electrochemical redox behavior of copper complexes of the TBTA derivative (2; E1/2 = 60 mV vs. Ag/AgCl), sulfonated bathophenanthroline (3; E1/2 = -60 mV), and sulfonated tris(benzimidazoylmethyl)amine (4; E1/2 ~ -70 mV), and showed catalytic turnover to be rapid relative to the voltammetry time scale. Under the influence of a -200 mV potential established using a reticulated vitreous carbon working electrode, CuSO4 and 3 formed a superior catalyst. Electrochemically-protected bioconjugations in air were performed using bacteriophage Qβ derivatized with azide moieties at surface lysine residues. The complete addressing of more than 600 reactive sites per particle was demonstrated within 12 hours of electrolysis with sub-stoichiometric quantities of Cu•3.
Keywords: azides, alkynes, click chemistry, cycloaddition, electrochemistry, bioconjugation
Introduction
The highly efficient and exquisitely selective copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) – the most widely recognized of the “click” reactions[1, 2] – has been rapidly adopted since its discovery[3, 4] for application in fields as diverse as surface science,[5-12] dendrimer synthesis,[13-17] polymer ligation,[18-25] combinatorial organic synthesis,[26-29] and bioconjugation.[30-38] Exemplifying its power is our use of the process to create polyvalent virus particles for diagnostic and therapeutic applications, requiring hundreds of attachment reactions to occur on each particle under mild and dilute reaction conditions.[39-41]
CuAAC requires that the copper catalyst, usually prepared with an appropriate chelating ligand, be maintained in the air-sensititve CuI oxidation state, usually by the use of an in situ reducing agent such as ascorbate or tris(2-carboxyethyl)phosphine (TCEP). The most commonly used ligand is tris((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)amine (TBTA, 1, Scheme 1), first reported by Fokin and coworkers to accelerate the reaction.[42] Partly because of its poor water solubility, bioconjugations using 1 are slow and require a large excess of substrates (typically low mM in concentration), as well as mM concentrations of CuI and an excess of reducing agent. The latter two components can be detrimental to bio-macromolecules: for example, ascorbate-mediated degradation of DNA[43] and copper-mediated generation of reactive oxygen species (via CuI or CuII) are potentiallly destructive side reactions,[44-46] and dehydroascorbate and other ascorbate byproducts can react with protein side chains.[47-49] Our initial solution to this problem was the identification of a more potent catalyst using sulfonated bathophenanthroline ligand 3,[50] a discovery we have optimized into a robust bioconjugation protocol.[34] However, a significant drawback of this system is its acute air sensitivity, thus requiring air-free techniques which can be difficult to execute (e.g., bubble degassing or freeze-pump-thawing may cause proteins to denature) when an inert-atmosphere glove box is unavailable. Further, despite being “catalytic,” efficient bioconjugations are generally only achieved in the presence of excess CuI complex. We introduce here a more water-soluble version of TBTA as well as the use of an electrochemical cell to generate and protect catalytically-active CuI-ligand species for CuAAC bioconjugation and synthetic coupling reactions, allowing demanding reactions to be performed on the bench under mild conditions with minimal effort to exclude air.
Scheme 1.
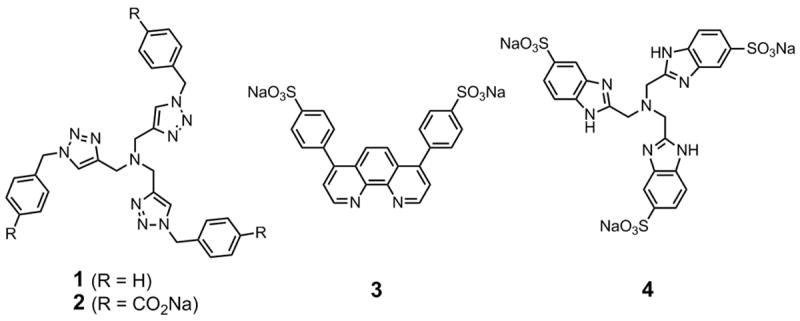
Accelerating ligands used for CuAAC reactions.
Results and Discussion
The poor water solubility of ligand 1 places some limits on its use in CuAAC reactions, particularly for bioconjugation. The p-carboxylate version 2 was therefore prepared and found to be substantially more soluble in water. Kinetic comparisons between 1 and 2 revealed the Cu•2 complex to be slightly more reactive than Cu•1 in a standard CuAAC reaction in organic solvent (data not shown). We therefore employed 2 as a surrogate for 1 in the bioconjugation experiments described below. We also included sulfonated tris(benzimidazoylmethyl)amine 4, a fully water-soluble example of the benzimidazole class of chelating ligands recently shown to provide catalysts of high activity.[51]
Cyclic voltammetry at 100 mV/s in buffer (10 mM HEPES pH 8 / 20% DMSO / 100 mM KPF6; Figure 1A) revealed half-wave potentials for CuI complexes of 3 and 4 (E1/2 ≈ −60-70 mV; all potentials vs. Ag/AgCl) to be approximately 100 mV more negative than Cu•2 (E1/2 = +60 mV), consistent with the electron-donating power of phenanthroline and benzimidazole relative to triazole.[52] A titration of peak current vs. metal:ligand ratio showed that the electroactive species have a 1:2 stoichiometry, consistent with the expected coordination environments (Supporting Information); when the chelating ligands are omitted, CuSO4 gives no observable current under these conditions in the potential window examined. Plots of cathodic peak current vs. (scan rate)1/2 for the copper-ligand complexes were linear, indicating diffusion controlled electron transfer with diffusion coefficients Do of 1.1×10−6 cm2/s2, 2.4×10−6 cm2/s2, and 2.4×10–6 cm2/s2 for Cu•22, Cu•32, and Cu•42, respectively (Supporting Information).
