

Abstract
Artemisinin-based combination therapy is exerting novel selective pressure upon populations of Plasmodium falciparum across Africa. Levels of resistance to non-artemisinin partner drugs differ among parasite populations, and so the artemisinins are not uniformly protected from developing resistance, already present in South East Asia. Here, we consider strategies for prolonging the period of high level efficacy of combination therapy for two particular endemicities common in Africa. Under high intensity transmission, two alternating first-line combinations, ideally with antagonistic selective effects on the parasite genome, are advocated for paediatric malaria cases. This leaves second-line and other therapies for adult cases, and for intermittent preventive therapy. The drug portfolio would be selected to protect the “premier” combination regimen from selection for resistance, while maximising impact on severe disease and mortality in children. In endemic areas subject to low, seasonal transmission of Plasmodium falciparum, such a strategy may deliver little benefit, as children represent a minority of cases. Nevertheless, the deployment of other drug-based interventions in low transmission and highly seasonal areas, such as mass drug administration aimed to interrupt malaria transmission, or intermittent preventive therapy, does provide an opportunity to diversify drug pressure. We thus propose an integrated approach to drug deployment, which minimises direct selective pressure on parasite populations from any one drug component. This approach is suitable for qualitatively and quantitatively different burdens of malaria, and should be supported by a programme of routine surveillance for emerging resistance.
INTRODUCTION
The efficacy of antimalarial drugs used for the treatment of uncomplicated cases of falciparum malaria declined sharply in the last decade of the twentieth century in sub-Saharan Africa; this had already been observed in malaria endemic areas in Asia in the 1980s. The current decade is seeing the continued widespread deployment of artemisinin-based combination therapy (ACT) as first-line therapies to replace those drugs that are judged to have reached an unacceptable level of treatment failure. The number of ACT regimens with proven efficacy and safety is small, and currently only artemether-lumefantrine (AL), sulphadoxine-pyrimethamine plus artesunate (SPAS) and amodiaquine plus artesunate (AQAS) are widely used in Africa. Other combinations in an advanced stage of development and which may soon be routinely deployed in some countries include dihydroartemisinin / piperaquine (DP) and pyronaridine/artesunate (PYAS). Whereas some of these partner drugs have not been used previously in Africa, both amodiaquine and the anti-folates have been widely used, and lumefantrine is related to other previously used drugs such as halofantrine. Thus some ACT regimens may be deployed in areas where the parasite population has had some previous exposure to one or more of the partner drugs (or chemically-related compounds), and may be expected to have some underlying tolerance of, or limited resistance to, the combination. Careful consideration is required to ensure that malaria treatment policy, in striving to deliver the best clinical outcome for the greatest majority of treated individuals, does not generate a level of selection that causes rapid spread of resistance and erosion of therapeutic efficacy.
It is important to clearly define our intended meaning for the terms efficacy and strength of selection for the argument to follow, as these are not congruent. Efficacy of a single agent or combination regimen is intended to mean the operationally measurable outcome of in vivo efficacy studies: the absence of clinical or parasitological (microscopically detected) recurrence during 28 days of follow-up in a cohort of treated patients taking observed treatment doses. Strength of selection exerted by a regimen is not usually directly measurable, but here will signify the relative transmission advantage of a parasite genotype harbouring a mutation or mutations conferring reduced sensitivity (or resistance) to the regimen of interest, compared to genotypes lacking that mutation or mutations. In some studies this can be estimated from single time-point measures (e.g. day 7 or day 14 post-treatment) of genotype prevalence in infected Anopheles mosquitoes fed on blood from treated, infected individuals (Bousema et al. 2006; Hallett et al. 2006). This approach does not fully capture the transmissibility of different genotypes over extended time periods following treatment. The magnitude of selection could be better envisaged as the difference between the proportion of all post-treatment transmission events with non-wild-type genotypes (at loci implicated in resistance to the regimen used) and the proportion of such genotypes in the pre-treatment population. As Hallett et al. (2006) and Mendez et al. (2007) have shown, selection for transmission of advantageous genotypes can occur even when drug efficacy appears good.
Recent studies in Cambodia demonstrate a measurable reduction in the sensitivity of P. falciparum to artemisinin monotherapy in some areas, although treatment responses for ACTs remain satisfactory (Noedl et al. 2008; Dondorp et al. 2009). These observations suggest both an urgent need to develop strategies to protect ACT worldwide from the effects of possible spread of such parasites, and the necessity of implementing suitable surveillance of ACT efficacy throughout malaria endemic regions. In sub-Saharan Africa, where artemisinin monotherapy trials have not been performed, and where efficacy of ACT remains good or very good, protection of the efficacy of drug combinations now in use must be the first priority, and thus the opportunity for artemisinin-resistant P. falciparum to enter, or arise in, Africa must be minimised. The importance of this task is further emphasised by the recent demonstration that use of parenteral artesunate in place of quinine is able to reduce the death rate among hospitalised African children with severe malaria (Dondorp et al. 2010). Among the possible strategies to protect current regimens from the development of parasite resistance are lengthening of the duration of ACT treatment and/or increasing the total dosage given, or using additional partner drugs with existing dual-component combinations, essentially to maintain effective parasiticidal blood levels of as many component drugs as possible for as long as possible. Early in its development, a newly arising form of resistant parasites is likely to remain sensitive to high blood concentrations of the drug, but enjoy an advantage once drug levels fall, at concentrations still lethal to the wild-type. In fact, even fully developed supposedly CQ-resistant genotypes in Africa have been shown to be sensitive to unorthodox CQ regimens that distribute higher total drug dosage using more frequent individual doses (Ursing et al. 2007, 2011). Regarding the former, extension of AL regimens from 4 days to 6 days in The Gambia reduced the rate of recurrent parasitaemia, and was associated with a demonstrable benefit in reduced gametocyte carriage (Sutherland et al. 2005). In Guinea-Bissau, higher doses of CQ given for longer (5 – 7 days) have been delivered safely by the use of split-doses. This regimen was associated with continued efficacy against P. falciparum carrying parasites with known CQ-resistant genotypes at the pfcrt and pfmdr1 loci, and a reduced selective advantage of these mutant genotypes. This appears to have contributed to a significant extension of the effective life of CQ in that country (Ursing et al. 2007, 2011). Similarly, increasing and extending mefloquine (MQ) dosage as part of the MQAS combination stopped the decline in efficacy of the combination that had been evident when lower doses were used in Thailand (Carrara et al. 2009). This may not necessarily mean that the selective advantage of resistant genotypes (in this case copy number amplification of the pfmdr1 locus) has been neutralised; it is important to emphasise that even where drug efficacy is acceptable, selection for advantageous genotypes will still occur (Hallet et al. 2006). As for implementing artemisinin-based regimens with two or more partner drugs, this option must at least be considered as a plausible strategy, by analogy with chemotherapeutics against other infections such as tuberculosis and HIV. However we will not focus on either of these strategies in the current paper.
An alternative way of protecting ACT is to devise an innovative approach to antimalarial drug deployment across endemic country health systems, with the intention of generating diverse, “polyvalent” drug pressure. This would be designed to prevent (or slow) the emergence of parasite genotypes with a survival advantage under the major treatment regime. As the pattern of resistance to partner drugs may vary across different endemic settings, so will the level of direct selection pressure on parasite populations that is exerted by the artemisinin components themselves. Such selection pressure will be strongest where the efficacy of the partner drugs offer less protection for the artemisinin; it follows that protection of partner drug efficacy is an important component of any strategy to sustain the useful life of ACT. Therefore, interventions can be planned in an integrated way within countries, and even within regional groupings of different nations, so as to maximise partner drug efficacy, and so minimise additional selection acting directly on the artemisinin component of ACT. Such rational, integrated planning might use not only the principal strategy of partner drug diversification, but also where possible the deployment of non-artemisinin regimens for intermittent preventive therapy and treatment of clinical malaria cases in semi-immune individuals.
The primary objective of such integration is to reduce the selection for parasites resistant to the first-line combination(s) in order to prolong the useful life of the artemisinin components. In this paper we propose that, in high transmission settings, rational deployment requires preservation of the front-line ACT treatment for children, who are at the most risk of severe, life-threatening malaria. Adult malaria cases can then be treated with either an alternative ACT or a non-ACT combination, and drug-based preventive interventions such as IPTp and IPTi could be restricted to alternative non-artemisinin combination regimens. Policies in each country would be coordinated with, and deliberately complementary to, the policies of other countries in the region, wherever possible. Secondly, in low transmission settings, with Eastern Sudan considered as a specific example, we argue that paediatric targeting would be of little benefit. Nevertheless, the interruption of transmission by an extended dry season each year offers particular opportunities for preventative use of antimalarial drugs. Finally, we explore the options for surveillance and monitoring suitable for African settings, and recommend a newly developed approach to detect early signs of emerging resistance to ACT-based antimalarial therapies in the field under different transmission settings.
Integrated Use of Antimalarial Drugs in Malaria Control
A National Malaria Control Programme manages a range of activities aimed at minimising malaria-related morbidity and mortality, and at reducing malaria transmission. Here, we will consider only those activities that employ antimalarial drugs. Undoubtedly the interaction between vector control or personal protection (e.g. impregnated bednets) and antimalarial-based strategies will have an important bearing on the possible emergence of ACT-resistant parasites, but is beyond the scope of the current discussion. These activities can be listed thus:
Chemotherapy for confirmed clinical malaria cases in children under 10 years
Chemotherapy for confirmed clinical malaria cases in people over 10 years
IPTi for protection of infants, linked to standard EPI visits
IPTc for protection of children aged 2 – 5 years (high transmission seasonal settings only) or IPTc for school-aged children
IPTp for pregnant women
Each of these 5 uses of drugs may put infecting malaria parasites under variable levels of selection pressure. This is due to :
- different amounts of transmission from one recipient group to the others,
- acquired immunity being more common in older groups and
- IPT being administered to people with lower overall parasite burdens than those presenting with clinical malaria.
Where drug is the principal effector of parasite clearance, as in young non-immune children, we would expect intense directional selection for resistant parasites following treatment. However, in semi-immune individuals parasite clearance is a collaboration between direct drug effect and immunity (Diallo et al. 2007) and thus the intensity of selection on resistance-associated loci will be reduced. The magnitude of this reduction will depend both on the prevalence of effective immunity in that treated group, level of drug coverage and the degree of advantage afforded by the parasite resistance mechanism. Further, survival of a (small) proportion of the parasite population is more likely with higher parasite densities, as evidenced by the relationship between parasite clearance time and starting parasitaemia in vivo (Dondorp et al. 2009; Beshir et al. 2010a). Therefore, it seems likely that selection intensity will be greater with drug use against clinical malaria episodes, in non-immune adults and children, than with drug use for prophylaxis, or for suppression of pre-existing, low density asymptomatic infection, as in semi-immune individuals. Although more work is required to determine whether this is the case, Dunyo et al. (2006), in a large cross-sectional survey, found that asymptomatic infections not only had lower asexual parasite densities, but lower gametocyte prevalence. Further, when these individuals were treated and followed for three months, the area under the gametocyte carriage curve was significantly smaller than among individuals initially with clinical (high parasite density) malaria treated with the same regimen, suggesting reduced asexual parasite survival among the former. (Nevertheless, asymptomatic individuals may represent a large proportion of infections in high transmission settings and so may constitute a significant source of drug-exposed parasites in some circumstances.) With these arguments in mind, we have therefore presented activities 1 to 5 in the order of expected decreasing intensity of drug pressure and thus decreasing potential for selection and transmission of drug resistance genotypes.
Some antimalarial drugs exert opposite directional selection on parasite genotypes
After taking into account the hierarchy of selection intensity discussed in the preceding paragraph, the framework we wish to propose is then under-pinned by already existing (and some yet to be gathered) knowledge of degrees of drug sensitivity afforded by different parasite genotypes at key loci known to influence treatment responses in vivo and in vitro. Table 1 lists existing evidence from African settings on directional selection exerted by antimalarial drugs of interest on two of the loci implicated in parasite response to treatment, pfmdr1 and pfcrt. Selective effects of piperaquine and pyronaridine in vivo remain unclear, although a recent report using a previously untested methodology found evidence of an association between carriage of pfmdr1 encoding a tyrosine at codon 1246 and treatment failure with DP in malaria patients in Papua New Guinea (Wong et al. 2011). Thus additional knowledge is needed. A particularly interesting feature is the directionally opposed selection exerted by amodiaquine and artemisinin / lumefantrine on both pfmdr1 and pfcrt (Humphreys et al. 2007; Sisowath et al. 2009). Thus integrated use of amodiaquine-containing combinations and AL against the same parasite population is predicted to provide a reduction of selective effect for both regimens. In east Africa, amodiaquine has reduced efficacy due to the relatively high prevalence of the pfmdr1 YYY haplotype, and the pfcrt IETSE haplotype (Table 1; Holmgren et al. 2007; Humphreys et al. 2007), and thus this strategy is very likely to be compromised. However in west Africa, amodiaquine – based regimens retain good efficacy (Ursing et al. 2007; Sirima et al. 2009) and a “longitudinal paired combinations” strategy utilising AL and ASAQ, as described below, could be deployed there.
TABLE 1.
Known selective effects of antimalarial drugs on resistance loci of P. falciparum of African origind.
| DRUG | Locus | pfmdr1 | pfcrt |
Other
loci |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Codon | 86 | 184 | 1246 | 74 | 75 | 76 | 220 | 271 | ||
|
| ||||||||||
| Wild-type | N | Y | D | M | N | K | A | Q | ||
| chloroquine | Y | F | - | I | E | T | S | E | ||
| amodiaquine | Y | Y | Y | I | E | T | S | E | ||
| lumefantrinea | N | F | D | M | N | K | A | Q | ||
| artemisininsa | N | F | D | M | N | K | A | Q | ||
| pyronaridine | - | - | - | - | - | - | - | - | ||
| piperaquineb | - | - | Y | - | - | T | - | - | ||
| mefloquinec | N | Y | D | - | - | - | - | - | ||
| sulfadoxine | none | none | none | none | none | none | none | none | pfdhps | |
| pyrimethamine | none | none | none | none | none | none | none | none | pfdhfr | |
Dash ( - ) indicates lack of relevant data.
The combination AL selects for these genotypes; available in vivo and in vitro data suggest both drug components may both contribute to this effect.
A single in vitro study published to date suggests piperaquine may select for pfcrt 76T (Muangnoicharoen et al. 2009), but this has not been shown in vivo. A single study provides some evidence for selection for the 1246Y allele of pfmdr1 (Wong et al. 2011).
There are no recorded cases of mefloquine treatment failure in Africa in parasites which exhibited increased pfmdr1 copy number, as has been frequently described from Asia.
Data summarized from the following studies: Humphreys et al. 2007; Sisowath et al. 2007, 2009; Dokmajilar et al. 2006; Duraisingh et al. 1997, 2000; Muangnoicharoen et al. 2009; Wong et al. 2011.
It has been argued that the recently reported presence in Angola of P. falciparum harbouring the SVMNT haplotype of pfcrt at codons 72-76 seriously threatens the usefulness of AQ-containing combinations across the continent (Sa and Twu, 2010). Although this is not yet a proven threat, as there are insufficient in vivo data to evaluate the efficacy and selective effect of AQ-containing combinations against parasites with the SVMNT haplotype, ASAQ performed less well than other ACT regimens in a recent study in Burma (Smtihuis et al. 2010), where SVMNT parasite are expected to occur. It is now important to investigate whether this haplotype enjoys a selective advantage under ASAQ in Asia and Africa. The poor penetration of this genotype into Africa suggests it does not enjoy a meaningful fitness advantage in the absence of high levels of AQ selection, and in the presence of acquired immunity to malaria in many individuals. In particular, parasites with this pfcrt genotype are not highly chloroquine resistant, and are reliant on the presence of particular haplotypes of pfmdr1 to survive CQ or AQ treatment (Sa et al. 2009). Together with the much higher levels of intra-host competition among parasite genotypes in African human populations, due to higher multiplicity of infection and enhanced immunity, this may explain the failure of SVMNT genotype to flourish in Africa as they have done in South America, The Philippines, India, Pakistan and Afghanistan (Ord et al. 2007; Beshir et al. 2010b). More in vivo efficacy data are needed to resolve this question.
Targetting paediatric malaria cases with longitudinally paired antimalarial combinations
As discussed above, treatment of young children with clinical malaria is the most likely activity to exert intense selection on P. falciparum genotypes at the population level in high transmission settings. Therefore we propose targeted paediatric therapy with a novel strategy consisting of two, alternating, highly effective combination antimalarial regimens (Table 2). These should be chosen such that, where possible, the non-artemisinin partners exert opposite directional selection on key resistance-associated loci. This strategy could, for example, deploy ASAQ as first-line treatment for children under 10 years presenting with confirmed P. falciparum malaria, but at the next episode experienced by that child, AL would be provided. For the third, fifth and seventh episodes of malaria ASAQ would again be administered, with AL given at the fourth and sixth. Thus in any one individual, each subsequent malaria episode is treated with a drug combination that should display good efficacy against any parasites that may have persisted, and been selected for a particular allele of pfmdr1, from the previous malaria episode. This alternative first-line regimen would also be used as “second-line” treatment administered due to clinical symptoms persisting or recurring within 28 days of treatment of a clinical malaria episode. Although efficacy is good in most of sub-Saharan Africa (Sirima et al. 2009), ASAQ performs less well in some countries (Zwang et al. 2009; Smithuis et al. 2010), and mefloquine-artesunate (MQAS) may be the best choice as an alternative. Caveats to be considered are the relatively high cost of mefloquine, and the need for pharmacovigilance in situations where subsequent regimens overlap closely; it is known that the administration of halofantrine in patients with persisting blood levels of mefloquine increases the risk of cardiotoxicity (Bouchaud et al. 2009). Although this alternating approach requires good record-keeping to maintain the pattern of drug alternation, these records will also provide useful information on the periodicity of malaria episodes requiring treatment in children, and thus could be used as part of ongoing routine surveillance of treatment efficacy. Thus implementation of such a strategy could be beneficially coupled to capacity development in the periphery of African health systems. This strategy specifically deploys, at the individual level, pairs of combination regimens for which some evidence of antagonistic selection on the parasite genome exists (based on previously identified markers of resistance such as pfmdr1). In such cases, it is expected to perform better than strategies using concurrent or time-revolving mixtures of drugs at the population level. When such antagonism does not exist, care must be taken when treating persistent infections with a different drug since this may enhance the development of multi-drug resistance. Further, if drug cross-resistance occurs as in the case of CQ and AQ, sequential use is very likely to lead to poor therapeutic outcomes for the second malaria episode. Unfortunately, in most clinical settings there are few choices available for second-line therapy.
TABLE 2.
Hypothetical treatment matrix for reduction of drug resistance selection pressure in high transmission settings in West Africa (where AQ and SP currently retain good efficacy).
| INDICATION | CHILDREN < 10 years | ADOLESCENTS, ADULTS | |
|---|---|---|---|
| Uncomplicated falciparum malaria |
Alternating for subsequent episodes |
|
SP-PQ |
| Treatment failure of above |
|
AL | |
| Severe hospitalized malaria |
|
||
Treating adults with malaria
Do African adults with uncomplicated symptomatic malaria need to be treated with ACT? In high transmission areas, where the prevalence of acquired immunity is significant, we would argue that they do not. Treating adults with an effective combination of two non-artemisinin partners would be cheaper and, due to the lower proportion of infected adults being treated, and the relatively important contribution of immunity to the treatment outcome, would reduce the selective pressure on P. falciparum populations towards the development of artemisinin resistance. One such combination likely to be very effective in West Africa is piperaquine plus SP, a regimen recently successfully trialled for use for IPTc in children from both Senegal (Cisse et al. 2009) and The Gambia (Bojang et al. 2010). Further, these three relatively long half-life drugs would offer each other mutual protection towards the right-hand end of the pharmacodynamic curve. This may only be prudent where the piperaquine-containing ACT DP was not in use as a first-line regimen for children.
Restricting therapeutics to malaria cases confirmed by diagnosis
The well documented pan-African spread of parasites resistant to CQ, SP or both was almost certainly assisted by selective pressure due to chemotherapy unnecessarily administered to febrile patients presumed to be suffering from clinical malaria. Uncertain diagnosis and over estimation of malaria in most African primary health-care settings can lead to frequent prescription of anti-malarials to both children and adults who do not have malaria (Reyburn et al. 2004), and substantially higher drug pressure than necessary. The selective pressure exerted by unwarranted therapy is likely to be substantially higher as parasites acquired subsequent to this treatment may emerge into the bloodstream and encounter falling, sub-therapeutic drug levels, particularly for component drugs with long half-lives. This can only be prevented by the appropriate use of diagnostics prior to treatment of every suspected case of clinical malaria. Curiously, there are theoretical reasons to argue that mis-use of SP in particular may have improved the rate of development of anti-parasite immunity in some settings (Sutherland et al. 2007; Gosling et al. 2008). However, the possible existence of such an unexpected benefit of improper drug use does not mean that there is no price to be paid in terms of avoidable drug selection pressure on the P. falciparum genome.
Where children are not the main group at risk – the example of eastern Sudan
As presented above, two considerations are important here: maximising drug efficacy at a given frequency of resistance, and reducing the selective advantage of resistant genotypes. The impact of P. falciparum drug resistance on the highly seasonal malaria transmission around the town of Gedarif, in eastern Sudan has been well studied for two decades (Bayoumi et al. 1989, 1993; Abdel-Muhsin et al. 2004; Adam et al, 2004; Babiker et al. 2005; Nassir et al. 2005; Al-Saai et al. 2009; Gadalla et al. 2010). Symptomatic malaria cases occur during a matter of weeks each year after brief seasonal rains, and may occur in all ages; with the majority of cases in older children and adults. Although asymptomatic chronic parasite carriage continues in a subset of people throughout the dry season, acquired immunity which protects against disease is not a prominent feature of populations in this setting. Thus, in contrast to higher transmission seasonal settings such as Burkina Faso (Diallo et al. 2007), a close correlation between carriage of parasites with drug resistant pfcrt or pfdhfr / pfdhps genotypes and treatment failure was observed for CQ and SP respectively during the 1990s and 2000s (Bayoumi et al. 1989; Babiker et al., 2001; Abdel-Muhsin et al., 2004; Al-Saai et al. 2009). In other words, the impact of drug resistance on treatment outcomes here is not softened by the helping hand of previously acquired immunity. An additional intervention option, of particular relevance in this setting due to the occurrence of strict seasonality, is mass drug administration (MDA) to reduce or eliminate the dry season parasite reservoir among asymptomatic carriers before the new malaria season begins (von Seidlein et al. 2003; El Sayed et al. 2007). Due to the high coverage of drug used, this strategy may be particularly vulnerable to rapid selection of resistant parasite genotypes, and must not overlap with the front line therapeutic regimens for clinical cases.
In such a setting then, it is particularly beneficial for chemotherapeutic interventions against P. falciparum to be designed to minimise the impact of selection for resistance. With this principle in mind, we suggest the following antimalarial drug deployment matrix for therapy in an intervention area such as the Gedarif region of Eastern Sudan:
| First-line therapy (uncomplicated, parasitologically-confirmed malaria, all ages) |
AL | |
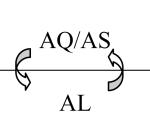
| Second-line therapy (for relapses, treatment failures, adverse reactions to AL) |
AQ/AS | |
| Parenteral therapy for severe malaria | AS i.v. or oral | (first-line) |
| QN i.v. or oral | (second-line) | |
| Preventative therapy | DP + SP | (MDA) |
To maximise the useful therapeutic life of the first-line therapy, we have avoided SP/AS, a credible candidate which is currently in use in Sudan, because of evidence that SP-resistance is already present in this area (Al-Saai et al. 2009), whereas AL efficacy is good (Elamin et al, 2010). Thus in a small proportion of treated cases, SP/AS would not be providing the full benefit of combination therapy. This could dangerously expose AS to new tolerant or resistant parasites should they arise in the population or be imported from elsewhere.
The second-line therapy is chosen to minimise the survival of any parasites persisting after treatment with AL. Recent studies have described the selective survival of parasites harbouring particular pfmdr1 mutations after AL treatment (Sisowath et al. 2007; Dokmalijar et al. 2006). However, Humphreys et al. (2007) demonstrated that AQ selects against these mutations, coding for the amino acids Gly (N), Phe (F) and Asp (D) at codons 86, 184 and 1246 of pfmdr1 (Table 1). Thus the use of AQ/AS may complement any previous AL treatment that did not clear parasitaemia, or at least failed to prevent a new infection with parasites carrying the NFD pfmdr1 genotype.
In-patient treatment for severe malaria in Africa is still usually provided by parenteral QN, although a recent multi-centre randomised trial found that intra-venous artesunate gives a small but significant reduction in mortality, from 10.9% to 8.5%, among severe malaria cases in hospitalised African children (Dondorp et al. 2010). Therefore we consider i.v. AS a preferable in-patient treatment. The numbers of severe, hospitalised malaria cases is a small proportion of total cases, and so the contribution of third-line treatment to overall drug pressure on the parasite population is negligible. A possible exception to this is in epidemic outbreaks, which may occur in areas of seasonal transmission following years of no/low rains and pause in annual transmission (Bayoumi et al., 1989).
Finally, preventive therapies in low intensity, highly seasonal settings such as Gedarif are probably restricted to MDA, as IPT for pregnant women or infants is not indicated where infection risk is so low. Although an argument can be made for the inclusion of primaquine in the MDA regimen, the risk of haemolysis requires screening for G6PD genotypes at risk, and so renders implementation difficult (Shekalage et al. 2010). Therefore we suggest DP (with reasonably good activity against developing gametocytes) plus SP to give a long pharmacodynamic tail comprising all three compounds. As SP resistant parasites are present in the study area, the impact on post-MDA parasite carriage needs to be closely monitored. A logistic difficulty with the DP plus SP regimen is the need to give multiple doses; compliance may fall if doses two and three of DP are not delivered, or at least observed, by health workers.
A crucial role for surveillance and monitoring
The rational deployment recommendations present above revolve around a single key imperative: to minimise the impact of resistant P. falciparum on the efficacy of artemisinin-containing combination regimens used to treat children with uncomplicated malaria only (high transmission areas) or uncomplicated malaria cases of all ages (low / seasonal transmission areas). Thus monitoring of antimalarial efficacy in children under 5 years or in cases of all ages, respectively, should be undertaken in these settings. When drug efficacy is high, passive monitoring, which normally entails recording cases of treatment failure that return to the health system requiring re-treatment, will provide little data of use. In fact only when substantial loss of efficacy has occurred will such an approach begin to indicate a measurable reduction in effectiveness of the malaria treatment programme. We , therefore, propose a simple approach to health-system based active monitoring that could be promoted throughout Africa. This is based on the collection of a pair of finger-prick filter-paper blood-spots at day 0 and day 2 (or day 3) of treatment i.e. immediately prior to treatment (normally a small portion of the diagnostic blood sample) and then 48 (or 72) hours later once treatment has finished. These blood-spots need only a few microlitres of blood and a simple dab of a pricked finger is sufficient. The paired day 0 and day 2 filter-papers are in-expensive, easily collected and can be stored desiccated at room temperature (but do require an additional contact with the patient). New methodologies now permit such paired samples to be used for quantitative analysis of parasite reduction rate by qPCR (Beshir et al. 2010a); this molecular test could easily be set up on a reference or regional lab basis and performed on a proportion of cases (perhaps 10-20%) for continuous monitoring of efficacy. Ideally, blood films would also be made on the additional visit and stored, but these would not normally need to be examined on the day of collection if the patient reports being well. For a small increase in capacity in the periphery, important monitoring information could thus be gathered in every health centre in Africa that dispenses antimalarial drugs. Together with the record-keeping required to operate the drug alternation system, this would greatly add to the surveillance capability of national malaria control programmes.
Concluding Remarks
Our approach assumes that parasites infecting hosts in the different compartments in the population targeted by the various uses of malaria drugs (children, symptomatic adults, infant and school-age recipients of IPT, pregnant women) are able to frequently transmit parasites to mosquitoes that will subsequently infect individuals in other compartments. In this way, parasites in a host from one compartment may be transmitted to a host in another compartment where the type of drug used differs, and their survival advantage may therefore be compromised, or even reversed in the case of antagonistic mechanisms of resistance. Thus the parasite population may be forced into a pattern of diversifying selection at key drug resistance loci, analogous to the selection exerted on parasite antigens by acquired immunity in people: the diversity of host immune responses leads to a balanced but dynamic diversity of alleles among genes encoding parasite proteins recognised by those responses that are protective (Polley et al. 2003; Weedall et al. 2007). Recent sequencing studies of pfcrt and pfmdr1 from African settings where monomorphic CQ drug pressure has been replaced by a diversity of drugs suggest that these two resistance-associated loci are now under diversifying selection (Sutherland & Polley, 2011; Gadalla et al. unpublished observations). Thus new mutations, currently at low frequency, may emerge and escalate in frequency. The response of parasites harbouring these newly recognised forms of pfcrt and pfmdr1 to the combination therapies currently being deployed across the African continent may be a crucial determinant of the long-term effectiveness of these regimens. The measures we have discussed do not require a substantial increase in programme costs, but may help sustain effectiveness for some extra years; we cannot dare to hope that current ACTs will retain good efficacy a decade or more hence. Thus an arsenal of new antimalarial drugs is desperately needed to replace the artemisinins and their current partners in the 2020’s.
Acknowledgements
This paper has its origins in a workshop on drug resistance in malaria, sponsored by the British Council and the International Atomic Energy Agency, held at the Tropical Medical Research Institute, Khartoum, in January 2007. We thank the British Society for Parasitology and the Royal Society of Tropical Medicine and Hygiene for supporting the presentation of this paper at the Autumn Symposium held at the University of Durham, in September 2010.
CJS is supported by the UK Health Protection Agency. HB is supported by the Sultan Qaboos University Research Fund. MM is supported by Wellcome Trust Project Grant 088634 and Wellcome Trust Core Programme Grant 077092 to the KEMRI-Wellcome Trust Research Programme. This paper is published with the permission of the Director of the Kenyan Medical Research Institute. Work in LR-C’s laboratory is supported by the Wellcome Trust, European Commission (TM-REST, MalSig), BBSRC and the International Atomic Energy Agency. BB el S is supported by the British Council and the International Atomic Energy Agency.
REFERENCES
- Abdel-Muhsin AM, Mackinnon MJ, Ali E, Nassir el-K. A., Suleiman S, Ahmed S, Walliker D, Babiker HA. Evolution of drug-resistance genes in Plasmodium falciparum in an area of seasonal malaria transmission in Eastern Sudan. Journal of Infectious Diseases. 2004;189:1239–1244. doi: 10.1086/382509. [DOI] [PubMed] [Google Scholar]
- Adam I, Osman ME, Elghzali G, Ahmed GI, Gustafssons LL, Elbashir MI. Efficacies of chloroquine, sulfadoxine-pyrimethamine and quinine in the treatment of uncomplicated Plasmodium falciparum malaria in eastern Sudan. Annals of Tropical Medicine and Parasitology. 2004;98:661–666. doi: 10.1179/000349804225021514. [DOI] [PubMed] [Google Scholar]
- Al-Saai S, Kheir A, Abdel-Muhsin AM, Al-Ghazali A, Nwakanma D, Swedberg G, Babiker HA. Distinct haplotypes of dhfr and dhps among Plasmodium falciparum isolates in an area of high level of sulfadoxine-pyrimethamine (SP) resistance in eastern Sudan. Infection, Genetics and Evolution. 2009;9:778–783. doi: 10.1016/j.meegid.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Babiker HA, Pringle S, Abdel-Muhsin A, Mackinnon M, Hunt P, Walliker D. High level chloroquine resistance in Plasmodium falciparum is associated with mutations in the chloroquine resistance transporter gene Pfcrt and the multi-drug resistance gene Pfmdr1. J. Infect Dis. 2001;183:1535–8. doi: 10.1086/320195. [DOI] [PubMed] [Google Scholar]
- Babiker HA, Satti G, Ferguson H, Bayoumi R, Walliker D. Drug resistant Plasmodium falciparum in an area of seasonal transmission. Acta Tropica. 2005;94:260–268. doi: 10.1016/j.actatropica.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Bayoumi RA, Babiker HA, Ibrahim SM, Ghalib HW, Saeed BO, Khider S, Elwasila M, Karim EA. Chloroquine-resistant Plasmodium falciparum in eastern Sudan. Acta Tropica. 1989;46:157–165. doi: 10.1016/0001-706x(89)90032-6. [DOI] [PubMed] [Google Scholar]
- Bayoumi RA, Creasey AM, Babiker HA, Carlton JM, Sultan AA, Satti G, Sohal AK, Walliker D, Jensen JB, Arnot DE. Drug response and genetic characterization of Plasmodium falciparum clones recently isolated from a Sudanese village. Transaction sof the Royal Society for Tropical Medicine and Hygiene. 1993;87:454–458. doi: 10.1016/0035-9203(93)90034-n. [DOI] [PubMed] [Google Scholar]
- Beshir K, Hallett RL, Eziefula AC, Bailey R, Watson J, Wright SG, Chiodini PL, Polley SD, Sutherland CJ. Measuring the efficacy of anti-malarial drugs in vivo: quantitative PCR measurement of parasite clearance. Malaria Journal. 2010a;9:312. doi: 10.1186/1475-2875-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beshir K, Sutherland CJ, Merinopoulos I, Durrani N, Leslie T, Rowland M, Hallett RL. Amodiaquine resistance in Plasmodium falciparum malaria in Afghanistan is associated with the pfcrt SVMNT allele at codons 72 to 76. Antimicrobial Agents and Chemotherapy. 2010b;54:3714–3716. doi: 10.1128/AAC.00358-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojang K, Akor F, Bittaye O, Conway D, Bottomley C, Milligan P, Greenwood BM. A randomised trial to compare the safety, tolerability and efficacy of three drug combinations for intermittent preventive treatment in children. PLoS One. 2010;5:e11225. doi: 10.1371/journal.pone.0011225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchaud O, Imbert P, Touze JE, Dodoo AN, Danis M, Legros F. Fatal cardiotoxicity related to halofantrine: a review based on a worldwide safety data base. Malaria Journal. 2009;8:289. doi: 10.1186/1475-2875-8-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousema JT, Schneider P, Gouagna LC, Drakeley CJ, Tostmann A, Houben R, Githure JI, Ord R, Sutherland CJ, Omar SA, Sauerwein RW. Moderate effect of artemisinin-based combination therapy on transmission of Plasmodium falciparum. Journal of Infectious Diseases. 2006;193:1151–1159. doi: 10.1086/503051. [DOI] [PubMed] [Google Scholar]
- Carrara VI, Zwang J, Ashley EA, Price RN, Stepniewska K, Barends M, Brockman A, Anderson T, McGready R, Phaiphun L, Proux S, van Vugt M, Hutagalung R, Lwin KM, Phyo AP, Preechapornkul P, Imwong M, Pukrittayakamee S, Singhasivanon P, White NJ, Nosten F. Changes in the treatment responses to artesunate-mefloquine on the northwestern border of Thailand during 13 years of continuous deployment. PLoS One. 2009;4:e4551. doi: 10.1371/journal.pone.0004551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse B, Cairns M, Faye E, NDiaye O, Faye B, Cames C, Cheng Y, NDiaye M, Lô AC, Simondon K, Trape JF, Faye O, NDiaye JL, Gaye O, Greenwood B, Milligan P. Randomized trial of piperaquine with sulfadoxine-pyrimethamine or dihydroartemisinin for malaria intermittent preventive treatment in children. PLoS One. 2009;4:e7164. doi: 10.1371/journal.pone.0007164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallo D, Sutherland C, Nebié I, Konaté A, Ord R, Pota H, Roper C, Ilboudo-Sanogo E, Greenwood B, Cousens S. Children in Burkina Faso protected by insecticide treated materials are able to clear drug resistant parasites better than unprotected children. Journal of Infectious Diseases. 2007;196:138–144. doi: 10.1086/518252. [DOI] [PubMed] [Google Scholar]
- Dokomajilar C, Nsobya SL, Greenhouse B, Rosenthal PJ, Dorsey G. Selection of Plasmodium falciparum pfmdr1 alleles following therapy with artemether-lumefantrine in an area of Uganda where malaria is highly endemic. Antimicrob Agents Chemother. 2006;50:1893–1895. doi: 10.1128/AAC.50.5.1893-1895.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp AM, Fanello CI, Hendriksen IC, Gomes E, Seni A, Chhaganlal KD, Bojang K, Olaosebikan R, Anunobi N, Maitland K, Kivaya E, Agbenyega T, Nguah SB, Evans J, Gesase S, Kahabuka C, Mtove G, Nadjm B, Deen J, Mwanga-Amumpaire J, Nansumba M, Karema C, Umulisa N, Uwimana A, Mokuolu OA, Adedoyin OT, Johnson WB, Tshefu AK, Onyamboko MA, Sakulthaew T, Ngum WP, Silamut K, Stepniewska K, Woodrow CJ, