

Abstract
Osteocytes are long-lived and far more numerous than the short-lived osteoblasts and osteoclasts. Immured within the lacunar-canalicular system and mineralized matrix, osteocytes are ideally located throughout bone to detect the need for, and accordingly choreograph, the bone regeneration process by independently controlling rate limiting steps of bone resorption and formation. Consistent with this role, emerging evidence indicates that signals arising from apoptotic and old/or dysfunctional osteocytes are seminal culprits in the pathogenesis of involutional, post-menopausal, steroid-, and immobilization-induced osteoporosis. Osteocyte-originated signals may also contribute to the increased bone fragility associated with bone matrix disorders like osteogenesis imperfecta, and perhaps the rapid reversal of bone turnover above baseline following discontinuation of anti-resorptive treatments, like denosumab.
Keywords: osteocytes, apoptosis, autophagy, involutional osteoporosis, post-menopausal osteoporosis, steroid-induced osteoporosis, osteogenesis imperfecta, anti-resorptive therapies
1. Introduction
“No man is an island, entire of itself; every man is a piece of the continent, a part of the main; if a clod bee washed away by the sea, Europe is the less, as well as if a promontory were, as well as if a manor of thy friends or of thine own were; any man’s death diminishes me, because I am involved in mankind. And therefore never send to know for whom the bell tolls; it tolls for thee.”
John Donne, Devotions upon Emergent Occasions, Meditation XVII, 1624.
Osteocytes are by far the more abundant cell type in bone: one thousand times more than osteoclasts and ten times more than osteoblasts [1]. In difference to these other two cell types which are short-lived and only transiently present on a small fraction of the bone surface, osteocytes are long-lived and deployed throughout the skeleton. In many regions of the skeleton, osteocytes persist until the bone in which they lie is resorbed, so that their lifespan is the same as of that bone. Because of their long lifespan, osteocytes are subjected to the adverse effects of the aging process and therefore are directly relevant to pathophysiology of the aging of the skeleton.
In a manner analogous to neuronal cells, osteocytes form an elaborate communication network with each other, endothelial cells of the bone vasculature, and cells of the bone surface and the marrow, via cytoplasmic process and gap junctions [2]. Because of these unique features, osteocytes are ideally suited to mediate the homeostatic adaptation of bone to mechanical forces. Detection of changes in strain by osteocytes leads to changes in osteoclast and/or osteoblast recruitment by mechanisms which are slowly emerging, that counteract the change in strain. Indeed, it is abundantly clear by now that osteocytes not only detect the need for bone regeneration, but they also choreograph the regeneration process by independently controlling rate limiting steps of bone resorption and formation [3] (Figure 1).
Figure 1.

Model depicting mechanisms by which osteocytes may independently control bone resorption and bone formation. Osteoclasts (OCs) and osteoblasts (OBs) within a cancellous bone BMU are shown as being derived from precursors (pOC and pOB). Osteocytes alter the rate of bone remodeling by controlling osteoclast formation via production of RANKL. Osteocytes also control the balance between formation and resorption by regulating osteoblast formation via production of sclerostin. (Reproduced from Jinhu Xiong and Charles A. O’Brien J Bone Miner Res. 2012;27:499–505)
Osteocytes, but not osteoblasts or their precursors, are the essential cellular sources of RANKL for osteoclast formation [4, 5]. This particular distinction is important since osteocytes are downstream from bone formation and therefore RANKL expression in this cell type cannot explain the coupling of bone formation to bone resorption. Presently, it remains unclear how osteocyte-derived RANKL reaches osteoclast progenitors to activate its receptor RANK. However, RANKL is produced as a membrane protein that can be shed to form a soluble form, sRANKL [6]. Therefore, osteocytes may control osteoclast formation via the release of sRANKL into the lacunar-canalicular system, which directly communicates with the bone marrow. A similar process is probably involved in the regulation of bone formation by osteocytes through the secretion of sclerostin and perhaps Dkk1 [7, 8]. An alternative possibility is that RANKL, attached to the membranes of dendritic osteocyte processes extending onto the bone surface, interacts directly with osteoclast progenitors.
In this brief review, we will summarize emerging evidence that signals arising from apoptotic and old/or dysfunctional osteocytes are important participants in the pathogenesis of the most common forms of osteoporosis. In addition we will discuss the possibility that osteocyte–derived signals may also contribute to the increased bone fragility associated with congenital matrix disorders like osteogenesis imperfecta, and perhaps the reversal of bone turnover following discontinuation of anti-resorptive therapies.
2. Involutional osteoporosis
Decreased bone mass is only one of many age-related mechanisms responsible for the increase of fracture risk in the elderly [1, 9]. Indeed, the risk of hip fracture between the ages of 50 and 80 years increases 30-fold [9, 10], but only a 4-fold increase can be accounted by the decline in BMD [11]. Age-related changes in the bone itself clearly contribute to the increase in fracture risk independently of BMD.
In many areas of the skeleton osteocytes survive until the bone in which they lie is resorbed, so that their lifespan is the same as the age of that bone. In areas of very low turnover, osteocytes may die by apoptosis, leaving lacunae which appear empty in histologic sections [12], but which may contain the remnants of osteocytes which have not undergone phagocytosis [13], but are lost during section preparation. Almost fifty years ago, Harold Frost was the first to conceive that the lifespan of osteocytes is relevant to the pathophysiology of osteoporosis [14]. He noted that osteocyte death in rib cortical bone, inferred from the presence of an empty lacuna, increased in prevalence with age and was followed by hypermineralization of perilacunar bone and later by filling of canaliculae with mineralized connective tissue. Such changes are collectively referred to as micropetrosis and probably lead to increased brittleness of bone [15]. From these and other data Frost estimated the natural lifespan of osteocytes at about 25 years [16]. It is generally believed that only in cortical bone would osteocytes be left undisturbed by remodeling long enough for this intrinsic lifespan to be exceeded, but this view disregards the effect of distance from the surface on the probability of remodeling, and hence mean bone age [17]. In human cancellous bone, this effect is minor in bone less than 50 μm from the surface, but as the depth increases this effect increases rapidly, so that bone more than 75 μm from the surface is essentially isolated from surface remodeling and will be at least as old as the years of age of the subject since skeletal maturity; since during growth trabeculae increase in thickness by remodeling with positive balance, the age of such bone could be even greater [18].
