

Abstract
The molecular mechanisms and genetic programs required for cancer metastasis are sometimes overlapping, but components are clearly distinct from those promoting growth of a primary tumor. Every sequential, rate-limiting step in the sequence of events leading to metastasis requires coordinated expression of multiple genes, necessary signaling events, and favorable environmental conditions or the ability to escape negative selection pressures. Metastasis suppressors are molecules that inhibit the process of metastasis without preventing growth of the primary tumor. The cellular processes regulated by metastasis suppressors are diverse and function at every step in the metastatic cascade. As we gain knowledge into the molecular mechanisms of metastasis suppressors and cofactors with which they interact, we learn more about the process, including appreciation that some are potential targets for therapy of metastasis, the most lethal aspect of cancer. Until now, metastasis suppressors have been described largely by their function. With greater appreciation of their biochemical mechanisms of action, the importance of context is increasingly recognized especially since tumor cells exist in myriad microenvironments. In this review, we assemble the evidence that selected molecules are indeed suppressors of metastasis, collate the data defining the biochemical mechanisms of action, and glean insights regarding how metastasis suppressors regulate tumor cell communication to–from microenvironments.
Keywords: Metastasis suppressor, Angiogenesis, Intravasation, Extravasation, Proliferation, Apoptosis, Cell migration
1. Introduction
Cancer metastasis is an arduous pathological process that is the major contributor to the morbidity and mortality of cancer patients (Eccles and Welch, 2007; Jemal et al., 2010). Upon diagnosis, a patient may feel that cancer has suddenly struck and their life has immediately changed. In reality, however, diagnosis follows a culmination of years—possibly decades—of alterations occurring at the genetic, molecular, cellular, tissue, and organismal levels. Fortunately, the processes of tumor formation and, particularly, metastasis are extremely inefficient and only small fractions of cells from a tumor mass actually overcome the many hurdles to grow at a distant site (Eccles and Welch, 2007; Fidler, 1973a; Weiss, 1990). To metastasize, expression of particular genetic programs is required by a tumor cell to enable the appropriate interactions with changing microenvironments to promote continued survival and proliferation at secondary sites. Understanding these genetic programs and how they affect cellular interactions and signaling cascades is key to understanding the complex process of metastasis.
The existence of tumor suppressors and oncogenes is now accepted as dogma and is well supported by experimental and clinical data. Genes involved in the promotion of metastasis at distinct stages of the disease are also well accepted. However, the hypothesis for the existence of molecules that inhibit the process of metastasis without preventing primary tumor growth was initially met with much skepticism as demonstrated by the three-time rejection of the manuscript reporting the first metastasis suppressor gene NM23 (Steeg, 2004b). Since that time, multiple labs, using many different model systems, have demonstrated the existence of a multitude of protein coding and noncoding genes that significantly reduce metastasis without preventing primary tumor formation. It is now understood that metastasis, the ultimate step in tumor progression, involves many pathological processes; and, just as there are several hallmarks of primary tumor formation (Hanahan and Weinberg, 2000), there also exist hallmarks of metastatic cells (Fig. 3.1). Inhibition of a single step in the metastatic cascade leads to suppression of metastasis (Bruns et al., 2000; Eccles and Welch, 2007; Fidler and Radinsky, 1996). In this chapter, the process of metastasis and the functionality of metastasis suppressing molecules are discussed with the objective that this information can be utilized to identify potential antimetastatic therapeutic strategies. Before discussing metastasis suppressors, it is first necessary to establish the context in which they function.
Figure 3.1.

Hallmarks of metastasis. The necessary traits to form a neoplasm are illustrated for metastasis as an adaptation of the hallmarks of cancer proposed by Hanahan and Weinberg (2000). Only subsets of neoplastic cells successfully invade and metastasize. To invade, cells must alter cell:cell and cell:matrix adhesion, reorganize the extracellular matrix, and become motile. Upon detachment from the primary tumor mass, cells must survive sheer stress and avoid detachment-induced cell death (anoikis). Then, following arrest or adhesion at secondary sites, disseminated cells modify the local microenvironment in order to enable continued growth. Environmental-alterations can be accomplished directly by tumor cells or through surrogates, such as inflammatory cells or mobilized bone marrow stem cell populations. While millions of cells initiate the process of dissemination, only a small fraction completes the process by proliferating to form a macroscopic metastatic mass.
1.1. Genesis of cancer and neoplastic progression
The evolution of a normal cell into a neoplastic cell with progression to a potentially lethal macroscopic metastatic mass is referred to as neoplastic progression or, in the vernacular, tumor progression (Foulds, 1954; Welch and Tomasovic, 1985). There have been several distinct models to depict the cellular mechanisms for this progression including linear and parallel progression models, mutation–selection theory, cancer stem cells, and derivatives of each (Brabletz et al., 2005; Fidler, 2003; Fidler et al., 2007; Klein, 2009; Talmadge and Fidler, 2010; Welch, 1989; Welch and Tomasovic, 1985; Wellner et al., 2009). One of the primary difficulties in constructing generalized model systems for the study of cancer has been the fact that cancer is a heterogeneous disease. As the disease progresses, heterogeneity also increases (Heppner, 1984; Nowell, 1976, 1986). In fact, metastatic cells are behaviorally distinct from cells remaining at the site of primary tumor origin (Steeg and Theodorescu, 2007). These behavioral differences arise at multiple levels including intrinsic cellular changes (genetic and epigenetic heterogeneity), from characteristics of the physical environment (positional heterogeneity; e.g., O2, pH, growth factors, cytokines, chemokines, etc.) and/or from transient events (temporal heterogeneity; e.g., stage of cell cycle, manipulation of the tumor; Nicolson, 1984; Rubin, 1990; Welch, 1989; Welch and Tomasovic, 1985). The intrinsic molecular mechanisms underlying phenotypic differences that characterize a metastatic cell are still being elucidated. However, appreciation for the interrelationships between the surrounding microenvironment and cancer cell-associated genes is increasing (Albini et al., 2007; Ben-Baruch, 2003; Bodenstine and Welch, 2008; Finger and Giaccia, 2010; Joyce and Pollard, 2009; Lin et al., 2009; Pietras and Ostman, 2010; Witz and Levy-Nissenbaum, 2006). Selective regulation of gene transcription also occurs through chemical modifications of DNA and chromatin. Epigenetic modifications are modulated, in part, by how cells interact with the microenvironment(s) in which they find themselves (Lin et al., 2009; Marusyk and Polyak, 2010).
Heterogeneity, for the most part, does not result from multicellular transformation. Data from isoenzyme patterns, karyotypes, and protein production all indicate that the vast majority of tumors are derived from a single cell (Frumkin et al., 2008; Heppner and Miller, 1998; Welch and Tomasovic, 1985). Likewise, analogous methods have been used to show that >90% of metastases are also the result of single-cell outgrowth (i.e., clonal origin) rather than emboli seeding various tissues (Jones et al., 2005, 2008; Talmadge et al., 1982; Wang et al., 2009a; Yamamoto et al., 2003).
Genetic instability may be the chief driver of heterogeneity during tumor progression by random (i.e., not sequentially acquired) generation of variants as described by the mutation–selection theory (Balmain, 2001; Boveri, 1914). However, there are others who advocate that metastatic ability may be a trait acquired early, or commensurate with, tumorigenesis (Bernards and Weinberg, 2002). Regardless, neoplastic cells are significantly more genetically unstable than normal counterparts as shown by fluctuation analyses for multiple genes and loci (Cifone and Fidler, 1981; Otto et al., 1989; Tlsty, 1990; Tlsty et al., 1989). As a result, progression is most often believed to occur as a result of mutation and coupled selection. Subpopulations of cells that have acquired the ability to migrate, invade, and colonize ectopic sites may have a selective advantage since these tumor cells “acquired” the ability to respond, adapt, and/or survive changing environments. Ultimately, with continued selection and variant generation, subpopulations of cells may acquire the ability to penetrate a basement membrane (i.e., invade). Invasion is the unequivocal hallmark that defines malignancy. It should be emphasized that tumor stage is typically measured in terms of the tumor mass and location, rather than individual cells within the mass. Grade is typically defined by the most malignant cells identified within a tumor. Even if the majority of individual cells within a neoplasm are indolent, the term malignant is applied even if a single cell has penetrated a basement membrane. Microdissection of tumor cells has identified chromosomal and genetic changes between subpopulations within a tumor mass (Frost et al., 2001; Steeg and Theodorescu, 2007). This information has been useful for the prediction of genetic underpinnings controlling tumorigenesis, invasiveness, and metastasis. However, it is important to note that adjacent, apparently normal cells also have evidence of genetic instability (Hida and Klagsbrun, 2005).
The complexity of tumor progression leading to a metastatic cell, as described above, shows—not surprisingly, given the numerous steps required to complete the process of metastasis—that a single genetic change is insufficient to accurately predict the likelihood of a lesion progressing to a metastatic phenotype. In fact, defined subsets of genes can be used as prognostic tools (Jorissen et al., 2009; Liu et al., 2007). While multiple genes are required for the progression from primary tumor formation to metastasis, expression of even a single gene that disrupts any of these events would have the ability to suppress metastasis. Although cofactors may be necessary for suppressor function, identification of metastasis suppressors is, overall, usually less technically challenging and easier to interpret than the identification of metastasis-promoting genes.
1.2. Distinctions between tumorigenicity, metastasis, and steps in metastasis
As alluded to, the cellular and molecular events along the progression of a tumor cell into a fully metastatic macroscopic lesion can be broken down into discrete steps. These steps are often discussed interchangeably, therefore, incorrectly. Thus, it is first critical to define metastasis. Doing so is necessary for two reasons. First, metastasis is both a verb and a noun. The process of metastasis (the verb) was defined above. And, the product of the process is a metastasis, the noun. Therefore, it is important to recognize the context in which the discussion of metastasis occurs. Second, the definitions provide the framework to understand the mechanisms involved and develop therapeutic strategies.
In recent years, five misconceptions regarding metastasis have crept into the scientific and medical literature (Welch, 2006, 2007). (1) Metastasis is an inherent property of cancer cells. (2) Metastasis and invasion are equivalent phenotypes. (3) Metastases arise only from cells disseminated via the blood or lymphatics. (4) Tumor cells at secondary sites are metastases. (5) Extravasated cells are metastases. By looking at the definition of metastasis and the mechanisms underlying the process of metastasis, we hope to dispel these misconceptions.
1.2.1. Tumorigenicity and metastasis
Usually, when a primary mass is apparent to the individual or the diagnosing physician, it often comprises at least 1010 cells based on the fact that a cubic centimeter of tissue contains ~109 cells (Tannock, 1983). Although histological analysis reveals these cells to be pleiomorphic and single-cell clones isolated from a tumor vary dramatically in terms of biological behavior, not all cells in a neoplasm are capable of completing the required steps for metastasis.
In their outstanding review, Hanahan and Weinberg described six hallmarks of cancer cells (Hanahan and Weinberg, 2000). Besides immortality (apparently limitless replicative potential), abnormal growth regulation (i.e., failure to respond to growth-inhibitory signals or hyperresponsiveness to progrowth signals), self-sufficient growth, evasion of apoptosis and sustained angiogenesis, invasion and metastasis were listed as distinguishing characteristics. Unfortunately, some have interpreted the list as meaning that all tumors are invasive and/or metastatic, which is certainly not true. Some tumors are highly aggressive and metastatic (e.g., small cell carcinoma of the lung, melanoma, pancreatic carcinoma), while others rarely metastasize despite being locally invasive (e.g., basal cell carcinomas of the skin, glioblastoma multiforme). Therefore, metastasis is not an inherent property of all neoplastic cells (Welch, 2007).
In fact, the process of metastasis begins before cells migrate from a primary tumor mass. Several groups have discovered that the presence of a tumor elicits mobilization of hematopoietic (Erier et al., 2009; Kaplan et al., 2005) and mesenchymal (Hurst and Welch, 2007; Karnoub et al., 2007; Kitamura et al., 2007; Ojalvo et al., 2010; Patsialou et al., 2009; Wyckoff et al., 2007; Yan et al., 2010) stem cells. Both cell types can facilitate tumor cell migration and invasion (Barkan et al., 2010; Ojalvo et al., 2010; Patsialou et al., 2009; Wyckoff et al., 2007) and reorganize tissues in order to manipulate a “niche” into which tumor cells migrate and/or proliferate (Psaila and Lyden, 2009).
1.2.2. Invasion, motility, and metastasis
In most textbooks, metastasis is described in terms of blood-borne (i.e., hematogenous) dissemination. However, secondary tumors can arise because tumor cells have migrated via lymphatics (i.e., lymph node metastases are extremely common in many carcinomas; Eccles et al., 2007; Nathanson, 2003); traversing body cavities (e.g., ovarian carcinoma cells most frequently establish secondary tumors by dissemination in the peritoneum while rarely forming metastases via hematogenous spread; Lengyel, 2010); along capillaries (i.e., many melanomas migrate along already-existing vessels; Lugassy et al., 2002, 2004, 2006; Shields et al., 2007); or along nerves (i.e., pancreatic and prostate carcinomas often exhibit perineural spread; Liebig et al., 2009). So, the route of dissemination is not inherent to a definition of metastasis (Eccles and Welch, 2007; Welch, 2006, 2007). Rather, development of a metastasis needs only incorporate spread of tumor cells to secondary sites.
Although proteolysis-dependent invasion is not an inherent requirement for all tumors to metastasize, it is required for the majority of cancers since physical barriers usually surround a tumor. Understanding the complexity of invasion is necessary to appreciate the mechanisms of many metastasis suppressor genes. Invasive cells have often acquired other traits necessary to metastasize, however, if an invasive cell cannot complete any other step in the metastatic cascade, it will not form a metastasis.
Invasion requires substantial changes of cell morphology and phenotype in addition to modifications of the surrounding environment. During invasion, three important processes are dynamically regulated, including adhesion, ECM reorganization, and motility (Liotta, 1992; Wolf and Friedl, 2006). Normally, epithelial cells form polarized sheets that are maintained by tight intercellular junctions and are anchored to basement membranes by hemidesmosomes, associated intermediate filaments, and integrins. Invading cells have altered cell–cell and cell–matrix adhesion that must be balanced. If a cell is too strongly adherent, it cannot move; and, like a person trying to walk or drive on ice, if to lose an adhesion, cells do not have the traction to move. The structural and functional proteins that regulate cell adhesion and migration are key downstream targets of oncogenes and tumor suppressor-controlled signaling pathways and provide insights into how oncogenic transformation results in progression to an invasive phenotype. Many of the proteins involved in tumor invasion also affect cell survival, growth, apoptosis and angiogenesis, and hallmarks of malignancy. This highlights the intricate network of interrelated pathways modulating cancer cell behavior.
Many dramatic changes in tumor cell morphology during invasion are reminiscent of a normal process that occurs during embryonic development (Hay, 2005; Thiery, 2002), known as epithelial-to-mesenchymal transition (EMT). The EMT describes conversion from an epithelial morphology to a nonpolarized, motile, spindle-shaped cell resembling a fibroblast (Polyak and Weinberg, 2009; Thiery, 2002; Thompson and Newgreen, 2005). EMT is associated with the loss of epithelial-specific E-cadherin from the adherens junctions, and a switch from the expression of keratins as the major intermediate filament to the mesenchymal intermediate filament, vimentin. EMT is influenced by the tumor microenvironment and has been observed primarily at the tumor stromal interface (Polyak and Weinberg, 2009; Thompson and Newgreen, 2005), but a role for EMT in cancer invasion is not universally observed (Cardiff, 2005, 2010; Tarin, 2005). A key regulator of EMT is transforming growth factor beta (TGF-β) signaling (Bierie and Moses, 2006a,b; Creighton et al., 2010; Heldin et al., 2009; Huber et al., 2005; Oft et al., 1998; Pardali and Moustakas, 2007) but other mediators include hepatocyte growth factor/scatter factor (HGF/SF; Yang et al., 2009), PI3 kinase signaling pathway (Pon et al., 2008), MAP kinases (Bakin et al., 2002; Janda et al., 2002), Sprouty4 (Tennis et al., 2010), and the transcriptional factors ZEB1 (Wellner et al., 2009), Twist and Snail (Moreno-Bueno et al., 2008; Onder et al., 2008). Other signaling pathways implicated in stem cell maintenance that are linked to EMT are Wnt (Debies et al., 2008; ten Berge et al., 2008), Notch (Sahlgren et al., 2008), and Hedgehog (Bailey et al., 2007). Tumor cells may also reverse the process and undergo a mesenchymal-to-epithelial transition (MET) in the absence of EMT-inducing signals (Chaffer et al., 2006; Hugo et al., 2007). This transient nature of EMT helps explain why metastatic cells morphologically resemble primary tumor cells despite the fact that they by necessity accomplished all the steps of the metastatic cascade.
Cells induced to undergo EMT not only exhibit enhanced motility but are resistant to apoptosis (especially anoikis), another key requirement for successful metastasis. However, some cancer cells use EMT-independent modes of migration, including collective and amoeboid (Yilmaz and Christofori, 2010). For example in a clever study, Tsuji and colleagues isolated two populations from a single tumor (Tsuji et al., 2008). One population, herein designated Cell-I, exhibited properties of EMT and was able to enter the vasculature (i.e., intravasate). Cell-I was, however, unable to form metastases if injected directly into the vascular compartment. The second population, herein designated Cell-II, displayed an epithelial morphology and was not able to enter the blood stream or metastasize when injected orthotopically. However, Cell-II would colonize tissues when directly injected into the vasculature. If Cell-I and Cell-II were coinjected orthotopically, both were found in metastases. Critical to this review, however, cells undergoing EMT were not themselves successful for metastasis. The authors demonstrated that cellular cooperation existed within the primary tumor and was critical to form metastatic lesions. This would suggest that tumor heterogeneity not only exists but may also be essential for tumor progression.
The extracellular matrix (ECM) provides scaffolding for cells and spatial cues that dictate cellular behavior (Barkan et al., 2010). Matrices are comprised of proteins, primarily triple-helical collagens, glycoproteins such as laminin and fibronectin, and proteoglycans (Catchpole, 1982; Engbring and Kleinman, 2003; Iozzo et al., 2009; Liotta, 1986; Timpl, 1993; Timpl and Aumailley, 1989). Basement membranes are specialized ECM that form barriers separating polarized epithelial, endothelial, and muscle cells from the underlying tissue. Interstitial matrices provide structural characteristics to connective tissues (Erler and Weaver, 2009). The molecular composition of ECM varies between tissues and organs, and provides important contextual information to cellular constituents (Egeblad et al., 2010). In addition, the ECM interacts with many secreted molecules to serve as a repository for regulatory proteins and growth factors (Rozario and DeSimone, 2010). Thus, cell:matrix interactions dictate survival, growth, differentiation, and migration. Correspondingly, selective proteolysis of ECM components leads to release of fragments collectively known as matrikines (Arroyo and Iruela-Arispe, 2010; Duca et al., 2004; Tran et al., 2004), that further regulate protein function and may be involved in cell signaling.
Adhesion of cells to matrix occurs primarily through a family of transmembrane glycoproteins known as integrins, which are heterodimers assembled as specific combinations of 18 alpha and 8 beta subunits (Desgrosellier and Cheresh, 2010; Shattil et al., 2010). Each heterodimer binds distinct, but sometimes overlapping, ECM components. Integrin–ECM binding may be either tumor-promoting or -inhibitory. During tumor progression, cancer cells tend to downregulate the integrins that mediate adhesion and induce maintenance of a quiescent, differentiated state while simultaneously upregulating integrins that promote survival, migration, and proliferation. Although there is a cell-type dependency on integrin function, generally integrins α2β1 and α3β1 are viewed as suppressors of tumor progression, while αvβ3, αvβ6, and α6β4 promote cellular proliferation and migration (Desgrosellier and Cheresh, 2010).
Integrins bidirectionally mediate signals so that changes in intracellular signaling pathways can modulate cellular adhesion (i.e., inside-out signaling); and, changes in cellular adhesion can alter cellular phenotype (i.e., outside-in signaling). Integrin–ECM interactions often modulate cell function by cooperative signaling with different growth factor receptors (Askari et al., 2010; Desgrosellier and Cheresh, 2010). Many cellular responses induced by activation of receptor tyrosine kinases are dependent upon proper cellular adhesion to ECM substrates in an integrin-dependent manner. Signaling in response to ECM interaction usually activates focal adhesion kinase (FAK) and nonreceptor tyrosine kinases of the Src-family.
The ECM can be remodeled by degradative enzymes that are produced by the tumor cells themselves and surrounding stromal cells (Bhowmick et al., 2004). These enzymes contribute to matrix degradation and facilitate tumor cell invasion. Proteolytic enzymes, representing virtually every class of proteases, have been implicated in tumor cell invasion (Boyd, 1996; Gabbert, 1985; Khokha and Denhardt, 1989; Liotta and Stetler-Stevenson, 1991; Nakajima and Chop, 1991; Nicolson, 1982b; Pauli et al., 1983; Roycik et al., 2009; Stracke et al., 1994). Tumor progression-associated proteases include, but are not limited to, serine proteinases (plasmin, plasminogen activator, seprase, hepsin, and several kallikreins), cysteine proteinases (e.g., cathepsin B), aspartyl proteinases (e.g., cathepsin D), and metal-dependent proteinases (e.g., matrix metalloproteinases—MMP and a disintegrin and metalloproteinases—ADAM families). Other matrix-degrading enzymes such as heparanase, an endoglycosidase that cleaves heparin sulfate proteoglycans, and hyaluronidase that cleaves hyaluronic acid, have also been causally associated with tumor progression and invasion (Nakajima et al., 1983; Sanderson et al., 2004; Vlodavsky et al., 1990, 2002).
Liotta and colleagues observed that metastatic potential correlates with the degradation of type IV collagen found predominantly in the basement membrane and focused attention on the metal-dependent type IV collagenases or gelatinases that are now recognized as MMP-2 and MMP-9 (Thorgeirsson et al., 1985; Turpeenniemi-Hujanen et al., 1985). Subsequently, many of the 23 members of the MMP family of matrix-degrading metalloproteinases have been associated with tumor progression (Nelson et al., 2000). Elevated MMP levels correlate with invasion, metastasis, and poor prognosis in many cancer types. Animal models provide evidence for a causal role for MMP activity in cancer progression (Coussens et al., 2001; McCawley and Matrisian, 2000; Sternlicht and Werb, 2001; Sternlicht et al., 1999). Additionally, the plasminogen activator/plasmin system has been causally implicated in cancer invasion, and urokinase plasminogen activator (uPA) and plasminogen activator inhibitor-1 (PAI-1) are validated prognostic and predictive markers for breast cancer (Andreasen et al., 1997; Carlsen et al., 1984; DeClerck et al., 1997; Hildenbrand et al., 2009).
Regulation of matrix proteolysis occurs at multiple levels. In addition to the expression of proteases themselves, many cells also produce endogenous inhibitors including the tissue inhibitors of metalloproteinases (TIMPs; Chirco et al., 2006), serine proteinase inhibitors (SERPINs Bailey et al., 2006), and cysteine protease inhibitors (CYSTATINs; Cox, 2009). Some inhibitors accumulate in high concentrations within the ECM and paradoxically exhibit tumor-promoting functions, including protease activation (Jiang et al., 2002). Conversion of pro-MMP-2 to active MMP-2 requires the activity of MT1-MMP (MMP-14), a transmembrane MMP that is activated intracellularly by the propeptidase family member furin, and TIMP-2 (Hernandez-Barrantes et al., 2000). The stoichiometry of each of these molecules is critical for proper function and regulation. Other proteolytic cascades are important for regulating protease activity during the degradation of ECM, including cathepsin(s) → uPA → plasmin → MMP (Affara et al., 2009). Each protease in this cascade can cleave ECM components; therefore, attribution of function requires detailed and systematic evaluation of each component in the cascade.
The original view that proteolytic enzymes function predominantly to remove physical ECM barriers has been expanded with the realization that proteolysis regulates multiple steps of tumor progression. For example, MMP substrates in the matrix or on the cell surface that modulate cellular growth, differentiation, apoptosis, angiogenesis, chemotaxis, and migration have been identified (Kessenbrock et al., 2010). The abundant evidence for a role of MMPs in tumor progression led to the design and testing of synthetic MMP inhibitors for cancer therapy. These inhibitors proved to be disappointingly ineffective in clinical trials (Coussens et al., 2002), results that have been explained by problems with inhibitor or clinical trial design, as well as a lack of understanding of the broad range of MMP activities resulting in both cancer-promoting and cancer-inhibitory effects (Kruger et al., 2010; Lopez-Otin and Matrisian, 2007).
In addition to ECM remodeling, cell locomotion occurs via coordinated polymerization and depolymerization of the actin cytoskeleton to extend pseudopodia at the leading edge of the cell, known as invadopodia (Buccione et al., 2009; Weaver, 2006), followed by contraction associated with disassembly of cell:matrix adhesive contacts at the trailing edge (Wolf and Friedl, 2006). Adhesion molecules, including several β1 integrins and CD44, and proteases, including MMP and ADAM, are an intricate part of the invadopodia. Inside the plasma membrane, invadopodia contain actin and actin assembly molecules as well as multiple signaling molecules, including FAK, Rac1, and synaptojanin 2; src associated proteins such as p130Cas and Tks5/FISH; and the small GTPases cdc42, Arf1 and Arf6 (Chuang et al., 2004; Guarino, 2010; Muralidharan-Chari et al., 2009; Seals et al., 2005; Tannock, 1983; Yamaguchi et al., 2005). Actin cytoskeletal reorganization involves the Arp2/3 complex and its regulators, WASP, cortactin, and the GTPase Rac (TenKlooster et al., 2006). Actin contractility is regulated by myosin light chain kinase and upstream small GTPases, in particular Rho and its effector ROCK (Kosako et al., 2000; Olson and Sahai, 2009). Many of these molecules have been targeted since invadopodia are implicated as key cellular structures that coordinate and regulate the process of invasion (Buccione et al., 2009; Poincloux et al., 2009; Weaver, 2006).
As alluded to above in the discussion of EMT, single cells migrate either with a spindle-shaped morphology, referred to as mesenchymal migration, or with the less adhesive ellipsoid shape used by leukocytes and Dictyostelium termed amoeboid migration (Wyckoff et al., 2006). Collective migration can occur when the cells retain cell:cell junctions and clusters of cells move in single file through a tissue (Sahai, 2005; Yilmaz and Christofori, 2010). It is noted, however, that the ability of cells to utilize amoeboid migration has been called into question since methods used for reconstitution of matrix resulted in inferior barriers and protein:protein interactions (Sabeh et al., 2009). Another mechanism by which cells traverse cellular barriers is termed entosis (Overholtzer et al., 2007). Briefly, tumor cells transit through other cells and emerge on the other side. Amazingly, many times neither cell is harmed during the process. Based upon some in vitro estimates, entosis can sometimes be quite common. However, the frequency in vivo has not been well studied.
Each type of motility is governed by a variety of cellular factors. Cellular motility is triggered by autocrine inducers of random movement (Jiang et al., 2006; Silletti et al., 1994). Tumor cells produce lysophospholipase D (autotaxin) which stimulates motility, as does lysophosphatidic acid (LPA; Liu et al., 2009; Stracke et al., 1992). LPA can be produced by autotaxin activity on lysophosphatidylcholine. Likewise, HGF/SF interacts with its receptor, c-met, to induce chemokinetic activity of epithelial cells, resulting in an invasive phenotype (Klominek et al., 1998). In fact, disruption of the HGF axis is currently the target of drug development against metastasis (Cecchi et al., 2010; Eder et al., 2009). Directional motility is a chemotactic (following a soluble concentration gradient) or haptotactic (following an insoluble concentration gradient) effect in response to a gradient of soluble or localized factors, respectively. Chemotaxis is often the result of growth factors such as insulin-like growth factor (IGF), and chemokines of the CCR and CXC families (Mantovani et al., 2010). Among the best studied CCR/CXC interactions in metastasis is cellular response to SDF1 (CXCL-12; stromal derived factor-1) as a ligand for the CXCR4 receptor (Gladson and Welch, 2008; Muller et al., 2001; Teicher and Fricker, 2010). SDF-1 levels are often high in tissues commonly colonized by tumor cells (e.g., lung, bone) that express abundant CXCR4. As with the HGF axis, inhibitors of CXCR4 are being studied in preclinical models and are showing efficacy in multiple tumor types (Kim et al., 2008; Richert et al., 2009). Haptotaxis is characterized as a response to gradients of ECM components such as laminin-5 and fibronectin, and can be modulated positively or negatively by proteolysis (McCarthy et al., 1985).
Even cells that have been selected for invasive and metastatic capacity exhibit low efficiency for developing metastasis, seldom exceeding 0.1%. Entry of cells into the blood stream (termed intravasation) is apparently not uncommon. In fact, more than a million cells per gram of tumor can be shed daily (Butler and Gullino, 1975). Tarin and colleagues illustrated metastatic inefficiency of hematogenous metastases using peritovenous (Levine) shunts to palliate ascites burden for patients suffering from various cancer types (Tarin et al., 1984). Although millions of tumor cells were directly deposited into the vena cava daily, the petients did not develop secondary blood-borne tumors with higher frequency.
The fate of already intravasated tumor cells is uncertain because of apparently contradictory experimental evidence. Using radiolabeled cells, Fidler et al. found that most do not survive (Fidler, 1970, 1973b; Fidler and Nicolson, 1977) because of hemodynamic sheer (Weiss, 1989, 1990; Weiss and Schmid-Schonbein, 1989; Weiss et al., 1985), anoikis (Kim et al., 1999; Phadke et al., 2008; Wong et al., 2001), or immune selection (Fidler, 1974; Gorelik et al., 1980; Hanna, 1985; North and Nicolson, 1985; Van Netten et al., 1993; Young and Newby, 1986). In contrast, using a fluorescent tag Naumov et al. (1999, 2002) showed that a majority of cells not only survived but also extravasated. Muschel et al. used intravital microscopy in lung and brain metastasis models to show that the majority of cells remained intravascular and began to proliferate (Carbonell et al., 2009; Wong et al., 2001). Their data illustrate how extravasation is not essential for successful establishment of a secondary mass. Plausible explanations for these dichotomous results include different cell monitoring methods (i.e., radiolabeling vs. fluorescent tagging), analysis of tumor cell behavior in two different tissues (i.e., lung vs. liver), and whether the studies were done completely in vivo versus ex vivo.
Critically, all of these observations highlight the importance of tumor–stromal interactions in the metastatic process and clearly demonstrate that a “one-size-fits-all” description of the metastatic process does not exist. For a cell to accomplish all these “steps” involved in invasion, specific genetic programs must be expressed and functional. Once again, it is stressed that inhibition of any of these requirements would render a cell less metastatic.
1.3. Organotropism of metastasis
Secondary tumors can arise because tumor cells have migrated via lymphatics (i.e., lymph node metastases are extremely common in many carcinomas), the blood vasculature, or across body cavities (e.g., ovarian carcinoma cells most frequently establish secondary tumors by dissemination in the peritoneum while rarely forming metastases via blood-borne routes). Lugassy and colleagues recently documented dissemination of melanoma cells along the space between endothelium and basement membrane (Lugassy et al., 2002, 2004, 2007). That is, the cells do not appear to enter the vascular lumen per se. The latter route of dissemination is reminiscent of perineural spread, which is common in pancreatic and prostatic carcinomas in which tumor cells migrate along nerve sheaths (Liebig et al., 2009). Thus, the route of dissemination is not inherent to a definition of metastasis. Nonetheless, the varying pathways to metastasis illustrate different barriers which tumor cells must surmount.
English surgeon Stephen Paget asked, “What is it that decides what organs shall suffer in a case of disseminated cancer?” (Paget, 1889). Upon reviewing autopsy records from 735 women with breast cancer, he recognized discrepancies between the blood supply going to specific organs and the frequency of metastasis to those organs. For example, despite abundant blood circulation to the heart, spleen, and kidney, breast cancers (indeed most cancers) infrequently colonize these tissues. Paget concluded that unequal distribution of metastases could not be exclusively explained by passive embolus arrest in the first capillaries encountered, as supported by the famed pathologists, Rudolph Virchow (Talmadge and Fidler, 2010; Virchow, 1858), Leonard Weiss (Bross and Blumenson, 1976; Weiss, 1979, 1992; Weiss and Ward, 1982), and James Ewing (Ewing, 1919). Autopsy results for patients succumbing to multiple types of cancer indeed show that most metastases are found in the first lymph node or capillary beds encountered by intravasated tumor cells (Gershenwald and Fidler, 2002; Hess et al., 2006; Park et al., 2009). However, there are several well-known examples of metastatic colonization patterns that simply cannot be explained (Table 3.1).
Table 3.1.
Patterns of clinical metastases that cannot be explained by circulatory patterns or mechanical lodgment of blood-borne tumor cells
| Cancer type | Common sites of metastasis |
|---|---|
| Bladder carcinoma | Bone, liver, brain |
| Breast adenocarcinoma | Bone, brain, adrenal gland |
| GI, Kruckenberg adenocarcinoma | Ovary, liver |
| Kidney, clear cell carcinoma | Bone, liver, thyroid |
| Lung, small cell carcinoma | Brain, liver, bone |
| Melanoma, cutaneous | Brain, liver, bowel |
| Melanoma, uveal | Liver |
| Neuroblastoma | Liver, adrenal gland |
| Prostate adenocarcinoma | Bone |
| Testicular carcinoma | Liver |
| Thyroid, follicular adenocarcinoma | Bone |
Throughout the latter half of the twentieth century, numerous studies supported a blending of the seed and soil and the mechanical hypotheses. As alluded above, many tumor cells can seed lots of tissues, most commonly at the first lymph node or capillary bed encountered. However, the capacity of cells to proliferate and complete the metastatic process is determined by the ability of tumor cells to respond to growth promoting while avoiding growth inhibitory signals.
2. Genetic Regulation of Metastasis
The field of metastasis genetics and the very existence of genes that control specifically metastasis have been called into question (Steeg, 2004a). Inarguably, functional data with the metastasis suppressor genes specifically control metastasis, not tumorigenicity. Some array data were interpreted to suggest that metastatic potential is inherent in tumor cells (Bernards and Weinberg, 2002), but the metastasis suppressor data argue against this interpretation (Eccles and Welch, 2007). Furthermore, recent deep sequencing studies in human pancreatic carcinomas and metastases revealed selective genetic changes consistent with the existence of specific metastasis-regulatory genes (Campbell et al., 2010; Yachida et al., 2010). These new data further show that presumably asynchronous metastases share some, but not all, of the same genetic changes, suggesting multiple pathways in which a cell could succeed in it's quest to metastasize. In general, metastasis-regulatory genes can be grouped into promoting and inhibiting classes. Recent findings have added a third group that can be thought of as the underlying background upon which the promoting and suppressing genes operate. Since the promoting and suppressing genes operate upon this background, we will begin the discussion of metastasis genes with them.
2.1. Quantitative trait loci (QTL)
Complex phenotypes or traits—like metastasis—logically involve contributions from numerous genes, both positive and negative for a phenotype (Cookson et al., 2009; Winter and Hunter, 2008). Analyses to ascribe involvement is challenging because the contribution of each gene is individually relatively small, making linkage challenging. Ultimately, even if each step in metastasis was governed by one gene, more than a dozen genetic changes would be implicated. In reality, as illustrated for adhesion, migration, and invasion, there are scores of genes involved for each.
Thus, in somewhat overly simplistic terms, each of the genes contributes to the quantity of metastases rather than qualitative determination of metastasis development. Kent Hunter and colleagues have tackled this challenging problem and collected some very important and revolutionary data that support the existence of metastasis genes using breeding strategies in mice. Using a transgene-induced mouse mammary tumor model (MMTV-PyMT, in which the polyoma middle T oncogene is driven by the murine mammary tumor virus promoter), mice were crossed with mice of varying genetic backgrounds. Significant differences in metastasis were found in the F1 progeny despite failure to alter tumor initiation or growth kinetics in some strains (Lifsted et al., 1998). Since all the mouse tumors were initiated by the same oncogenic event, differences in metastasis and gene expression were most readily explained by genetic background. His data reinforced a notion introduced earlier—gene context is an important parameter in determining metastatic potential. Although this review is focused upon metastasis suppressors, appreciation of QTL and metastasis-promoting molecules is essential to understand structure–function relationships of the metastasis suppressors.
2.2. Pro-metastatic genes
It is clear that subsets of tumor cells are endowed with capabilities not present in their nonmetastatic counterparts. It follows, then, that metastatic cells turn on genes that promote metastasis. However, it is difficult to identify prometastatic genes because the ability to metastasize requires a cell to accomplish numerous tasks in multiple different microenvironments. Therefore, experimental studies are prone to false-negative studies for metastasis-promoting genes. More accurately, metastasis-promoting genes should probably be designated metastasis efficiency-enhancing genes.
Despite these caveats, mutated ras expressed in NIH-3T3 cells can confer tumorigenicity and metastatic capacity (Bondy et al., 1985; Chambers et al., 1990). Likewise, introduction of mutant MEK mutants—which mimic constitutively activated MEK—also render NIH-3T3 cells tumorigenic and metastatic (Welch et al., 2000). While this is true in experimental models using fibroblasts, the ability of Ras or MEK to transform and induce progression in all cell types remains to be determined. Together, these studies implicate signaling through the Ras-Raf-Mek-Erk pathway in metastasis. However, cross-talk to and from this signaling cascade affects numerous downstream mediators, thereby providing a plausible mechanism for coordinated expression of the multiple molecules necessary for metastasis. Similarly, Kang et al. (2003) and Minn et al. (2005) in the laboratory of Joan Massague have studied gene expression patterns that begin to explain organotropism of metastasis. Both found that coordinated expression of multiple genes is required for bone and lung metastasis, respectively. Interestingly, many of the genes implicated in both metastatic sites are downstream of TGF-β, a well-known promoter of tumor invasion and EMT as discussed above.
As discussed above with studies identifying QTL, context is critical. This concept is readily apparent when considering the role(s) of TGF-β in the development of multiple carcinomas. In normal breast, TGF-β is generally growth inhibitory (Nam et al., 2008; Wakefield and Stuelten, 2007); however, sometime during tumor progression, there is a paradoxical switch in which malignant behavior is promoted (Welch et al., 1990). Although numerous studies have focused on this phenomenon, the precise molecular mechanisms remain elusive.
2.3. Metastasis suppressor genes and methods to identify metastasis suppressors
Any single gene that disrupts a necessary biological process involved in the metastatic cascade could suppress metastasis. Since metastases develop only from neoplastic cells, tumor suppressors will also, by definition, suppress metastasis. However, we distinguish metastasis suppressors by their ability to inhibit metastasis without preventing primary tumor formation. (Note: some metastasis suppressors can delay tumor growth, but do not prevent tumor growth.) Metastasis suppressors have been found in virtually all cellular compartments and have a wide range of functions including cell adhesion, cell–cell communication, signaling, cell invasion, transcriptional regulation, etc. Below is a summary of metastasis suppressors grouped in broad functional categories. Table 3.2 provides a quick summary of the key points and Fig. 3.2 depicts key pathways involved.
Table 3.2.
Metastasis suppressors and proposed mechanisms
| Metastasis suppressor | Chromosomal location | Proposed mechanism(s) of action | Cellular localizationa | Step(s) in metastasis inhibited |
|---|---|---|---|---|
| BRMS1 | 11q13.1–ql3.2 | Transcriptional regulation via interaction with SIN3:HDAC complexes; downregulates PtdIns(4,5)P2 | N, some C | Multiple; colonization |
| Caspase 8 | 2q33–q45 | Induction of apoptosis if cells bind to unliganded integrins | C | Transport |
| E-cadherin | 16q22 | Cell:cell interactions | M | EMT; invasion |
| N-cadherin | 8q11.2 | Cell:cell interactions | M | EMT; invasion |
| Cadherin-11 | 16q22.1 | Cell:cell, cell:matrix interactions | M | EMT; invasion |
| CD44 | 11p13 | Hyaluronic acid receptor; osteopontin receptor stem cell marker (selected) | M | Migration |
| DCC | 18q21.3 | Regulates cytoskeletal organization; regulates MAPK signaling | C | Transport; migration |
| DLC1 | 8p22–p21.3 | RhoGTPase activating protein; regulates cytoskeletal structure | C | Motility; migration; invasion |
| DRG1 | 8q24.3 | Unknown | C, some N | Angiogenesis; colonization (?); intravasation (?) |
| GAS1 | 9q21.3–q22 | Inhibit cell cycle | N, some C | Unknown |
| Gelsolin | 9q33 | Regulates cytoskeletal structure; reduces motility | C | Motility; migration |
| HUNK | 21q22.1 | Protein kinase | C | Migration; invasion |
| KAI1 | 11p11.2 | Interacts with endothelial DARC to induce apoptosis | M | Intravasation; transport |
| KISS1 (kisspeptins) | 1q32 | Maintains dormancy at secondary sites | S | Colonization |
| KISS1R | 19p13.3 | G-protein coupled receptor | M | Colonizaton |
| KLF17 | 1p34.1 | Transcription | N | Invasion; EMT |
| LSD1 | 1p36.12 | Chromatin remodeling | N | Invasion |
| MKK4 | 17p11.2 | Stress-activated MAPK signaling | C | Colonization; migration |
| MKK7 | 19p13.3–p13.2 | Stress-activated MAPK signaling | C | Colonization; migration |
| p38 | 6p21.3–p21.2 | Stress-activated MAPK signaling | C | Colonization; migration |
| Nm23 | 17q22 | Phosphorylates KSR to prevent downstream activation of MAPK pathways | C, some N | Migration; colonization |
| OGR1 | 14q31 | GPCR signaling | M | Migration |
| RhoGDI2 | 12p12.3 | Regulates Rho; negatively alters endothelin 1 and neuromedin U expression | C | Migration; colonization |
| RKIP | 12q24.23 | Competitive inhibitor of RAF1–MEK interactions | C | Migration; invasion |
| RRM1 | 11p15.5 | Increases PTEN expression; decreases FAK phosphorylation | C | Motility; invasion |
| SSeCKS | 6q24–q25.1 | Scaffold protein for PKA and PKC; inhibits osteopontin, VEGF expression; up regulates vasostatin | C | Angiogenesis; migration |
| TIMPs | Multiple | Inhibit metalloproteinases; signaling | C, S, M | Angiogenesis; migration; invasion; transport |
Nuclear (N), cytoplasmic (C), membrane (M), secreted (S).
Figure 3.2.

Metastasis suppressors exist in every cellular compartment and in the extracellular milieu. Predominantly nuclear (blue), cytoplasmic/signaling (purple), membrane (red), or extracellular (green) molecules are shown, some with key interacting molecules/complexes (gray). Based upon current knowledge regarding location and function, inferences regarding mechanism of action are described in the text.
Metastasis suppressor genes have mostly been identified by first comparing loss of heterozygosity (LOH) and karyotypic abnormalities in different stage human cancers. Then, microcell-mediated chromosomal transfer (MMCT) was used to introduce individual chromosomes thought to encode one or more metastasis suppressors. This method has been the most lucrative and proved successful for the discovery of metastasis suppressors on chromosomes 1, 2, 7, 8, 10–13, 16, 17, and 20. Individual genes have also been successfully identified by subtractive hybridization, differential display, comparative genomic hybridization (CGH), microdissection, real-time RT-PCR, microarray, and proteomic approaches (Rinker-Schaeffer et al., 2006; Vaidya and Welch, 2007). Details for the discovery of individual genes are discussed in Section 3.
More recently discovered metastasis suppressors are the direct result of improved techniques to identify functions associated with metastasis. Genome-wide shRNA screens were used to identify Growth Arrest-Specific 1 (GAS1) and Krüppel-Like transcriptional Factor 17 (KLF17) as melanoma and breast cancer metastasis suppressors, respectively (Gobeil et al., 2008; Gumireddy et al., 2009). In their study, Gobeil et al. discovered 22 genes in which shRNA knockdown resulted in an increase in metastasis using the highly metastatic B16–F10 murine melanoma cell line. They focused on GAS1 since it was substantially down-regulated in the B16-F10 cells. It is presently unclear why the identified genes in their screen did not overlap with already known metastasis suppressor genes.
In the study by Gumireddy et al. (2009), an shRNA library and in vivo screen in which the nonmetastatic 168FARN breast cancer cells that metastasized were selected. RNAi for the KLF17 gene was identified and subsequently chosen for more detailed studies. KLF17 was found to bind to the promoter region of inhibitor of differentiation 1 (Id1) leading to inhibition of invasion and EMT. Improved techniques such as these shRNA screens should increase our discovery of metastasis suppressor genes. Questions regarding specific cell line or model systems may become crucial to our understanding of how context-dependent factors play a major role in suppressor function.
3. Functionally Validated Metastasis Suppressor Genes
3.1. Transcriptional regulators
3.1.1. BRMS1
Because metastasis requires the coordinated expression of particular genes at multiple steps, a key regulatory molecule would be one that functions by regulating metastasis-associated gene transcription. Breast cancer metastasis suppressor-1 (BRMS1) alters the expression of multiple metastasis-associated genes including osteopontin (OPN; Hedley et al., 2008; Samant et al., 2007; Shevde et al., 2006), uPA; (Cicek et al., 2005, 2009), fascin (Zhang et al., 2006), epidermal growth factor receptor (EGFR; Hurst et al., 2008; Vaidya et al., 2008), CXCR4 (Yang et al., 2008), as well as coordinately regulating many metastamiR (Edmonds et al., 2009a,b; Hurst et al., 2009a). These genes are associated with metastasis at many different steps. Likewise, BRMS1 affects multiple phenotypes implicated in cancer metastasis (Phadke et al., 2008), including restoration of homotypic (Saunders et al., 2001; Shevde et al., 2002) and heterotypic (Kapoor et al., 2004) gap junctional intercellular communication, inhibition of migration and invasion, promotion of anoikis, and differential modulation of growth factor signaling. Additionally, the selective downregulation of phosphoinositide phosphatidylinositol (4,5) bisphosphate (PtdIns(4,5)P2) has been demonstrated (Champine et al., 2007; DeWald et al., 2005; Vaidya et al., 2008) that may have dramatic signaling effects in response to the microenvironment. In vivo experiments have demonstrated that BRMS1 inhibits several steps of metastasis, including the ultimate step, colonization at the secondary site (Phadke et al., 2008).
BRMS1 was originally identified by analysis of differentially expressed genes in the metastatic breast carcinoma cell line MDA-MB-435 (Chambers, 2009; Grijalva et al., 2003; Hollestelle and Schutte, 2009; Montel et al., 2009) following MMCT of neomycin-tagged chromosome 11 (Seraj et al., 2000). Differential display was used to compare chromosome 11-containing with parental cells, which led to the identification and cloning of BRMS1, which was subsequently mapped to 11q13.1–q13.2. It was then directly transfected into metastatic breast cancer cell lines that express no detectable levels of BRMS1 transcript and using xenograft (Seraj et al., 2000) and syngeneic (Samant et al., 2002, 2006) mammary tumor models was found to significantly suppress metastasis. Since that time, multiple labs using several different model systems have found that BRMS1 suppresses metastasis of melanoma (Shevde et al., 2002), ovarian (Zhang et al., 2006), and nonsmall cell lung carcinomas (Smith et al., 2009) in addition to breast carcinoma.
Determining the mechanism of action for BRMS1 has occurred in a somewhat circuitous manner. Protein sequence homology provided few clues regarding possible mechanisms. So, protein:protein interaction studies using yeast two-hybrid genetic screens and coprecipitations were undertaken. Almost simultaneously, both approaches identified a direct interacting partner for BRMS1, Rb-binding protein-1 (RBBP1) which is now known as AT rich interacting domain 4A (ARID4A) (Hurst et al., 2008; Meehan et al., 2004). Also, directly binding to BRMS1 is suppressor of defective silencing-3, SUDS3 (a.k.a. mSDS3 or SAP45; Hurst et al., 2008; Meehan et al., 2004; Silveira et al., 2009). Both ARID4A and SUDS3 are components of the SIN3 histone deacetylase chromatin remodeling complexes. Other groups studying SIN3 complexes and associated proteins have identified BRMS1 by mass spectrometry, affinity purification, and coimmunoprecipitation (Doyon et al., 2006; Le Guezennec et al., 2006; Nikolaev et al., 2004; Shiio et al., 2006; Smith et al., 2010).
When BRMS1 is present in complexes with the Gal4 promoter–luciferase reporter, transcriptional repression is observed (Hurst et al., 2008; Meehan et al., 2004; Silveira et al., 2009). However, mRNA expression arrays reveal a complex pattern of>500 nonrandom expression changes (Champine et al., 2007; Cicek et al., 2005). Cicek et al. were the first to demonstrate selective differential expression of proteins in BRMS1-expressing breast cancer cells using 2D gel-electrophoresis (Cicek et al., 2004), and recently Rivera et al., used a similar approach in melanoma cells to identify differentially expressed proteins (Rivera et al., 2007). Some differentially expressed proteins were identified in both studies, for example, annexins, and glutathione-S-transferases, but overlap was not predominant probably because BRMS1 regulation may be cell-type dependent. By both proteomic and genomic discovery approaches, BRMS1 regulates genes involved in lipid metabolism and transport, secretion, and cellular architecture.
To date, however, whether gene regulation effects are direct versus indirect has not been clearly demonstrated. Jones et al. (Liu et al., 2006; Smith et al., 2009) identified interaction of BRMS1 with the p65 subunit of the NFκB transcription factor. Presumably, recruitment of SIN3∷HDAC complexes reduces NFκB activity, a finding that has been observed using reporter assays. Inhibition of NFκB activity through recruitment of HDAC1 has been observed by different laboratories (Cicek et al., 2005, 2009; Samant et al., 2007). Although it is likely that BRMS1 will interact with other transcription factors, their identities, if any, have not yet been determined.
Mutational analysis of BRMS1 has determined that direct ARID4A and SUDS3 interactions are not essential for metastasis suppression (Hurst et al., 2008; Silveira et al., 2009). Different domains of the BRMS1 protein bind each molecule; however, BRMS1 remained associated with SIN3 and HDAC1/2. Interestingly, disruption of each direct BRMS1 interaction alters the gene expression profiles of cells reexpressing the BRMS1 mutants (Hurst et al., 2008; Silveira et al., 2009). These findings reveal that the mix-and-match nature of the SIN3∷HDAC∷BRMS1 complexes determines the expression of individual genes. In fact, Smith et al. showed that HDAC inhibitors could differentially cause modification to the SIN∷HDAC complex composition (Smith et al., 2010).
Clinical studies with BRMS1 have been relatively inconsistent with regard to patient survival and metastasis correlations. The inconsistencies are thought to be primarily because most clinical studies measured mRNA expression, but BRMS1 mRNA and protein do not always correlate (Hurst et al., 2009c). Also, BRMS1 protein is sensitive to proteasome degradation and is stabilized by the heat shock protein HSP90 (Hurst et al., 2006), highlighting the importance of measuring protein levels. Ultimately, simply measuring protein levels for a protein that functions differentially depending upon its interaction partners may be moot. Nonetheless, BRMS1 protein expression using IHC is predictive for survival and metastasis development in subsets of breast (Frolova et al., 2009; Hicks et al., 2006) and nonsmall cell lung carcinomas (Smith et al., 2009).
3.1.2. CRSP3 and TXNIP
Two additional transcriptional regulators, CRSP3 (a.k.a. cofactor required for SP1 activity; DRIP130, Vitamin D regulatory interacting protein 130) and TXNIP (a.k.a. thioredoxin interacting protein; TBP2, thioredoxin binding protein 2; VDUP, vitamin-D3 upregulated protein) have been identified as metastasis suppressors (Goldberg et al., 2003). Both molecules have been studied in regard to their redox regulation and/or signaling in addition to their apparent association with the vitamin D transcription complex. CRSP3, which maps to chromosome 6q23.2, upregulates TXNIP, which maps to chromosome 1q, which, in turn, regulates the KISS1 metastasis suppressor (see Section 3.3.3).
3.1.3. LSD1
Lysine-Specific Demethylase 1 (LSD1) is an amine oxidase catalyzing the demethylation of histone proteins and has been shown to be a component of many chromatin remodeling complexes including CoREST (Lee et al., 2005; Shi et al., 2005), CtBP (Wang et al., 2007), and other HDAC containing complexes (You et al., 2001). More recently, it was found to be an integral component of the Nucleosome Remodeling and Deacetylase (NuRD) protein complex to inhibit invasion and suppress metastasis in breast cancer model systems (Wang et al., 2009b).
LSD1 was found to be downregulated in breast carcinomas and expression was inversely correlated with TGFβ1. Interestingly, the NuRD complexes have also been implicated in promoting metastasis as several studies show important functions associated with histone deacetylation for the metastasis-associated proteins (MTA; Bagheri-Yarmand et al., 2004; Nicolson et al., 2003; Ohshiro et al., 2010; Toh et al., 1994, 2004). Analogous to the complexes formed by BRMS1, the paradigm supported by LSD1 is that the function of chromatin remodeling complexes is clearly dependent on the specific composition of each complex.
3.2. Posttranscriptional regulators
It is clear that metastasis is regulated by the expression of genes necessary for phenotypes required for each step in the cascade and transcriptional regulation of metastasis-associated genes is one key mechanism to inhibit or promote metastasis. The majority of proteins in a cell are also regulated posttranscriptionally and this serves as yet another level for controlling metastasis. It has recently been shown that several microRNA (miRNA) genes significantly influence several steps in the metastatic cascade that have now been given the term metastamiR. Other molecules that regulate signaling in response to the microenvironment or affect adhesion to the microenvironment may dramatically inhibit metastasis.
3.2.1. MetastamiR and noncoding RNA
With the initial discovery of miRNA in the control of the timing of Caenorhabditis elegans larval development (Lee et al., 1993), they were identified in plant (Park et al., 2002) and mammalian cells less than a decade later (Wightman et al., 1993). These small RNA genes are typically transcribed by RNA polymerase II to the pri-miRNA that adopts a characteristic hairpin loop structure. They are further processed to pre-miRNA by the RNAse 3 Drosha and exported to the cytoplasm by Exportin 5 where the enzyme Dicer processes the hairpin to a mature 18–26 nucleotide miRNA that associates with the RNA-induced silencing complex (RISC). Both Drosha and Dicer form complexes with proteins containing dsRNA-binding domains. The Drosha partner is DiGeorge syndrome critical region gene 8 (DGCR8) and the Dicer partner is TAR RNA binding protein (TRBP).
The latest release of the miRBase database has catalogued >1000 miRNA in humans. Each mature miRNA (19–24 nt) complements the 3′-UTR of mRNA. Moreover, microRNA can regulate the translation of hundreds of genes through sequence-specific binding to mRNA depending on sequence complementarity will result in the inhibition of translation and/or degradation of target mRNAs (Stefani and Slack, 2008). However, some microRNA upregulate some genes by direct and indirect mechanisms. As a result of such promiscuity, it is perhaps not surprising that a single metastamiR might regulate metastasis similarly to a transcription factor that exerts its effect on multiple mRNA or proteins.
Altered regulation of miRNA expression exerts profound effects on cell phenotypes. Soon after their discovery in mammalian cells, miRNA were reported to play key roles in cancer, recurrence, development of metastases, and/or survival (Edmonds et al., 2009b; Hurst et al., 2009b; Nicoloso et al., 2009). At least, a dozen miRNA have been shown to promote or inhibit metastasis in experimental models and that number will likely grow even further because >20 more have been shown to impact critical steps in the metastatic cascade, such as EMT, apoptosis, and angiogenesis. Typically, metastamiR were discovered using in vitro screens for individual steps in metastasis including proliferation, EMT, adhesion, migration, invasion, apoptosis, and/or angiogenesis. As mentioned previously, a critical point to validate a miRNA as a bona fide metastasis suppressor is to perform in vivo assays.
The first suppressing metastamiR was identified by Tavazoie et al., who compared miRNA expression in metastatic variants derived from the human breast carcinoma cell line, MDA-MB-231 (Tavazoie et al., 2008). They identified six miRNAs with low relative expression in the metastatic cells. Three of these, miR-335, -126, and -206, suppressed metastasis in vivo; however, miR-126 also inhibited cell proliferation and tumorigenesis, removing it from the metastasis suppressor category, by definition. Both miR-335 and -206 inhibited invasion and migration in vitro. miR-335 targets SOX4 (SRY-box containing transcription factor), PTPRN2 (receptor type tyrosine protein phosphatase), MERTK (c-Mer tyrosine kinase), and possibly TNC (tenascin C). Additionally, inhibition of SOX4 or TNC by shRNA inhibited invasion in vitro and metastasis in vivo. Their findings elegantly demonstrate how a single miRNA could impact several downstream pathways by arborizing signaling pathway components. There was also a clinical association of miR-335 expression with metastasis-free survival in a set of 20 primary breast tumor samples.
Several groups had shown roles for miR-146 in inflammation through regulation of NFκB (O'Connell et al., 2010). Although the miR-146a and -146b genes are encoded on different chromosomes, their mature sequence differs by only two nucleotides at the 3′ region. So their mRNA targets are predicted to overlap significantly. Indeed, both miR-146a and -146b inhibit invasion and migration of breast cancer cells by downregulating NFκB by targeting IRAK1 and TRAF6 (Bhaumik et al., 2008). These studies were extended in vivo by demonstrating miR-146a and -146b suppressed metastasis that may involve targeting of EGFR (Hurst et al., 2009a) or ROCK1 (Nicoloso et al., 2009), both of which are involved in promoting invasion and metastasis. In clinical samples, miR-146a expression is inversely correlated with prostate cancer progression, further supporting a metastasis suppressor function for this metastamiR (Lin et al., 2008).
While inhibition of any step in the metastasis cascade precludes metastasis, a single metastamiR could result in more robust inhibition of the metastatic process by targeting multiple steps. Evidence to support this conclusion comes from studies with miR-31, which inhibits invasion, anoikis, and colonization leading to a 95% reduction in lung metastasis in an orthotopic model of breast cancer (Valastyan et al., 2009, 2010). Additionally, miR-31 levels were lower in a pilot study of breast cancer patients with metastasis.
MetastamiR are not limited to suppressors of metastasis. miR-10b was the first metastamiR discovered by Ma et al. (2007a). They hypothesized that certain miRNA could regulate specific stages of tumor progression and found that miR-10b was highly expressed only in metastatic breast cancer cell lines compared to primary human mammary epithelial or spontaneously immortalized cells. After showing that miR-10b enhanced migration and invasion in vitro and metastasis in vivo, they identified a pathway where the prometastatic gene TWIST1 upregulates miR-10b that targets HOXD10 leading to an increase in RHOC. Additionally, RTQ with 23 primary breast tumors was used to show a general increase in miR-10b expression in patients with metastasis.
Interestingly, the BRMS1 metastasis suppressor that regulates miR-146a/b also regulates TWIST, miR-10b, and RhoC expression (Edmonds et al., 2009a). Whether the regulation of these genes by BRMS1 is direct or indirect is still not known. Regardless, the data all point to common pathways impacted by these metastasis-regulatory molecules (Fig. 3.3).
Figure 3.3.
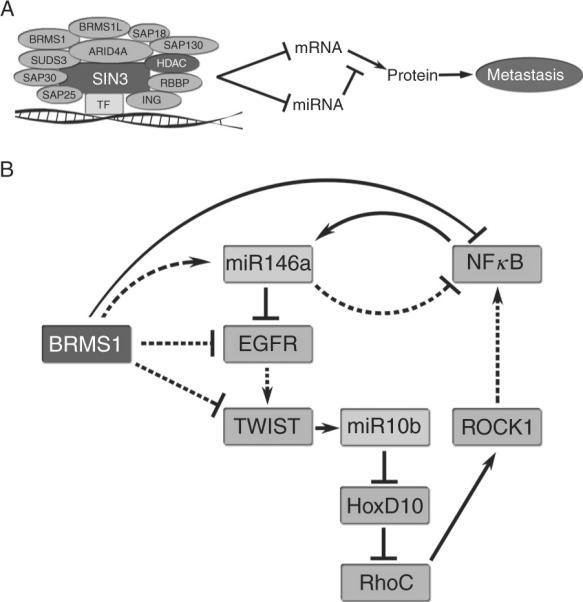
Metastasis suppressor networks. Metastasis suppressors may function together to inhibit metastasis-associated networks at multiple levels. (A) BRMS1 is part of a chromatin remodeling complex that represses transcription. This leads to direct repression of gene transcription and indirect repression through downregulation of upstream repressors. Additionally, noncoding genes including miRNA are repressed leading to altered translation of mRNA. (B) BRMS1 regulates the expression of EGFR by downregulation of transcription and by increased expression of miR-146a that targets EGFR. This leads to decreased levels of TWIST1 and the metastasis-promoting metastamiR, miR-10b. Translation of the transcription factor, HoxD10, is then increased with a subsequent decrease in RhoC and ROCK1. This pathway feeds back through NFκB. Dotted lines indicate indirect mechanisms or multiple steps not shown and solid lines indicate direct regulation.
Huang and colleagues transduced nonmetastatic MCF7 human breast cancer cells with an miRNA expression library and screened the transductants using a transwell migration assay (Huang et al., 2008). Both miR-373 and -520c promoted migration and were subsequently found to increase in vivo metastasis, at least in part, by targeting the hyaluronate receptor and stem cell marker, CD44. Clinically, miR-373 expression was higher in lymph-node metastasis compared with the primary tumors from 11 pairs of matched samples.
Invasion and migration are increased while apoptosis is decreased by miR-21 expression in breast, colon, and glioma (Asangani et al., 2008; Gabriely et al., 2008; Zhu et al., 2008). miR-21 targets TPM1 (tropomyosin 1), PDCD4 (programmed cell death 4), and regulators of MMP. miR-143 and miR-182 promoted hepatocellular carcinoma and melanoma metastasis, respectively (Segura et al., 2009; Zhang et al., 2009b). miR-143 is upregulated by NFκB and decreases adhesion. miR-182's effects can be reversed by reexpression of MITF (microphthalmia-associated transcription factor M) or FOXO3. miRNA-182 is part of a cluster (miR-183-96-182). Many miRNA are encoded as genetically linked clusters (perhaps operons?) and are expressed as a single pri-miRNA. As a result, it is not always possible to distinguish biological effects that are the result of a single miRNA or the collective actions of multiple miRNA. Since many experimental studies manipulate single members of a cluster, interactions or feedback mechanisms may be missed if the cluster expression is not taken fully into account.
3.2.2. MetastamiR pathways, concepts, and future directions
While metastamiR have only been recognized for slightly more than 3 years, the rapid discovery of this important family of molecules is impressive. MetastamiR are components of complex pathways and are often expressed downstream of pro- or antimetastatic signals, including pathways regulated by NFκB, EGFR, TWIST1, BRMS1, ZEB1/2, and HIF1α. Unfortunately, understanding the mechanisms by which miRNA are regulated is still in its infancy.
Interestingly, positive and negative feedback loops have been found whereby the upstream effectors are themselves targets of the miRNA that they regulate (Aguda et al., 2008; Bracken et al., 2008; Castellano et al., 2009; Cheloufi et al., 2010; Taganov et al., 2006; Wellner et al., 2009). This implies an important role for metastamiR in modulating key signaling pathways involved in tumorigenicity and metastasis. Because of their position as nodes within signaling pathways and their promiscuity with regard to downstream targets, each metastamiR can (and probably does) amplify pro- and antimetastatic signaling events. It is likely that metastamiR regulation of these signaling events is context dependent, relying on microenvironmental cues in both directions. We predict that yet-to-be-discovered cofactors will lead to specificity of miRNA effects on selected pathways; however, their existence is speculation at this time. We find ourselves in the midst of a revolution with regard to the biochemical and molecular regulation of cancer metastasis. Old notions of equating tumorigenicity with metastasis have to be discarded. There are clear distinctions between the phenotypes; biologically, biochemically, and genetically. Understanding the interrelationships between regulatory genes and gene products and how these are modulated by the microenvironmental context is beginning to unravel the complex tapestry that is cancer metastasis.
During the course of assembling references for this chapter, the expanding complexity of miRNA and more specifically, metastamiR, exploded. As targets are assigned to individual miRNA, one must now take into account previously ignored pseudogenes. Poliseno et al. showed that the pseudogene, PTENP1, was biologically active by virtue of regulating cellular levels of the tumor suppressor PTEN (Poliseno et al., 2010). Although PTENP1 is not translated into protein due to a missense mutation of the initiator methionine codon, it still possesses a 3′ UTR with high homology to PTEN. As a result, increased expression of PTENP1 serves as a “decoy” for PTEN targeting miRNA and leads to increased translation and protein levels of biologically functional PTEN. Therefore, pseudogenes are now going to have to be considered when analyzing various functions of miRNA.
Future studies regarding noncoding RNA involved in metastasis will not be limited to miRNA. A recent report describes the large intergenic noncoding RNA (lincRNA) HOTAIR that promotes metastasis (Gupta et al., 2010). HOTAIR associates with the chromatin remodeling complexes Polycomb Repressive Complex 2 (PRC2) and LSD1 and alters the methylation pattern on histone lysine residues, specifically methylation of H3K27 and demethylation of H3K4 (Gupta et al., 2010; Tsai et al., 2010). This leads to epigenetic changes in gene expression that are favorable for metastasis. Another recent report identified p53 as a mediator of many lincRNA including lincRNA-p21 (Huarte et al., 2010) demonstrating the likelihood of identifying many more metastasis-associated lincRNA.
3.3. Regulators of cellular communication
In retrospect, it seems obvious that the cell surface would be a key site for critical molecules involved in cancer metastasis since tumor cells encounter numerous different microenvironments during their journey. Three lines of evidence have been used to support the involvement of cell-surface molecules in the process of metastasis. The first is that enzymatic modification of cell-surface components can alter adhesion, survival in the circulation, and arrest at secondary sites (Hagmar and Norrby, 1973; Welch, 1997; Welch et al., 1994a). The second is involved in biosynthetic modification of surface glycoproteins and glycolipids (Gasic and Gasic, 1962; Irimura et al., 1981; Shaikh et al., 2008). The third has involved transfer of cell-surface molecules from metastatic to nonmetastatic cells with a corresponding enhancement of metastatic efficiency (Legrue, 1982; Poste and Nicolson, 1980; Poste et al., 1980). There are abundant more examples for each of these experimental strategies. Readers are referred to several excellent reviews for additional details (Geiger and Peeper, 2009; Lu and Kang, 2009; Nicolson, 1982a, 1988a,b). The examples listed below focus exclusively on metastasis suppressor genes that are found on the cell-surface or cell-cell junctions.
3.3.1. Cell-surface receptors and junctions
3.3.1.1.CD44
CD44 is a transmembrane glycoprotein that binds ECM components such as hyaluronic acid and the prometastatic factor, osteopontin (Underhill, 1992). CD44 is proposed to modulate adhesion, lymphocyte homing, and activation (Kallakury et al., 1996) and maps to 11p13 (Rudy et al., 1993). In clinical samples, there is a correlative loss of CD44 expression in high-grade tumors and metastases (Kallakury et al., 1996). Depending on the type of cancer, cell line used, and the model being evaluated, CD44 expression can increase tumorigenicity and metastatic potential or function as a metastasis suppressor (Kallakury et al., 1996; Rudy et al., 1993). In recent years, many cancer researchers have become enamored by the cancer stem cell theory or the cancer progenitor cell theory (Brabletz et al., 2005). Briefly, the theory proposes that migrating cells with properties similar to stem cells—capacity to self renew for extended times, ability to regenerate a mixed population of cells with both specialized and unspecialized properties. Since the majority of metastases are clonal in origin yet heterogeneous by the time overt, macroscopic metastases are diagnosed, there are abundant similarities. Several laboratories have indicated that CD44 surface expression is a marker for cancer stem cells (Bauerschmitz et al., 2008; Sackstein et al., 2008; Tang et al., 2007), further raising questions regarding the role(s) of CD44 in metastatic behavior.
Ambiguity regarding metastasis-promoting or -suppressing effects by CD44 probably rests in the high degree of posttranscriptional and splicing variation that occur in different cell types. As a result, some of the splice variants may have different functions from others. Until reagents are developed and the cell-specific changes are categorized, the issue cannot be resolved.
3.3.1.2. E-cadherin
Epithelial cell–cell interactions are mediated primarily by cadherins, transmembrane glycoproteins that form Ca+2-dependent homotypic complexes (Harris and Tepass, 2010). For many tumor types, loss of E-cadherin occurs during EMT and correlates with increased invasion and metastasis. Reexpression in experimental models can block invasion. Taken together, these observations suggest that loss of E-cadherin is causative for invasion. E-cadherin loss occurs because of transcriptional repression and proteolytic degradation (Jeanes et al., 2008; Li et al., 2004; Onder et al., 2008; Van Roy and Berx, 2008). The zinc finger transcriptional repressors Snail and Slug, in particular, have been implicated in regulating EMT by virtue of their ability to repress E-cadherin transcription. Cadherins are regulated by catenins (α, β, γ, and p120 catenins), cytoplasmic proteins that functionally link the cadherin complex to the actin cytoskeleton. βb-catenin is both a cell adhesion protein and a transcription factor. In addition to its role in adherens junctions, β-catenin participates in canonical Wnt signaling (Behrens, 1999; Giles et al., 2003), a signaling pathway implicated in development and cancer. E-cadherin levels and function are also disrupted by loss of p120 catenin, which may also contribute to metastasis.
E-cadherin is not the only cell:cell adhesion molecule associated with invasion and metastasis. Another member of the immunoglobulin cell adhesion molecule (Ig-CAM) family, NCAM, is downregulated in several tumor types. NCAM loss increases the ability of tumor cells to disseminate in some tumor types (Crnic et al., 2004). Still other Ig-CAMs, such as DCC, CEACAM1, and Mel-CAM, have reduced expression in some cancers. Please note: not all cell:cell adhesion molecules can be viewed as potential invasion suppressors. Several adhesion molecules, such as L1, CEA, and ALCAM (Cavallaro and Christofori, 2004), are overexpressed in advanced cancers. Additionally, N-cadherin promotes cell motility (Hazan et al., 2000; Nieman et al., 1999). This complexity may be explained by (in)direct signaling functions for these molecules that are distinct from their roles in cell:cell adhesion (Behrens, 1999; Jeanes et al., 2008; Van Roy and Berx, 2008). Because of the interrelatedness of proliferation and invasion, adhesion and growth effects and the complexity of tumor tissue (including complexity that still exists in well-defined experimental models), it is not always possible to distinguish the myriad functions of so-called adhesion molecules.
Another cadherin implicated as a metastasis suppressor is N-cadherin, which when overexpressed in the LM8 osteosarcoma line, inhibited pulmonary metastasis (Kashima et al., 2003). However, there are contradictory data showing that N-cadherin can increase aggressiveness and metastasis in breast and melanoma cell lines (Hazan et al., 2000; Li et al., 2001). Clearly, more work will be required to understand how cadherins play a role in metastasis suppression. Nonetheless, it is clear that different cadherins will play distinct roles in different tissues. This highlights an emerging theme in the metastasis suppressor field—context is critical.
3.3.1.3. KAI1
Kang-Ai1 (Chinese for anticancer; a.k.a. CD82/C33/TIP30) was first identified in AT3.1 and AT6.1 rat Dunning prostate cancer cells (Dong et al., 1995). Similar to the story for BRMS1 discovery, human chromosome 11 was introduced by MMCT because previous work had implicated chromosomal aberrations in late-stage prostate carcinoma. Chromosome 11 hybrids significantly blocked metastasis without preventing primary tumor formation, indicating the presence of one or more metastasis suppressors. Positional cloning mapped KAI1 to 11p11.2 (Dong et al., 1995; Ichikawa et al., 1992). Subsequent experiments demonstrated that KAI1 inhibits metastasis of breast and melanoma cell lines (Phillips et al., 1998; Wei et al., 1996; Yang et al., 1997). Consistent with its role as a metastasis suppressor, KAI1 expression is frequently downregulated during prostate (Dong et al., 1995), breast (Phillips et al., 1998; Yang et al., 2000), colorectal (Lombardi et al., 1999), ovarian (Liu et al., 2000), cervical (Liu et al., 2000, 2001), oral (Farhadieh et al., 2004), and nonsmall cell lung (Goncharuk et al., 2004) cancers.
Regulation of KAI1 is complex and is still being elucidated. KAI1 expression is positively regulated by p53, junB, and AP2 (Marreiros et al., 2003) is induced following etoposide treatment through a p53 and c-Jun pathways (Mashimo et al., 2000); appears to be dependent upon Tip60 and β-catenin–reptin complexes (Kim et al., 2005a); and, can be induced by protein kinase C (PKC); Akita et al., 2000). As with many of the other metastasis suppressor genes, KAI1 is associated with abundant methylation of CpG islands in the promoter; however, treatment of cells with 5-aza-2-deoxycytidine (which prevents cytosine methylation) or trichostatin-A (which inhibits histone deacetylation) failed to upregulate KAI1 (Sekita et al., 2001), suggesting other regulatory controls.
Since KAI1 is a member of the tetraspanin superfamily, much of the regulation of, and mechanistic insights regarding, KAI1 are inferred from studying the role of tetraspanin family members in T-cells. Consistent with a role in metastasis, KAI1 and other tetraspanins are thought to stabilize molecular networks regulating motility, invasion, and other cellular processes (Hemler, 2005; Longo et al., 2001; Sridhar and Miranti, 2006; Sugiura and Berditchevski, 1999; Zhou et al., 2004a), many of which are already associated with metastasis.
Tetraspanins, including KAI1, interact with a multitude of other signaling molecules. KAI1 interacts with other tetraspanins, immunoglobulins, integrins, and histocompatibility molecules (Angelisova et al., 1994; Delaguillaumie et al., 2002, 2004; Horvath et al., 1998; Lee et al., 2004; Mannion et al., 1996; Mashimo et al., 2000; Shibagaki et al., 1999; Szollosi et al., 1996; Zhang et al., 2003). Palmitoylated KAI1 can directly interact with EGF receptor (Odintsova et al., 2003), which instigates EGFR endocytosis and migration signals. When palmitoylation was disrupted, motility and invasion were disrupted, suggesting that KAI1 might suppress metastasis by controlling cellular responses to external signals.
The most convincing studies suggesting a mechanism of action for KAI1 involve discovery that KAI1 directly interacts with a cell-surface molecule, DARC (Duffy antigen receptor for chemokines/gp-FY) on vascular endothelial cell surfaces. Watabe and colleagues, in a series of systematic and elegant studies obtained data supporting a model in which KAI1–DARC interaction induces tumor cell senescence via induction of the cyclin kinase inhibitor p21WAF1 (Bandyopadhyay et al., 2006). Curiously, senescence occurred even in the absence of KAI1-dependent activation of DARC signaling (Horuk et al., 1993). Collectively, their model proposes that KAI1-expressing cancer cells grow and invade locally, but upon intravasation and interaction with DARC-expressing endothelial cells, the tumor cells cannot complete subsequent steps in the metastatic cascade.
3.3.1.4. KISS1R
Although discussed in more detail below in the context of Section 3.3.3, the KISS1 receptor (KISS1R, a.k.a. GPR54, AXOR12, hOT7T175) appears to be critical for metastasis suppression in some tumor cells. Briefly, KISS1R is a G-protein-coupled receptor that is expressed almost ubiquitously at low levels, but is abundantly present in specialized neurons located within the hypothalamus, pituitary, and arcuate nucleus, where it is responsible for regulating pubertal development in the hypothalamic-pituitary-gonadal access (Beck and Welch, 2010; Colledge, 2009; Hameed and Dhillo, 2010; Oakley et al., 2009). Three laboratories had identified an orphan GPCR (Kotani et al., 2001; Muir et al., 2001; Ohtaki et al., 2001), which was subsequently shown to bind internal fragments derived from the KISS1 metastasis suppressor protein. Ohtaki et al. showed that overexpression of KISS1R in B16 melanoma cells diminished metastasis when mice were treated with KISS1-derived polypeptides (Ohtaki et al., 2001). However, an autocrine loop has not been formally established in any cell line that has not been transfected with the receptor (Beck and Welch, 2010).
In studies attempting to characterize whether KISS1-or KISS1-derived peptide(a.k.a. kisspeptins) secretion was required for metastasis suppression, a surprising finding was that none of the cell lines that were suppressed for metastasis following transfection and reexpression of KISS1 possessed detectable levels of the receptor, arguing that an autocrine loop was not responsible in the majority of cases. Thus, we speculated that paracrine signaling to surrounding stroma might be responsible for the metastasis suppressing effects of KISS1 (Beck and Welch, 2010; Nash and Welch, 2006). Beck and colleagues recently demonstrated that primary cultures derived from skin and lung differentially expressed KISS1R. Moreover, the primary cultures from lung secrete growth inhibitory signals more abundantly than the skin primary cultures (Beck, B.H. and Welch, D.R., unpublished observations). These findings illustrate how melanoma cells expressing KISS1 might be able to grow in the skin, but fail to grow after they have already disseminated (Nash et al., 2007).
3.3.1.5. OGR1
Another GPCR, ovarian cancer G-protein coupled receptor (OGR1, a.k.a. GPR58), when overexpressed in PC3 prostate cancer cells did not inhibit tumor cell proliferation and significantly inhibited metastasis to multiple organs (Singh et al., 2007). To date, only preliminary mechanisms have been proposed regarding OGR1 function and these have been based upon the roles of related family members in mediating the functions of lysophospholipids (Xu, 2002).
Functionally, OGR1 regulates endothelial barrier function, proliferation, and tube formation as well as T-cell migration, glucocorticoid-induced thymocyte apoptosis, and globoid cell formations (Im et al., 2001; Kim et al., 2005b; Qiao et al., 2006; Radu et al., 2005; Tosa et al., 2003). Interestingly, OGR1 and other family members also exhibit proton sensing properties. Overexpression of OGR1 increased the levels of Gαi1 transcription and translocation to the cell membrane concomitant with secretion of a hydrophobic factor which appears to be important for OGR1 antimetastatic actions (Singh et al., 2007).
3.3.1.6. DCC1
Deleted in colon cancer (a.k.a. UNC-40 or Frazzled) was first described as a tumor suppressor in colorectal cancer (Fearon et al., 1990). However, expression loss in late-stage cancers led to studies regarding its potential role in metastasis (Iino et al., 1994; Itoh et al., 1993; Kikuchi-Yanoshita et al., 1992; Ookawa et al., 1993). In esophageal (Miyake et al., 1994), bladder (Brewster et al., 1994), neuroblastoma (Ikeami et al., 1985; Reale et al., 1996), and glioma (Reyes-Mugica et al., 1997), DCC expression is lower in lymph-node metastases, invading, and disseminated cells. And further correlation has been described in an experimental model using Madin-Darby canine kidney cells, transfection with DCC resulted in decreased lymph-node and lung metastasis without impairing growth at the site of injection (Rodrigues et al., 2007). However, the results are not entirely clear because a tumor cell marker (i.e., luciferase) was significantly reduced despite overall tumor size appearing to be identical. This leaves open the possibility that a tumor suppressing effect was somehow mast by other cells that had been recruited to the site of tumor cell injection.
The mechanism of action for DCC is largely unclear because it has been implicated in so many diverse functions, such as axon guidance. Among the mechanisms by which DCC is thought to direct cellular movement is by induction of the apoptosome (Forcet et al., 2001). However, induction of apoptosis in metastatic cells has not been measured to the best of our knowledge.
3.3.2. Intracellular signaling molecules
Once cells have received a signal from the milieu, they must interpret and transmit appropriate signals to alter tumor cell function. The majority of metastasis suppressors identified to date are involved in signal transduction.
3.3.2.1. RKIP
Raf Kinase Inhibitor Protein was discovered as a metastasis suppressor gene in prostate cells (Fu et al., 2003; Keller et al., 2004). Clinical data only shows a correlative relationship of RKIP as a metastasis suppressor gene in breast cancer, that is, expressed in primary tumors, but absent in matched lymph-node metastases (Hagan et al., 2005).
RKIP binds directly to Raf, inhibiting MEK1 activation with a concomitant decrease of downstream signaling. Since MEK and RKIP compete to bind to RAF, the presumed mechanism of action is the regulation of ERK signaling. Interestingly, RKIP appears to selectively regulate Raf1, but not Braf (Trakul et al., 2005). This latter observation suggests that RKIP exerts antimetastatic effects only in certain cell types, but this has not been formally tested. Recently, in immune cells, RKIP has been implicated in NFκB and Snail signaling (Wu and Bonavida, 2009), which suggests that it plays roles in EMT and associated phenotypes.
Dangi-Garimella and colleagues recently showed that RKIP suppresses a metastasis signaling cascades involving the microRNA LIN28 and let-7 (Dangi-Garimella et al., 2009). They showed that inhibition of invasion by RKIP involves reduced MAPK signaling and decreased transcription of LIN28 by Myc. Correspondingly, reductions in LIN28 lead to enhanced let-7 processing and inhibition of the chromatin remodeling protein HMG2A, which is involved in regulating prometastatic genes such as Snail.
3.3.2.2. Nm23
Nm23 (nonmetastatic clone #23) was the first metastasis suppressor discovered. It was isolated from a differential colony hybridization screen using murine K1735 melanoma cell lines (Steeg et al., 1988). Since that time, seven other Nm23 family members have been identified, but only Nm23-H1 and Nm23-H2 have been demonstrated to suppress metastasis in experimental models (Lacombe et al., 2000). Metastasis suppression has been observed in multiple tumor types (Freije et al., 1998; Marshall et al., 2010).
Since it was the first discovered, Nm23 has been studied much more extensively than other metastasis suppressors in clinical samples. Generally, with the exception of neuroblastoma, Nm23 expression is inversely correlated with poor survival and tumor grade for breast, gastric, ovarian, non-small cell lung, hepatocellular, oral squamous cell carcinomas, and melanoma (Guan-Zhen et al., 2007; Hartsough and Steeg, 2000; Katakura et al., 2002; Mao et al., 2001; Niu et al., 2002; Terasaki-Fukuzawa et al., 2002; Wang et al., 2004). It is important to acknowledge that not all studies evaluating Nm23 in clinical specimens have revealed prognostic value. While this can be explained away because of tumor heterogeneity, technical issues have also contributed to muddying the literature. Antibody specificity is called into question for virtually all commercially available antibodies/antisera (Bordeaux et al., 2010); so, readers are cautioned to be skeptical unless data validating antibody specificity is provided.
Transfection of Nm23 reduces motility in response to multiple growth factors in vitro (Kantor et al., 1993; Leone et al., 1993; Suzuki et al., 2004). Horak et al. showed that Nm23 downregulates the LPA receptor, EDG2, and the HGF, c-Met (Horak et al., 2007a,b). EDG2 reexpression in Nm23-expressing cells completely restored motility in Nm23-H1-expressing cells while c-Met reexpression only partially restored motility, indicating that EDG2 regulation is closely associated with Nm23-induced metastasis suppression.
Using coimmunoprecipitation and yeast two-hybrid genetic analyses, Nm23 has been found to interact with Tiam1 (Kuppers et al., 2005), Rad (Tseng et al., 2001; Zhu et al., 1999), glyceraldehyde 3-phosphate dehydrogenase (Engel et al., 1998), vimentin (Pinon et al., 1999), various G-proteins (Kimura and Shimada, 1990), casein kinase 2 (CK2) (Biondi et al., 1996), and numerous other proteins (Salerno et al., 2003). Together, these findings implicate Nm23 in an extensive array of cytoskeletal organizing and signaling pathways. However, the physiological relevance of many interactors remains speculative because Nm23 is “sticky.” Nm23 definitely directly interacts with and phosphorylates kinase suppressor of Ras (KSR) at Ser392, possibly altering KSR binding to other proteins and preventing downstream activation of the MAPK pathway. This hypothesis is strengthened by the observation that Nm23-H1 transfectants show reduced basal and stimulated MAPK phosphorylation (Hartsough et al., 2002).
Four distinct activities have been reported for Nm23—NDP kinase (Biggs et al., 1990), histidine kinase (Freije et al., 1997), exonuclease (Ma et al., 2004), and maintenance of genomic stability (Kaetzel et al., 2009). Yet, it is still somewhat unclear which of these plays the critical role in suppressing cancer metastasis. NDP kinase- and exonuclease-disrupting mutants still suppress metastasis (to varying degrees), suggesting complex and overlapping roles in metastasis regulation (Kaetzel et al., 2006; MacDonald et al., 1993). Moreover, the activities associated with them vary by cell type, making extrapolation to other tumor histologic types risky.
Another approach to elucidate the mechanism of action for Nm23 has been to explore the proteome before and after Nm23 expression (Lee et al., 2009). Interestingly, pathway analysis revealed that posttranscriptional processing of RNA was the most commonly affected. Overexpression of Gemin5, which is involved in RNA splicing, affected the splicing patterns of several motility-, invasion-, and metastasis-associated gene-encoded RNA. Since it has been estimated that the human genome encodes approximately 30,000 genes with several hundred thousand potential splice variants (Black, 2000), this finding highlights the depressingly daunting task for sorting out the myriad changes that occur in metastatic cells.
Patricia Steeg and her laboratory continue to take the lead with regard to identifying ways in which metastasis suppressors, specifically Nm23, could help in the clinical management of cancer metastasis. Beyond the potential as a prognostic marker, they have shown that restored expression of a metastasis suppressor is a potentially viable therapeutic option (Marshall et al., 2010). The rationale for their strategy is based upon observations that most, if not all, metastasis suppressors are infrequently mutated. Rather, their expression is downregulated in advanced cancers (Steeg, 2006; Steeg and Theodorescu, 2007). Therefore, administration of agents that selectively induce metastasis suppressor expression could be a therapeutic option. After promoter analysis, they proceeded to show that both dexamethasone and medroxyprogesterone acetate (MPA) induce Nm23 expression in vitro. Furthermore MPA-treated mice had significantly fewer metastases (Ouatas et al., 2002; Palmieri et al., 2005). These provocative and enticing experiments lead the way toward possible use of metastasis suppressor expression through external drug administration.
3.3.2.3. JNKK1/MKK7/p38
By combining MMCT and positional cloning, the Rinker-Schaeffer and Yamada laboratories identified JNKK1/MKK4 (SEK1/MEK4/MAP2K4) as a metastasis suppressor in prostate (Kim et al., 2001) and subsequently ovarian cancer (Yamada et al., 2002). Consistent with its role as a metastasis suppressor, expression has been found to be lower in poor prognosis patients with pancreatic, breast, or gastric cancers (Cunningham et al., 2006; Stark et al., 2004; Xin et al., 2004). However, clinical and experimental measurements of JNKK1 mRNA or protein expression have not always been consistent nor yielded similar conclusions. Expression is often higher in some high-grade tumor types or can promote tumorigenicity (Finegan and Tournier, 2010; Kim et al., 2001; Lotan et al., 2007). However, as emphasized by Taylor et al. (Taylor et al., 2008a,b), mere measurement of signaling protein levels is looking at the wrong parameter. Measurement of activation state (in this case phosphorylation status) is more relevant, and informative.
In an experimental xenograft metastasis model of ovarian cancer, JNKK1/MKK4 activity correlated with growth arrest, but did not increase apoptosis (Lotan et al., 2008). In this model, JNKK1/MKK4 acted through the p38 arm of the SAPK pathway (Hickson et al., 2006). In a prostate cancer model, MKK7, a specific activator of JNK, caused reduced overt metastases more than 90%, compared with controls, just as was observed when the same AT6.1 cells were transfected with JNKK1/MKK4 alone (Vander Griend et al., 2005). However, ectopic expression of MKK6, a specific activator of p38, did not affect metastasis in the prostate model (Vander Griend et al., 2005).
Studies related to the role(s) of JNKK1/MKK4 in metastasis are challenging long-held notions that metastatic cells are selected for a universal ability to override growth inhibitory signals. Instead, the JNKK1/MKK4 data suggest that there may exert a reversible cell cycle arrest that occurs with signaling (in)activation state (Lotan et al., 2008). The very nature of the experimental models in which a small fraction of initially suppressed cells escape growth inhibition will be useful for defining the signals which enforce or oppose dormancy and, in a more clinically relevant way, contribute to understanding how disseminated cells may break dormancy to form macroscopic metastases.
The careful, methodical studies of Rinker-Schaeffer and colleagues deserve reemphasis. Simplistic “-omic” measurements can, and often do, mislead. Moreover, the context of the cells and the stimuli impinging upon the tumor cell will alter the signaling cascades. Therefore, interpretation of all known variables and use of well-defined, specific reagents to measure the relevant activation states is essential.
3.3.2.4. RhoGDI2
RhoGDI2 is a member of a family of molecules that bind to Rho GTPases, sequester them in the cytosol keeping Rho proteins in the GDP-bound or inactive state. They do so by precluding Rho interaction with GTPase activating proteins (GAP) and guanine nucleotide exchange factors (GEF) (Ellenbroek and Collard, 2007; Karlsson et al., 2009). RhoGDI2 is a metastasis suppressor in bladder cancers (Theodorescu et al., 2004) and Hodgkin's lymphoma (Ma et al., 2007b), but also been shown to promote metastasis in other cancers (Hu et al., 2007; Tapper et al., 2001; Wang et al., 2005; Zhang, 2006, 2005, 2009c).