Figure 1.
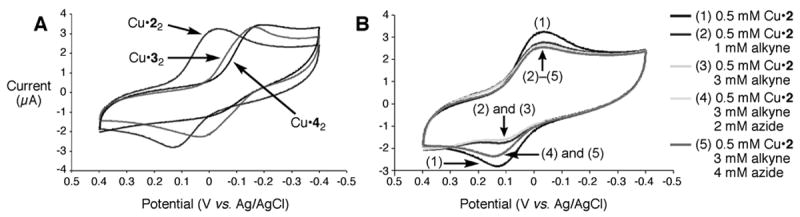
(A) Cyclic voltammograms of the copper-ligand complexes (0.5 mM CuSO4, 1 mM ligand) recorded with a glassy carbon electrode at 100 mV/s in 10 mM HEPES buffer (pH 8.0), containing 20% DMSO and 0.1 M KPF6. (B) Cyclic voltammograms in the presence of the indicated reagents, added in the order indicated; conditions as in part (A).
The electrochemical activity of the copper-ligand complexes suggested that the active CuI catalysts for solution-phase CuAAC reactions could be generated by bulk electrolysis, following studies at electrode surfaces by Collman, Chidsey, and coworkers.[53] The ability of each complex to mediate the reaction was first evaluated using benzyl azide and phenylacetylene as substrates, with concentrations approximating those typically used for CuAAC bioconjugation reactions. Electrolyses were performed in a two-compartment cell with a glass frit separating the Pt mesh counter electrode from the Ag/AgCl reference and reticulated vitreous carbon working electrodes. Product formation was monitored and quantified using LC/MS with an internal standard.
Each electrochemical reaction was conducted by first electrolyzing a buffered solution of CuSO4 and ligand (1:2 molar ratio) at a potential just negative of the cathodic peak in the appropriate cyclic voltammogram to reduce the metal complex to the active CuI oxidation state. During this period the current decayed with time, reaching a small steady value after 30 minutes (Supporting Information). For Cu•32, the total number of electrons passed during this period (approximately 0.96 coulombs) was found to be approximately four times the molar amount of O2 expected to be dissolved in an aqueous solution of that volume (0.27 mM), consistent with the expected four-electron reduction to water mediated by the CuI. complex.[54] Solutions of phenylacetylene and benzyl azide in DMSO were then added and the reaction was monitored with maintenance of the initial applied potential. No effort was made to exclude air from the reactions other than to cap the working compartments of the cell with septa when not adding or withdrawing material.
Table 1 lists the applied potentials and yields for the electrochemical reactions, and for analogous reactions performed using sodium ascorbate as the reductant. No significant triazole formation occurred in the absence of copper or in the presence of only CuII (entries 1-3). As noted previously,[50] even in the presence of ascorbate, CuI without accelerating ligands was insufficiently active to provide substantial amounts of triazole at the dilute concentrations tested (entries 4, 8, 9). Cu•3 proved to be the most potent system under the electrochemical conditions, yielding complete conversion within 15 minutes (entry 11 vs. 10 and 12). In contrast, ligands 2 and 4 provided better yields in the presence of ascorbate. This is due to the much faster reaction of Cu•3 with atmospheric oxygen, thereby depleting the ascorbate pool and deactivating the catalyst within several minutes, as indicated by the yields and the loss of the characteristic color of the CuI complex of 3. In separate experiments, Cu•3 also provided a rare example of catalytic performance under the dilute conditions typical of bioconjugation reactions: 200 μM Cu•3 with 5 mM benzyl azide and 5 mM phenylacetylene produced the 1,4-triazole product in 90% yield after 3 hours.
Table 1.
Results from the reaction shown in Equation 1 as a function of ligand and applied electrochemical potential. The reported yields were obtained by quantitative LC-MS analysis of aliquots removed at the indicated times, and are the average of three independent runs (error ±5%).

| |||||
|---|---|---|---|---|---|
| Entry | Reductant | Copper | Ligand | % Yield 15 min | % Yield 60 min |
| 1 | E, none [a] | None | none | 1 | 1 |
| 2 | none | CuSO4 | none | 3 | 3 |
| 3 | E, none [a] | CuSO4 | any | 3 | 3 |
| 4 | Ascorbate [b] | CuSO4 | none | 4 | 8 |
| 5 | Ascorbate [b] | CuSO4 | 2 | 82 | 97 |
| 6 | Ascorbate [b] | CuSO4 | 3 | 68 | 68 |
| 7 | Ascorbate [b] | CuSO4 | 4 | 98 | 99 |
| 8 | E, -50mV | CuSO4 | none | 3 | 5 |
| 9 | E, -200mV | CuSO4 | none | 4 | 8 |
| 10 | E, -50mV | CuSO4 | 2 | 58 | 84 |
| 11 | E, -200mV | CuSO4 | 3 | 98 | 99 |
| 12 | E, -200mV | CuSO4 | 4 | 6 | 42 |
In the electrochemical cell with electrodes disconnected.
1 mM sodium ascorbate.
Electrochemical monitoring also proved to be informative about the reaction mechanism. In the presence of ligand 2 and phenylacetylene, the oxidation half-wave of the Cu•2 complex was diminished relative to the reduction wave (Figure 1B), suggesting that formation of the CuI acetylide complex is fast and that the redox potential of this species lies outside of the electrochemical window. When benzyl azide was added, the oxidation wave returned to give the reversible couple characteristic of [Cu•2]I/II, at scan rates up to 500 mV/s. This behavior indicates that the triazole-forming catalytic turnover steps are also fast relative to scan rate, establishing Cu•2 as the resting state of the catalyst under these turnover conditions.
We tested the highly active Cu•3 electrochemical system in a bioconjugation reaction involving bacteriophage Qβ, an icosahedral virus comprised of 180 copies of a 14.1 kDa coat protein.[55] We have previously employed the non-infectious capsid as a robust scaffold for the display of gadolinium complexes, carbohydrates, and other species,[40, 41] prepared with the use of [Cu•32]OTf under strict inert atmosphere conditions in a glove box. The polyvalent CuAAC substrate 6 was prepared by acylation of surface lysine and N-terminal amine groups (4 per subunit; 720 per particle) with a large excess of azido N-hydroxysuccinimide ester 5 (Scheme 2). The 4-pentynoyl amide of selenomethionine (7) was used as the alkyne component to give polytriazole adduct 8. After purification, the amount of selenium attached to the protein was measured by inductively coupled plasma optical emission spectroscopy (ICP-OES). This technique replaces our normal use of dyes such as fluorescein, which can suffer from batch-to-batch differences in purity of the commercial reagent, occasional changes in molar absorptivity upon protein attachment, and particle instability for some capsids.[56] Although ICP-OES is not as convenient as UV-visible absorption spectroscopy, it is highly sensitive and has the advantage of being unaffected by the chemical environment of the probe element(s).
Scheme 2.
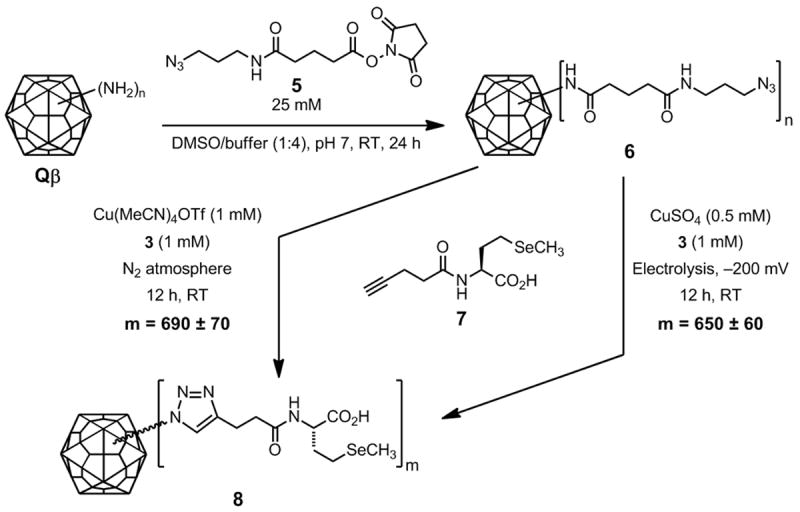
Electrochemically protected CuAAC bioconjugation with substrates 6 (2 mg/mL, approximately 400 μM in azide) and 7 (2.5 mM).
The electrochemical activity of the copper-ligand complexes suggested that the active CuI catalysts for solution-phase CuAAC reactions could be generated by bulk electrolysis, following studies at electrode surfaces by Collman, Chidsey, and coworkers.[51] The ability of each complex to mediate the reaction was first evaluated using benzyl azide and phenylacetylene as substrates, with concentrations approximating those typically used for CuAAC bioconjugation reactions. Electrolyses were performed in a two-compartment cell with a glass frit separating the Pt mesh counter electrode from the Ag/AgCl reference and reticulated vitreous carbon working electrodes. Product formation was monitored and quantified using LC/MS with an internal standard.
Each electrochemical reaction was conducted by first electrolyzing a buffered solution of CuSO4 and ligand (1:2 molar ratio) at a potential just negative of the cathodic peak in the appropriate cyclic voltammogram to reduce the metal complex to the active CuI oxidation state. During this period the current decayed with time, reaching a small steady value after 30 minutes (Supporting Information). For Cu•32, the total number of electrons passed during this period (approximately 0.96 coulombs) was found to be approximately four times the molar amount of O2 expected to be dissolved in an aqueous solution of that volume (0.27 mM), consistent with the expected four-electron reduction to water mediated by the CuI. complex.[49] Solutions of phenylacetylene and benzyl azide in DMSO were then added and the reaction was monitored with maintenance of the initial applied potential. No effort was made to exclude air from the reactions other than to cap the working compartments of the cell with septa when not adding or withdrawing material.
Table 1 lists the applied potentials and yields for the electrochemical reactions, and for analogous reactions performed using sodium ascorbate as the reductant. No significant triazole formation occurred in the absence of copper or in the presence of only CuII (entries 1-3). As noted previously,[46] even in the presence of ascorbate, CuI without accelerating ligands was insufficiently active to provide substantial amounts of triazole at the dilute concentrations tested (entries 4, 8, 9). Cu•3 proved to be the most potent system under the electrochemical conditions, yielding complete conversion within 15 minutes (entry 11 vs. 10 and 12). In contrast, ligands 2 and 4 provided better yields in the presence of ascorbate. This is due to the much faster reaction of Cu•3 with atmospheric oxygen, thereby depleting the ascorbate pool and deactivating the catalyst within several minutes, as indicated by the yields and the loss of the characteristic color of the CuI complex of 3. In separate experiments, Cu•3 also provided a rare example of catalytic performance under the dilute conditions typical of bioconjugation reactions: 200 μM Cu•3 with 5 mM benzyl azide and 5 mM phenylacetylene produced the 1,4-triazole product in 90% yield after 3 hours.
Electrochemical monitoring also proved to be informative about the reaction mechanism. In the presence of ligand 2 and phenylacetylene, the oxidation half-wave of the Cu•2 complex was diminished relative to the reduction wave (Figure 1B), suggesting that formation of the CuI acetylide complex is fast and that the redox potential of this species lies outside of the electrochemical window. When benzyl azide was added, the oxidation wave returned to give the reversible couple characteristic of [Cu•2]I/II, at scan rates up to 500 mV/s. This behavior indicates that the triazole-forming catalytic turnover steps are also fast relative to scan rate, establishing Cu•2 as the resting state of the catalyst under these turnover conditions.
We tested the highly active Cu•3 electrochemical system in a bioconjugation reaction involving bacteriophage Qβ, an icosahedral virus comprised of 180 copies of a 14.1 kDa coat protein.[55] We have previously employed the non-infectious capsid as a robust scaffold for the display of gadolinium complexes, carbohydrates, and other species,[40, 41] prepared with the use of [Cu•32]OTf under strict inert atmosphere conditions in a glove box. The polyvalent CuAAC substrate 6 was prepared by acylation of surface lysine and N-terminal amine groups (4 per subunit; 720 per particle) with a large excess of azido N-hydroxysuccinimide ester 5 (Scheme 2). The 4-pentynoyl amide of selenomethionine (7) was used as the alkyne component to give polytriazole adduct 8. After purification, the amount of selenium attached to the protein was measured by inductively coupled plasma optical emission spectroscopy (ICP-OES). This technique replaces our normal use of dyes such as fluorescein, which can suffer from batch-to-batch differences in purity of the commercial reagent, occasional changes in molar absorptivity upon protein attachment, and particle instability for some capsids.[56] Although ICP-OES is not as convenient as UV-visible absorption spectroscopy, it is highly sensitive and has the advantage of being unaffected by the chemical environment of the probe element(s).
The benchmark for comparison is our previously established CuAAC bioconjugation method using the CuI complex Cu(MeCN)4OTf (0.5 mM) and 3 (1 mM) in a nitrogen-atmosphere glovebox:[34] under these conditions, 690±60 molecules of 7 were attached, representing complete loading of the particle. The electrochemical reaction shown in Scheme 2 was initiated by pre-reduction of a 1:2 mixture of CuSO4:3 at −200 mV for 30 minutes to form the active CuI catalyst. This species provided a convenient visual report on its oxidation state as the CuI complex of 3 is deep green, whereas the CuII complex is pale yellow. The virus-azide and 7 were then added, and the electrochemical potential (and characteristic color of CuI•3) was maintained throughout the course of the overnight reaction. The product virus-like particles were purified by ultracentrifugation through a sucrose gradient, isolation of the band corresponding to intact capsids, concentration by ultrapelleting, and resuspension in the desired buffer. Good yields (>70%) of labeled particles were obtained, bearing 650±60 SeMet-alkynes per capsid, within experimental error of the glovebox reaction. When catalytic quantities of Cu•32 precatalyst (0.25 mM) were used, comparable loadings were obtained after 12 h (620±60 per particle). The decrease in required copper-ligand concentration is generally to be desired, as it facilitates purification and minimizes potentially detrimental protein-catalyst interactions (although we find that the presence of the chelating ligand shields most proteins from copper complexation). The recovered Qβ protein was exclusively found to be in the form of intact icosahedral capsids, as shown by size-exclusion chromatography (Superose 6) and transmission electron microscopy (Supporting Information).
Conclusions
The oxidized products from ascorbic acid such as dehydroascorbate are potentially reactive with biomacromolecules.[43, 48, 49] The use of ascorbate as a reducing agent for the generation of the active CuI oxidation state for CuAAC reactions may therefore not be desired for bioconjugation applications.[57] The electrochemical protocol described here provides an efficient and facile way to perform CuAAC reactions when chemical reducing agents cannot be employed, eliminating the need for manipulations under inert atmosphere as well as simplifying product purification. This is especially important for the highly active complex of Cu and bathophenanthroline ligand 3, which provides superior performance in bioconjugation at the cost of extra air sensitivity brought about by its more reducing CuI/II redox potential.[34] We also highlight the use of two other water-soluble accelerating ligands, including a more hydrophilic version of the popular tris(triazolyl)methyl TBTA structure. The successful bulk syntheses of both small molecule and larger biomolecule triazole conjugates using these ligands, under the protective umbrella of a reducing electrochemical potential, provides a broadly applicable method for CuAAC ligation in aqueous environments.
Experimental Section
Ligands and Linker 4
Compounds 1,[42] 4,[51] and 5 [30] were prepared as previously reported; 3 is commercially available. Note: We find occasional batches of 3 to be contaminated with substantial quantities of NMR-silent material (presumably inorganic salts) as received. The problem can be detected by quantitative NMR (adding a known quantity of an internal standard), and can be resolved by recrystallization. Ligand 2 was prepared by the reaction of tripropargylamine with p-azidomethylbenzoic acid under similar conditions as reported for 1. After the azide-alkyne cycloaddition reaction was completed the highly water soluble carboxylate salt form of the ligand was precipitated by acidification with glacial acetic acid. If further purification is desired, the material can be boiled in methanol or dissolved in aqueous base and re-precipitated with acetic acid. Synthetic details can be found in the Supporting Information.
Protein
Expression and purification of the Qβ coat protein from a recombinant plasmid has been previously described.[58] Briefly, a 135-amino acid version of the Qβ coat protein gene was cloned into the vector pQE-60 and expressed under IPTG control in M15MA cells in SOB media. After expression, collected cells were lysed by sonication and lysozyme treatment and then centrifuged to remove insoluble cell components. The virus-like particles were precipitated from the resulting supernatant using 8% PEG 8000. Following further centrifugation, the isolated pellet was resuspended in 0.1M potassium phosphate pH 7.0. The capsid then underwent a final purification by ultracentrifugation through a 10-40% sucrose gradient followed by ultrapelleting and resuspension in 0.1M potassium phosphate pH 7.0. Qβ concentrations were determined using the Modified Lowry Protein Assay (Pierce).
Qβ bearing azides at surface-exposed lysine residues (6) was prepared by incubating a 10 mg/mL solution of Qβ with 25 mM 5 (35-fold excess with respect to protein subunit) in 0.1 M potassium phosphate buffer (pH 7) with 20% DMSO for 12 hours. The derivatized virus was separated from excess reagent by ultracentrifugation using a 10-40% sucrose gradient, isolating the protein band corresponding to intact virions, and concentrating by subsequent ultrapelleting and solvation in 20 mM HEPES buffer pH 8. Mass recoveries of derivatized virus were typically 60-80%. Size-exclusion fast protein liquid chromatography (FPLC, Superose-6 column) indicated that >95% of the recovered virus was composed of intact particles.
For the reference bioconjugation reaction, a solution of Qβ-azide 6 (2.0 mg/mL, 140 μM in protein subunits, 560 μM in azide groups), and alkyne 7 (2.5 mM) was prepared under nitrogen atmosphere in a glove box (Vacuum Atmospheres, Inc.). A solution of Cu(MeCN)4(OTf) and ligand 3 in a 1:2 molar ratio was added to initiate the reaction, which had a final volume of 1 mL, and a final copper concentration of 0.5 mM. The reaction was agitated by gentle tumbling overnight at room temperature under nitrogen, and the resulting conjugate 8 was purified and characterized as above. Reaction yields were quantified by comparing protein concentration (Lowry assay) to the concentration of Se determined by ICP-OES, calibrating with reference standards (10-10,000 ppb Se) in the presence of a constant quantity of YbCl3 as internal standard. The error limits given for loading values are derived from repeated independent experiments and reflect mostly the uncertainties in protein concentration determined by the Lowry assay.
Cyclic voltammetry
Electrochemical measurements were performed with an Epsilon workstation (Bioanalytical Systems, Inc., BAS), using glassy carbon working (area 0.07 cm2) and Ag/AgCl reference electrodes. The surface of the working electrode was prepared by polishing with a 0.3 μm alumina slurry followed by brief sonication in water and drying in air. Voltammetry experiments were performed in a two-compartment cell with a Luggin capillary separating the working electrode and platinum wire counter electrode from the reference. All voltammetry experiments were performed under argon in thoroughly degassed buffer (100 mM KPF6, 10 mM HEPES, pH 8, 20% DMSO).