Even in normal human iliac cancellous bone, a substantial proportion is more than 20 years old [17] so it is not surprising that osteocyte death (inferred from an empty lacuna) has been demonstrated. In superficial bone (<25 μm from the surface) obtained from healthy women the density (number/unit area) of total lacunae, osteocytes and empty lacunae does not change with age because bone in which some osteocytes have died is replaced by bone containing a full complement of osteocytes [12]. The data suggest that rather than having a fixed lifespan, osteocytes die by a stochastic process occurring at a fractional rate of about 2.5%/y. Consequently, in deep bone (more than 45 μm from the surface) that is rarely or never remodeled, osteocyte density declines exponentially with age, approaching an asymptotic value which at age 75 is about 40% of the value at age 20 [19]. Evidently osteocyte death is dependent on the age of the bone, not on the age of the subject. In deep bone total lacunar density is lower than in superficial bone and falls substantially with age, so that micropetrosis, the only process that could lead to obliteration of lacunae, occurs in cancellous as well as in cortical bone [12]. There is a close spatial relationship between empty lacunae and microscopic fatigue damage [20] but it is unclear which occurs first, although both are the expected consequences of excessive bone age [21].
3. Contribution of osteocyte death to increased bone fragility with old age
The age related decline in osteocyte number is accompanied by reduced bone strength, both in patients with vertebral fracture [22] and in healthy mice [23]. Notably, ablation of osteocytes in young mice recapitulates at least some of the effects of old age on bone and rapidly leads to decreased bone strength, microfractures, and osteoporosis [24]. Several mechanisms likely contribute to this relationship, and increased release of RANKL triggered by the death of osteocytes may be one of them. Consistent with this, reduced osteocyte density in central cancellous bone is associated with increased surface remodeling [19], which is an independent contributor to bone fragility [25]. Additionally, osteocyte death with advancing age leads to a decline in bone vascularity and hydration, which reduces bone strength by mechanisms not yet fully understood, but which probably include changes in crystallinity and promotion of micropetrosis [15, 26]. Conversely, protection of osteocytes from the adverse affect of aging on their apoptosis in the mouse maintains bone crystallinity, vasculature volume, circulation of interstitial fluid, and strength [26].
4. Osteocytes and cortical porosity
The term osteoporosis, coined by the French pathologist Jean Georges Chretien Frederic Martin Lobstein ‘the Younger’ (1777–1835), is derived from the Greek words osteo (bone) and poros (little hole), referring to the cavities which were observed by the pathologist in certain patients’ bones [27]. An increase in cortical porosity with age was first noted in histologic studies reported by Amprino and Bairati in 1936 in an Italian publication, and radiologically by Meema in 1969 [28]. One of the authors of the present article (MP) was the first to explicitly identify increased cortical porosity as another factor in bone fragility [29]. He noted the similarity of bone fragility occurring during the high turnover of the adolescent growth spurt and early post-menopause. This argument was further developed in a later paper [30]. Until recently, the contribution of cortical porosity to the decreased bone strength and increased fragility had been underappreciated because of technical limitations for its detection. All these years, much more attention was placed instead on the more readily measurable bone mineral density (BMD). It was not until the recent development of high resolution peripheral CT that it was shown for the first time that cortical porosity may be responsible for a substantial amount of the loss of bone and the resultant structural deterioration that occurs after the age of 65 [31, 32].
Recent studies from the laboratory of Robert Jilka have provided genetic evidence for a cause-and-effect relationship between the apoptosis of osteocytes and cortical porosity [33]. In this work, the investigators used mice that are deficient for both Bak and Bax, two genes indispensable for apoptosis, selectively in osteoblasts and osteocytes. In two different models (one using an osteocalcin Cre deleter, and the other using an osterix Cre deleter), there was a substantial increase in trabecular bone mass that was maintained up to 22 months of age. This finding provides the first genetic demonstration that attenuation of osteoblast apoptosis can indeed increase the working lifespan of this cell type and lead to bone anabolism - a concept suggested by several earlier studies [34, 35]. Unexpectedly, however, the Bak/Bax-deficient mice also exhibited a 7-fold increase in pore formation throughout the femoral cortex by 22 months of age, increased expression of RANKL and numerous osteoclasts within the pores. Notably, at the same age, cortical porosity in the wild type controls was limited to the distal metaphysis. More strikingly, in both Bak/Bax-deficient and wild type mice, the pores were adjacent to dysmorphic osteocytes, the number of which was far greater in the Bak/Bax-deficient mice. These findings demonstrate that abrogation of apoptosis prolongs the working lifespan of short-lived osteoblasts, but it exaggerates the adverse effects of aging on long-lived cortical osteocytes. Moreover, they strongly suggest that distress signals from age-damaged osteocytes and a focal increase of RANKL production are the likely culprits of cortical porosity.
5. Failure of osteocyte autophagy with age
“Autophagy”, the Greek word for self-eating, is a highly conserved process that is mediated by lysosomes and is critical for cellular defense against stressful stimuli. Autophagy is particularly important for terminally differentiated long-lived cells, such as osteocytes, in which damaged components are not diluted by cell replication and need to be eliminated before becoming toxic. During authophagy, large structures such as organelles (i.e. mitochondria) and unwanted or damaged proteins are recycled. During periods of starvation, autophagy provides amino acids that are essential for survival. Autophagy is stimulated by numerous stressors including oxidative stress, impaired mitochondria function, hypoxia, as well as caloric restriction [36–38]. Autophagy prevents diseases. Failure of autophagy (i.e. the inability to mount an autophagic response), on the other hand, is responsible for the ultimate demise of long-lived, post-mitotic cells in many organs, including brain, heart, muscle, and kidney. Indeed, autophagy failure is linked to degenerative diseases of the nervous system, like Alzheimer’s and Parkinson’s disease, as well as heart failure and cancer [39].