In human bladder cancer, RhoGDI2 levels inversely correlate with development of metastatic disease. In fact, RhoGDI2 is an independent prognostic marker of recurrence following radical cystectomy (Theodorescu et al., 2004). However, approximately one-third of patients with high levels of RhoGDI2 protein develop metastases, suggesting that other mechanisms might regulate the metastasis suppressor functions of RhoGDI2, such as phosphorylation, protein complex partners, protein turnover, and subcellular localization.
RhoGDI2 has a relatively modest effect on RhoGTPase function; however, RhoGDI2 binds with Rac1 (Moissoglu et al., 2009), which can itself exert antimetastatic actions (Uhlenbrock et al., 2004), presumably by altering cytoskeletal structure and organization. However, when the associations of RhoGDI2 and the oncogene Src are taken into account, such as rare concurrent decreased levels, involvement of RhoGDI2 in Src signaling becomes an enticing possible mechanism of action (Wu et al., 2009). Src phosphorylation is known to modulate RhoGDI1- and RhoGDI2- RhoGTPase complex formation (Dermardirossian et al., 2006). Theodorescu and colleagues suggest that specific phosphorylation of RhoGDI2 by Src at Tyr153 may affect the metastasis suppressor function by specifically altering membrane-bound Rac1 (Wu et al., 2009).
3.3.2.5. DLC1
Deleted in liver cancer 1 (a.k.a. Rho-GAP 7 or START domain containing protein 12) was discovered in hepatocellular carcinoma (Yuan et al., 1998). Like RKIP and RhoGDI2, regulation of GTPase activity is presumably the mechanism by which DLC1 inhibits metastasis (Goodison et al., 2005; Yuan et al., 2003), although this has yet to be proven. To term DLC1, a metastasis suppressor may be a misnomer because it can also act as a tumor suppressor, depending upon the cell line to which it is introduced (Yuan et al., 2003, 2004; Zhou et al., 2004b).
Even clinically, DLC1 gene expression is so commonly decreased in many human cancers (Durkin et al., 2007; Yuan et al., 2003) that its function is more likely as a tumor suppressor than as a metastasis suppressor. Yet, a recent report suggests that reduced expression in renal cell cancers correlates with increased invasion and poor prognosis (Zhang et al., 2009a). Thus, while DLC1 satisfies the definition of a metastasis suppressor in some cell lines and models, it does not in others.
3.3.2.6. DRG1
Drg1 (a.k.a. cap43/rit42/RTP/Ndrg1/TDD5) was discovered in colon carcinoma (Kurdistani et al., 1998) (van Belzen et al., 1997) and is found mostly in the cytoplasm. Following DNA damage, Drg1 expression increases and it translocates and accumulates in the nucleus (Kim et al., 2004; Kurdistani et al., 1998; Piquemal et al., 1999). Drg1 was first identified as a tumor suppressor in human bladder and pancreatic cancers (Kurdistani et al., 1998), but overexpression in breast, colon, and prostate cancer cell lines suppressed metastasis without suppressing tumorigenicity (Guan et al., 2000), a pattern which is supported in limited clinical studies in breast, prostate, and liver cancers (Bandyopadhyay et al., 2003; Chua et al., 2007).
DRG-1 appears to be downstream of many cancer- and metastasis-associated signaling pathways, such as p53 (Kim et al., 2004; Kurdistani et al., 1998), PI3K/PTEN (Bandyopadhyay et al., 2004), PKC (Fujii et al., 2008), hypoxia (Agarwala et al., 2000; Bandyopadhyay et al., 2003; Park et al., 2000), and in breast and prostate tumors, estrogens and androgens (Ulrix et al., 1999). Downstream mediators include VEGF and IL8 which are both involved in angiogenesis (Maruyama et al., 2006). Taken together, these findings suggest that DRG1 might control metastasis by controlling intravasation due to vessel integrity or by controlling colonization.
3.3.2.7. Gelsolin
As discussed previously, cytoskeletal organization plays a major role in the ability of a cell to migrate. Gelsolin is a major actin-binding protein that has been shown to suppress metastasis in B16-BL6 mouse melanoma cells (Fujita et al., 2001). Depending on the cell line and model system, gelsolin may play either tumor suppressive or metastasis suppressive roles (Tanaka et al., 1995). More recently, gelsolin expression was correlated with metastatic potential in human colon adenocarcinoma cells (Litwin et al., 2009). Gelsolin is a member of a superfamily of calcium-dependent actin-binding proteins, and in response to extracellular stimuli, gelsolin modulates the reorganization of the actin cytoskeleton (Dos Remedios et al., 2003). This regulation of cytoskeletal changes is most likely one of the key mechanisms for its ability to suppress metastasis.
3.3.2.8. SSeCKS/GRAVIN/AKAP12
Src is a well-known oncogene that alters cell signaling and cytoskeletal organization by direct phosphorylation of protein substrates and through transcriptional regulation of tumor-promoting or -suppressing genes (Dehbi and Bedard, 1992; Frame, 2004; Jove and Hanafusa, 1987). Src-suppressed protein Kinase C Substrate (SSeCKS) is the rodent ortholog of human GRAVIN and is an important regulator of cell signaling and cytoskeletal dynamics (Chapline et al., 1998; Gelman et al., 1998; Lin et al., 1996; Nelson et al., 1999). It was discovered using PCR-based subtractive hybridization in NIH3T3 mouse fibroblasts and was found to be suppressed by oncogenic src, ras, fos, and myc (Lin et al., 1996). Reexpression of SSeCKS in rat prostatic cancer MATLyLu cells resulted in significantly reduced lung metastasis in nude mouse models with only slightly decreased growth of the primary subcutaneous tumors (Su et al., 2006; Xia et al., 2001). Clinically, SseCKS/GRAVIN/AKAP12 is expressed in benign and well-differentiated prostate carcinomas, but not in highly aggressive and undifferentiated prostate lesions (Xia et al., 2001).
SSeCKS is phosphorylated by PKC and dynamically regulates cytoskeletal architecture by binding F-actin resulting in modulation of signaling pathways (Gelman et al., 1998; Lin et al., 1996). SSeCKS is also known as A-Kinase Anchor Protein 12 (AKAP12) since it has the ability to scaffold PKA through a C-terminal RII subunit binding motif (Nauert et al., 1997). In its dephosphorylated state, SSeCKS acts as a scaffolding protein where it binds signaling molecules such as PKC, PKA, calmodulin, and cyclins. Upon mitogenic signaling, SSeCKS is phosphorylated and translocates to the perinuclear membrane releasing signaling molecules to mediate changes within a cell (Lin et al., 2000). These changes induced from extracellular growth factors and adhesion of integrins to the ECM through SSeCKS has a significant impact on migration. Focal adhesions are also impacted by SSeCKS. Focal adhesions are formed at the plasma membrane when receptors bind ECM proteins and induce clustering of actin containing protein complexes leading to the recruitment and activation of the protein tyrosine kinase, FAK (Zhao and Guan, 2009). Phosphorylation of SSeCKS through stimulation by multiple growth factors has been shown to be FAK-dependent through mediation of an unidentified FAK induced kinase (Xia and Gelman, 2002). This FAK-dependent phosphorylation of SSeCKS not only induces its release of signaling molecules, but reduces its F-actin-binding capability. Thus, SSeCKS acts to sequester growth factors and binds F-actin when in its dephosphorylated state during G0 and early G1 phases of the cell cycle. But upon mitogenic signaling SSeCKS relinquishes actin binding and allows induction of signaling cascades leading to changes in the cytoskeleton.
In addition to cytoskeletal organization changes, SSeCKS also has been correlated with decreased levels of proangiogenic factors including HIF1, VEGF, FGF-7, angiopoietin, tenascin C, PDGF-R, and OPN and an increase in antiangiogenic factors vasostatin and col18 (Su et al., 2006; Xia et al., 2001). More recently, SSeCKS significantly decreased invasion through Matrigel that correlated with decreased MMP-2 levels and suppression of serum-induced activation of PKC-Raf/MEK/ERK pathway (Su et al., 2010). In that study, podosome formation was inhibited independent of the actin cytoskeleton, but inhibition of MEK/ERK activation required actin cytoskeletal remodeling. Taken together, SSeCKS suppression of metastasis appears to involve multiple mechanisms that impact cell motility, invasion, and angiogenesis.
3.3.2.9. HUNK
The Hormonally Upregulated Neu-associated Kinase (HUNK) was recently identified as a suppressor of breast cancer metastasis by blocking actin polymerization leading to reduced cell motility (Quintela-Fandino et al., 2010), although it has previously been shown to promote metastasis (Wertheim et al., 2009). These discrepancies are likely the result of, as yet unresolved, context-dependent factors. HUNK was previously shown to be a kinase essential for mammary tumor metastasis through the restoration of invasion and migration (Wertheim et al., 2009). More recently, it has been shown to inactivate cofilin-1 (CFL-1) by preventing the binding of protein phosphatase 2-A (PP2A) to CFL-1 (Quintela-Fandino et al., 2010). In the latter report, no kinase activity was detected. Additional studies will be required to identify specific context-dependent functions of HUNK in metastasis.
3.3.2.10. Caspase 8
Death receptor-activated apoptotic pathways function by activation of caspase 8, which maps to human chromosome 2q33–q34. Addition of TNFα and FAS ligand activates caspase 8 by activating death domains which, in turn, cleave procaspase 8. Activated caspase 8 cleaves caspase 3, leading to apoptosis. Using a neuroblastoma model, Stupack et al. reexpressed caspase and found significant suppression of metastasis (Stupack et al., 2006). Subsequent analysis of relapsed glioblastoma multiforme tissues revealed hypermethylation and silencing of the caspase 8 gene (Martinez et al., 2007). Although not metastasis per se, the trend in glioblastoma is supportive of a metastasis-associated role.
Paradoxically, caspase 8 can promote migration in cells in which apoptotic machinery is compromised (Barbero et al., 2009). Caspase 8 was recruited to migration machinery following integrin ligation. While activity is not required for caspase 8-enhanced cell migration, association with FAK and calpain 2 was thought to be integrally involved. Thus, caspase 8 exemplifies the differential roles that a single molecule may play in cellular behavior, depending upon other molecules with which it interacts. Additionally, caspase 8's role in anoikis is intriguing. Stupack has elegantly demonstrated that anoikis need not be exclusively the result of no adhesion. But rather, anoikis can be induced if cells adhere to a nonpreferred substrate (Cheresh and Stupack, 2002; Stupack and Cheresh, 2002). This observation has led to the hypothesis that analogous pathways might be involved in organotropism or failure to thrive following dissemination and arrest (Lahti et al., 2006).
3.3.2.11. Ribonucleotide reductase M1
The ribonucleotide reductase M1 (RRM1) gene maps to chromosome 11 and its reexpression in the murine lung carcinoma cell line, Line 1, met the functional definition for a metastasis suppressor (Gautam and Bepler, 2006). Genome comparisons showed that LOH was observed in approximately half of informative lung tumor specimens, but mutation was not observed (Pitterle et al., 1999), but correlation with human lung cancer metastasis was not determined. RRM1 converts ribonucleotides to deoxyribonucleotides for DNA synthesis, suggesting that it may regulate dNTP pools which, in turn, would determine proliferative capacity. However, this hypothesis has not been tested. Interestingly, RRM1 has been found to upregulate PTEN and reduce FAK phosphorylation. In addition, RRM2, a related ribonucleotide reductase has been studied with regard to cellular responsiveness to DNA damage and repair. Whether similar functions might alter tumor aggressiveness has not yet, to our knowledge, been tested.
3.3.3. Extracellular signaling molecules and KISS1/kisspeptins
Metastasis is clearly determined, to a sizable extent, by tumor-host interactions, that is, the microenvironment participates in the induction and selective proliferation of malignant cells. Host physiology can foster or reject neoplastic cells. It stands to reason, then, that some metastasis suppressors might work because they render the ectopic microenvironment less hospitable. The most obvious example of cell-cell signaling relates to inflammatory cells that infiltrate the tissue adjoining a tumor or disseminated cell in response to secreted cytokines, chemokines, or hormones. The immune populations can produce pro- or antitumor molecules that alter the capacity of tumor cells to migrate, invade, or survive (Fridlender et al., 2009; Mantovani et al., 2006). However, it is imperative that other stromal populations not be ignored in the interplay.
The milieu surrounding a tumor cell undoubtedly explains metastatic organotropism. Yet, despite more than a century of attempts, clear-cut biochemical explanations still do not exist. Nonetheless, some clues may be forthcoming from understanding the mechanisms of action of extracellular metastasis suppressors.
High-frequency deletions or rearrangements involving chromosome 6q in late-stage melanoma prompted introduction of full-length chromosome 6 into the human metastatic melanoma cell line C8161 by MMCT (Welch et al., 1994b). KISS1 was subsequently identified using subtractive hybridization between metastatic and nonmetastatic cell line variants (Lee and Welch, 1997a; Lee et al., 1996, 1997). Transfection of full-length KISS1 cDNA into melanoma (Lee and Welch, 1997a; Lee et al., 1996, 1997), breast carcinoma (Lee and Welch, 1997b), ovarian (Jiang et al., 2005), and pancreatic adenocarcinoma (McNally et al., 2010) cell lines suppressed metastasis in spontaneous and experimental metastasis assays.
Unexpectedly, the KISS1 gene mapped to the long arm of chromosome 1. Using cDNA microarrays and chromosome 6 MMCT donors with defined deletions on the long arm of chromosome 6, Goldberg et al. found that KISS1 was regulated by TXNIP and CRSP3 (see Section 3.1.2; Goldberg et al., 2003). The KISS1 gene encodes a 154 amino acid protein but full-length KISS1 is rarely detectable since the nascent protein is proteolytically processed into numerous polypeptides, termed kisspeptins (Kotani et al., 2001; Ohtaki et al., 2001). An internal 54-amino acid polypeptide, termed kisspeptin (KP)-54 or metastin, binds to the KISS1 receptor (Kotani et al., 2001; Muir et al., 2001; Ohtaki et al., 2001). KISS1R expression is highest in placenta, pituitary gland, pancreas, brain, and spinal cord (Kotani et al., 2001; Muir et al., 2001), while KISS1 expression is more restricted, located primarily in the placenta, pancreas, kidney, and the arcuate nucleus of the hypothalamus (Lee et al., 1996; Muir et al., 2001; Ohtaki et al., 2001).
KP processing is still not understood (Nash and Welch, 2006; Nash et al., 2007). However, KISS1 protein is thought to be cleaved by furins or prohormone convertases (Kotani et al., 2001; Ohtaki et al., 2001) based upon the amino acids at the ends of the KP. The preponderance of primary literature on KISS1 and KISS1R relates to the involvement of these molecules is in pubertal development (Colledge, 2009; Gianetti and Seminara, 2008; Roa et al., 2008; Tena-Sempere, 2008). Since this is not the focus of this chapter, those aspects of KISS1 biology are not highlighted here.
Clinical reports from a variety of tumor types generally support a positive correlation between KISS1 expression, and metastasis-free survival and other progression-associated phenotypes (Beck and Welch, 2010; Dhar et al., 2004; Guan-Zhen et al., 2007; Hata et al., 2007; Ikeguchi et al., 2004; Katagiri et al., 2009; Kostadima et al., 2007; Martin et al., 2005; Masui et al., 2004; Nagai et al., 2009; Prentice et al., 2007; Shirasaki et al., 2001; Stark et al., 2004). The key exception has been a positive correlation with hepatocellular carcinoma progression (Ikeguchi et al., 2003; Schmid et al., 2007), but a recent immunohistochemical study suggests that KISS1 might be significantly inversely associated with stage and intrahepatic metastasis from HCC (Shengbing et al., 2009). As described above, many of these studies measured only mRNA expression using in situ hybridization or PCR-based methods. The former are less ambiguous than studies in which stromal cells contaminate the cell preparation, making it impossible to judge the origins of KISS1 or KISS1R. mRNA measurements were required because of difficulties in generating specific antibodies. Most commercially available antibodies used have not been validated or the data have not been published (Beck and Welch, 2010; Bordeaux et al., 2010). Since KISS1 is processed and (presumably, though not formally proven) not all KP are biologically active, nonprotein-based studies should be interpreted with caution. To the best of our knowledge, no one has yet evaluated KISS1 → KP processing in clinical samples.
Surprisingly, based upon the nature of the experimental studies showing KISS1 functionality as a metastasis suppressor, many tumor cells suppressed by KISS1 reexpression do not express KISS1R (Nash et al., 2007). This has led us to postulate a paracrine feedback loop in which KISS1/KP secreted by tumor cells acts upon one or more stromal populations which, in turn, respond with growth-inhibitory factors (Beck and Welch, 2010; Nash and Welch, 2006).
4. Conclusions and Perspectives
The process of metastasis is obviously very complex and involves intrinsic and extrinsic factors. In this chapter, we have focused on genetic changes, specifically metastasis suppressors, in tumor cells, but as the data were collated and as the field matured, awareness that the function of metastasis regulatory genes were not functioning autonomously became acute. Even with incomplete knowledge regarding the interrelationships of tumor cells with their microenvironments, some patterns are emerging. The basic observation that neoplastic cells expressing metastasis suppressors grow at orthotopic sites but fail to successfully complete the metastatic process illustrates that the mechanisms of action for the suppressors are context sensitive. Although many of the metastasis suppressors inhibit metastasis in different histio types, most have not been extensively evaluated (largely because there are so few metastasis models). Several examples of metastasis suppressors showing opposite effects (e.g., worse prognosis or more aggressive behaviors for Nm23 in neuroblastoma or KISS1 in hepatocellular carcinoma) further highlight that the cellular backgrounds are crucial variables for understanding mechanism.
Metastasis suppressor mutations are rare. Instead, expression is downregulated in advanced cancers. Notwithstanding the apparent position downstream of some oncogenic signaling pathways which may be complicit in driving tumor progression toward metastasis, the undeniable inference is that the expression of metastasis suppressors can change under varying circumstances. Among the potentially altered queues are the cofactors with which many metastasis suppressor interact. Such cofactors can interact directly (e.g., BRMS1 with components of SIN3:HDAC complexes), indirectly as part of quaternary protein complexes (e.g., LSD1 with NuRD chromatin remodeling complex components or Nm23 as part of MAPK scaffolding) or indirectly as nodes within signaling cascades (e.g., SSeCKs or JNKK1 receiving and passing along signaling).
If we couple the observations involving metastasis suppressors with literature emphasizing that three-dimensional architecture (Debnath and Brugge, 2005; Nam et al., 2010) and cellular tension (Butcher et al., 2009) alter cellular behavior and the irrefutable fact that in vitro assays are inadequate to study metastasis, then a major priority needs to be developing experimental manipulable assays that more faithfully model different microenvironments in which tumor cells find themselves. Some recent advances in recapitulating elements of more complex in vivo milieus are chipping away at this problem (Mendoza et al., 2010), but more concerted efforts are needed.
This basic research effort has practical applications related to metastasis treatment. Recall that all treatments beyond surgical removal of the primary tumor are designed to prevent new or eliminate already established metastases. Since the disease is systemic, therapy must also be systemic. Unfortunately, most cancer treatments are extremely toxic; contribute to a poor quality of life during the therapy; and are only modestly effective. The natural products represented by metastasis suppressors might offer some opportunity for therapeutic development and improvements in one or more of these shortcomings. Of course, the usual caveats regarding gene replacement, drug targeting, and other pharmacokinetic parameters must be taken into account. However, use of intrinsic molecules would appear to offer some advantages.
Two potential treatments invoking metastasis suppressors include restoration of expression (Marshall et al., 2009; Ouatas et al., 2003; Palmieri et al., 2006) or to utilize the metastasis suppressor gene product(s) directly (Beck and Welch, 2010; Nash and Welch, 2006). The latter, if the kisspeptins remain as nontoxic as they currently appear in other clinical settings (Mead et al., 2007; Niida et al., 2006; Ramaswamy et al., 2007), look promising, especially with regard to the potential utility against seeded, but not yet overt metastases. Our current understanding of KISS1 is that it actually targets stromal populations, which means that the treatment would be directed to supposedly more genetically stable cell populations, which is good. However, similar thought processes were involved when designing antiangiogenic drugs (Folkman, 2006). The assumption of genetic stability may not have been entirely accurate (Hida and Klagsbrun, 2005; Hida et al., 2004). Moreover, relatively incomplete knowledge of full interplay between tumor cells and endothelial cells probably contributed to the unexpectedly enhanced metastatic progression observed in anti-VEGFR-treated mice (Ebos et al., 2009; Paez-Ribes et al., 2009). In addition to highlighting deficiencies in current knowledge related to tumor–stromal interactions, those two papers illustrate the essential need to incorporate metastasis studies into preclinical testing of anticancer agents (Steeg et al., 2009). Regardless, the clinical need is enormous and unmet. The daunting complexity of the metastasis suppressor function upon which genetic background and extrinsic signals are superimposed should not paralyze nor discourage. We hope that, by assembling the facts, a level of clarity and perspective have emerged that will facilitate more rapid translation into clinical practice.
ACKNOWLEDGMENTS
We are grateful to many colleagues who have shared insights with us during the writing of this chapter. Although we have attempted to be thorough, there may be some whose work was not cited. For that we apologize. We dedicate this chapter to the memories of our mothers, who died from cancers, and for the inspiration both still give us. Work from the Welch and Hurst laboratories has been generously funded by the National Cancer Institute (CA062168; CA087728, CA134981, CA089019), U.S. Army Medical Research and Materiel Command, the National Foundation for Cancer Research, and Susan G. Komen for the Cure.