Bulk electrode-driven CuAAC
Electrolyses were conducted in a two-compartment cell in air with a glass frit separating the platinum gauze counter electrode from the reticulated vitreous carbon working and Ag/AgCl reference electrodes. For CuAAC of phenylacetylene and benzyl azide, reactions were initiated by placing into the working chamber 10 mL of 0.1 mM CuSO4 with ligands 2, 3, or 4, and 1-benzyl-4-phenyl-1,2,3-triazole-d7 (internal standard) in buffer (100 mM KPF6, 25 mM HEPES, pH 8, 20% DMSO). Electrolyses at the appropriate potential (Table 1) were then conducted for 30 minutes. Solutions of phenylacetylene and benzyl azide in DMSO were then added to final concentrations of 0.15 mM and 0.9 mM, respectively, and the applied potential was maintained throughout the reaction. 5 μL aliquots were taken every 15 minutes and diluted with 1.5 mL of ethanol for LC-MS analysis on an Agilent 1100 (G1946D) instrument, equipped with a 35 mm Agilent Zorbax 1.8 micron SB-C18 column. The elution solvent for the detection of 1-benzyl-4-phenyl-1,2,3-triazole was 55:45 H2O:CH3CN, with 0.5% trifluoroacetic acid. Detection was performed in single ion mode (SIM) with 1-benzyl-4-phenyl-1,2,3-triazole-d7 as internal reference for all experiments.
Bioconjugation reactions were conducted in air by first placing into the working chamber 8 mL of 1:2 CuSO4:3 (0.5 mM with respect to copper) in buffer (100 mM KPF6, 25mM HEPES, pH 8). Electrolysis was then performed for 30 minutes to generate the active CuI oxidation state. A total of 2 mL of virus and substrate solutions were added, resulting in final concentrations of 2 mg/mL virus and 2.5 mM 7. The chambers of the cell were capped loosely by septa when reagents were not being introduced or withdrawn. The electrochemical potential was applied during the 12-hour reaction period, with a low, steady-state passage of current noted throughout. The current was presumably due to the reduction of CuII to CuI, the former being generated by oxidation of reduced metal by oxygen diffusing into the reaction mixture. No traces of reactive oxygen species such as peroxide, superoxide, or singlet oxygen were detected by standard tests, consistent with the known electrochemically-driven four-electron reduction of O2 to water by Cu-phenanthroline.[54] Furthermore, no decomposition of ligands, substrates, or products was observed by LC-MS. Virus particles were purified from the reaction mixture and analyzed as described above.
Supplementary Material
Acknowledgments
This work was supported by The Skaggs Institute for Chemical Biology, the NIH (CA112075), and the David and Lucille Packard Foundation. AKU is supported by a postdoctoral fellowship from the CIHR. We thank Duane Prasuhn and Dr. Sejin Lee for selenomethionine derivative 7, Valentin Rodionov for ligand 4, Stanislav Presolski for useful discussions, and Dr. So-Hye Cho for the transmission electron micrographs shown in Supporting Information.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 2.Hawker CJ, Fokin VV, Finn MG, Sharpless KB. Aust J Chem. 2007;60:381–383. [Google Scholar]
- 3.Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3062. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 4.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 5.Evans RA. Aust J Chem. 2007;60:384–395. [Google Scholar]
- 6.Meng J-C, Averbuj C, Lewis WG, Siuzdak G, Finn MG. Angew Chem Int Ed. 2004;43:1255–1260. doi: 10.1002/anie.200352803. [DOI] [PubMed] [Google Scholar]
- 7.Lummerstorfer T, Hoffmann H. J Phys Chem B. 2004;108:39663–33966. [Google Scholar]
- 8.Collman JP, Devaraj NK, Chidsey CED. Langmuir. 2004;20:1051–1053. doi: 10.1021/la0362977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Binder WH, Kluger C, Josipovic M, Straif CJ, Friedbacher G. Macromolecules. 2006;39:8092–8101. [Google Scholar]
- 10.Chen GJ, Tao L, Mantovani G, Ladmiral V, Burt DP, Macpherson JV, Haddleton DM. Soft Matter. 2006;3:732–739. doi: 10.1039/b618325e. [DOI] [PubMed] [Google Scholar]
- 11.Prakash S, Long TM, Selby JC, Moore JS, Shannon MA. Anal Chem. 2006;79:1661–1667. doi: 10.1021/ac061824n. [DOI] [PubMed] [Google Scholar]
- 12.Collman JP, Devaraj NK, Eberspacher TPA, Chidsey CED. Langmuir. 2006;22:2457–2464. doi: 10.1021/la052947q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu P, Feldman AK, Nugent AK, Hawker CJ, Scheel A, Voit B, Pyun J, Fréchet JMJ, Sharpless KB, Fokin VV. Angew Chem Int Ed. 2004;43:3928–3932. doi: 10.1002/anie.200454078. [DOI] [PubMed] [Google Scholar]
- 14.Malkoch M, Schleicher K, Drockenmuller E, Hawker CJ, Russell TP, Wu P, Fokin VV. Macromolecules. 2005;38:3663–3678. [Google Scholar]
- 15.Wu P, Malkoch M, Hunt J, Vestberg R, Kaltgrad E, Finn MG, Fokin VV, Sharpless KB, Hawker CJ. Chem Commun. 2005:5775–5777. doi: 10.1039/b512021g. [DOI] [PubMed] [Google Scholar]
- 16.Joralemon MJ, O’Reilly RK, Matson JB, Nugent AK, Hawker CJ, Wooley KL. Macromolecules. 2005;38:5436–5443. [Google Scholar]
- 17.Ornelas C, Aranzaes JR, Cloutet E, Alves S, Astruc D. Angew Chem Int Ed. 2007;46:872–877. doi: 10.1002/anie.200602858. [DOI] [PubMed] [Google Scholar]
- 18.Sen Gupta S, Raja KS, Kaltgrad E, Strable E, Finn MG. Chem Commun. 2005:4315–4317. doi: 10.1039/b502444g. [DOI] [PubMed] [Google Scholar]
- 19.Lutz J-F, Börner HG, Weichenhan K. Macromol Rapid Commun. 2005;26:514–518. [Google Scholar]
- 20.Tsarevsky NV, Sumerlin BS, Matyjaszewski K. Macromolecules. 2005;38:3558–3561. [Google Scholar]
- 21.Dirks AJ, Van Berkel SS, Hatzakis NS, Opsteen JA, van Delft FL, Cornelissen JJLM, Rowan AE, van Hest JCM, Rutjes FPJT, Nolte RJM. Chem Commun. 2005:4172–4174. doi: 10.1039/b508428h. [DOI] [PubMed] [Google Scholar]
- 22.Ladmiral V, Mantovani G, Clarkson GJ, Cauet S, Irwin JL, Haddleton DM. J Am Chem Soc. 2006;128:4823–4830. doi: 10.1021/ja058364k. [DOI] [PubMed] [Google Scholar]
- 23.Quémener D, Davis TP, Barner-Kowollik C, Stenzel MH. Chem Commun. 2006:5051–5053. doi: 10.1039/b611224b. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Riva R, Jerome R, Lecomte P. Macromolecules. 2007;40:824–831. [Google Scholar]
- 25.Lutz J-F. Angew Chem Int Ed. 2007;46:1018–1025. doi: 10.1002/anie.200604050. [DOI] [PubMed] [Google Scholar]
- 26.Fazio F, Bryan MC, Blixt O, Paulson JC, Wong C-H. J Am Chem Soc. 2002;124:14397–14402. doi: 10.1021/ja020887u. [DOI] [PubMed] [Google Scholar]
- 27.Van der Peet P, Gannon CT, Walker I, Dinev Z, Angelin M, Tam S, Ralton JE, McConville MJ, Williams SJ. ChemBioChem. 2006;7:1384–1391. doi: 10.1002/cbic.200600159. [DOI] [PubMed] [Google Scholar]
- 28.Goess BC, Hannoush RN, Chan LK, Kirchhausen T, Shair MD. J Am Chem Soc. 2006;128:5391–5403. doi: 10.1021/ja056338g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie J, Seto CT. Bioorg Med Chem. 2007;15:458–473. doi: 10.1016/j.bmc.2006.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. J Am Chem Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 31.Speers AE, Adam GC, Cravatt BF. J Am Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 32.Link AJ, Tirrell DA. J Am Chem Soc. 2003;125:11164–11165. doi: 10.1021/ja036765z. [DOI] [PubMed] [Google Scholar]
- 33.Speers AE, Cravatt BF. Chem Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 34.Sen Gupta S, Kuzelka J, Singh P, Lewis WG, Manchester M, Finn MG. Bioconjugate Chem. 2005;16:1572–1579. doi: 10.1021/bc050147l. [DOI] [PubMed] [Google Scholar]
- 35.Beatty KE, Xie F, Wang Q, Tirrell DA. J Am Chem Soc. 2005;127:14150–14151. doi: 10.1021/ja054643w. [DOI] [PubMed] [Google Scholar]
- 36.Sieber SA, Niessen S, Hoover HS, Cravatt BF. Nat Chem Biol. 2006;2:274–281. doi: 10.1038/nchembio781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin P-C, Ueng S-H, Tseng M-C, Ko J-L, Huang K-T, Yu S-C, Adak AK, Chen Y-J, Lin C-C. Angew Chem Int Ed. 2006;45:4286–4290. doi: 10.1002/anie.200600756. [DOI] [PubMed] [Google Scholar]
- 38.Hsu T-L, Hanson SR, Kishikawa K, Wang S-K, Sawa M, Wong C-H. Proc Natl Acad Sci USA. 2007;104:2614–2619. doi: 10.1073/pnas.0611307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rae CS, Khor IW, Wang Q, Destito G, Gonzalez MJ, Singh PR, Thomas DE, Estrada MN, Powell E, Finn MG, Manchester M. Virology. 2005;343:224–235. doi: 10.1016/j.virol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 40.Prasuhn J, D E, Yeh RM, Obenaus A, Manchester M, Finn MG. Chem Commun. 2007:1269–1271. doi: 10.1039/b615084e. [DOI] [PubMed] [Google Scholar]
- 41.Kaltgrad E, Sen Gupta S, Punna S, Huang C-Y, Chang A, Wong C-H, Finn MG, Blixt O. ChemBioChem. 2007;8:1455–1462. doi: 10.1002/cbic.200700225. [DOI] [PubMed] [Google Scholar]
- 42.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 43.Liu P-Y, Jiang N, Zhang J, Wei X-H, Lin H-H, Yu X-Q. Chem Biodiversity. 2006;3:958–965. doi: 10.1002/cbdv.200690104. [DOI] [PubMed] [Google Scholar]
- 44.Brewer GJ. Exp Biol Med. 2007;232:323–335. [PubMed] [Google Scholar]
- 45.Halliwell B, Gutteridge JMC. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alemón-Medina R, Breña-Valle M, Muñoz-Sánchez JL, Gracia-Mora MI, Ruiz-Azuara L. Cancer Chemother Pharmacol. 2007;60:219–228. doi: 10.1007/s00280-006-0364-9. [DOI] [PubMed] [Google Scholar]
- 47.Nagaraj RH, Sell DR, Prabhakaram M, Ortwerth BJ, Monnier VM. Proc Natl Acad Sci U S A. 1991;88:10257–10261. doi: 10.1073/pnas.88.22.10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagaraj RH, Sell DR, Prabhakaram M, Ortwerth BJ, Monnier VM. Proc Natl Acad Sci U S A. 1991;88:10257–10261. doi: 10.1073/pnas.88.22.10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ortwerth BJ, Monnier VM. In: Vitamin C in Health and Disease. Packer L, Fuchs J, editors. Marcel Decker, Inc.; New York: 1997. pp. 123–142. [Google Scholar]
- 50.Lewis WG, Magallon FG, Fokin VV, Finn MG. J Am Chem Soc. 2004;126:9152–9153. doi: 10.1021/ja048425z. [DOI] [PubMed] [Google Scholar]
- 51.Rodionov VO, Presolski S, Gardinier S, Lim Y-H, Finn MG. J Am Chem Soc. 2007;129:12696–12704. doi: 10.1021/ja072678l. [DOI] [PubMed] [Google Scholar]
- 52.The pKa of the conjugate acids of 1,2,3-triazoles of this type are approximately 0.1-1.0 (Abboud JLM, Foces-Foces C, Notario R, Trifonov RE, Volovodenko AP, Ostrovskii VA, Alkorta I, Elguero J. Eur J Org Chem. 2001:3013–3024.), whereas 1,2-phenanthroline (pKa = 4.3; Albert A, Goldacre R, Phillips J. J Chem Soc. 1948:2240–2249.) and benzimidazoles (pKa ≈ 6.3; Langner R, Zundel G. Can J Chem. 2001;79:1376–1380.; Huyskens PL, Cleuren W, Franz M, Vuylsteke MA. J Phys Chem. 1980;84:2748–2751.) are better bases.
- 53.Devaraj NK, Dinolfo PH, Chidsey CED, Collman JP. J Am Chem Soc. 2006;128:1794–1795. doi: 10.1021/ja058380h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu SJ, Huang CH, Chang CC. Mat Chem Phys. 2003;82:551–556. [Google Scholar]
- 55.Liljas L, Golmohammadi R. Structure. 1996;4:543–554. doi: 10.1016/s0969-2126(96)00060-3. [DOI] [PubMed] [Google Scholar]
- 56.For example, we have found cowpea mosaic virus to be destabilized by the covalent conjugation of some dyes and ferrocenyl derivatives to the exterior surface, and hepatitis B virus capsid to be broken up by the attachment of more than one florescein molecule for every two protein subunits in a position-selective manner. We believe that tethered compounds that are flat and hydrophobic can wedge themselves into the hydrophobic interfaces between subunits that are largely responsible for holding capsids together, thereby destabilizing the overall structure. These observations will be reported in detail elsewhere.
- 57.Ascorbate is well tolerated when the protein is especially stable; for example, see Bruckman M, Kaur G, Lee LA, Xie F, Sepulveda J, Breitenkamp R, Zhang X, Joralemon M, Russell TP, Emrick T, Wang Q. ChemBioChem. 2008 doi: 10.1002/cbic.200700559. in press.
- 58.Strable E, Prasuhn DE, Jr, Udit AK, Brown S, Link AJ, Ngo JT, Lander G, Quispe J, Potter CS, Carragher B, Tirrell DA, Finn MG. Bioconjugate Chem. 2008 doi: 10.1021/bc700390r. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou Z, Fahrni CJ. J Am Chem Soc. 2004;126:8862–8863. doi: 10.1021/ja049684r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.