Failure of osteocyte autophagy, resulting from long-term increase in oxidative stress, may be yet another age-related mechanism contributing to the pathogenesis of involutional osteoporosis. Indeed, a compromised autophagic response by osteocytes and the resulting apoptosis could well be the trigger of pathologic remodeling and the increased cortical porosity associated with old age. It was recently shown in vitro that osteocyte autophagy increases in response to stresses, such as growth factor starvation, hypoxia, and glucocorticoids [40]; and that increased autophagy plays a protective role against glucocorticoid-induced cell death [41]. In addition, unpublished work from Dr. O’Brien’s laboratory in our group has revealed that expression of autophagy-related genes is lower in the cortical bone of 20-month-old, compared with 6-month-old, mice [40]. This is associated with an increase in the amount of mitochondrial DNA, which is a consequence of reduced turnover of mitochondria. To directly address whether autophagy is functionally important in osteocytes, O’Brien and colleagues went on to delete ATG7, a gene that is essential for autophagy, from osteocytes using the DMP1-Cre transgene, which is active predominantly in osteocytes. Similar to the results from aged mice, the reduction in autophagy in the conditional KO mice was associated with an increase in the levels of mitochondrial DNA isolated from cortical bone. Moreover, BMD was significantly lower, compared to littermate controls, in the spines and femurs of both male and female conditional KO mice beginning at 4 months of age and continuing to at least 6 months of age without differences in body weight between genotypes. Micro-CT analysis revealed low cancellous bone volume, low cortical thickness, and high cortical porosity in 6-month-old conditional KO mice compared with control littermates. At this age, osteoclast number, osteoblast number, bone formation rate, and wall width were also lower in the conditional KO mice. In addition, oxidative stress was higher in the bones of conditional KO mice as measured by reactive oxygen species levels in the bone marrow of the femur and by p66shc phosphorylation in vertebral bone. Strikingly, all these changes including this low bone mass and low rate of bone turnover are similar to those observed in aged wild type mice. Hence, these results demonstrate that loss of autophagy in osteocytes leads to low bone mass associated with low bone remodeling and suggest that a decline in osteocyte autophagy with age contributes to the low bone mass associated with aging.
6. Post-menopausal osteoporosis
The decline of estrogen production at menopause leads to loss of bone that is associated with an increase in the bone remodeling rate, increased osteoclast and osteoblast numbers, and increased resorption and formation, albeit unbalanced. Conversely, estrogen replacement decreases bone resorption, restrains the rate of bone remodeling, and helps to maintain a focal balance between bone formation and resorption. These effects result from influences on the birth rate and lifespan of osteoclast and osteoblast lineage cells that are mediated via direct actions of estrogens on these two cell types as well as indirect actions mediated via cytokines produced by osteocytes, osteoblastic/marrow stromal cells, and perhaps T and B lymphocytes, macrophages, and dendritic cells [42–44].
Consistent with evidence for a direct anti-apoptotic effect of estrogens on osteocytes [45, 46], estrogen deficiency increases the prevalence of osteocyte apoptosis in both cancellous and cortical bone in animals and humans [47–49]. Importantly, in a recent study in mice, the increased osteocyte apoptosis caused by loss of estrogens was regional, rather than uniform, in the bone cortex; and the location of the apoptotic osteocytes was tightly correlated with the areas where endocortical resorption was subsequently activated [50]. This observation is in line with the idea that apoptotic osteocytes, or more likely the live neighbors of the dead cells, produce factors that increase osteoclast formation and recruitment in their vicinity, leading to the resorption and thereby removal of the dead cells. These latest findings together with the demonstration of the essential role of osteocyte-derived RANKL in osteoclastogenesis and bone remodeling, also support the long-held view that at least some remodeling is a targeted, as opposed to stochastic, process in which cells that can sense mechanical damage and/or the death of their neighbors are beacons for the excavation and repair of a specified area of bone and the removal of the dead cells [3].
7. Glucocorticoid- induced osteoporosis
Glucocorticoid excess, similar to the osteoporosis of old age, decreases bone strength disproportionately to its adverse effect on bone mass; and frequently presents with fractures as early as three months following the initiation of therapy with these agents and before any detectable decline on bone mass [51]. Extensive evidence implicates an increase in the prevalence of osteocyte apoptosis in the rapid increase of fracture incidence in patients with this condition [52].
Glucocorticoids strongly and rapidly stimulate osteoblast and osteocyte apoptosis directly, and also suppresses osteoblastogenesis while increasing the lifespan of osteoclasts [13, 53–55]. Glucocorticoids, like loss of mechanical strain, increases osteocyte apoptosis by interfering with focal adhesion kinase-mediated survival signals, thereby leading to anoikis [56]. Endogenous glucocorticoid production and sensitivity to the effects of glucocorticoids increase with age, apparently contributing to the effects of old age in the development of osteoporosis. Increased sensitivity to glucocorticoids is the result of increased bone expression of 11beta-hydroxysteroid dehydrogenase (11beta-HSD) type 1, the enzyme that activates glucocorticoids. In both aging and pharmacologic hyperglucocorticoidism, increased apoptosis of osteocytes can account for the loss of bone strength that occurs before the loss of bone mineral density and the mismatch between bone mineral density and the risk of fracture in patients with glucocorticoid-induced osteoporosis [26, 57, 58]. Specifically, and similar to aging, in glucocorticoid excess the volume of the bone vasculature and solute transport from the peripheral circulation to the lacunar-canalicular system is decreased, leading to decreased skeletal hydration [26]. The molecular underpinning of these effects is decreased angiogenesis resulting from decreased VEGF production by osteoblasts/osteocytes, as well as decreased VEGF action. These changes represent a chain of interconnected pathogenetic mechanisms as indicated by anatomical evidence that the cellular processes of osteocytes are in direct contact with the bone vasculature. Together with evidence that dehydration of bone decreases strength, these new insights reveal that endogenous glucocorticoids increase skeletal fragility in old age as a result of cell autonomous effects on osteocytes leading to interconnected decrements in bone angiogenesis, vasculature volume, and osteocyte-lacunar-canalicular fluid.
8. Immobilization-induced osteoporosis
The mechanostat function of the osteocyte is dependent on interaction with the ECM. Osteocytes interact with the extracellular matrix (ECM) in the pericellular space through discrete sites in their membranes, which are enriched in integrins and vinculin [59, 60], as well as through transverse elements that tether osteocytes to the canalicular wall [61]. Fluid movement in the canaliculi resulting from mechanical loading might induce ECM deformation, shear stress, and/or tension in the tethering elements. The resulting changes in circumferential strain in osteocyte membranes might be converted into intracellular signals by integrin clustering and integrin interactions with cytoskeletal and catalytic proteins at focal adhesions [62, 63]. Mechanical forces transduce signals through integrins and a signalsome comprising actin filaments, microtubules, the focal adhesion kinase FAK, and Src kinases, resulting in activation of the ERK pathway and attenuation of osteocyte apoptosis [64]. Similarly, physiological levels of mechanical strain imparted by pulsatile fluid flow prevent apoptosis of cultured osteocytes [65].
Conversely, reduced mechanical forces increase the prevalence of osteocyte apoptosis [66]. Within 3 days of tail suspension, mice or rats exhibit an increased incidence of osteocyte apoptosis in both trabecular and cortical bone. This change is followed 2 weeks later by increased osteoclast numbers and cortical porosity, reduced trabecular and cortical width, and decreased spinal bone mineral density and vertebral strength. Importantly, whereas in ambulatory animals apoptotic osteocytes are randomly distributed, in the unloaded rodents apoptotic osteocytes are preferentially sequestered in endosteal cortical bone, the site that is subsequently resorbed. Moreover, treatment with an apoptosis inhibitor blocks both the unloading-induced apoptosis and the increased intracortical resorption [67]. The effect of unloading on osteocyte apoptosis and bone resorption is reproduced in transgenic mice in which osteocytes are refractory to glucocorticoid action, indicating that stress and hypercortisolemia cannot account for these effects [66]. Hence, diminished mechanical forces eliminate signals that maintain osteocyte viability, thereby leading to apoptosis. Intriguingly, a ligand-independent function of the estrogen receptor is indispensable for mechanically-induced ERK activation and attenuation of osteocyte apoptosis [68], consistent with reports that mice lacking the estrogen receptors α and β exhibit poor osteogenic response to loading [69].
The mechanism(s) by which reduced mechanical forces trigger osteocyte apoptosis remain unclear, but a decrease in nitric oxide (NO) and prostaglandins are most likely involved [70]. In support of this view, mechanical stimulation increases the production of NO by osteocytes [71–73]. Mechanical stimulation of chicken and canine bone also increases the production of prostaglandin E2 (PGE2) [74, 75], an agent with known antiapoptotic properties [76].
9. A role of osteocytes in diseases of abnormal bone matrix
Signals for osteocyte survival must also be provided by the ECM itself. In agreement with this view, loss of survival signals from the ECM causes osteoblastic cell apoptosis or “anoikis”, and neutralizing antibodies to the ECM protein fibronectin induce osteoblast apoptosis [77]. In addition, transgenic mice expressing collagenase-resistant collagen type-I exhibit a striking increase in the prevalence of osteocyte and osteoblast apoptosis [78]. Collectively, these lines of evidence suggest that exposure of cryptic sites of ECM proteins by matrix metalloproteinases is required for the maintenance of cell-ECM interactions that result in “outside-in” integrin signaling that preserves osteocyte viability. This scenario is consistent with in vitro evidence that physiological levels of mechanical strain promote survival of osteocytes via an integrin-mediated mechanism [64]. In view of this, it is reasonable to suspect that osteocytes residing in the bone of patients with matrix abnormalities sense the defective matrix and generate signals to repair it, futile as this response may be. One such condition may well be osteogenesis imperfect (OI).
OI is a genetic disorder characterized by increased bone fragility and low bone mass. Although mutations affecting collagen type I are responsible for the disease in most patients, the mechanisms by which the genetic defects cause abnormal bone development have not been well characterized. One possibility is that abnormal ECM leads to misperception of strain by osteocytes such that the mechanostat setpoint is too high [79]. Nevertheless, quantitative static and dynamic histomorphometric analysis of tetracycline-labeled iliac bone biopsies from children with OI types I, III, and IV revealed that surface-based parameters of bone remodeling are increased in all OI types, indicating increased recruitment of remodeling units [80]. Consistent with this, the thickening of secondary trabeculae by remodeling is severely compromised. Also long bones are narrower because periosteal apposition is reduced.
10. Above baseline reversal of bone turnover following discontinuation of antiresorptive treatments
Commonly used drugs for the treatment of osteoporosis are anti-resorptive agents that slow remodeling in proportion to their anti-resorptive efficacy. The emergence of serious side effects with the long term use of such agents, however, has raised major concerns about the optimal duration of such therapies and their long term consequences [81–83]. Moreover, in all instances, their effects are transient and in the case of the newer and more effective ones, have in fact a rapid resolution-of-effect with the potential for increasing fracture risk [84]. Considering the preceding discussion it is inexorable that if necessary remodeling is postponed, microdamage and osteocyte death or dysfunction will accumulate, so that a period of catch up is required to restore microdamage burden to normal.
11. Concluding thoughts
In keeping with the literary analogy, considerable evidence indicates that the bell tolls for dead or dysfunctional osteocytes, alerting the rest of the osteocyte network to the need for removal of the dead cell bodies and/or the repair of the damaged matrix (Figure 2). Removal of dead osteocytes by osteoclastic resorption is the only option for cells that are entombed within mineral and are therefore inaccessible to phagocytosis by ordinary macrophages. The molecular means by which dead or unhealthy osteocytes convey their distress signals to their alive, healthy neighbors remains completely unknown, but efforts to uncover them seem to us a very fruitful area of inquiry. Mechanical loading stimulates Wnt signaling and thereby osteoblastogenesis and bone formation in healthy bone, by decreasing the production of the osteocyte-derived Wnt antagonist sclerostin [85–87]. At this stage, it is unclear whether osteocyte death or dysfunction also lead to sclerostin-dependent changes in osteoblastogenesis and bone formation, but reports of changes in the circulating levels of serum sclerostin in some disease states has raised this possibility [88]. Nonetheless, a lot more mechanistic studies and vigorous validation of the sclerostin assays and the biological relevance of circulating sclerostin to bone metabolism, if any, is needed before firm conclusions can be drawn about the putative contribution of dead or dysfunctional osteocytes to compromised bone formation in pathologic states [89]. Albeit, the evidence that dysapoptotic osteocytes (i.e. osteocytes unable to undergo apoptosis because of lack of Bak and Bax) cause cortical porosity and lack of osteocyte autophagy recapitulates the low bone turnover phenotype of old age in the murine skeleton suggests that such efforts will be worthwhile [33, 40].
Figure 2.
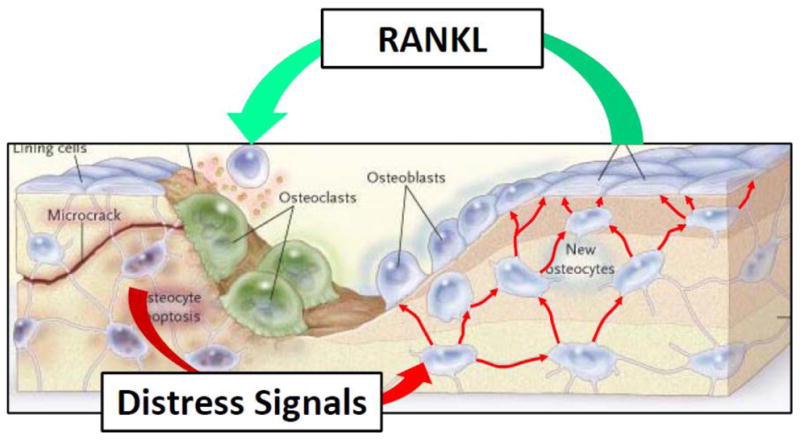
Illustration of the concept that the neighbors of apoptotic osteocytes (caused by microcracks in the particular instance depicted in the model) receive and propagate distress signals (red arrow) that trigger the process of the removal of dead osteocytes and the abnormal matrix that surrounds them. Red arrows connecting osteocytes imply that such signals travel through the lacunocanalicular system to reach the bone surface and thereby target the area in need of damage repair. RANKL is shown as a representative of probably several additional mediators of the repair process. (Modified from Seeman, E. and Delmas, D.P. NEJM 2006;354:2250–2261)
Acknowledgments
SCM’s research is supported by the National Institutes of Health (P01 AG13918); the Department of Veterans Affairs (Merit Review); and Tobacco Settlement funds provided by the University of Arkansas for Medical Sciences. The authors wish to thank Leah Elrod for assistance in the preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Manolagas SC, Parfitt AM. What old means to bone. Trends Endocrinol Metab. 2010;21:369–74. doi: 10.1016/j.tem.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–38. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manolagas SC. Perspective: Choreography from the tomb: an emerging role of dying osteocytes in the purposeful, and perhaps not so purposeful, targeting of bone remodeling. BoneKey-Osteovision. 2006;3:5–14. doi: 10.1138/20060193. [DOI] [Google Scholar]
- 4.Xiong J, Onal M, Jilka RL, Weinstein RS, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nature Medicine. 2011;17:1235–41. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–4. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 6.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–76. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 7.Van Bezooijen RL, Roelen BA, Visser A, van dW-P, de WE, Karperien M, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–14. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24:1651–61. doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- 9.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31:266–300. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hui SL, Slemenda CW, Johnston CC., Jr Age and bone mass as predictors of fracture in a prospective study. J Clin Invest. 1988;81:1804–9. doi: 10.1172/JCI113523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanis JA, Borgstrom F, De LC, Johansson H, Johnell O, Jonsson B, et al. Assessment of fracture risk. Osteoporos Int. 2005;16:581–9. doi: 10.1007/s00198-004-1780-5. [DOI] [PubMed] [Google Scholar]
- 12.Qiu S, Rao DS, Palnitkar S, Parfitt AM. Age and distance from the surface but not menopause reduce osteocyte density in human cancellous bone. Bone. 2002;31:313–8. doi: 10.1016/s8756-3282(02)00819-0. [DOI] [PubMed] [Google Scholar]
- 13.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–82. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frost HM. In vivo osteocyte death. J Bone Joint Surg [Am] 1960;42:138–43. [PubMed] [Google Scholar]
- 15.Frost HM. Micropetrosis. J Bone Joint Surg [Am] 1960;42A:144–50. [PubMed] [Google Scholar]
- 16.Frost HM. In: Bone Remodeling Dynamics. Thomas CC, editor. Springfield; 1963. [Google Scholar]
- 17.Parfitt AM, Kleerekoper M, Villanueva AR. Increased bone age: Mechanisms and consequences. In: Christianson C, Johnsen C, Riis BJ, editors. Osteoporosis. Copenhagen: Osteopress ApS; 1987. pp. 301–8. [Google Scholar]
- 18.Parfitt AM, Travers R, Rauch F, Glorieux FH. Structural and cellular changes during bone growth in healthy children. Bone. 2000;27:487–94. doi: 10.1016/s8756-3282(00)00353-7. [DOI] [PubMed] [Google Scholar]
- 19.Qiu S, Rao DS, Palnitkar S, Parfitt AM. Relationships between osteocyte density and bone formation rate in human cancellous bone. Bone. 2002;31:709–11. doi: 10.1016/s8756-3282(02)00907-9. [DOI] [PubMed] [Google Scholar]
- 20.Qiu S, Rao DS, Fyhrie DP, Palnitkar S, Parfitt AM. The morphological association between microcracks and osteocyte lacunae in human cortical bone. Bone. 2007:10–5. doi: 10.1016/j.bone.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 21.Parfitt AM. Skeletal heterogeneity and the purposes of bone remodeling: implications for the understanding of osteoporosis. In: Marcus R, Feldman D, Nelson D, Rosen C, editors. Osteoporosis. San Diego, CA, USA: Elsevier; 2007. pp. 71–89. [Google Scholar]
- 22.Qiu S, Rao DS, Palnitkar S, Parfitt AM. Reduced iliac cancellous osteocyte density in patients with osteoporotic vertebral fracture. J Bone Miner Res. 2003;18:1657–63. doi: 10.1359/jbmr.2003.18.9.1657. [DOI] [PubMed] [Google Scholar]
- 23.Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282:27285–97. doi: 10.1074/jbc.M702810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–75. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Heaney RP. Is the paradigm shifting? Bone. 2003;33:457–65. doi: 10.1016/s8756-3282(03)00236-9. [DOI] [PubMed] [Google Scholar]
- 26.Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O’Brien C, et al. Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in aged mice. Aging Cell. 2010;9:147–61. doi: 10.1111/j.1474-9726.2009.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schapira D, Schapira C. Osteoporosis: the evolution of a scientific term. Osteoporos Int. 1992;2:164–7. doi: 10.1007/BF01623921. [DOI] [PubMed] [Google Scholar]
- 28.Meema HE, Meema S. Cortical bone mineral density versus cortical thickness in the diagnosis of osteoporosis: a roentgenologic-densitometric study. J Am Geriatr Soc. 1969;17:120–41. doi: 10.1111/j.1532-5415.1969.tb03167.x. [DOI] [PubMed] [Google Scholar]
- 29.Parfitt AM. The two faces of growth: benefits and risks to bone integrity. Osteoporos Int. 1994;4:382–98. doi: 10.1007/BF01622201. [DOI] [PubMed] [Google Scholar]
- 30.Parfitt AM. Cortical porosity in postmenopausal and adolescent wrist fractures. In: Uhthoff H, Jaworski Z, editors. Current Concepts of Bone Fragility. Berlin Heidelberg: Springer-Verlag; 1986. pp. 167–72. [Google Scholar]
- 31.Zebaze RM, Ghasem-Zadeh A, Bohte A, Iuliano-Burns S, Mirams M, Price RI, et al. Intracortical remodelling and porosity in the distal radius and post-mortem femurs of women: a cross-sectional study. Lancet. 2010;375:1729–36. doi: 10.1016/S0140-6736(10)60320-0. [DOI] [PubMed] [Google Scholar]
- 32.Nicks KM, Amin S, Atkinson EJ, Riggs BL, Melton LJ, III, Khosla S. Relationship of age to bone microstructure independent of areal bone mineral density. J Bone Miner Res. 2012;27:637–44. doi: 10.1002/jbmr.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jilka RL, Deloose A, Climer L, Berryhill S, O’Brien C, Weinstein R, et al. Deletion of the pro-apoptotic proteins Bax and Bak from osteoblasts and osteocytes increases bone mass, and retards the loss of cancellous bone but dramatically accelerates cortical porosity in aged mice. Journal of Bone & Mineral Research. 2011;26(Suppl 1):S68. [Google Scholar]
- 34.Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104:439–46. doi: 10.1172/JCI6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jilka RL, Weinstein RS, Parfitt AM, Manolagas SC. Quantifying Osteoblast and Osteocyte Apoptosis: Challenges and Rewards. J Bone Miner Res. 2007;22:1492–501. doi: 10.1359/jbmr.070518. [DOI] [PubMed] [Google Scholar]
- 36.Cuervo AM. Calorie restriction and aging: the ultimate “cleansing diet”. J Gerontol A Biol Sci Med Sci. 2008;63:547–9. doi: 10.1093/gerona/63.6.547. [DOI] [PubMed] [Google Scholar]
- 37.Pursiheimo JP, Rantanen K, Heikkinen PT, Johansen T, Jaakkola PM. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene. 2009;28:334–44. doi: 10.1038/onc.2008.392. [DOI] [PubMed] [Google Scholar]
- 38.Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: crosstalk and redox signalling. Biochem J. 2012;441:523–40. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ventruti A, Cuervo AM. Autophagy and neurodegeneration. Curr Neurol Neurosci Rep. 2007;7:443–51. doi: 10.1007/s11910-007-0068-5. [DOI] [PubMed] [Google Scholar]
- 40.Zhao H, Xiong J, Onal M, Cazer P, Weinstein R, Manolagas S, et al. Osteocyte autophagy declines with age in mice and suppression of autophagy decreases bone mass. J Bone Min Res. 2011;26(Suppl 1):S13. [Google Scholar]
- 41.Xia X, Kar R, Gluhak-Heinrich J, Yao W, Lane NE, Bonewald LF, et al. Glucocorticoid-induced autophagy in osteocytes. J Bone Miner Res. 2010;25:2479–88. doi: 10.1002/jbmr.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–37. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- 43.Riggs BL, Khosla S, Melton LJ., III Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev. 2002;23:279–302. doi: 10.1210/edrv.23.3.0465. [DOI] [PubMed] [Google Scholar]
- 44.Manolagas SC, Kousteni S, Jilka RL. Sex steroids and bone. Recent Prog Horm Res. 2002;57:385–409. doi: 10.1210/rp.57.1.385. [DOI] [PubMed] [Google Scholar]
- 45.Kousteni S, Bellido T, Plotkin LI, O’Brien CA, Bodenner DL, Han K, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–30. [PubMed] [Google Scholar]
- 46.Marathe N, Rangaswami H, Zhuang S, Boss GR, Pilz RB. Pro-survival effects of 17beta-estradiol on osteocytes are mediated by nitric oxide/cGMP via differential actions of cGMP-dependent protein kinases I and II. J Biol Chem. 2012;287:978–88. doi: 10.1074/jbc.M111.294959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomkinson A, Reeve J, Shaw RW, Noble BS. The death of osteocytes via apoptosis accompanies estrogen withdrawal in human bone. J Clin Endocrinol Metab. 1997;82:3128–35. doi: 10.1210/jcem.82.9.4200. [DOI] [PubMed] [Google Scholar]
- 48.Kousteni S, Chen J-R, Bellido T, Han L, Ali AA, O’Brien C, et al. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science. 2002;298:843–6. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- 49.Tomkinson A, Gevers EF, Wit JM, Reeve J, Noble BS. The role of estrogen in the control of rat osteocyte apoptosis. J Bone Miner Res. 1998;13:1243–50. doi: 10.1359/jbmr.1998.13.8.1243. [DOI] [PubMed] [Google Scholar]
- 50.Emerton KB, Hu B, Woo AA, Sinofsky A, Hernandez C, Majeska RJ, et al. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone. 2010;46:577–83. doi: 10.1016/j.bone.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manolagas SC. Corticosteroids and fractures: a close encounter of the third cell kind [editorial] J Bone Miner Res. 2000;15:1001–5. doi: 10.1359/jbmr.2000.15.6.1001. [DOI] [PubMed] [Google Scholar]
- 52.Weinstein RS, Manolagas SC. Apoptosis and osteoporosis. Am J Med. 2000;108:153–64. doi: 10.1016/s0002-9343(99)00420-9. [DOI] [PubMed] [Google Scholar]
- 53.Weinstein RS, Nicholas RW, Manolagas SC. Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip. J Clin Endocrinol Metab. 2000;85:2907–12. doi: 10.1210/jcem.85.8.6714. [DOI] [PubMed] [Google Scholar]
- 54.Weinstein RS, Chen JR, Powers CC, Stewart SA, Landes RD, Bellido T, et al. Promotion of osteoclast survival and antagonism of bisphosphonate-induced osteoclast apoptosis by glucocorticoids. J Clin Invest. 2002;109:1041–8. doi: 10.1172/JCI14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jia D, O’Brien CA, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoclasts to increase their life span and reduce bone density. Endocrinology. 2006;147:5592–9. doi: 10.1210/en.2006-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plotkin LI, Manolagas SC, Bellido T. Glucocorticoids Induce Osteocyte Apoptosis by Blocking Focal Adhesion Kinase-mediated Survival: Evidence for Inside-Out Signaling Leading to Anoikis. J Biol Chem. 2007;282:24120–30. doi: 10.1074/jbc.M611435200. [DOI] [PubMed] [Google Scholar]
- 57.Almeida M, Han L, Ambrogini E, Weinstein RS, Manolagas SC. Glucocorticoids and tumor necrosis factor (TNF) alpha increase oxidative stress and suppress WNT signaling in osteoblasts. J Biol Chem. 2011;286:44326–35. doi: 10.1074/jbc.M111.283481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weinstein RS. Glucocorticoid-induced bone disease. N Engl J Med. 2011;365:62–70. doi: 10.1056/NEJMcp1012926. [DOI] [PubMed] [Google Scholar]
- 59.Gohel AR, Hand AR, Gronowicz GA. Immunogold localization of β1-integrin in bone: Effect of glucocorticoids and insulin-like growth factor I on integrins and osteocyte formation. J Histochem Cytochem. 1995;43:1085–96. doi: 10.1177/43.11.7560891. [DOI] [PubMed] [Google Scholar]
- 60.Aarden EM, Nijweide PJ, Plas A, Alblas MJ, Mackie EJ, Horton MA, et al. Adhesive properties of isolated chick osteocytes in vitro. Bone. 1996;18:305–13. doi: 10.1016/8756-3282(96)00010-5. [DOI] [PubMed] [Google Scholar]
- 61.You LD, Weinbaum S, Cowin SC, Schaffler MB. Ultrastructure of the osteocyte process and its pericellular matrix. Anat Rec A Discov Mol Cell Evol Biol. 2004;278:505–13. doi: 10.1002/ar.a.20050. [DOI] [PubMed] [Google Scholar]
- 62.Clark EA, Brugge JS. Integrins and signal transduction pathways: The road taken. Science. 1995;268:233–9. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 63.Giancotti FG. Integrin signaling: specificity and control of cell survival and cell cycle progression. Curr Opin Cell Biol. 1997;9:691–700. doi: 10.1016/s0955-0674(97)80123-8. [DOI] [PubMed] [Google Scholar]
- 64.Plotkin LI, Mathov I, Aguirre JI, Parfitt AM, Manolagas SC, Bellido T. Mechanical stimulation prevents osteocyte apoptosis: requirement of integrins, Src kinases, and ERKs. Am J Physiol Cell Physiol. 2005;289:C633–C643. doi: 10.1152/ajpcell.00278.2004. [DOI] [PubMed] [Google Scholar]
- 65.Bakker A, Klein-Nulend J, Burger E. Shear stress inhibits while disuse promotes osteocyte apoptosis. Biochem Biophys Res Commun. 2004;320:1163–8. doi: 10.1016/j.bbrc.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 66.Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res. 2006;21:605–15. doi: 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- 67.Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J Bone Miner Res. 2009;24:597–605. doi: 10.1359/JBMR.081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aguirre JI, Plotkin LI, Gortazar AR, Millan MM, O’Brien CA, Manolagas SC, et al. A novel ligand-independent function of the estrogen receptor is essential for osteocyte and osteoblast mechanotransduction. J Biol Chem. 2007;282:25501–8. doi: 10.1074/jbc.M702231200. [DOI] [PubMed] [Google Scholar]
- 69.Lee K, Jessop H, Suswillo R, Zaman G, Lanyon L. Endocrinology: bone adaptation requires oestrogen receptor-alpha. Nature. 2003;424:389. doi: 10.1038/424389a. [DOI] [PubMed] [Google Scholar]
- 70.Burger EH, Klein-Nulend J, Smit TH. Strain-derived canalicular fluid flow regulates osteoclast activity in a remodelling osteon--a proposal. J Biomech. 2003;36:1453–9. doi: 10.1016/s0021-9290(03)00126-x. [DOI] [PubMed] [Google Scholar]
- 71.Zaman G, Pitsillides AA, Rawlinson SC, Suswillo RF, Mosley JR, Cheng MZ, et al. Mechanical strain stimulates nitric oxide production by rapid activation of endothelial nitric oxide synthase in osteocytes. J Bone Miner Res. 1999;14:1123–31. doi: 10.1359/jbmr.1999.14.7.1123. [DOI] [PubMed] [Google Scholar]
- 72.Klein-Nulend J, Semeins CM, Ajubi NE, Nijweide PJ, Burger EH. Pulsating fluid flow increases nitric oxide (NO) synthesis by osteocytes but not periosteal fibroblasts--correlation with prostaglandin upregulation. Biochem Biophys Res Commun. 1995;217:640–8. doi: 10.1006/bbrc.1995.2822. [DOI] [PubMed] [Google Scholar]
- 73.Pitsillides AA, Rawlinson SC, Suswillo RF, Bourrin S, Zaman G, Lanyon LE. Mechanical strain-induced NO production by bone cells: a possible role in adaptive bone (re)modeling? FASEB J. 1995;9:1614–22. doi: 10.1096/fasebj.9.15.8529841. [DOI] [PubMed] [Google Scholar]
- 74.Ajubi NE, Klein-Nulend J, Nijweide PJ, Vrijheid-Lammers T, Alblas MJ, Burger EH. Pulsating fluid flow increases prostaglandin production by cultured chicken osteocytes--a cytoskeleton-dependent process. Biochem Biophys Res Commun. 1996;225:62–8. doi: 10.1006/bbrc.1996.1131. [DOI] [PubMed] [Google Scholar]
- 75.Rawlinson SCF, El-Haj AJ, Minter SL, Tavares IA, Bennett A, Lanyon LE. Loading-related increases in prostaglandin production in cores of adult canine cancellous bone in vitro: A role for prostacyclin in adaptive bone remodeling. J Bone Miner Res. 1991;6:1345–51. doi: 10.1002/jbmr.5650061212. [DOI] [PubMed] [Google Scholar]
- 76.Kitase Y, Barragan L, Qing H, Kondoh S, Jiang JX, Johnson ML, et al. Mechanical induction of PGE2 in osteocytes blocks glucocorticoid-induced apoptosis through both the beta-catenin and PKA pathways. J Bone Miner Res. 2010;25:2657–68. doi: 10.1002/jbmr.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Globus RK, Doty SB, Lull JC, Holmuhamedov E, Humphries MJ, Damsky CH. Fibronectin is a survival factor for differentiated osteoblasts. J Cell Sci. 1998;111:1385–93. doi: 10.1242/jcs.111.10.1385. [DOI] [PubMed] [Google Scholar]
- 78.Zhao W, Byrne MH, Wang Y, Krane SM. Osteocyte and osteoblast apoptosis and excessive bone deposition accompany failure of collagenase cleavage of collagen. J Clin Invest. 2000;106:941–9. doi: 10.1172/JCI10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frost HM. Bone “mass” and the “mechanostat”: a proposal. Anat Rec. 1987;219:1–9. doi: 10.1002/ar.1092190104. [DOI] [PubMed] [Google Scholar]
- 80.Rauch F, Travers R, Parfitt AM, Glorieux FH. Static and dynamic bone histomorphometry in children with osteogenesis imperfecta. Bone. 2000;26:581–9. doi: 10.1016/s8756-3282(00)00269-6. [DOI] [PubMed] [Google Scholar]
- 81.Shane E, Burr D, Ebeling PR, Abrahamsen B, Adler RA, Brown TD, et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2010;25:2267–94. doi: 10.1002/jbmr.253. [DOI] [PubMed] [Google Scholar]
- 82.Sambrook PN, Ebeling P. Osteonecrosis of the jaw. Curr Rheumatol Rep. 2008;10:97–101. doi: 10.1007/s11926-008-0018-5. [DOI] [PubMed] [Google Scholar]
- 83.Estrogen and progestogen use in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause. 2010;17:242–55. doi: 10.1097/gme.0b013e3181d0f6b9. [DOI] [PubMed] [Google Scholar]
- 84.Bauer DC. Discontinuation of odanacatib and other osteoporosis treatments: here today and gone tomorrow? J Bone Miner Res. 2011;26:239–41. doi: 10.1002/jbmr.335. [DOI] [PubMed] [Google Scholar]
- 85.Robinson JA, Chatterjee-Kishore M, Yaworsky PJ, Cullen DM, Zhao W, Li C, et al. Wnt/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem. 2006;281:31720–8. doi: 10.1074/jbc.M602308200. [DOI] [PubMed] [Google Scholar]
- 86.Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–75. doi: 10.1074/jbc.M705092200. [DOI] [PubMed] [Google Scholar]
- 87.Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50:209–17. doi: 10.1016/j.bone.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Modder UI, Hoey KA, Amin S, McCready LK, Achenbach SJ, Riggs BL, et al. Relation of age, gender, and bone mass to circulating sclerostin levels in women and men. J Bone Miner Res. 2011;26:373–9. doi: 10.1002/jbmr.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yavropoulou MP, van Lierop AH, Hamdy NA, Rizzoli R, Papapoulos SE. Serum sclerostin levels in Paget’s disease and prostate cancer with bone metastases with a wide range of bone turnover. Bone. 2012;51:153–7. doi: 10.1016/j.bone.2012.04.016. [DOI] [PubMed] [Google Scholar]