REFERENCES
- Affara NI, Andreu P, Coussens LM. Delineating protease functions during cancer development. Methods Mol. Biol. 2009;539:1–32. doi: 10.1007/978-1-60327-003-8_1. [DOI] [PubMed] [Google Scholar]
- Agarwala KL, Kokame K, Kato H, Miyata T. Phosphorylation of RTP, an ER stress-responsive cytoplasmic protein. Biochem. Biophys. Res. Commun. 2000;272:641–647. doi: 10.1006/bbrc.2000.2833. [DOI] [PubMed] [Google Scholar]
- Aguda BD, Kim Y, Piper-Hunter MG, Friedman A, Marsh CB. MicroRNA regulation of a cancer network: consequences of the feedback loops involving miR-17–92, E2F, and Myc. Proc. Natl. Acad. Sci. USA. 2008;105:19678–19683. doi: 10.1073/pnas.0811166106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita H, Iizuka A, Hashimoto Y, Kohri K, Ikeda K, Nakanishi M. Induction of KAI-1 expression in metastatic cancer cells by phorbol esters. Cancer Lett. 2000;153:79–83. doi: 10.1016/s0304-3835(00)00352-9. [DOI] [PubMed] [Google Scholar]
- Albini A, Mirisola V, Pfeffer U. Metastasis signatures: genes regulating tumor–microenvironment interactions predict metastatic behavior. Cancer Metastasis Rev. 2007;27:75–83. doi: 10.1007/s10555-007-9111-x. [DOI] [PubMed] [Google Scholar]
- Andreasen PA, Kjoller L, Christensen L, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: a review. Int. J. Cancer. 1997;72:1–22. doi: 10.1002/(sici)1097-0215(19970703)72:1<1::aid-ijc1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Angelisova P, Hilgert I, Horejsi V. Association of four antigens of the tetraspans family (CD37, CD53, TAPA-1, and R2/C33) with MHC class II glycoproteins. Immunogenetics. 1994;39:249–256. doi: 10.1007/BF00188787. [DOI] [PubMed] [Google Scholar]
- Arroyo AG, Iruela-Arispe ML. Extracellular matrix, inflammation, and the angiogenic response. Cardiovasc. Res. 2010;86:226–235. doi: 10.1093/cvr/cvq049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani IA, Rasheed SAK, Nikolova DA, Leupold JH, Colburn NH, Post S, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- Askari JA, Tynan CJ, Webb SED, Martin-Fernandez ML, Ballestrem C, Humphries MJ. Focal adhesions are sites of integrin extension. J. Cell Biol. 2010;188:891–903. doi: 10.1083/jcb.200907174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri-Yarmand R, Talukder AH, Wang RA, Vadlamudi RK, Kumar R. Metastasis-associated protein 1 deregulation causes inappropriate mammary gland development and tumorigenesis. Development. 2004;131:3469–3479. doi: 10.1242/dev.01213. [DOI] [PubMed] [Google Scholar]
- Bailey CM, Khalkhali-Ellis Z, Seftor EA, Hendrix MJC. Biological functions of maspin. J. Cell. Physiol. 2006;209:617–624. doi: 10.1002/jcp.20782. [DOI] [PubMed] [Google Scholar]
- Bailey JM, Singh PK, Hollingsworth MA. Cancer metastasis facilitated by developmental pathways: sonic hedgehog, notch, and bone morphogenic proteins. J. Cell. Biochem. 2007;102:829–839. doi: 10.1002/jcb.21509. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGFβ-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 2002;115:3193–3206. doi: 10.1242/jcs.115.15.3193. [DOI] [PubMed] [Google Scholar]
- Balmain A. Cancer genetics: from Boveri and Mendel to microarrays. Nat. Rev. Cancer. 2001;1:77–82. doi: 10.1038/35094086. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S, Pai SK, Gross SC, Hirota S, Hosobe S, Miura K, et al. The Drg-1 gene suppresses tumor metastasis in prostate cancer. Cancer Res. 2003;63:1731–1736. [PubMed] [Google Scholar]
- Bandyopadhyay S, Pai SK, Hirota S, Hosobe S, Tsukada T, Miura K, et al. PTEN up-regulates the tumor metastasis suppressor gene Drg-1 in prostate and breast cancer. Cancer Res. 2004;64:7655–7660. doi: 10.1158/0008-5472.CAN-04-1623. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S, Zhan R, Chaudhuri A, Watabe M, Pai SK, Hirota S, et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat. Med. 2006;12:933–938. doi: 10.1038/nm1444. [DOI] [PubMed] [Google Scholar]
- Barbero S, Mielgo A, Torres V, Teitz T, Shields DJ, Mikolon D, et al. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res. 2009;69:3755–3763. doi: 10.1158/0008-5472.CAN-08-3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur. J. Cancer. 2010;46:1181–1188. doi: 10.1016/j.ejca.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerschmitz GJ, Ranki T, Kangasniemi L, Ribacka C, Eriksson M, Porten M, et al. Tissue-specific promoters active in CD44+CD24−/low breast cancer cells. Cancer Res. 2008;68:5533–5539. doi: 10.1158/0008-5472.CAN-07-5288. [DOI] [PubMed] [Google Scholar]
- Beck BH, Welch DR. The KISS1 metastasis suppressor: a good night kiss for disseminated cancer cells. Eur. J. Cancer. 2010;46:1283–1289. doi: 10.1016/j.ejca.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J. Cadherins and catenins: role in signal transduction and tumor progression. Cancer Metastasis Rev. 1999;18:15–30. doi: 10.1023/a:1006200102166. [DOI] [PubMed] [Google Scholar]
- Ben-Baruch A. Host microenvironment in breast cancer development: inflammatory cells, cytokines and chemokines in breast cancer progression: reciprocal tumor–microenvironment interactions. Breast Cancer Res. 2003;5:31–36. doi: 10.1186/bcr554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernards R, Weinberg RA. Metastasis genes: a progression puzzle. Nature. 2002;418:823. doi: 10.1038/418823a. [DOI] [PubMed] [Google Scholar]
- Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene. 2008;42:5643–5647. doi: 10.1038/onc.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierie B, Moses HL. TGF-β and cancer. Cytokine Growth Factor Rev. 2006a;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Bierie B, Moses HL. TGFβ: the molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer. 2006b;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- Biggs J, Hersperger E, Steeg PS, Liotta LA, Shearn A. A Drosophila gene that is homologous to a mammalian gene associated with tumor metastasis codes for a nucleoside diphosphate kinase. Cell. 1990;63:933–940. doi: 10.1016/0092-8674(90)90496-2. [DOI] [PubMed] [Google Scholar]
- Biondi RM, Engel M, Sauane M, Welter C, Issinger OG, Jimenez de AL, et al. Inhibition of nucleoside diphosphate kinase activity by in vitro phosphorylation by protein kinase CK2. Differential phosphorylation of NDP kinases in HeLa cells in culture. FEBS Lett. 1996;399:183–187. doi: 10.1016/s0014-5793(96)01299-9. [DOI] [PubMed] [Google Scholar]
- Black DL. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000;103:367–370. doi: 10.1016/s0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Bodenstine TM, Welch DR. Metastasis suppressors and the tumor microenvironment. Cancer Microenviron. 2008;1:1–11. doi: 10.1007/s12307-008-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy GP, Wilson S, Chambers AF. Experimental metastatic ability of H-ras-transformed NIH3T3 cells. Cancer Res. 1985;45:6005–6009. [PubMed] [Google Scholar]
- Bordeaux J, Welsh A, Agarwal S, Killiam E, Baquero M, Hanna J, et al. Antibody validation. Biotechniques. 2010;48:197–209. doi: 10.2144/000113382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri T. Zur frage der Entstehung maligner tumoren. Gustav Fisher; Jena: 1914. [Google Scholar]
- Boyd D. Invasion and metastasis. Cancer Metastasis Rev. 1996;15:77–89. doi: 10.1007/BF00049488. [DOI] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat. Rev. Cancer. 2005;5:744–749. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial–mesenchymal transition. Cancer Res. 2008;68:7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- Brewster SF, Browne S, Brown KW. Somatic allelic loss at the DCC, APC, nm23-H1 and p53 tumor suppressor gene loci in human prostatic carcinoma. J. Urol. 1994;151:1073–1077. doi: 10.1016/s0022-5347(17)35186-8. [DOI] [PubMed] [Google Scholar]
- Bross IDJ, Blumenson LE. Metastatic sites that produce generalized cancer: identification and kinetics of generalizing sites. In: Weiss L, editor. Fundamental Aspects of Metastasis. North Holland, Amsterdam: 1976. pp. 359–376. [Google Scholar]
- Bruns CJ, Solorzano CC, Harbison MT, Ozawa S, Tsan R, Fan D, et al. Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma. Cancer Res. 2000;60:2926–2935. [PubMed] [Google Scholar]
- Buccione R, Caldieri G, Ayala I. Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev. 2009;28:137–149. doi: 10.1007/s10555-008-9176-1. [DOI] [PubMed] [Google Scholar]
- Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat. Rev. Cancer. 2009;9:108–122. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler TP, Gullino PM. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975;35:512–516. [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal SA, LeRoy C, Jia M, Menzies A, Butler AP, Teague JW, Griffin CA, Burton J, Swerdlow H, Quail MA, Stratton MR, Iacobuzio-Donahue CA, Futreal PA. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonell WS, Ansorge O, Sibson N, Muschel R. The vascular basement membrane as “soil” in brain metastasis. PLoS ONE. 2009;4:e5857. doi: 10.1371/journal.pone.0005857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardiff RD. Epithelial to mesenchymal transition tumors: fallacious or snail's pace? Clin. Cancer Res. 2005;11:8534–8537. doi: 10.1158/1078-0432.CCR-05-2250. [DOI] [PubMed] [Google Scholar]
- Cardiff RD. The pathology of EMT in mouse mammary tumorigenesis. J. Mammary Gland Biol. Neoplasia. 2010;15:225–233. doi: 10.1007/s10911-010-9184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsen SA, Ramshaw IA, Warrington RC. Involvement of plasminogen activator production with tumor metastasis. Cancer Res. 1984;44:3012–3016. [PubMed] [Google Scholar]
- Castellano L, Giamas G, Jacob J, Coombes RC, Lucchesi W, Thiruchelvam P, et al. The estrogen receptor-alpha-induced microRNA signature regulates itself and its transcriptional response. Proc. Natl. Acad. Sci. USA. 2009;106:15732–15737. doi: 10.1073/pnas.0906947106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchpole HR. Connective tissue, basement membrane, extracellular matrix. In: Ioachim HL, editor. Pathobiology Annual. Raven Press; New York: 1982. pp. 1–33. [PubMed] [Google Scholar]
- Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signalling pathway in cancer. Eur. J. Cancer. 2010;46:1260–1270. doi: 10.1016/j.ejca.2010.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006;66:11271–11278. doi: 10.1158/0008-5472.CAN-06-2044. [DOI] [PubMed] [Google Scholar]
- Chambers AF. MDA-MB-435 and M14 cell lines: identical but not M14 melanoma? Cancer Res. 2009;69:5292–5293. doi: 10.1158/0008-5472.CAN-09-1528. [DOI] [PubMed] [Google Scholar]
- Chambers AF, Denhardt GH, Wilson SM. ras-Transformed NIH3T3 cell lines, selected for metastatic ability in chick embryos, have increased proportions of p21-expressing cells and are metastatic in nude mice. Invasion Metastasis. 1990;10:225–240. [PubMed] [Google Scholar]
- Champine PJ, Michaelson J, Weimer B, Welch DR, DeWald DB. Micro-array analysis reveals potential mechanisms of BRMS1-mediated metastasis suppression. Clin. Exp. Metastasis. 2007;24:551–565. doi: 10.1007/s10585-007-9092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapline C, Cottom J, Tobin H, Hulmes J, Crabb J, Jaken S. A major, transformation-sensitive PKC-binding protein is also a PKC substrate involved in cyto-skeletal remodeling. J. Biol. Chem. 1998;273:19482–19489. doi: 10.1074/jbc.273.31.19482. [DOI] [PubMed] [Google Scholar]
- Cheloufi S, Dos Santos CO, Chong MMW, Hannon G. A dicer-independent miRNA biogenesis pathway that requires ago catalysis. Nature. 2010;465:584–589. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheresh DA, Stupack DG. Integrin-mediated death: an explanation of the integrin-knockout phenotype? Nat. Med. 2002;8:193–194. doi: 10.1038/nm0302-193. [DOI] [PubMed] [Google Scholar]
- Chirco R, Liu XW, Jung KK, Kim HR. Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev. 2006;25:99–113. doi: 10.1007/s10555-006-7893-x. [DOI] [PubMed] [Google Scholar]
- Chua MS, Sun H, Cheung ST, Mason V, Higgins J, Ross DT, et al. Overexpression of NDRG1 is an indicator of poor prognosis in hepatocellular carcinoma. Mod. Pathol. 2007;20:76–83. doi: 10.1038/modpathol.3800711. [DOI] [PubMed] [Google Scholar]
- Chuang YY, Tran NL, Rusk N, Nakada M, Berens ME, Symons M. Role of synaptojanin 2 in glioma cell migration and invasion. Cancer Res. 2004;64:8271–8275. doi: 10.1158/0008-5472.CAN-04-2097. [DOI] [PubMed] [Google Scholar]
- Cicek M, Samant RS, Kinter M, Welch DR, Casey G. Identification of metastasis-associated proteins through protein analysis of metastatic MDA-MB-435 and metastasis-suppressed BRMS1 transfected-MDA-MB-435 cells. Clin. Exp. Metastasis. 2004;21:149–157. doi: 10.1023/b:clin.0000024729.19084.f0. [DOI] [PubMed] [Google Scholar]
- Cicek M, Fukuyama R, Welch DR, Sizemore N, Casey G. Breast cancer metastasis suppressor 1 inhibits gene expression by targeting nuclear factor-kB activity. Cancer Res. 2005;65:3586–3595. doi: 10.1158/0008-5472.CAN-04-3139. [DOI] [PubMed] [Google Scholar]
- Cicek M, Fukuyama R, Cicek MS, Sizemore S, Welch DR, Sizemore N, et al. BRMS1 contributes to the negative regulation of uPA gene expression through recruitment of HDAC1 to the NF-kappaB binding site of the uPA promoter. Clin. Exp. Metastasis. 2009;26:229–237. doi: 10.1007/s10585-009-9235-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifone MA, Fidler IJ. Increasing metastatic potential is associated with increasing genetic instability of clones isolated from murine neoplasms. Proc. Natl. Acad. Sci. USA. 1981;78:6949–6952. doi: 10.1073/pnas.78.11.6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge WH. Kisspeptins and GnRH neuronal signalling. Trends Endocrinol. Metab. 2009;20:115–121. doi: 10.1016/j.tem.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Shapiro SD, Soloway PD, Werb Z. Models for gain-of-function and loss-of-function of MMPs. Transgenic and gene targeted mice. Methods Mol. Biol. 2001;151:149–179. doi: 10.1385/1-59259-046-2:149. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B, Matrisian LM. Cancer therapy—matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science (Washington, D.C.) 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- Cox JL. Cystatins and cancer. Front. Biosci. 2009;14:463–474. doi: 10.2741/3255. [DOI] [PubMed] [Google Scholar]
- Creighton CJ, Chang JC, Rosen JM. Epithelial–mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J. Mammary Gland Biol. Neoplasia. 2010;15:253–260. doi: 10.1007/s10911-010-9173-1. [DOI] [PubMed] [Google Scholar]
- Crnic I, Strittmatter K, Cavallaro U, Kopfstein L, Jussila L, Alitalo K, et al. Loss of neural cell adhesion molecule induces tumor metastasis by up-regulating lymphangiogenesis. Cancer Res. 2004;64:8630–8638. doi: 10.1158/0008-5472.CAN-04-2523. [DOI] [PubMed] [Google Scholar]
- Cunningham SC, Kamangar F, Kim MP, Hammoud S, Haque R, Iacobuzio-Donahue C, et al. MKK4 status predicts survival after resection of gastric adenocarcinoma. Arch. Surg. 2006;141:1095–1099. doi: 10.1001/archsurg.141.11.1095. [DOI] [PubMed] [Google Scholar]
- Dangi-Garimella S, Yun J, Eves EM, Newman M, Erkeland SJ, Hammond SM, et al. Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. EMBO J. 2009;28:347–358. doi: 10.1038/emboj.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debies MT, Gest SA, Mathers JL, Mikse OR, Leonard TL, Moody SE, et al. Tumor escape in a Wnt1-dependent mouse breast cancer model is enabled by p19 (Arf)/p53 pathway lesions but not p16(Ink4a) loss. J. Clin. Investig. 2008;118:51–63. doi: 10.1172/JCI33320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- DeClerck YA, Imren S, Montgomery AP, Mueller BM, Reisfeld RA, Laug WE. Proteases and protease inhibitors in tumor progression. Adv. Exp. Med. Biol. 1997;425:89–97. doi: 10.1007/978-1-4615-5391-5_9. [DOI] [PubMed] [Google Scholar]
- Dehbi M, Bedard PA. Regulation of gene expression in oncogenically transformed cells. Biochem. Cell Biol. 1992;70:980–997. doi: 10.1139/o92-142. [DOI] [PubMed] [Google Scholar]
- Delaguillaumie A, Lagaudriere-Gesbert C, Popoff MR, Conjeaud H. Rho GTPases link cytoskeletal rearrangements and activation processes induced via the tetraspanin CD82 in T lymphocytes. J. Cell Sci. 2002;115:433–443. doi: 10.1242/jcs.115.2.433. [DOI] [PubMed] [Google Scholar]
- Delaguillaumie A, Harriague J, Kohanna S, Bismuth G, Rubinstein E, Seigneuret M, et al. Tetraspanin CD82 controls the association of cholesterol-dependent microdomains with the actin cytoskeleton in T lymphocytes: relevance to co-stimulation. J. Cell Sci. 2004;117:5269–5282. doi: 10.1242/jcs.01380. [DOI] [PubMed] [Google Scholar]
- Dermardirossian C, Rocklin G, Seo JY, Bokoch GM. Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Mol. Biol. Cell. 2006;17:4760–4768. doi: 10.1091/mbc.E06-06-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat. Rev. Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWald DB, Torabinejad J, Samant RS, Johnston D, Erin N, Shope JC, et al. Metastasis suppression by breast cancer metastasis suppressor 1 involves reduction of phosphoinositide signaling in MDA-MB-435 breast carcinoma cells. Cancer Res. 2005;65:713–717. [PubMed] [Google Scholar]
- Dhar DK, Naora H, Kubota H, Maruyama R, Yoshimura H, Tonomoto Y, et al. Downregulation of KiSS-1 expression is responsible for tumor invasion and worse prognosis in gastric carcinoma. Int. J. Cancer. 2004;111:868–872. doi: 10.1002/ijc.20357. [DOI] [PubMed] [Google Scholar]
- Dong JT, Lamb PW, Rinker-Schaeffer CW, Vukanovic J, Ichikawa T, Isaacs JT, et al. KAI1, a metastasis suppressor gene for prostate cancer on human chromo-some 11p11.2. Science (Washington, D.C.) 1995;268:884–886. doi: 10.1126/science.7754374. [DOI] [PubMed] [Google Scholar]
- Dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, et al. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol. Rev. 2003;83:433–473. doi: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W, et al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell. 2006;21:51–64. doi: 10.1016/j.molcel.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Duca L, Floquet N, Alix AJ, Haye B, Debelle L. Elastin as a matrikine. Crit. Rev. Oncol. Hematol. 2004;49:235–244. doi: 10.1016/j.critrevonc.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Durkin ME, Yuan BZ, Zhou X, Zimonjic DB, Lowy DR, Thorgeirsson SS, et al. DLC-1: a Rho GTPase-activating protein and tumour suppressor. J. Cell. Mol. Med. 2007;11:1185–1207. doi: 10.1111/j.1582-4934.2007.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos JML, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742–1757. doi: 10.1016/S0140-6736(07)60781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles S, Paon L, Sleeman J. Lymphatic metastasis in breast cancer: importance and new insights into cellular and molecular mechanisms. Clin. Exp. Metastasis. 2007;24:619–636. doi: 10.1007/s10585-007-9123-5. [DOI] [PubMed] [Google Scholar]
- Eder JP, Vande Woude GF, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin. Cancer Res. 2009;15:2207–2214. doi: 10.1158/1078-0432.CCR-08-1306. [DOI] [PubMed] [Google Scholar]
- Edmonds MD, Hurst DR, Vaidya KS, Stafford LJ, Chen D, Welch DR. Breast cancer metastasis suppressor 1 (BRMS1) coordinately regulates metastasis-associated microRNA expression. Int. J. Cancer. 2009a;125:1778–1785. doi: 10.1002/ijc.24616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonds MD, Hurst DR, Welch DR. Linking metastasis suppression with metastamiR regulation. Cell Cycle. 2009b;8:2673–2675. doi: 10.4161/cc.8.17.9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev. Cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenbroek SIJ, Collard JG. Rho GTPases: functions and association with cancer. Clin. Exp. Metastasis. 2007;24:657–672. doi: 10.1007/s10585-007-9119-1. [DOI] [PubMed] [Google Scholar]
- Engbring JA, Kleinman HK. The basement membrane matrix in malignancy. J. Pathol. 2003;200:465–470. doi: 10.1002/path.1396. [DOI] [PubMed] [Google Scholar]
- Engel M, Seifert M, Theisinger B, Seyfert U, Welter C. Glyceraldehyde-3-phosphate dehydrogenase and Nm23-H1/nucleoside diphosphate kinase A—two old enzymes combine for the novel Nm23 protein phosphotransferase function. J. Biol. Chem. 1998;273:20058–20065. doi: 10.1074/jbc.273.32.20058. [DOI] [PubMed] [Google Scholar]
- Erier JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erler JT, Weaver VM. Three-dimensional context regulation of metastasis. Clin. Exp. Metastasis. 2009;26:35–49. doi: 10.1007/s10585-008-9209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing J. Neoplastic Disease. W.B. Saunders; Philadelphia: 1919. [Google Scholar]
- Farhadieh RD, Smee R, Ow K, Yang JL, Russell PJ, Crouch R, et al. Down-regulation of KAI1/CD82 protein expression in oral cancer correlates with reduced disease free survival and overall patient survival. Cancer Lett. 2004;213:91–98. doi: 10.1016/j.canlet.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Cho KR, Nigro JM, Kern SE, Simons JW, Ruppert JM, et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science (Washington, D.C.) 1990;247:49–56. doi: 10.1126/science.2294591. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125I-5-iodo-2'-deoxyuridine. J. Natl. Cancer Inst. 1970;45:773–782. [PubMed] [Google Scholar]
- Fidler IJ. Selection of successive tumor lines for metastasis. Nat. New Biol. 1973a;242:148–149. doi: 10.1038/newbio242148a0. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. The relationship of embolic heterogeneity, number size and viability to the incidence of experimental metastasis. Eur. J. Cancer. 1973b;9:223–227. doi: 10.1016/s0014-2964(73)80022-2. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. Immune stimulation–inhibition of experimental cancer metastasis. Cancer Res. 1974;34:491–498. [PubMed] [Google Scholar]
- Fidler IJ. The pathogenesis of cancer metastasis: the `seed and soil' hypothesis revisited. Nat. Rev. Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- Fidler IJ, Nicolson GL. Fate of recirculating B16 melanoma metastatic variant cells in parabiotic syngeneic recipients. J. Natl. Cancer Inst. 1977;58:1867–1872. doi: 10.1093/jnci/58.6.1867. [DOI] [PubMed] [Google Scholar]
- Fidler IJ, Radinsky R. Search for genes that suppress cancer metastasis. J. Natl. Cancer Inst. 1996;88:1700–1703. doi: 10.1093/jnci/88.23.1700. [DOI] [PubMed] [Google Scholar]
- Fidler IJ, Kim SI, Langley RR. The role of the organ microenvironment in the biology and therapy of cancer metastasis. J. Cell. Biochem. 2007;101:927–936. doi: 10.1002/jcb.21148. [DOI] [PubMed] [Google Scholar]
- Finegan KG, Tournier C. The mitogen-activated protein kinase kinase 4 has a pro-oncogenic role in skin cancer. Cancer Res. 2010;70:5797–5806. doi: 10.1158/0008-5472.CAN-09-3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010;29:285–293. doi: 10.1007/s10555-010-9224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis. Annu. Rev. Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- Forcet C, Ye X, Granger L, Corset V, Shin H, Bredesen DE, et al. The dependence receptor DCC (deleted in colorectal cancer) defines an alternative mechanism for caspase activation. Proc. Natl. Acad. Sci. USA. 2001;98:3416–3421. doi: 10.1073/pnas.051378298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulds L. The experimental study of tumor progression: a review. Cancer Res. 1954;14:327–339. [PubMed] [Google Scholar]
- Frame MC. Newest findings on the oldest oncogene; how activated src does it. J. Cell Sci. 2004;117:989–998. doi: 10.1242/jcs.01111. [DOI] [PubMed] [Google Scholar]
- Freije JMP, Blay P, MacDonald NJ, Manrow RE, Steeg PS. Site-directed mutation of Nm23-H1. Mutations lacking motility suppressive capacity upon transfection are deficient in histidine-dependent protein phosphotransferase pathways in vitro. J. Biol. Chem. 1997;272:5525–5532. doi: 10.1074/jbc.272.9.5525. [DOI] [PubMed] [Google Scholar]
- Freije JM, MacDonald NJ, Steeg PS. Nm23 and tumour metastasis: basic and translational advances. Biochem. Soc. Symp. 1998;63:261–271. [PubMed] [Google Scholar]
- Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng GJ, Ling LN, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova N, Edmonds MD, Bodenstine TM, Seitz R, Johnson MR, Feng R, et al. A shift from nuclear to cytoplasmic breast cancer metastasis suppressor 1 expression is associated with highly proliferative estrogen receptor-negative breast cancers. Tumor Biol. 2009;30:148–159. doi: 10.1159/000228908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost AR, Eltoum I, Siegal GP. Laser capture microdissection. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Protocols in Molecular Biology. John Wiley and Sons; New York: 2001. pp. 25A.1.1–25A.1.24. [Google Scholar]
- Frumkin D, Wasserstrom A, Itzkovitz S, Stern T, Harmelin A, Eilam R, et al. Cell lineage analysis of a mouse tumor. Cancer Res. 2008;68:5924–5931. doi: 10.1158/0008-5472.CAN-07-6216. [DOI] [PubMed] [Google Scholar]
- Fu Z, Smith PC, Zhang L, Rubin MA, Dunn RL, Yao Z, et al. Effects of Raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J. Natl. Cancer Inst. 2003;95:878–889. doi: 10.1093/jnci/95.12.878. [DOI] [PubMed] [Google Scholar]
- Fujii T, Yokoyama G, Takahashi H, Toh U, Kage M, Ono M, et al. Preclinical and clinical studies of novel breast cancer drugs targeting molecules involved in protein kinase C signaling, the putative metastasis-suppressor gene cap43 and the Y-box binding protein-1. Curr. Med. Chem. 2008;15:528–537. doi: 10.2174/092986708783769759. [DOI] [PubMed] [Google Scholar]
- Fujita H, Okada F, Hamada J, Hosokawa M, Moriuchi T, Koya RC, et al. Gelsolin functions as a metastasis suppressor in B16–BL6 mouse melanoma cells and requirement of the carboxyl-terminus for its effect. Int. J. Cancer. 2001;93:773–780. doi: 10.1002/ijc.1413. [DOI] [PubMed] [Google Scholar]
- Gabbert H. Mechanisms of tumor invasion: evidence from in vivo observations. Cancer Metastasis Rev. 1985;4:293–309. doi: 10.1007/BF00048094. [DOI] [PubMed] [Google Scholar]
- Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, Linsley PS, et al. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol. Cell. Biol. 2008;28:5369–5380. doi: 10.1128/MCB.00479-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasic G, Gasic T. Removal of sialic acid from the cell coat in tumor cells and vascular endothelium, and its effects on metastasis. Proc. Natl. Acad. Sci. USA. 1962;48:1172–1177. doi: 10.1073/pnas.48.7.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam A, Bepler G. Suppression of lung tumor formation by the regulatory subunit of ribonucleotide reductase. Cancer Res. 2006;66:6497–6502. doi: 10.1158/0008-5472.CAN-05-4462. [DOI] [PubMed] [Google Scholar]
- Geiger TR, Peeper DS. Metastasis mechanisms. Biochim. Biophys. Acta. 2009;1796:293–308. doi: 10.1016/j.bbcan.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Gelman IH, Lee K, Tombler E, Gordon R, Lin X. Control of cytoskeletal architecture by the src-suppressed C kinase substrate, SSeCKS. Cell Motil. Cytoskeleton. 1998;41:1–17. doi: 10.1002/(SICI)1097-0169(1998)41:1<1::AID-CM1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Gershenwald JE, Fidler IJ. Cancer: targeting lymphatic metastasis. Science (Washington, D.C.) 2002;296:1811–1812. doi: 10.1126/science.10731318. [DOI] [PubMed] [Google Scholar]
- Gianetti E, Seminara S. Kisspeptin and KISS1R: a critical pathway in the reproductive system. Reproduction. 2008;136:295–301. doi: 10.1530/REP-08-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles RH, vanEs JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta Rev. Cancer. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- Gladson CL, Welch DR. New insights into the role of CXCR4 in prostate cancer metastasis. Cancer Biol. Ther. 2008;7:1849–1851. doi: 10.4161/cbt.7.11.7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeil S, Zhu XC, Doillon CJ, Green MR. A genome-wide shRNA screen identifies GAS1 as a novel melanoma metastasis suppressor gene. Genes Dev. 2008;22:2932–2940. doi: 10.1101/gad.1714608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg SF, Miele ME, Hatta N, Takata M, Paquette-Straub CA, Freedman LP, et al. Melanoma metastasis suppression by chromosome 6: evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res. 2003;63:432–440. [PubMed] [Google Scholar]
- Goncharuk VN, del-Rosario A, Kren L, Anwar S, Sheehan CE, Carlson JA, et al. Co-downregulation of PTEN, KAI-1, and nm23-H1 tumor/metastasis suppressor proteins in non-small cell lung cancer. Ann. Diagn. Pathol. 2004;8:6–16. doi: 10.1016/j.anndiagpath.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Goodison S, Yuan G, Sloan D, Kim R, Li C, Popescu NC, et al. The RhoGAP protein DLC-1 functions as a metastasis suppressor in breast cancer cells. Cancer Res. 2005;65:6042–6053. doi: 10.1158/0008-5472.CAN-04-3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik E, Segal S, Feldman M. Control of lung metastasis progression in mice: role of growth kinetics of 3LL Lewis lung carcinoma and host immune reactivity. J. Natl. Cancer Inst. 1980;65:1257–1264. [PubMed] [Google Scholar]
- Grijalva R, Yang W, Zhou X, Bar-Eli M, Mills GB, Yu D. Lineage infidelity of MDA-MB-435 Cells: expression of melanocyte proteins in a breast cancer cell line. Proc. Am. Assoc. Cancer Res. 2003;44:3155. doi: 10.1158/0008-5472.CAN-3299-2. [DOI] [PubMed] [Google Scholar]
- Guan RJ, Ford HL, Fu Y, Li Y, Shaw LM, Pardee AB. Drg-1 as a differentiation-related, putative metastatic suppressor gene in human colon cancer. Cancer Res. 2000;60:749–755. [PubMed] [Google Scholar]
- Guan-Zhen Y, Ying C, Can-Rong N, Guo-Dong W, Jian-Xin Q, Jie-Jun W. Reduced protein expression of metastasis-related genes (nm23, KISS1, KAI1 and p53) in lymph node and liver metastases of gastric cancer. Int. J. Exp. Pathol. 2007;88:175–183. doi: 10.1111/j.1365-2613.2006.00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarino M. Src signaling in cancer invasion. J. Cell. Physiol. 2010;223:14–26. doi: 10.1002/jcp.22011. [DOI] [PubMed] [Google Scholar]
- Gumireddy K, Li AP, Gimotty PA, Klein-Szanto AJ, Showe LC, Katsaros D, et al. KLF17 is a negative regulator of epithelial–mesenchymal transition and metastasis in breast cancer. Nat. Cell Biol. 2009;11:1297–1304. doi: 10.1038/ncb1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan S, Al-Mulla F, Mallon E, Oien K, Ferrier R, Gusterson B, et al. Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clin. Cancer Res. 2005;11:7392–7397. doi: 10.1158/1078-0432.CCR-05-0283. [DOI] [PubMed] [Google Scholar]
- Hagmar B, Norrby K. Influence of cultivation, trypsinization and aggregation on the transplantability of melanoma B16 cells. Int. J. Cancer. 1973;11:663–675. doi: 10.1002/ijc.2910110317. [DOI] [PubMed] [Google Scholar]
- Hameed S, Dhillo WS. Biology of kisspeptins. Kallmann Syndrome and Hypogonadotropic Hypogonadism. 2010;39:25–36. doi: 10.1159/000312691. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanna N. The role of natural killer cells in the control of tumor growth and metastasis. Biochim. Biophys. Acta. 1985;780:213–226. doi: 10.1016/0304-419x(85)90004-6. [DOI] [PubMed] [Google Scholar]
- Harris TJC, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat. Rev. Mol. Cell Biol. 2010;11:502–514. doi: 10.1038/nrm2927. [DOI] [PubMed] [Google Scholar]
- Hartsough MT, Steeg PS. Nm23/nucleoside diphosphate kinase in human cancers. J. Bioenerg. Biomembr. 2000;32:301–308. doi: 10.1023/a:1005597231776. [DOI] [PubMed] [Google Scholar]
- Hartsough MT, Morrison DK, Salerno M, Palmieri D, Ouatas T, Mair M, et al. Nm23-H1 metastasis suppressor phosphorylation of kinase suppressor of ras via a histidine protein kinase pathway. J. Biol. Chem. 2002;277:32389–32399. doi: 10.1074/jbc.M203115200. [DOI] [PubMed] [Google Scholar]
- Hata K, Dhar DK, Watanabe Y, Nakai H, Hoshiai H. Expression of metastin and a G-protein-coupled receptor (AXOR12) in epithelial ovarian cancer. Eur. J. Cancer. 2007;43:1452–1459. doi: 10.1016/j.ejca.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev. Dyn. 2005;233:706–720. doi: 10.1002/dvdy.20345. [DOI] [PubMed] [Google Scholar]
- Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J. Cell Biol. 2000;148:779–790. doi: 10.1083/jcb.148.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedley BD, Welch DR, Allan AL, Al-Katib W, Dales DW, Postenka CO, et al. Downregulation of osteopontin contributes to metastasis suppression by breast cancer metastasis suppressor 1. Int. J. Cancer. 2008;123:526–534. doi: 10.1002/ijc.23542. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Lanstrom M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial–mesenchymal transition. Curr. Opin. Cell Biol. 2009;21:166–176. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- Hemler ME. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005;6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- Heppner GH. Tumor heterogeneity. Cancer Res. 1984;44:2259–2265. [PubMed] [Google Scholar]
- Heppner GH, Miller FR. The cellular basis of tumor progression. Int. Rev. Cytol. 1998;177:1–56. doi: 10.1016/s0074-7696(08)62230-5. [DOI] [PubMed] [Google Scholar]
- Hernandez-Barrantes S, Toth M, Bernardo MM, Yurkova M, Gervasi DC, Raz Y, et al. Binding of active (57 kDa) membrane type 1-matrix metalloproteinase (MT1-MMP) to tissue inhibitor of metalloproteinase (TIMP)-2 regulates MT1-MMP processing and pro-MMP-2 activation. J. Biol. Chem. 2000;275:12080–12089. doi: 10.1074/jbc.275.16.12080. [DOI] [PubMed] [Google Scholar]
- Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R, et al. Metastatic patterns in adenocarcinoma. Cancer. 2006;106:1624–1633. doi: 10.1002/cncr.21778. [DOI] [PubMed] [Google Scholar]
- Hicks DG, Yoder BJ, Short S, Tarr S, Prescott N, Crowe JP, et al. Loss of BRMS1 protein expression predicts reduced disease-free survival in hormone receptor negative and HER2 positive subsets of breast cancer. Clin. Cancer Res. 2006;12:6702–6708. doi: 10.1158/1078-0432.CCR-06-0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson JA, Huo D, Vander Griend DJ, Lin A, Rinker-Schaeffer CW, Yamada SD. The p38 Kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Res. 2006;66:2264–2270. doi: 10.1158/0008-5472.CAN-05-3676. [DOI] [PubMed] [Google Scholar]
- Hida K, Klagsbrun M. A new perspective on tumor endothelial cells: unexpected chromosome and centrosome abnormalities. Cancer Res. 2005;65:2507–2510. doi: 10.1158/0008-5472.CAN-05-0002. [DOI] [PubMed] [Google Scholar]
- Hida K, Hida Y, Amin DN, Flint AF, Panigrahy D, Morton CC, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004;64:8249–8255. doi: 10.1158/0008-5472.CAN-04-1567. [DOI] [PubMed] [Google Scholar]
- Hildenbrand R, Schaaf A, Dorn-Beineke A, Allgayer H, Sutterlin M, Marx A, et al. Tumor stroma is the predominant uPA-, uPAR-, PAI-1-expressing tissue in human breast cancer: prognostic impact. Histol. Histopathol. 2009;24:869–877. doi: 10.14670/HH-24.869. [DOI] [PubMed] [Google Scholar]
- Hollestelle A, Schutte M. Comment Re: MDA-MB-435 and M14 cell lines: identical but not M14 melanoma? Cancer Res. 2009;69:7893. doi: 10.1158/0008-5472.CAN-09-2396. [DOI] [PubMed] [Google Scholar]
- Horak CE, Lee JH, Elkahloun AG, Boissan M, Dumont S, Maga TK, et al. Nm23-H1 suppresses tumor cell motility by down-regulating the lysophosphatidic acid receptor EDG2. Cancer Res. 2007a;67:7238–7246. doi: 10.1158/0008-5472.CAN-07-0962. [DOI] [PubMed] [Google Scholar]
- Horak CE, Mendoza A, Vega-Valle E, Albaugh M, Graff-Cherry C, McDermott WG, et al. Nm23-H1 suppresses metastasis by inhibiting expression of the lysophosphatidic acid receptor EDG2. Cancer Res. 2007b;67:11751–11759. doi: 10.1158/0008-5472.CAN-07-3175. [DOI] [PubMed] [Google Scholar]
- Horuk R, Chitnis CE, Darbonne WC, Colby TJ, Rybicki A, Hadley TJ, et al. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science (Washington, D.C.) 1993;261:1182–1184. doi: 10.1126/science.7689250. [DOI] [PubMed] [Google Scholar]
- Horvath G, Serru V, Clay D, Billard M, Boucheix C, Rubinstein E. CD19 is linked to the integrin-associated tetraspans CD9, CD81, and CD82. J. Biol. Chem. 1998;273:30537–30543. doi: 10.1074/jbc.273.46.30537. [DOI] [PubMed] [Google Scholar]
- Hu LD, Zou HF, Zhan SX, Cao KM. Biphasic expression of RhoGDI2 in the progression of breast cancer and its negative relation with lymph node metastasis. Oncol. Rep. 2007;17:1383–1389. [PubMed] [Google Scholar]
- Huang QH, Gumireddy K, Schrier M, LeSage C, Nagel R, Nair S, et al. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat. Cell Biol. 2008;10:202–210. doi: 10.1038/ncb1681. [DOI] [PubMed] [Google Scholar]
- Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber MA, Kraut N, Beug H. Molecular requirements for epithelial–mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, et al. Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J. Cell. Physiol. 2007;213:374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- Hurst DR, Welch DR. A MSC-ing link in metastasis? Nat. Med. 2007;13:1289–1291. doi: 10.1038/nm1107-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst DR, Mehta A, Moore BP, Phadke PA, Meehan WJ, Accavitti MA, et al. Breast cancer metastasis suppressor 1 (BRMS1) is stabilized by the Hsp90 chaperone. Biochem. Biophys. Res. Commun. 2006;348:1429–1435. doi: 10.1016/j.bbrc.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst DR, Xie Y, Vaidya KS, Mehta A, Moore BP, Accavitti-Loper MA, et al. Alterations of BRMS1-ARID4A interaction modify gene expression but still suppress metastasis in human breast cancer cells. J. Biol. Chem. 2008;283:7438–7444. doi: 10.1074/jbc.M709446200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst DR, Edmonds MD, Scott GK, Benz CC, Vaidya KS, Welch DR. Breast cancer metastasis suppressor 1 BRMS1 up-regulates miR-146 that suppresses breast cancer metastasis. Cancer Res. 2009a;69:1279–1283. doi: 10.1158/0008-5472.CAN-08-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst DR, Edmonds MD, Welch DR. Metastamir: the field of metastasis-regulatory microRNA is spreading. Cancer Res. 2009b;69:7495–7498. doi: 10.1158/0008-5472.CAN-09-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst DR, Xie Y, Edmonds MD, Welch DR. Multiple forms of BRMS1 are differentially expressed in the MCF10 isogenic breast cancer progression model. Clin. Exp. Metastasis. 2009c;26:89–96. doi: 10.1007/s10585-008-9216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa T, Ichikawa Y, Dong J, Hawkins AL, Griffin CA, Isaacs WB, et al. Localization of metastasis suppressor gene(s) for prostatic cancer to the short arm of human chromosome 11. Cancer Res. 1992;52:3486–3490. [PubMed] [Google Scholar]
- Iino H, Fukayama M, Maeda Y, Koike M, Mori T, Takahashi T, et al. Molecular genetics for clinical management of colorectal carcinoma. 17p, 18q, and 22q loss of heterozygosity and decreased DCC expression are correlated with the metastatic potential. Cancer. 1994;73:1324–1331. doi: 10.1002/1097-0142(19940301)73:5<1324::aid-cncr2820730503>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Ikeami M, Mizuno D, Yamazaki M. Drug-dependent cellular cytotoxicity mediated by polymorphonuclear leukocytes. Jpn. J. Cancer Res. (GANN) 1985;76:637–643. [PubMed] [Google Scholar]
- Ikeguchi M, Hirooka Y, Kaibara N. Quantitative reverse transcriptase polymerase chain reaction analysis for KiSS-1 and orphan G-protein-coupled receptor (hOT7T175) gene expression in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2003;129:531–535. doi: 10.1007/s00432-003-0469-z. [DOI] [PubMed] [Google Scholar]
- Ikeguchi M, Yamaguchi K, Kaibara N. Clinical significance of the loss of KiSS-1 and orphan G-protein-coupled receptor (hOT7T175) gene expression in esophageal squamous cell carcinoma. Clin. Cancer Res. 2004;10:1379–1383. doi: 10.1158/1078-0432.ccr-1519-02. [DOI] [PubMed] [Google Scholar]
- Im DS, Heise CE, Nguyen T, O'Dowd BF, Lynch KR. Identification of a molecular target of psychosine and its role in globoid cell formation. J. Cell Biol. 2001;153:429–434. doi: 10.1083/jcb.153.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo RV, Zoeller JJ, Nystrom A. Basement membrane proteoglycans: modulators par excellence of cancer growth and angiogenesis. Mol. Cells. 2009;27:503–513. doi: 10.1007/s10059-009-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimura T, Gonzales R, Nicolson GL. Effects of tunicamycin on B16 metastatic melanoma cell surface glycoproteins and blood-borne arrest and survival properties. Cancer Res. 1981;41:3411–3418. [PubMed] [Google Scholar]
- Itoh F, Hinoda Y, Ohe M, Ohe Y, Ban T, Endo T, et al. Decreased expression of DCC mRNA in human colorectal cancers. Int. J. Cancer. 1993;53:260–263. doi: 10.1002/ijc.2910530215. [DOI] [PubMed] [Google Scholar]
- Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, et al. Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J. Cell Biol. 2002;156:299–313. doi: 10.1083/jcb.200109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008;27:6920–6929. doi: 10.1038/onc.2008.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J. Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Jiang YF, Goldberg ID, Shi YE. Complex roles of tissue inhibitors of metalloproteinases in cancer. Oncogene. 2002;21:2245–2252. doi: 10.1038/sj.onc.1205291. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Berk M, Singh LS, Tan HY, Yin LH, Powell CT, et al. KiSS1 suppresses metastasis in human ovarian cancer via inhibition of protein kinase C alpha. Clin. Exp. Metastasis. 2005;22:369–376. doi: 10.1007/s10585-005-8186-4. [DOI] [PubMed] [Google Scholar]
- Jiang WG, Raz A, Douglas-Jones A, Mansel RE. Expression of autocrine motility factor (AMF) and its receptor, AMFR, in human breast cancer. J. Histochem. Cytochem. 2006;54:231–241. doi: 10.1369/jhc.5A6785.2005. [DOI] [PubMed] [Google Scholar]
- Jones TD, Carr MD, Eble JN, Wang MS, Lopez-Beltran A, Cheng L. Clonal origin of lymph node metastases in bladder carcinoma. Cancer. 2005;104:1901–1910. doi: 10.1002/cncr.21466. [DOI] [PubMed] [Google Scholar]
- Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl. Acad. Sci. USA. 2008;105:4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorissen RN, Gibbs P, Christie M, Prakash S, Lipton L, Desai J, et al. Metastasis-associated gene expression changes predict poor outcomes in patients with Dukes stage B and C colorectal cancer. Clin. Cancer Res. 2009;15:7642–7651. doi: 10.1158/1078-0432.CCR-09-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jove R, Hanafusa H. Cell transformation by the viral src oncogene. Annu. Rev. Cell Biol. 1987;3:31–56. doi: 10.1146/annurev.cb.03.110187.000335. [DOI] [PubMed] [Google Scholar]
- Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat. Rev. Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaetzel DM, Zhang QB, Yang MM, McCorkle JR, Ma DQ, Craven RJ. Potential roles of 3'′5' exonuclease activity of NM23-H1 in DNA repair and malignant progression. J. Bioenerg. Biomembr. 2006;38:163–167. doi: 10.1007/s10863-006-9040-3. [DOI] [PubMed] [Google Scholar]
- Kaetzel DM, McCorkle JR, Novak M, Yang MM, Jarrett SG. Potential contributions of antimutator activity to the metastasis suppressor function of NM23-H1. Mol. Cell. Biochem. 2009;329:161–165. doi: 10.1007/s11010-009-0108-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallakury BV, Yang F, Figge J, Smith KE, Kausik SJ, Tacy NJ, et al. Decreased levels of CD44 protein and mRNA in prostate carcinoma. Correlation with tumor grade and ploidy. Cancer. 1996;78:1461–1469. doi: 10.1002/(sici)1097-0142(19961001)78:7<1461::aid-cncr13>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Kang YB, Siegel PM, Shu WP, Drobnjak M, Kakonen SM, Cordón-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Kantor JD, McCormick B, Steeg PS, Zetter BR. Inhibition of cell motility after nm23 transfection of human and murine tumor cells. Cancer Res. 1993;53:1971–1973. [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor P, Saunders MM, Li Z, Zhou Z, Schaeffer N, Kunze EL, et al. Breast cancer metastatic potential: correlation with increased heterotypic gap junctional intercellular communication between breast cancer cells and osteoblastic cells. Int. J. Cancer. 2004;111:693–697. doi: 10.1002/ijc.20318. [DOI] [PubMed] [Google Scholar]
- Karlsson R, Pedersen ED, Wang Z, Brakebusch C. Rho GTPase function in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer. 2009;1796:91–98. doi: 10.1016/j.bbcan.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- Kashima T, Nakamura K, Kawaguchi J, Takanashi M, Ishida T, Aburatani H, et al. Overexpression of cadherins suppresses pulmonary metastasis of osteosarcoma in vivo. Int. J. Cancer. 2003;104:147–154. doi: 10.1002/ijc.10931. [DOI] [PubMed] [Google Scholar]
- Katagiri F, Nagai K, Kida A, Tomita K, Oishi S, Takeyama M, et al. Clinical significance of plasma metastin level in pancreatic cancer patients. Oncol. Rep. 2009;21:815–819. [PubMed] [Google Scholar]
- Katakura H, Tanaka F, Oyanagi H, Miyahara R, Yanagihara K, Otake Y, et al. Clinical significance of nm23 expression in resected pathologic-stage I, non-small cell lung cancer. Ann. Thorac. Surg. 2002;73:1060–1064. doi: 10.1016/s0003-4975(01)03597-4. [DOI] [PubMed] [Google Scholar]
- Keller ET, Fu Z, Yeung K, Brennan M. Raf kinase inhibitor protein: a prostate cancer metastasis suppressor gene. Cancer Lett. 2004;207:131–137. doi: 10.1016/j.canlet.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokha R, Denhardt DT. Matrix metalloproteinases and tissue inhibitor of metalloproteinases: a review of their role in tumorigenesis and tissue invasion. Invasion Metastasis. 1989;9:391–405. [PubMed] [Google Scholar]
- Kikuchi-Yanoshita R, Konishi M, Fukunari H, Tanaka K, Miyaki M. Loss of expression of the DCC gene during progression of colorectal carcinomas in familial adenomatous polyposis and non-familial adenomatous polyposis patients. Cancer Res. 1992;52:3801–3803. [PubMed] [Google Scholar]
- Kim HRC, Lin HM, Biliran H, Raz A. Cell cycle arrest and inhibition of anoikis by galectin-3 in human breast epithelial cells. Cancer Res. 1999;59:4148–4154. [PubMed] [Google Scholar]
- Kim HL, Van der Griend DJ, Yang X, Benson DA, Dubauskas Z, Yoshida BA, et al. Mitogen-activated protein kinase kinase 4 metastasis suppressor gene expression is inversely related to histological pattern in advancing human prostatic cancers. Cancer Res. 2001;61:2833–2837. [PubMed] [Google Scholar]
- Kim KT, Ongusaha PP, Hong YK, Kurdistani SK, Nakamura M, Lu KP, et al. Function of Drg1/Rit42 in p53-dependent mitotic spindle checkpoint. J. Biol. Chem. 2004;279:38597–38602. doi: 10.1074/jbc.M400781200. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kim B, Cai L, Choi HJ, Ohgi KA, Tran C, et al. Transcriptional regulation of a metastasis suppressor gene by Tip60 and β-catenin complexes. Nature. 2005a;434:921–926. doi: 10.1038/nature03452. [DOI] [PubMed] [Google Scholar]
- Kim KS, Ren J, Jiang Y, Ebrahem Q, Tipps R, Cristina K, et al. GPR4 plays a critical role in endothelial cell function and mediates the effects of sphingosylphosphorylcholine. FASEB J. 2005b;19:819–821. doi: 10.1096/fj.04-2988fje. [DOI] [PubMed] [Google Scholar]
- Kim SY, Lee CH, Midura BV, Yeung C, Mendoza A, Hong SH, et al. Inhibition of the CXCR4/CXCL12 chemokine pathway reduces the development of murine pulmonary metastases. Clin. Exp. Metastasis. 2008;25:201–211. doi: 10.1007/s10585-007-9133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura N, Shimada N. Evidence for complex formation between GTP binding protein(Gs) and membrane-associated nucleoside diphosphate kinase. Biochem. Biophys. Res. Commun. 1990;168:99–106. doi: 10.1016/0006-291x(90)91680-q. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H, et al. SMAD4-deficient intestinal tumors recruit CCR1(+) myeloid cells that promote invasion. Nat. Genet. 2007;39:467–475. doi: 10.1038/ng1997. [DOI] [PubMed] [Google Scholar]
- Klein CA. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer. 2009;9:302–312. doi: 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- Klominek J, Baskin B, Liu Z, Hauzenberger D. Hepatocyte growth factor/scatter factor stimulates chemotaxis and growth of malignant mesothelioma cells through c-met receptor. Int. J. Cancer. 1998;76:240–249. doi: 10.1002/(sici)1097-0215(19980413)76:2<240::aid-ijc12>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Kosako H, Yoshida T, Matsumura F, Ishizaki T, Narumiya S, Inagaki M. Rho-kinase/ROCK is involved in cytokinesis through the phosphorylation of myosin light chain and not ezrin/radixin/moesin proteins at the cleavage furrow. Oncogene. 2000;19:6059–6064. doi: 10.1038/sj.onc.1203987. [DOI] [PubMed] [Google Scholar]
- Kostadima L, Pentheroudakis G, Pavlidis N. The missing kiss of life: transcriptional activity of the metastasis suppressor gene KiSS1 in early breast cancer. Anticancer Res. 2007;27:2499–2504. [PubMed] [Google Scholar]
- Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E, et al. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J. Biol. Chem. 2001;276:34631–34636. doi: 10.1074/jbc.M104847200. [DOI] [PubMed] [Google Scholar]
- Krüger A, Kates RE, Edwards DR. Avoiding spam in the proteolytic internet: Future strategies for anti-metastatic MMP inhibition. Biochim. et Biophys. Acta-Molec. Cell Res. 2010;1803:95–102. doi: 10.1016/j.bbamcr.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Kuppers DA, Lan K, Knight JS, Robertson ES. Regulation of matrix metalloproteinase 9 expression by Epstein–Barr virus nuclear antigen 3C and the suppressor of metastasis Nm23-H1. J. Virol. 2005;79:9714–9724. doi: 10.1128/JVI.79.15.9714-9724.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdistani SK, Arizti P, Reimer CL, Sugrue MM, Aaronson SA, Lee SW. Inhibition of tumor cell growth by RTP/rit42 and its responsiveness to p53 and DNA damage. Cancer Res. 1998;58:4439–4444. [PubMed] [Google Scholar]
- Lacombe ML, Milon L, Munier A, Mehus JG, Lambeth DO. The human Nm23/nucleoside diphosphate kinases. J. Bioenerg. Biomembr. 2000;32:247–258. doi: 10.1023/a:1005584929050. [DOI] [PubMed] [Google Scholar]
- Lahti JM, Teitz T, Stupack DG. Does integrin-mediated cell death confer tissue tropism in metastasis? Cancer Res. 2006;66:5981–5984. doi: 10.1158/0008-5472.CAN-06-0131. [DOI] [PubMed] [Google Scholar]
- Le Guezennec X, Vermeulen M, Stunnenberg HG. Molecular characterization of Sin3 PAH-domain interactor specificity and identification of PAH partners. Nucleic Acids Res. 2006;34:3929–3937. doi: 10.1093/nar/gkl537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-H, Welch DR. Identification of highly expressed genes in metastasis-suppressed chromosome 6/human malignant melanoma hybrid cells using subtractive hybridization and differential display. Int. J. Cancer. 1997a;71:1035–1044. doi: 10.1002/(sici)1097-0215(19970611)71:6<1035::aid-ijc20>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Lee J-H, Welch DR. Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res. 1997b;57:2384–2387. [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Lee J-H, Miele ME, Hicks DJ, Phillips KK, Trent JM, Weissman BE, et al. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J. Natl. Cancer Inst. 1996;88:1731–1737. doi: 10.1093/jnci/88.23.1731. [DOI] [PubMed] [Google Scholar]
- Lee J-H, Miele ME, Hicks DJ, Phillips KK, Trent JM, Weissman BE, et al. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene [erratum] J. Natl. Cancer Inst. 1997;89:1549. doi: 10.1093/jnci/88.23.1731. [DOI] [PubMed] [Google Scholar]
- Lee JH, Park SR, Chay KO, Seo YW, Kook H, Ahn KY, et al. KAI1 COOH-terminal interacting tetraspanin (KITENIN), a member of the tetraspanin family, interacts with KAI1, a tumor metastasis suppressor, and enhances metastasis of cancer. Cancer Res. 2004;64:4235–4243. doi: 10.1158/0008-5472.CAN-04-0275. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- Lee JH, Marshall JC, Steeg PS, Horak CE. Altered gene protein and expression by Nm23-H1 in metastasis suppression. Mol. Cell. Biochem. 2009;329:141–148. doi: 10.1007/s11010-009-0124-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrue SJ. 1-Butanol extraction and subsequent reconstitution of membrane components which mediate metastatic phenotype. Cancer Res. 1982;42:2126–2134. [PubMed] [Google Scholar]
- Lengyel E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010;177:1053–64. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone A, Flatow U, VanHoutte K, Steeg PS. Transfection of human nm23-H1 into the human MDA-MB-435 breast carcinoma cell line: effects on tumor metastatic potential, colonization and enzymatic activity. Oncogene. 1993;8:2325–2333. [PubMed] [Google Scholar]
- Li G, Satyamoorthy K, Herlyn M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001;61:3819–3825. [PubMed] [Google Scholar]
- Li LC, Okino ST, Dahiya R. DNA methylation in prostate cancer. Biochim. Biophys. Acta Rev. Cancer. 2004;1704:87–102. doi: 10.1016/j.bbcan.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Liebig C, Ayala G, Wilks JA, Berger DH, Albo D. Perineural invasion in cancer: a review of the literature. Cancer. 2009;115:3379–3391. doi: 10.1002/cncr.24396. [DOI] [PubMed] [Google Scholar]
- Lifsted T, Le Voyer T, Williams M, Muller W, Klein-Szanto A, Buetow KH, et al. Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression. Int. J. Cancer. 1998;77:640–644. doi: 10.1002/(sici)1097-0215(19980812)77:4<640::aid-ijc26>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Lin X, Tombler E, Nelson PJ, Ross M, Gelman IH. A novel src- and ras-suppressed protein kinase C substrate associated with cytoskeletal architecture. J. Biol. Chem. 1996;271:28430–28438. doi: 10.1074/jbc.271.45.28430. [DOI] [PubMed] [Google Scholar]
- Lin X, Nelson P, Gelman IH. SSeCKS, a major protein kinase C substrate with tumor suppressor activity, regulates G(1)–>S progression by controlling the expression and cellular compartmentalization of cyclin D. Mol. Cell. Biol. 2000;20:7259–7272. doi: 10.1128/mcb.20.19.7259-7272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SL, Chiang A, Chang D, Ying SY. Loss of mir-146a function in hormone-refractory prostate cancer. RNA. 2008;14:417–424. doi: 10.1261/rna.874808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HJL, Zuo T, Chao JR, Peng ZG, Asamoto LK, Yamashita SS, et al. Seed in soil, with an epigenetic view. Biochim. Biophys. Acta Gen. Subj. 2009;1790:920–924. doi: 10.1016/j.bbagen.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta LA. Tumor invasion and metastases—role of the extracellular matrix: Rhoads Memorial Award lecture. Cancer Res. 1986;46:1–7. [PubMed] [Google Scholar]
- Liotta LA. Cancer cell invasion and metastasis. Sci. Am. 1992;266:54–63. doi: 10.1038/scientificamerican0292-54. [DOI] [PubMed] [Google Scholar]
- Liotta LA, Stetler-Stevenson WG. Tumor invasion and metastasis: an imbalance of positive and negative regulation. Cancer Res. 1991;51:5054s–5059s. [PubMed] [Google Scholar]
- Litwin M, Mazur AJ, Nowak D, Mannherz HG, Malicka-Blaszkiewicz M. Gelsolin in human colon adenocarcinoma cells with different metastatic potential. Acta Biochim. Pol. 2009;56:739–743. [PubMed] [Google Scholar]
- Liu FS, Dong JT, Chen JT, Hsieh YT, Ho ES, Hung MJ. Frequent down-regulation and lack of mutation of the KAI1 metastasis suppressor gene in epithelial ovarian carcinoma. Gynecol. Oncol. 2000;78:10–15. doi: 10.1006/gyno.2000.5801. [DOI] [PubMed] [Google Scholar]
- Liu FS, Chen JT, Dong JT, Hsieh YT, Lin AJ, Ho ESC, et al. KAI1 metastasis suppressor gene is frequently down-regulated in cervical carcinoma. Am. J. Pathol. 2001;159:1629–1634. doi: 10.1016/s0002-9440(10)63009-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Smith PW, Jones DR. Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. Mol. Cell. Biol. 2006;26:8683–8696. doi: 10.1128/MCB.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Wang XH, Chen GY, Dalerba P, Gurney A, Hoey T, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J. Med. 2007;356:217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- Liu SY, Umezu-Goto M, Murph M, Lu YL, Liu WB, Zhang F, et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell. 2009;15:539–550. doi: 10.1016/j.ccr.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi DP, Geradts J, Foley JF, Chiao C, Lamb PW, Barrett JC. Loss of KAI1 expression in the progression of colorectal cancer. Cancer Res. 1999;59:5724–5731. [PubMed] [Google Scholar]
- Longo N, Yanez-Mo M, Mittelbrunn M, de la Rosa G, Munoz ML, Sanchez-Madrid F, et al. Regulatory role of tetraspanin CD9 in tumor-endothelial cell interaction during transendothelial invasion of melanoma cells. Blood. 2001;98:3717–3726. doi: 10.1182/blood.v98.13.3717. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Matrisian LM. Tumour microenvironment—opinion—emerging roles of proteases in tumour suppression. Nat. Rev. Cancer. 2007;7:800–808. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- Lotan TL, Lyon M, Huo D, Taxy JB, Brendler C, Foster BA, et al. Up-regulation of MKK4, MKK6 and MKK7 during prostate cancer progression: an important role for SAPK signalling in prostatic neoplasia. J. Pathol. 2007;212:386–394. doi: 10.1002/path.2194. [DOI] [PubMed] [Google Scholar]
- Lotan T, Hickson J, Souris J, Huo D, Taylor J, Li T, et al. c-Jun NH2-terminal kinase activating kinase 1/mitogen-activated protein kinase kinase 4-mediated inhibition of SKOV3ip. 1 ovarian cancer metastasis involves growth arrest and p21 up-regulation. Cancer Res. 2008;68:2166–2175. doi: 10.1158/0008-5472.CAN-07-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Kang YB. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009;69:8536–8539. doi: 10.1158/0008-5472.CAN-09-2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugassy C, Kleinman HK, Fernandez PM, Patierno SR, Webber MM, Ghanem G, et al. Human melanoma cell migration along capillary-like structures in vitro: a new dynamic model for studying extravascular migratory metastasis. J. Investig. Dermatol. 2002;119:703–704. doi: 10.1046/j.1523-1747.2002.01857.x. [DOI] [PubMed] [Google Scholar]
- Lugassy C, Kleinman HK, Engbring JA, Welch DR, Harms JF, Rufner R, et al. Pericyte-like location of GFP-tagged melanoma cells: ex vivo and in vivo studies of extravascular migratory metastasis. Am. J. Pathol. 2004;164:1191–1198. doi: 10.1016/S0002-9440(10)63207-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugassy C, Vernon SE, Busam K, Engbring JA, Welch DR, Poulos EG, et al. Angiotropism of human melanoma: studies involving in transit and other cutaneous metastases and the chicken chorioallantoic membrane: implications for extravascular melanoma invasion and metastasis. Am. J. Dermatopathol. 2006;28:187–193. doi: 10.1097/00000372-200606000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugassy C, Kleinman HK, Vernon SE, Welch DR, Barnhill RL. C16 laminin peptide increases angiotropic extravascular migration of human melanoma cells in a shell-less chick chorioallantoic membrane assay. Br. J. Dermatol. 2007;157:780–782. doi: 10.1111/j.1365-2133.2007.08120.x. [DOI] [PubMed] [Google Scholar]
- Ma DQ, McCorkle JR, Kaetzel DM. The metastasis suppressor NM23-H1 possesses 3'–5' exonuclease activity. J. Biol. Chem. 2004;279:18073–18084. doi: 10.1074/jbc.M400185200. [DOI] [PubMed] [Google Scholar]
- Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007a;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- Ma L, Xu G, Sotnikova A, Szczepanowski M, Giefing M, Krause K, et al. Loss of expression of LyGDI (ARHGDIB), a rho GDP-dissociation inhibitor, in Hodgkin lymphoma. Br. J. Haematol. 2007b;139:217–223. doi: 10.1111/j.1365-2141.2007.06782.x. [DOI] [PubMed] [Google Scholar]
- MacDonald NJ, de la Rosa A, Benedict MA, Freije JM, Krutsch H, Steeg PS. A serine phosphorylation of Nm23, and not its nucleoside diphosphate kinase activity, correlates with suppression of tumor metastatic potential. J. Biol. Chem. 1993;268:25780–25789. [PubMed] [Google Scholar]
- Mannion BA, Berditchevski F, Kraeft SK, Chen LB, Hemler ME. Transmembrane-4 superfamily proteins CD81 (TAPA-1), CD82, CD63, and CD53 specifically associated with integrin alpha 4 beta 1 (CD49d/CD29) J. Immunol. 1996;157:2039–2047. [PubMed] [Google Scholar]
- Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315–322. doi: 10.1007/s10555-006-9001-7. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Savino B, Locati M, Zammataro L, Allavena P, Bonecchi R. The chemokine system in cancer biology and therapy. Cytokine Growth Factor Rev. 2010;21:27–39. doi: 10.1016/j.cytogfr.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Mao H, Liu H, Fu X, Fang Z, Abrams J, Worsham MJ. Loss of nm23 expression predicts distal metastases and poorer survival for breast cancer. Int. J. Oncol. 2001;18:587–591. doi: 10.3892/ijo.18.3.587. [DOI] [PubMed] [Google Scholar]
- Marreiros A, Czolij R, Yardley G, Crossley M, Jackson P. Identification of regulatory regions within the KAI1 promoter: a role for binding of AP1, AP2 and p53. Gene. 2003;302:155–164. doi: 10.1016/s0378-1119(02)01101-0. [DOI] [PubMed] [Google Scholar]
- Marshall JC, Lee JH, Steeg PS. Clinical–translational strategies for the elevation of Nm23-H1 metastasis suppressor gene expression. Mol. Cell. Biochem. 2009;329:115–120. doi: 10.1007/s11010-009-0116-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JC, Collins J, Marino N, Steeg P. The Nm23-H1 metastasis suppressor as a translational target. Eur. J. Cancer. 2010;46:1278–1282. doi: 10.1016/j.ejca.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TA, Watkins G, Jiang WG. KiSS-1 expression in human breast cancer. Clin. Exp. Metastasis. 2005;22:503–511. doi: 10.1007/s10585-005-4180-0. [DOI] [PubMed] [Google Scholar]
- Martinez R, Setien F, Voelter C, Casado S, Quesada MP, Schackert G, et al. CpG island promoter hypermethylation of the pro-apoptotic gene caspase-8 is a common hallmark of relapsed glioblastoma multiforme. Carcinogenesis. 2007 doi: 10.1093/carcin/bgm014. doi:10.1093/carcin/bgm014. [DOI] [PubMed] [Google Scholar]
- Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta Rev. Cancer. 2010;1805:105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama Y, Ono M, Kawahara A, Yokoyama T, Basaki Y, Kage M, et al. Tumor growth suppression in pancreatic cancer by a putative metastasis suppressor gene Cap43/NDRG1/Drg-1 through modulation of angiogenesis. Cancer Res. 2006;66:6233–6242. doi: 10.1158/0008-5472.CAN-06-0183. [DOI] [PubMed] [Google Scholar]
- Mashimo T, Bandyopadhyay S, Goodarzi G, Watabe M, Pai SK, Gross SC, et al. Activation of the tumor metastasis suppressor gene, KAI1, by etoposide is mediated by p53 and c-Jun genes. Biochem. Biophys. Res. Commun. 2000;274:370–376. doi: 10.1006/bbrc.2000.3139. [DOI] [PubMed] [Google Scholar]
- Masui T, Doi R, Mori T, Toyoda E, Koizumi M, Kami K, et al. Metastin and its variant forms suppress migration of pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2004;315:85–92. doi: 10.1016/j.bbrc.2004.01.021. [DOI] [PubMed] [Google Scholar]
- McCarthy JB, Basara ML, Palm SL, Sas DF, Furcht LT. The role of cell adhesion proteins—laminin and fibronectin—in the movement of malignant and metastatic cells. Cancer Metastasis Rev. 1985;4:125–152. doi: 10.1007/BF00050692. [DOI] [PubMed] [Google Scholar]
- McCawley LJ, Matrisian LM. Matrix metalloproteinases: multifunctional contributors to tumor progression. Mol. Med. Today. 2000;6:149–156. doi: 10.1016/s1357-4310(00)01686-5. [DOI] [PubMed] [Google Scholar]
- McNally LR, Welch DR, Beck BH, Stafford LJ, Long JW, Sellers JC, et al. KISS1 over-expression suppresses metastasis of pancreatic adenocarcinoma in a xenograft mouse model. Clin. Exp. Metastasis. 2010 doi: 10.1007/s10585-010-9349-5. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead EJ, Maguire JJ, Kuc RE, Davenport AP. Kisspeptins are novel potent vasoconstrictors in humans, with a discrete localization of their receptor, G protein-coupled receptor 54, to atherosclerosis-prone vessels. Endocrinology. 2007;148:140–147. doi: 10.1210/en.2006-0818. [DOI] [PubMed] [Google Scholar]
- Meehan WJ, Samant RS, Hopper JE, Carrozza MJ, Shevde LA, Workman JL, et al. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J. Biol. Chem. 2004;279:1562–1569. doi: 10.1074/jbc.M307969200. [DOI] [PubMed] [Google Scholar]
- Mendoza A, Hong SH, Osborne T, Khan MA, Campbell K, Briggs J, et al. Modeling metastasis biology and therapy in real time in the mouse lung. J. Clin. Investig. 2010;120:2979–2988. doi: 10.1172/JCI40252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu WP, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake S, Nagai K, Yoshino K, Oto M, Endo M, Yuasa Y. Point mutations and allelic deletion of tumor suppressor gene DCC in human esophageal squamous cell carcinomas and their relation to metastasis. Cancer Res. 1994;54:3007–3010. [PubMed] [Google Scholar]
- Moissoglu K, McRoberts KS, Meier JA, Theodorescu D, Schwartz MA. Rho GDP dissociation inhibitor 2 suppresses metastasis via unconventional regulation of RhoGTPases. Cancer Res. 2009;69:2838–2844. doi: 10.1158/0008-5472.CAN-08-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montel V, Suzuki M, Galloy C, Mose ES, Tarin D. Expression of melanocyte-related genes in human breast cancer and its implications. Differentiation. 2009;78:283–291. doi: 10.1016/j.diff.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27:6958–6969. doi: 10.1038/onc.2008.346. [DOI] [PubMed] [Google Scholar]
- Muir AI, Chamberlain L, Elshourbagy NA, Michalovich D, Moore DJ, Calamari A, et al. AXOR12: a novel human G protein-coupled receptor, activated by the peptide KiSS-1. J. Biol. Chem. 2001;276:28969–28975. doi: 10.1074/jbc.M102743200. [DOI] [PubMed] [Google Scholar]
- Muller A, Homey B, Soto H, Ge NF, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- Muralidharan-Chari V, Hoover H, Clancy J, Schweitzer J, Suckow MA, Schroeder V, et al. ADP-ribosylation factor 6 regulates tumorigenic and invasive properties in vivo. Cancer Res. 2009;69:2201–2209. doi: 10.1158/0008-5472.CAN-08-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai K, Doi R, Katagiri F, Ito T, Kida A, Koizumi M, et al. Prognostic value of metastin expression in human pancreatic cancer. J. Exp. Clin. Cancer Res. 2009;28:28–29. doi: 10.1186/1756-9966-28-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M, Chop AM. Tumor invasion and extracellular matrix degradative enzymes: regulation of activity by organ factors. Semin. Cancer Biol. 1991;2:178–196. [PubMed] [Google Scholar]
- Nakajima M, Irimura T, Di Ferranti N, Di Ferranti D, Nicolson GL. Heparan sulfate degradation: relation to tumor invasive and metastatic properties of mouse B16 melanoma sublines. Science (Washington, D.C.) 1983;220:611–613. doi: 10.1126/science.6220468. [DOI] [PubMed] [Google Scholar]
- Nam JS, Terabe M, Mamura M, Kang NJ, Chae H, Stuelten C, et al. An anti-transforming growth factor β antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 2008;68:3835–3843. doi: 10.1158/0008-5472.CAN-08-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam JM, Onodera Y, Bissell MJ, Park CC. Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin α5β1 and fibronectin. Cancer Res. 2010;70:5238–5248. doi: 10.1158/0008-5472.CAN-09-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash KT, Welch DR. The KISS1 metastasis suppressor: mechanistic insights and clinical utility. Front. Biosci. 2006;11:647–659. doi: 10.2741/1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash KT, Phadke PA, Navenot J-M, Hurst DR, Accavitti-Loper MA, Sztul E, et al. KISS1 metastasis suppressor secretion, multiple organ metastasis suppression, and maintenance of tumor dormancy. J. Natl. Cancer Inst. 2007;99:309–321. doi: 10.1093/jnci/djk053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson SD. Insights into the mechanisms of lymph node metastasis. Cancer. 2003;98:413–423. doi: 10.1002/cncr.11464. [DOI] [PubMed] [Google Scholar]
- Nauert JB, Klauck TM, Langeberg LK, Scott JD. Gravin, an autoantigen recognized by serum from myasthenia gravis patients, is a kinase scaffold protein. Curr. Biol. 1997;7:52–62. doi: 10.1016/s0960-9822(06)00027-3. [DOI] [PubMed] [Google Scholar]
- Naumov GN, Wilson SM, MacDonald IC, Schmidt EE, Morris VL, Groom AC, et al. Cellular expression of green fluorescent protein, coupled with high-resolution in vivo videomicroscopy, to monitor steps in tumor metastasis. J. Cell Sci. 1999;112:1835–1842. doi: 10.1242/jcs.112.12.1835. [DOI] [PubMed] [Google Scholar]
- Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N, Nadkarni KV, Wilson SM, et al. Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res. 2002;62:2162–2168. [PubMed] [Google Scholar]
- Nelson PJ, Moissoglu K, Vargas J, Jr., Klotman PE, Gelman IH. Involvement of the protein kinase C substrate, SSeCKS, in the actin-based stellate morphology of mesangial cells. J. Cell Sci. 1999;112:361–370. doi: 10.1242/jcs.112.3.361. [DOI] [PubMed] [Google Scholar]
- Nelson AR, Fingleton B, Rothenberg ML, Matrisian LM. Matrix metalloproteinases: biologic activity and clinical implications. J. Clin. Oncol. 2000;18:1135–1149. doi: 10.1200/JCO.2000.18.5.1135. [DOI] [PubMed] [Google Scholar]
- Nicoloso MS, Spizzo R, Shimizu M, Rossi S, Calin GA. MicroRNAs—the micro steering wheel of tumour metastases. Nat. Rev. Cancer. 2009;9:293–302. doi: 10.1038/nrc2619. [DOI] [PubMed] [Google Scholar]
- Nicolson GL. Cancer metastasis. Organ colonization and the cell-surface properties of malignant cells. Biochim. Biophys. Acta. 1982a;695:113–176. doi: 10.1016/0304-419x(82)90020-8. [DOI] [PubMed] [Google Scholar]
- Nicolson GL. Cell surface properties of metastatic tumor cells. In: Liotta LA, Hart IR, editors. Tumor Invasion and Metastasis. Martinus Nijhoff; The Hague: 1982b. pp. 57–79. [Google Scholar]
- Nicolson GL. Generation of phenotypic diversity and progression in metastatic tumor cells. Cancer Metastasis Rev. 1984;3:25–42. doi: 10.1007/BF00047691. [DOI] [PubMed] [Google Scholar]
- Nicolson GL. Cancer metastasis: tumor cell and host organ properties important in metastasis to secondary sites. Biochim. Biophys. Acta. 1988a;948:175–224. doi: 10.1016/0304-419x(88)90010-8. [DOI] [PubMed] [Google Scholar]
- Nicolson GL. Organ specificity of tumor metastasis: role of preferential adhesion, invasion and growth of malignant cells at specific secondary sites. Cancer Metastasis Rev. 1988b;7:143–188. doi: 10.1007/BF00046483. [DOI] [PubMed] [Google Scholar]
- Nicolson GL, Nawa A, Toh Y, Taniguchi S, Nishimori K, Moustafa A. Tumor metastasis-associated human MTA1 gene and its MTA1 protein product: role in epithelial cancer cell invasion, proliferation and nuclear regulation. Clin. Exp. Metastasis. 2003;20:19–24. doi: 10.1023/a:1022534217769. [DOI] [PubMed] [Google Scholar]
- Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ. N-cadherin motility in human breast cancer cells regardless of their E-cadherin expression. J. Cell Biol. 1999;147:631–643. doi: 10.1083/jcb.147.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida A, Wang ZX, Tomita K, Oishi S, Tamamura H, Otaka A, et al. Design and synthesis of downsized metastin (45–54) analogs with maintenance of high GPR54 agonistic activity. Bioorg. Med. Chem. 2006;16:134–137. doi: 10.1016/j.bmcl.2005.09.054. [DOI] [PubMed] [Google Scholar]
- Nikolaev AY, Papanikolaou NA, Li M, Qin J, Gu W. Identification of a novel BRMS1-homologue protein p40 as a component of the mSin3A/p33(ING1b)/HDAC1 deacetylase complex. Biochem. Biophys. Res. Commun. 2004;323:1216–1222. doi: 10.1016/j.bbrc.2004.08.227. [DOI] [PubMed] [Google Scholar]
- Niu Y, Fu X, Lv A, Fan Y, Wang Y. Potential markers predicting distant metastasis in axillary node-negative breast carcinoma. Int. J. Cancer. 2002;98:754–760. doi: 10.1002/ijc.10136. [DOI] [PubMed] [Google Scholar]
- North SM, Nicolson GL. Effect of host immune status on the spontaneous metastasis of cloned cell lines of the 13762NF rat mammary adenocarcinoma. Br. J. Cancer. 1985;52:747–755. doi: 10.1038/bjc.1985.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell P. The clonal evolution of tumor cell populations. Science (Washington, D.C.) 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- Nowell PC. Mechanisms of tumor progression. Cancer Res. 1986;46:2203–2207. [PubMed] [Google Scholar]
- Oakley AE, Clifton DK, Steiner RA. Kisspeptin signaling in the brain. Endocr. Rev. 2009;30:713–743. doi: 10.1210/er.2009-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- Odintsova E, Voortman J, Gilbert E, Berditchevski F. Tetraspanin CD82 regulates compartmentalisation and ligand-induced dimerization of EGFR. J. Cell Sci. 2003;116:4557–4566. doi: 10.1242/jcs.00793. [DOI] [PubMed] [Google Scholar]
- Oft M, Heider KH, Beug H. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr. Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- Ohshiro K, Rayala SK, Wigerup C, Pakala SB, Natha RS, Gururaj AE, et al. Acetylation-dependent oncogenic activity of metastasis-associated protein 1 co-regulator. EMBO Rep. 2010;11:691–7. doi: 10.1038/embor.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, Kanehashi K, et al. Metastasis suppressor gene KiSS1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613–617. doi: 10.1038/35079135. [DOI] [PubMed] [Google Scholar]
- Ojalvo LS, Whittaker CA, Condeelis JS, Pollard JW. Gene expression analysis of macrophages that facilitate tumor invasion supports a role for Wnt-signaling in mediating their activity in primary mammary tumors. J. Immunol. 2010;184:702–712. doi: 10.4049/jimmunol.0902360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MF, Sahai E. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis. 2009;26:273–287. doi: 10.1007/s10585-008-9174-2. [DOI] [PubMed] [Google Scholar]
- Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- Ookawa K, Sakamoto M, Hirohashi S, Yoshida Y, Sugimura T, Terada M, et al. Concordant p53 and DCC alterations and allelic losses on chromosomes 13q and 14q associated with liver metastases of colorectal carcinoma. Int. J. Cancer. 1993;53:382–387. doi: 10.1002/ijc.2910530307. [DOI] [PubMed] [Google Scholar]
- Otto E, McCord S, Tlsty TD. Increased incidence of CAD gene amplification in tumorigenic rat lines as an indicator of genomic instability of neoplastic cells. J. Biol. Chem. 1989;264:3390–3396. [PubMed] [Google Scholar]
- Ouatas T, Clare SE, Hartsough MT, De L, Steeg PS. MMTV-associated transcription factor binding sites increase nm23-H1 metastasis suppressor gene expression in human breast carcinoma cell lines. Clin. Exp. Metastasis. 2002;19:35–42. doi: 10.1023/a:1013897022827. [DOI] [PubMed] [Google Scholar]
- Ouatas T, Halverson D, Steeg PS. Dexamethasone and medroxyprogesterone acetate elevate Nm23-H1 metastasis suppressor expression in metastatic human breast carcinoma cells via glucocorticoid receptor-dependent, transcriptional and post-transcriptional mechanisms: new uses for old compounds. Clin. Cancer Res. 2003;9:3763–3772. [PubMed] [Google Scholar]
- Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131:966–979. doi: 10.1016/j.cell.2007.10.040. [DOI] [PubMed] [Google Scholar]
- Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–573. [PubMed] [Google Scholar]
- Palmieri D, Halverson DO, Ouatas T, Horak CE, Salerno M, Johnson J, et al. Medroxyprogesterone acetate elevation of Nm23-H1 metastasis suppressor expression in hormone receptor-negative breast cancer. J. Natl. Cancer Inst. 2005;97:632–642. doi: 10.1093/jnci/dji111. [DOI] [PubMed] [Google Scholar]
- Palmieri D, Horak CE, Lee JH, Halverson DO, Steeg PS. Translational approaches using metastasis suppressor genes. J. Bioenerg. Biomembr. 2006;38:151–161. doi: 10.1007/s10863-006-9039-9. [DOI] [PubMed] [Google Scholar]
- Pardali K, Moustakas A. Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim. Biophys. Acta Rev. Cancer. 2007;1775:21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Park H, Adams MA, Lachat P, Bosman F, Pang SC, Graham CH. Hypoxia induces the expression of a 43-kDa protein (PROXY-1) in normal and malignant cells. Biochem. Biophys. Res. Commun. 2000;276:321–328. doi: 10.1006/bbrc.2000.3475. [DOI] [PubMed] [Google Scholar]
- Park W, Li J, Song R, Messing J, Chen X. CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr. Biol. 2002;12:1484–1495. doi: 10.1016/s0960-9822(02)01017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Karesen R, Naume B, Synnestvedt M, Beraki E, Sauer T. The prognostic impact of occult nodal metastasis in early breast carcinoma. Breast Cancer Res. Treat. 2009;118:57–66. doi: 10.1007/s10549-009-0340-2. [DOI] [PubMed] [Google Scholar]
- Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 2009;69:9498–9506. doi: 10.1158/0008-5472.CAN-09-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli BU, Schwartz DE, Thonar EJM, Kuettner KE. Tumor invasion and host cellular matrix. Cancer Metastasis Rev. 1983;2:129–152. doi: 10.1007/BF00048966. [DOI] [PubMed] [Google Scholar]
- Phadke PA, Vaidya KS, Nash KT, Hurst DR, Welch DR. BRMS1 suppresses breast cancer experimental metastasis to multiple organs by inhibiting several steps of the metastatic process. Am. J. Pathol. 2008;172:809–817. doi: 10.2353/ajpath.2008.070772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips KK, White AE, Hicks DJ, Welch DR, Barrett JC, Wei LL, et al. Correlation between reduction of metastasis in the MDA-MB-435 model system and increased expression of the Kai-1 protein. Mol. Carcinog. 1998;21:111–120. [PubMed] [Google Scholar]
- Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp. Cell Res. 2010;316:1324–1331. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- Pinon VP, Millot G, Munier A, Vassy J, Linares-Cruz G, Capeau J, et al. Cytoskeletal association of the A and B nucleoside diphosphate kinases of interphasic but not mitotic human carcinoma cell lines: specific nuclear localization of the B subunit. Exp. Cell Res. 1999;246:355–367. doi: 10.1006/excr.1998.4318. [DOI] [PubMed] [Google Scholar]
- Piquemal D, Joulia D, Balaguer P, Basset A, Marti J, Commes T. Differential expression of the RTP/Drg1/Ndr1 gene product in proliferating and growth arrested cells. Biochim. Biophys. Acta. 1999;1450:364–373. doi: 10.1016/s0167-4889(99)00056-7. [DOI] [PubMed] [Google Scholar]
- Pitterle DM, Kim YC, Jolicoeur EM, Cao Y, O'Briant KC, Bepler G. Lung cancer and the human gene for ribonucleotide reductase subunit M1 (RRM1) Mamm. Genome. 1999;10:916–922. doi: 10.1007/s003359901114. [DOI] [PubMed] [Google Scholar]
- Poincloux R, Lizarraga F, Chavrier P. Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J. Cell Sci. 2009;122:3015–3024. doi: 10.1242/jcs.034561. [DOI] [PubMed] [Google Scholar]
- Poliseno L, Salmena L, Zhang JW, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- Pon YL, Zhou HY, Cheung ANY, Ngan HYS, Wong AST. p70 S6 kinase promotes epithelial to mesenchymal transition through snail induction in ovarian cancer cells. Cancer Res. 2008;68:6524–6532. doi: 10.1158/0008-5472.CAN-07-6302. [DOI] [PubMed] [Google Scholar]
- Poste G, Nicolson GL. Arrest and metastasis of blood-borne tumor cells are modified by fusion of plasma membrane vesicles from highly metastatic cells. Proc. Natl. Acad. Sci. USA. 1980;77:399–403. doi: 10.1073/pnas.77.1.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poste G, Lyon NC, Macander P, Porter CW, Reeve P, Bachmeyer H. Liposome-mediated transfer of integral membrane glycoproteins into the plasma membrane of cultured cells. Exp. Cell Res. 1980;129:393–408. doi: 10.1016/0014-4827(80)90508-x. [DOI] [PubMed] [Google Scholar]
- Prentice LM, Klausen C, Kalloger S, Kobel M, McKinney S, Santos JL, et al. Kisspeptin and GPR54 immunoreactivity in a cohort of 518 patients defines favourable prognosis and clear cell subtype in ovarian carcinoma. BMC Med. 2007;5:33. doi: 10.1186/1741-7015-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat. Rev. Cancer. 2009;9:285–293. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J, Huang F, Naikawadi RP, Kim KS, Said T, Lum H. Lysophosphatidylcholine impairs endothelial barrier function through the G protein-coupled receptor GPR4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;291:L91–L101. doi: 10.1152/ajplung.00508.2005. [DOI] [PubMed] [Google Scholar]
- Quintela-Fandino M, Arpaia E, Brenner D, Goh T, Yeung FA, Blaser H, et al. HUNK suppresses metastasis of basal type breast cancers by disrupting the interaction between PP2A and cofilin-1. Proc. Natl. Acad. Sci. USA. 2010;107:2622–2627. doi: 10.1073/pnas.0914492107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu CG, Nijagal A, McLaughlin J, Wang L, Witte ON. Differential proton sensitivity of related G protein-coupled receptors T cell death-associated gene 8 and G2A expressed in immune cells. Proc. Natl. Acad. Sci. USA. 2005;102:1632–1637. doi: 10.1073/pnas.0409415102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Seminara SB, Pohl CR, DiPietro MJ, Crowley WF, Plant TM. Effect of continuous intravenous administration of human metastin 45–54 on the neuroendocrine activity of the hypothalamic-pituitary-testicular axis in the adult male rhesus monkey (Macaca mulatta) Endocrinology. 2007;148:3364–3370. doi: 10.1210/en.2007-0207. [DOI] [PubMed] [Google Scholar]
- Reale MA, Reyes-Mugica M, Pierceall WE, Rubinstein MC, Hedrick L, Cohn SL, et al. Loss of DCC expression in neuroblastoma is associated with disease dissemination. Clin. Cancer Res. 1996;2:1097–1102. [PubMed] [Google Scholar]
- Reyes-Mugica M, Rieger-Christ K, Ohgaki H, Ekstrand BC, Helie M, Kleinman G, et al. Loss of DCC expression and glioma progression. Cancer Res. 1997;57:382–386. [PubMed] [Google Scholar]
- Richert MM, Vaidya KS, Mills CN, Wong D, Korz W, Hurst DR, et al. Inhibition of CXCR4 by CTCE-9908 inhibits breast cancer metastasis to lung and bone. Oncol. Rep. 2009;21:761–767. [PubMed] [Google Scholar]
- Rinker-Schaeffer CW, O'Keefe JP, Welch DR, Theodorescu D. Metastasis suppressor proteins: discovery, molecular mechanisms and clinical application. Clin. Cancer Res. 2006;12:3382–3389. doi: 10.1158/1078-0432.CCR-06-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera J, Megias D, Bravo J. Proteomics-based strategy to delineate the molecular mechanisms of the metastasis suppressor gene BRMS1. J. Proteome Res. 2007;6:4006–4018. doi: 10.1021/pr0703167. [DOI] [PubMed] [Google Scholar]
- Roa J, Aguilar E, Dieguez C, Pinilla L, Tena-Sempere M. New frontiers in kisspeptin/GPR54 physiology as fundamental gatekeepers of reproductive function. Front. Neuroendocrinol. 2008;29:48–69. doi: 10.1016/j.yfrne.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Rodrigues S, DeWever O, Bruyneel E, Rooney RJ, Gespach C. Opposing roles of netrin-1 and the dependence receptor DCC in cancer cell invasion, tumor growth and metastasis. Oncogene. 2007;26:5615–5625. doi: 10.1038/sj.onc.1210347. [DOI] [PubMed] [Google Scholar]
- Roycik MD, Fang X, Sang QXA. A fresh prospect of extracellular matrix hydrolytic enzymes and their substrates. Curr. Pharm. Des. 2009;15:1295–1308. doi: 10.2174/138161209787846676. [DOI] [PubMed] [Google Scholar]
- Rozario T, DeSimone DW. The extracellular matrix in development and morphogenesis: a dynamic view. Dev. Biol. 2010;341:126–140. doi: 10.1016/j.ydbio.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin H. The significance of biological heterogeneity. Cancer Metastasis Rev. 1990;9:1–20. doi: 10.1007/BF00047585. [DOI] [PubMed] [Google Scholar]
- Rudy W, Hoffman M, Schwartz-Albiez R, Zoller M, Keider K-H, Ponta H, et al. The two major CD44 proteins expressed on a metastatic rat tumor cell line are derived from different splice variants: each one individually suffices to confer metastatic behavior. Cancer Res. 1993;53:1262–1268. [PubMed] [Google Scholar]
- Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus-independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J. Cell Biol. 2009;185:11–19. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackstein R, Merzaban JS, Cain DW, Dagia NM, Spencer JA, Lin CP, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 2008;14:181–187. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- Sahai E. Mechanisms of cancer cell invasion. Curr. Opin. Genet. Dev. 2005;15:87–96. doi: 10.1016/j.gde.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA. 2008;105:6392–6397. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salerno M, Ouatas T, Palmieri D, Steeg PS. Inhibition of signal transduction by the nm23 metastasis suppressor: possible mechanisms. Clin. Exp. Metastasis. 2003;20:3–10. doi: 10.1023/a:1022578000022. [DOI] [PubMed] [Google Scholar]
- Samant RS, Debies MT, Shevde LA, Verderame MF, Welch DR. Identification and characterization of murine ortholog (Brms1) of breast cancer metastasis suppressor 1 (BRMS1) Int. J. Cancer. 2002;97:15–20. doi: 10.1002/ijc.1569. [DOI] [PubMed] [Google Scholar]
- Samant RS, Debies MT, Hurst DR, Moore BP, Shevde LA, Welch DR. Suppression of murine mammary carcinoma metastasis by the murine ortholog of breast cancer metastasis suppressor 1 (Brms1) Cancer Lett. 2006;235:260–265. doi: 10.1016/j.canlet.2005.04.032. [DOI] [PubMed] [Google Scholar]
- Samant RS, Clark DW, Fillmore RA, Cicek M, Metge BJ, Chandramouli KH, et al. Breast cancer metastasis suppressor 1 (BRMS1) inhibits osteopontin transcription by abrogating NF-kappaB activation. Mol. Cancer. 2007;6:6. doi: 10.1186/1476-4598-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson RD, Yang Y, Suva LJ, Kelly T. Heparan sulfate proteoglycans and heparanase—partners in osteolytic tumor growth and metastasis. Matrix Biol. 2004;23:341–352. doi: 10.1016/j.matbio.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Saunders MM, Seraj MJ, Li ZY, Zhou ZY, Winter CR, Welch DR, et al. Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Res. 2001;61:1765–1767. [PubMed] [Google Scholar]
- Schmid K, Wang XW, Haitel A, Sieghart W, Peck-Radosavljevic M, Bodingbauer M, et al. KiSS-1 overexpression as an independent prognostic marker in hepatocellular carcinoma: an immunohistochemical study. Virchows Arch. 2007;450:143–149. doi: 10.1007/s00428-006-0352-9. [DOI] [PubMed] [Google Scholar]
- Seals DF, Azucena EF, Pass I, Tesfay L, Gordon R, Woodrow M, et al. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell. 2005;7:155–165. doi: 10.1016/j.ccr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Segura MF, Hanniford D, Menendez S, Reavie L, Zou X, Alvarez-Diaz S, et al. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc. Natl. Acad. Sci. USA. 2009;106:1814–1819. doi: 10.1073/pnas.0808263106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekita N, Suzuki H, Ichikawa T, Kito H, Akakura K, Igarashi T, et al. Epigenetic regulation of the KAI1 metastasis suppressor gene in human prostate cancer cell lines. Jpn. J. Cancer Res. (GANN) 2001;92:947–951. doi: 10.1111/j.1349-7006.2001.tb01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000;60:2764–2769. [PubMed] [Google Scholar]
- Seraj MJ, Harding MA, Gildea JJ, Welch DR, Theodorescu D. The relationship of BRMS1 and RhoGDI2 gene expression to metastatic potential in lineage related human bladder cancer cell lines. Clin. Exp. Metastasis. 2001;18:519–525. doi: 10.1023/a:1011819621859. [DOI] [PubMed] [Google Scholar]
- Shaikh FM, Seales EC, Clem WC, Hennessy KM, Zhuo Y, Bellis SL. Tumor cell migration and invasion are regulated by expression of variant integrin glycoforms. Exp. Cell Res. 2008;314:2941–2950. doi: 10.1016/j.yexcr.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shengbing Z, Feng LJ, Bin W, Lingyun G, Aimin H. Expression of KiSS-1 gene and its role in invasion and metastasis of human hepatocellular carcinoma. Anat. Rec. 2009;292:1128–1134. doi: 10.1002/ar.20950. [DOI] [PubMed] [Google Scholar]
- Shevde LA, Samant RS, Goldberg SF, Sikaneta T, Alessandrini A, Donahue HJ, et al. Suppression of human melanoma metastasis by the metastasis suppressor gene, BRMS1. Exp. Cell Res. 2002;273:229–239. doi: 10.1006/excr.2001.5452. [DOI] [PubMed] [Google Scholar]
- Shevde LA, Samant RS, Paik JC, Metge BJ, Chambers AF, Casey G, et al. Osteopontin knockdown suppresses tumorigenicity of human metastatic breast carcinoma. Clin. Exp. Metastasis. 2006;23:123–133. doi: 10.1007/s10585-006-9013-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Shibagaki N, Hanada K, Yamashita H, Shimada S, Hamada H. Overexpression of CD82 on human T cells enhances LFA-1/ICAM-1-mediated cell–cell adhesion: functional association between CD82 and LFA-1 in T cell activation. Eur. J. Immunol. 1999;29:4081–4091. doi: 10.1002/(SICI)1521-4141(199912)29:12<4081::AID-IMMU4081>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Shields JD, Emmett MS, Dunn DBA, Joory KD, Sage LM, Rigby H, et al. Chemokine-mediated migration of melanoma cells towards lymphatics—a mechanism contributing to metastasis. Oncogene. 2007;26:2997–3005. doi: 10.1038/sj.onc.1210114. [DOI] [PubMed] [Google Scholar]
- Shiio Y, Rose DW, Aur R, Donohoe S, Aebersold R, Eisenman RN. Identification and characterization of SAP25, a novel component of the mSin3 corepressor complex. Mol. Cell. Biol. 2006;26:1386–1397. doi: 10.1128/MCB.26.4.1386-1397.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaki F, Takata M, Hatta N, Takehara K. Loss of expression of the metastasis suppressor gene KiSS1 during melanoma progression and its association with LOH of chromosome 6q16.3-q23. Cancer Res. 2001;61:7422–7425. [PubMed] [Google Scholar]
- Silletti S, Timar J, Honn KV, Raz A. Autocrine motility factor induces differential 12-lipoxygenase expression and activity in high- and low-metastatic K1735 melanoma cell variants. Cancer Res. 1994;54:5752–5756. [PubMed] [Google Scholar]
- Silveira AC, Hurst DR, Vaidya KS, Ayer DE, Welch DR. Over-expression of the BRMS1 family member SUDS3 does not suppress metastasis of human cancer cells. Cancer Lett. 2009;276:32–37. doi: 10.1016/j.canlet.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh LS, Berk M, Oates R, Zhao ZW, Tan HY, Jiang Y, et al. Ovarian cancer G protein-coupled receptor 1, a new metastasis suppressor gene in prostate cancer. J. Natl. Cancer Inst. 2007;99:1313–1327. doi: 10.1093/jnci/djm107. [DOI] [PubMed] [Google Scholar]
- Smith PW, Liu Y, Siefert SA, Moskaluk CA, Petroni GR, Jones DR. Breast cancer metastasis suppressor 1 (BRMS1) suppresses metastasis and correlates with improved patient survival in non-small cell lung cancer. Cancer Lett. 2009;276:196–203. doi: 10.1016/j.canlet.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KT, Martin-Brown SA, Florens L, Washburn MP, Workman JL. Deacetylase inhibitors dissociate the histone-targeting ING2 subunit from the Sin3 complex. Chem. Biol. 2010;17:65–74. doi: 10.1016/j.chembiol.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar SC, Miranti CK. Tetraspanin KAI1/CD82 suppresses invasion by inhibiting integrin-dependent crosstalk with c-Met receptor and Src kinases. Oncogene. 2006;25:2367–2378. doi: 10.1038/sj.onc.1209269. [DOI] [PubMed] [Google Scholar]
- Stark AM, Tongers K, Maass N, Mehdorn HM, Held-Feindt J. Reduced metastasis-suppressor gene mRNA-expression in breast cancer brain metastases. J. Cancer Res. Clin. Oncol. 2004;131:191–198. doi: 10.1007/s00432-004-0629-9. [DOI] [PubMed] [Google Scholar]
- Steeg PS. Metastasis suppressor genes—art. J. Natl. Cancer Inst. 2004a;96:E4. doi: 10.1093/jnci/djh107. no. E4. [DOI] [PubMed] [Google Scholar]
- Steeg PS. Perspectives on classic article: metastasis suppressor genes. J. Natl. Cancer Inst. 2004b;96:E4. doi: 10.1093/jnci/djh107. [DOI] [PubMed] [Google Scholar]
- Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat. Med. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- Steeg PS, Theodorescu D. Metastasis: a therapeutic target for cancer. Nat. Clin. Pract. Oncol. 2007;5:206–219. doi: 10.1038/ncponc1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeg PS, Bevilacqua G, Kopper L, Thorgeirsson UP, Talmadge JE, Liotta LA, et al. Evidence for a novel gene associated with low tumor metastatic potential. J. Natl. Cancer Inst. 1988;80:200–204. doi: 10.1093/jnci/80.3.200. [DOI] [PubMed] [Google Scholar]
- Steeg PS, Anderson RL, Bar-Eli M, Chambers AF, Eccles SA, Hunter K, et al. Preclinical drug development must consider the impact on metastasis. Clin. Cancer Res. 2009;15:4529–4530. doi: 10.1158/1078-0432.CCR-09-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat. Rev. Mol. Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracke ML, Krutzsch HC, Unsworth EJ, Arestad A, Cioce V, Schiffmann E, et al. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992;267:2524–2529. [PubMed] [Google Scholar]
- Stracke ML, Murata J, Aznavoorian S, Liotta LA. The role of the extracellular matrix in tumor cell metastasis. In Vivo. 1994;8:49–58. [PubMed] [Google Scholar]
- Stupack DG, Cheresh DA. Get a ligand, get a life: integrins, signaling and cell survival. J. Cell Sci. 2002;115:3729–3738. doi: 10.1242/jcs.00071. [DOI] [PubMed] [Google Scholar]
- Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, et al. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 2006;439:95–99. doi: 10.1038/nature04323. [DOI] [PubMed] [Google Scholar]
- Su B, Zheng Q, Vaughan MM, Bu Y, Gelman IH. SSeCKS metastasis-suppressing activity in MatLyLu prostate cancer cells correlates with vascular endothelial growth factor inhibition. Cancer Res. 2006;66:5599–5607. doi: 10.1158/0008-5472.CAN-05-4123. [DOI] [PubMed] [Google Scholar]
- Su B, Bu Y, Engelberg D, Gelman IH. SSeCKS/Gravin/AKAP12 inhibits cancer cell invasiveness and chemotaxis by suppressing a protein kinase C-Raf/MEK/ERK pathway. J. Biol. Chem. 2010;285:4578–4586. doi: 10.1074/jbc.M109.073494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Berditchevski F. Function of alpha3beta1-tetraspanin protein complexes in tumor cell invasion. Evidence for the role of the complexes in production of matrix metalloproteinase 2 (MMP-2) J. Cell Biol. 1999;146:1375–1389. doi: 10.1083/jcb.146.6.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki E, Ota T, Tsukuda K, Okita A, Matsuoka K, Murakami M, et al. nm23-H1 reduces in vitro cell migration and the liver metastatic potential of colon cancer cells by regulating myosin light chain phosphorylation. Int. J. Cancer. 2004;108:207–211. doi: 10.1002/ijc.11546. [DOI] [PubMed] [Google Scholar]
- Szollosi J, Horejsi V, Bene L, Angelisova P, Damjanovich S. Supramolecular complexes of MHC class I, MHC class II, CD20, and tetraspan molecules (CD53, CD81, and CD82) at the surface of a B cell line JY. J. Immunol. 1996;157:2939–2946. [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–5669. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmadge JE, Wolman SR, Fidler IJ. Evidence for the clonal origin of spontaneous metastases. Science (Washington, D.C.) 1982;217:361–363. doi: 10.1126/science.6953592. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Mullauer L, Ogiso Y, Fujita H, Moriya S, Furuuchi K, et al. Gelsolin: a candidate for suppressor of human bladder cancer. Cancer Res. 1995;55:3228–3232. [PubMed] [Google Scholar]
- Tang DG, Patrawala L, Calhoun T, Bhatia B, Choy G, Schneider-Broussard R, et al. Prostate cancer stem/progenitor cells: identification, characterization, and implications. Mol. Carcinog. 2007;46:1–14. doi: 10.1002/mc.20255. [DOI] [PubMed] [Google Scholar]
- Tannock IF. Biology and tumor growth. Hosp. Pract. (Off. Ed.) 1983;18:81–93. doi: 10.1080/21548331.1983.11702514. [DOI] [PubMed] [Google Scholar]
- Tapper J, Kettunen E, El-Rifai W, Seppala M, Andersson LC, Knuutila S. Changes in gene expression during progression of ovarian carcinoma. Cancer Genet. Cytogenet. 2001;128:1–6. doi: 10.1016/s0165-4608(01)00386-7. [DOI] [PubMed] [Google Scholar]
- Tarin D. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005;65:5996–6000. doi: 10.1158/0008-5472.CAN-05-0699. [DOI] [PubMed] [Google Scholar]
- Tarin DT, Price JE, Kettlewell MGW, Souter RG, Vass ACR, Crossley B. Mechanisms of human tumor metastasis studied in patients with peritoneovenous shunts. Cancer Res. 1984;44:3584–3592. [PubMed] [Google Scholar]
- Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang QQ, Bos PD, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–152. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J, Hickson J, Lotan T, Yamada DS, Rinker-Schaeffer C. Using metastasis suppressor proteins to dissect interactions among cancer cells and their micro-environment. Cancer Metastasis Rev. 2008a;27:67–73. doi: 10.1007/s10555-007-9106-7. [DOI] [PubMed] [Google Scholar]
- Taylor JL, Szmulewitz RZ, Lotan T, Hickson J, Griend DV, Yamada SD, et al. New paradigms for the function of JNKK1/MKK4 in controlling growth of disseminated cancer cells. Cancer Lett. 2008b;272:12–22. doi: 10.1016/j.canlet.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010;16:2927–2931. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- ten Berge D, Koole W, Fuerer C, Fish M, Eroglu E, Nusse R. Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell. 2008;3:508–518. doi: 10.1016/j.stem.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tena-Sempere M. Timeline: the role of kisspeptins in reproductive biology. Nat. Med. 2008;14:1196. doi: 10.1038/nm1108-1196. [DOI] [PubMed] [Google Scholar]
- TenKlooster JP, Evers EE, Janssen L, Machesky LM, Michiels F, Hordijk P, et al. Interaction between Tiam1 and the Arp2/3 complex links activation of Rac to actin polymerization. Biochem. J. 2006;397:39–45. doi: 10.1042/BJ20051957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennis MA, Van Scoyk MM, Freeman SV, Vandervest KM, Nemenoff RA, Winn RA. Sprouty-4 inhibits transformed cell growth, migration and invasion, and epithelial-mesenchymal transition, and is regulated by Wnt7A through PPAR(Y) in non-small cell lung cancer. Mol. Cancer Res. 2010;8:833–843. doi: 10.1158/1541-7786.MCR-09-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki-Fukuzawa Y, Kijima H, Suto A, Takeshita T, Iezumi K, Sato S, et al. Decreased nm23 expression, but not Ki-67 labeling index, is significantly correlated with lymph node metastasis of breast invasive ductal carcinoma. Int. J. Mol. Med. 2002;9:25–29. [PubMed] [Google Scholar]
- Theodorescu D, Sapinoso LM, Conaway MR, Oxford G, Hampton GM, Frierson HF., Jr. Reduced expression of metastasis suppressor RhoGDI2 is associated with decreased survival for patients with bladder cancer. Clin. Cancer Res. 2004;10:3800–3806. doi: 10.1158/1078-0432.CCR-03-0653. [DOI] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- Thompson EW, Newgreen DF. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res. 2005;65:5991–5995. doi: 10.1158/0008-5472.CAN-05-0616. [DOI] [PubMed] [Google Scholar]
- Thorgeirsson UP, Turpeenniemi-Hujanen T, Williams JE, Westin EH, Heilman CA, Talmadge JE, et al. NIH/3T3 cells transfected with human tumor DNA containing activated ras oncogenes express the metastatic phenotype in nude mice. Mol. Cell. Biol. 1985;5:259–262. doi: 10.1128/mcb.5.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpl R. Proteoglycans of basement membranes. Experientia. 1993;49:417–428. doi: 10.1007/BF01923586. [DOI] [PubMed] [Google Scholar]
- Timpl R, Aumailley M. Biochemistry of basement membranes. Adv. Nephrol. 1989;18:59–76. [PubMed] [Google Scholar]
- Tlsty TD. Normal diploid human and rodent cells lack a detectable frequency of gene amplification. Proc. Natl. Acad. Sci. USA. 1990;87:3132–3136. doi: 10.1073/pnas.87.8.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlsty TD, Margolin BH, Lum K. Differences in the rates of gene amplification in nontumorigenic and tumorigenic cell lines as measured by Luria-Delbruck fluctuation analysis. Proc. Natl. Acad. Sci. USA. 1989;86:9441–9445. doi: 10.1073/pnas.86.23.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh Y, Pencil SD, Nicolson GL. A novel candidate metastasis-associated gene, mta1, differentially expressed in highly metastatic mammary adenocarcinoma cell lines. cDNA cloning, expression, and protein analyses. J. Biol. Chem. 1994;269:22958–22963. [PubMed] [Google Scholar]
- Toh Y, Ohga T, Endo K, Adachi E, Kusumoto H, Haraguchi M, et al. Expression of the metastasis-associated MTA1 protein and its relationship to deacetylation of the histone H4 in esophageal squamous cell carcinomas. Int. J. Cancer. 2004;110:362–367. doi: 10.1002/ijc.20154. [DOI] [PubMed] [Google Scholar]
- Tosa N, Murakami M, Jia WY, Yokoyama M, Masunaga T, Iwabuchi C, et al. Critical function of T cell death-associated gene 8 in glucocorticoid-induced thymocyte apoptosis. Int. Immunol. 2003;15:741–749. doi: 10.1093/intimm/dxg070. [DOI] [PubMed] [Google Scholar]
- Trakul N, Menard RE, Schade GR, Qian Z, Rosner MR. Raf kinase inhibitory protein regulates Raf-1 but not B-Raf kinase activation. J. Biol. Chem. 2005;280:24931–24940. doi: 10.1074/jbc.M413929200. [DOI] [PubMed] [Google Scholar]
- Tran KT, Griffith L, Wells A. Extracellular matrix signaling through growth factor receptors during wound healing. Wound Repair Regen. 2004;12:262–268. doi: 10.1111/j.1067-1927.2004.012302.x. [DOI] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science (Washington, D.C.) 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng YH, Vicent D, Zhu JH, Niu YL, Adeyinka A, Moyers JS, et al. Regulation of growth and tumorigenicity of breast cancer cells by the low molecular weight GTPase Rad and Nm23. Cancer Res. 2001;61:2071–2079. [PubMed] [Google Scholar]
- Tsuji T, Ibaragi S, Shima K, Hu MG, Katsurano M, Sasaki A, et al. Epithelial-mesenchymal transition induced by growth suppressor p12(CDK2-AP1) promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res. 2008;68:10377–10386. doi: 10.1158/0008-5472.CAN-08-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turpeenniemi-Hujanen T, Thorgeirsson UP, Hart IR, Grant SS, Liotta LA. Expression of collagenase IV (basement membrane collagenase) activity in murine tumor cell hybrids that differ in metastatic potential. J. Natl. Cancer Inst. 1985;75:99–103. [PubMed] [Google Scholar]
- Uhlenbrock K, Eberth A, Herbrand U, Daryab N, Stege P, Meier F, et al. The RacGEF Tiam1 inhibits migration and invasion of metastatic melanoma via a novel adhesive mechanism. J. Cell Sci. 2004;117:4863–4871. doi: 10.1242/jcs.01367. [DOI] [PubMed] [Google Scholar]
- Ulrix W, Swinnen JV, Heyns W, Verhoeven G. The differentiation-related gene 1, Drg1, is markedly upregulated by androgens in LNCaP prostatic adenocarcinoma cells. FEBS Lett. 1999;455:23–26. doi: 10.1016/s0014-5793(99)00845-5. [DOI] [PubMed] [Google Scholar]
- Underhill C. CD44: the hyaluronan receptor. J. Cell Sci. 1992;103(Pt 2):293–298. doi: 10.1242/jcs.103.2.293. [DOI] [PubMed] [Google Scholar]
- Vaidya KS, Welch DR. Metastasis suppressors and their roles in breast carcinoma. J. Mammary Gland Biol. Neoplasia. 2007;12:175–190. doi: 10.1007/s10911-007-9049-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya KS, Harihar S, Stafford LJ, Hurst DR, Hicks DG, Casey G, et al. Breast cancer metastasis suppressor-1 differentially modulates growth factor signaling. J. Biol. Chem. 2008;283:28354–28360. doi: 10.1074/jbc.M710068200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valastyan S, Benaich N, Chang A, Reinhardt F, Weinberg RA. Concomitant suppression of three target genes can explain the impact of a microRNA on metastasis. Genes Dev. 2009;23:2592–2597. doi: 10.1101/gad.1832709. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Valastyan S, Chang A, Benaich N, Reinhardt F, Weinberg RA. Concurrent suppression of integrin alpha5, radixin, and RhoA phenocopies the effects of miR-31 on metastasis. Cancer Res. 2010;70:5147–5154. doi: 10.1158/0008-5472.CAN-10-0410. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- van Belzen N, Dinjens WN, Diesveld MP, Groen NA, van der Made AC, Nozawa Y, et al. A novel gene which is up-regulated during colon epithelial cell differentiation and down-regulated in colorectal neoplasms. Lab. Investig. 1997;77:85–92. [PubMed] [Google Scholar]
- Van Netten JP, Ashmead BJ, Parker RL, Thornton IG, Fletcher C, Cavers D, et al. Macrophage-tumor cell associations: a factor in metastasis of breast cancer? J. Leukoc. Biol. 1993;54:360–362. doi: 10.1002/jlb.54.4.360. [DOI] [PubMed] [Google Scholar]
- Van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell. Mol. Life Sci. 2008;65:3756–3788. doi: 10.1007/s00018-008-8281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Griend DJ, Kocherginsky M, Hickson JA, Stadler WM, Lin AN, Rinker-Schaeffer CW. Suppression of metastatic colonization by the context-dependent activation of the c-jun NH2-terminal kinase kinases JNKK1/MKK4 and MKK7. Cancer Res. 2005;65:10984–10991. doi: 10.1158/0008-5472.CAN-05-2382. [DOI] [PubMed] [Google Scholar]
- Virchow R. Cellular pathologie. Nutr. Rev. 1858;47:23–25. doi: 10.1111/j.1753-4887.1989.tb02747.x. [DOI] [PubMed] [Google Scholar]
- Vlodavsky I, Korner G, Ishai-Michaeli R, Baskhin P, Bar-Shavit R, Fuks Z. Extracellular matrix-resident growth factors and enzymes: possible involvement in tumor metastasis and angiogenesis. Cancer Metastasis Rev. 1990;9:203–226. doi: 10.1007/BF00046361. [DOI] [PubMed] [Google Scholar]
- Vlodavsky I, Goldshmidt O, Zcharia E, Atzmon R, Rangini-Guatta Z, Elkin M, et al. Mammalian heparanase: involvement in cancer metastasis, angiogenesis and normal development. Semin. Cancer Biol. 2002;12:121–129. doi: 10.1006/scbi.2001.0420. [DOI] [PubMed] [Google Scholar]
- Wakefield LM, Stuelten C. Keeping order in the neighborhood: new roles for TGFP in maintaining epithelial homeostasis. Cancer Cell. 2007;12:293–295. doi: 10.1016/j.ccr.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Wang YF, Chow KC, Chang SY, Chiu JH, Tai SK, Li WY, et al. Prognostic significance of nm23-HI expression in oral squamous cell carcinoma. Br. J. Cancer. 2004;90:2186–2193. doi: 10.1038/sj.bjc.6601808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882–887. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- Wang XY, Wang MS, MacLennan GT, Abdul-Karim FW, Eble JN, Jones TD, et al. Evidence for common clonal origin of multifocal lung cancers. J. Natl. Cancer Inst. 2009a;101:560–570. doi: 10.1093/jnci/djp054. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang H, Chen YP, Sun YM, Yang F, Yu WH, et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009b;138:660–672. doi: 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin. Exp. Metastasis. 2006;23:97–105. doi: 10.1007/s10585-006-9014-1. [DOI] [PubMed] [Google Scholar]
- Wei LL, Yang X, Phillips KK, Weissman BE, Welch DR. Analysis of KAI-1 mRNA expression in human breast cancer cell lines. Proc. Am. Assoc. Cancer Res. 1996;37:529. [Google Scholar]
- Weiss L. Dynamic aspects of cancer cell populations in metastasis. Am. J. Pathol. 1979;97:601–608. [PMC free article] [PubMed] [Google Scholar]
- Weiss L. Biomechanical destruction of cancer cells in metastasis. Clin. Exp. Metastasis. 1989;7:483–491. doi: 10.1007/BF01753809. [DOI] [PubMed] [Google Scholar]
- Weiss L. Metastatic inefficiency. Adv. Cancer Res. 1990;54:159–211. doi: 10.1016/s0065-230x(08)60811-8. [DOI] [PubMed] [Google Scholar]
- Weiss L. Comments on hematogenous metastatic patterns in humans as revealed by autopsy. Clin. Exp. Metastasis. 1992;10:191–199. doi: 10.1007/BF00132751. [DOI] [PubMed] [Google Scholar]
- Weiss L, Schmid-Schonbein GW. Biomechanical interactions of cancer cells with the microvasculature during metastasis. Cell Biophys. 1989;14:187–215. doi: 10.1007/BF02797133. [DOI] [PubMed] [Google Scholar]
- Weiss L, Ward PM. Arrest and retention of circulating cancer cells in the lungs of animals with defined metastatic status. Cancer Res. 1982;42:1898–1903. [PubMed] [Google Scholar]
- Weiss L, Dimitrov DS, Angelova M. The hemodynamic destruction of intravascular cancer cells in relation to myocardial metastasis. Proc. Natl. Acad. Sci. USA. 1985;82:5737–5741. doi: 10.1073/pnas.82.17.5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch DR. Factors involved in the development and maintenance of tumor heterogeneity. In: Abraham S, editor. Carcinogenesis and Dietary Fat. Kluwer Academic Publishers; Boston: 1989. pp. 279–301. [Google Scholar]
- Welch DR. Technical considerations for studying cancer metastasis in vivo. Clin. Exp. Metastasis. 1997;15:272–306. doi: 10.1023/a:1018477516367. [DOI] [PubMed] [Google Scholar]
- Welch DR. Defining a cancer metastasis. In: American Association for Cancer Research, editor. AACR Education Book 2006. AACR; Philadelphia: 2006. pp. 111–115. [Google Scholar]
- Welch DR. Do we need to redefine a cancer metastasis and staging definitions? Breast Dis. 2007;26:3–12. doi: 10.3233/bd-2007-26102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch DR, Tomasovic SP. Implications of tumor progression on clinical oncology. Clin. Exp. Metastasis. 1985;3:151–188. doi: 10.1007/BF01786761. [DOI] [PubMed] [Google Scholar]
- Welch DR, Aeed PA, Earhart RH, McClure SA. Evidence for paracrine regulation of experimental metastasis in 13762NF rat mammary adenocarcinoma cell clones. Anticancer Res. 1994a;14:1743–1752. [PubMed] [Google Scholar]
- Welch DR, Chen P, Miele ME, McGary CT, Bower JM, Weissman BE, et al. Microcell-mediated transfer of chromosome 6 into metastatic human C8161 melanoma cells suppressesmetastasis but does not inhibit tumorigenicity. Oncogene. 1994b;9:255–262. [PubMed] [Google Scholar]
- Welch DR, Fabra A, Nakajima M. Transforming growth factor beta stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proceedings of the National Academy of Science (USA) 1990;87:7678–7682. doi: 10.1073/pnas.87.19.7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch DR, Sakamaki T, Pioquinto R, Leonard TO, Goldberg SF, Hon Q, et al. Transfection of constitutively active Mek1 confers tumorigenic and metastatic potentials to NIH3T3 cells. Cancer Res. 2000;60:1552–1556. [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- Wertheim GBW, Yang TW, Pan TC, Ramne A, Liu ZD, Gardner HP, et al. The Snf1-related kinase, Hunk, is essential for mammary tumor metastasis. Proc. Natl. Acad. Sci. USA. 2009;106:15855–15860. doi: 10.1073/pnas.0906993106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- Winter SF, Hunter KW. Mouse modifier genes in mammary tumorigenesis and metastasis. J. Mammary Gland Biol. Neoplasia. 2008;13:337–342. doi: 10.1007/s10911-008-9089-1. [DOI] [PubMed] [Google Scholar]
- Witz IP, Levy-Nissenbaum O. The tumor microenvironment in the post-PAGET era. Cancer Lett. 2006;242:1–10. doi: 10.1016/j.canlet.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Wolf K, Friedl P. Molecular mechanisms of cancer cell invasion and plasticity. Br. J. Dermatol. 2006;154:11–15. doi: 10.1111/j.1365-2133.2006.07231.x. [DOI] [PubMed] [Google Scholar]
- Wong CW, Lee A, Shientag L, Yu J, Dong Y, Kao G, et al. Apoptosis: an early event in metastatic inefficiency. Cancer Res. 2001;61:333–338. [PubMed] [Google Scholar]
- Wu K, Bonavida B. The activated NF-kappa B-Snail-RKIP circuitry in cancer regulates both the metastatic cascade and resistance to apoptosis by cytotoxic drugs. Crit. Rev. Immunol. 2009;29:241–254. doi: 10.1615/critrevimmunol.v29.i3.40. [DOI] [PubMed] [Google Scholar]
- Wu YM, Moissogiu K, Wang H, Wang XJ, Frierson HF, Schwartz MA, et al. Src phosphorylation of RhoGDI2 regulates its metastasis suppressor function. Proc. Natl. Acad. Sci. USA. 2009;106:5807–5812. doi: 10.1073/pnas.0810094106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS, Sahai E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr. Biol. 2006;16:1515–1523. doi: 10.1016/j.cub.2006.05.065. [DOI] [PubMed] [Google Scholar]
- Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- Xia W, Gelman IH. Mitogen-induced, FAK-dependent tyrosine phosphorylation of the SSeCKS scaffolding protein. Exp. Cell Res. 2002;277:139–151. doi: 10.1006/excr.2002.5560. [DOI] [PubMed] [Google Scholar]
- Xia W, Unger P, Miller L, Nelson J, Gelman IH. The Src-suppressed C kinase substrate, SSeCKS, is a potential metastasis inhibitor in prostate cancer. Cancer Res. 2001;61:5644–5651. [PubMed] [Google Scholar]
- Xin W, Yun KJ, Ricci F, Zahurak M, Qiu W, Su GH, et al. MAP2K4/ MKK4 expression in pancreatic cancer: genetic validation of immunohistochemistry and relationship to disease course. Clin. Cancer Res. 2004;10:8516–8520. doi: 10.1158/1078-0432.CCR-04-0885. [DOI] [PubMed] [Google Scholar]
- Xu Y. Sphingosylphosphorylcholine and lysophosphatidylcholine: G protein-coupled receptors and receptor-mediated signal transduction. Biochim. Biophys. Acta. 2002;1582:81–88. doi: 10.1016/s1388-1981(02)00140-3. [DOI] [PubMed] [Google Scholar]
- Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, Iacobuzio-Donahue CA. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada SD, Hickson JA, Hrobowski Y, Vander Griend DJ, Benson D, Montag A, et al. Mitogen-activated protein kinase kinase 4 (MKK4) acts as a metastasis suppressor gene in human ovarian carcinoma. Cancer Res. 2002;62:6717–6723. [PubMed] [Google Scholar]
- Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J. Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N, Yang M, Jiang P, Xu MX, Tsuchiya H, Tomita K, et al. Determination of clonality of metastasis by cell-specific color-coded fluorescent-protein imaging. Cancer Res. 2003;63:7785–7790. [PubMed] [Google Scholar]
- Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, Chytil A, et al. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010;70:6139–6149. doi: 10.1158/0008-5472.CAN-10-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XH, Welch DR, Phillips KK, Weissman BE, Wei LL. KAI1, a putative marker for metastatic potential in human breast cancer. Cancer Lett. 1997;119:149–155. doi: 10.1016/s0304-3835(97)00273-5. [DOI] [PubMed] [Google Scholar]
- Yang X, Wei L, Tang C, Slack R, Montgomery E, Lippman M. KAI1 protein is down-regulated during the progression of human breast cancer. Clin. Cancer Res. 2000;6:3424–3429. [PubMed] [Google Scholar]
- Yang J, Zhang B, Lin Y, Yang Y, Liu X, Lu F. Breast cancer metastasis suppressor 1 inhibits SDF-1alpha-induced migration of non-small cell lung cancer by decreasing CXCR4 expression. Cancer Lett. 2008;269:46–56. doi: 10.1016/j.canlet.2008.04.016. [DOI] [PubMed] [Google Scholar]
- Yang JB, Price MA, Li GY, Bar-Eli M, Salgia R, Jagedeeswaran R, et al. Melanoma proteoglycan modifies gene expression to stimulate tumor cell motility, growth, and epithelial-to-mesenchymal transition. Cancer Res. 2009;69:7538–7547. doi: 10.1158/0008-5472.CAN-08-4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol. Cancer Res. 2010;8:629–642. doi: 10.1158/1541-7786.MCR-10-0139. [DOI] [PubMed] [Google Scholar]
- You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc. Natl. Acad. Sci. USA. 2001;98:1454–1458. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MR, Newby M. Differential induction of suppressor macrophages by cloned Lewis lung carcinoma variants in mice. J. Natl. Cancer Inst. 1986;77:1255–1260. [PubMed] [Google Scholar]
- Yuan BZ, Miller MJ, Keck CL, Zimonjic DB, Thorgeirsson SS, Popescu NC. Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res. 1998;58:2196–2199. [PubMed] [Google Scholar]
- Yuan BZ, Zhou X, Durkin ME, Zimonjic DB, Gumundsdottir K, Eyfjord JE, et al. DLC-1 gene inhibits human breast cancer cell growth and in vivo tumorigenicity. Oncogene. 2003;22:445–450. doi: 10.1038/sj.onc.1206064. [DOI] [PubMed] [Google Scholar]
- Yuan BZ, Jefferson AM, Baldwin KT, Thorgeirsson SS, Popescu NC, Reynolds SH. DLC-1 operates as a tumor suppressor gene in human non-small cell lung carcinomas. Oncogene. 2004;23:1405–1411. doi: 10.1038/sj.onc.1207291. [DOI] [PubMed] [Google Scholar]
- Zhang B. Rho GDP dissociation inhibitors as potential targets for anticancer treatment. Drug Resist. Updat. 2006;9:134–141. doi: 10.1016/j.drup.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Zhang XA, Lane WS, Charrin S, Rubinstein E, Liu L. EWI2/PGRL associates with the metastasis suppressor KAI1/CD82 and inhibits the migration of prostate cancer cells. Cancer Res. 2003;63:2665–2674. [PubMed] [Google Scholar]
- Zhang BL, Zhang YQ, Dagher MC, Shacter E. Rho GDP dissociation inhibitor protects cancer cells against drug-induced apoptosis. Cancer Res. 2005;65:6054–6062. doi: 10.1158/0008-5472.CAN-05-0175. [DOI] [PubMed] [Google Scholar]
- Zhang S, Lin QD, Di W. Suppression of human ovarian carcinoma metastasis by the metastasis-suppressor gene, BRMS1. Int. J. Gynecol. Cancer. 2006;16:522–531. doi: 10.1111/j.1525-1438.2006.00547.x. [DOI] [PubMed] [Google Scholar]
- Zhang T, Zheng J, Liu C, Lu Y. Expression of DLC-1 in clear cell renal cell carcinoma: prognostic significance for progression and metastasis. Urol. Int. 2009a;82:380–387. doi: 10.1159/000218524. [DOI] [PubMed] [Google Scholar]
- Zhang XY, Liu SR, Hu TS, Liu SP, He Y, Sun SH. Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology. 2009b;50:490–499. doi: 10.1002/hep.23008. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Rivera Rosado LA, Moon SY, Zhang B. Silencing of D4-GDI inhibits growth and invasive behavior in MDA-MB-231 cells by activation of Racdependent p38 and JNK signaling. J. Biol. Chem. 2009c;284:12956–12965. doi: 10.1074/jbc.M807845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- Zhou B, Liu L, Reddivari M, Zhang XA. The palmitoylation of metastasis suppressor KAI1/CD82 is important for its motility- and invasiveness-inhibitory activity. Cancer Res. 2004a;64:7455–7463. doi: 10.1158/0008-5472.CAN-04-1574. [DOI] [PubMed] [Google Scholar]
- Zhou X, Thorgeirsson SS, Popescu NC. Restoration of DLC-1 gene expression induces apoptosis and inhibits both cell growth and tumorigenicity in human hepatocellular carcinoma cells. Oncogene. 2004b;23:1308–1313. doi: 10.1038/sj.onc.1207246. [DOI] [PubMed] [Google Scholar]
- Zhu JH, Tseng YH, Kantor JD, Rhodes CJ, Zetter BR, Moyers JS, et al. Interaction of the Ras-related protein associated with diabetes Rad and the putative tumor metastasis suppressor NM23 provides a novel mechanism of GTPase regulation. Proc. Natl. Acad. Sci. USA. 1999;96:14911–14918. doi: 10.1073/pnas.96.26.14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu SM, Wu HL, Wu FT, Nie DT, Sheng SJ, Mo YY. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008;18:350–359. doi: 10.1038/cr.2008.24. [DOI] [PubMed] [Google Scholar]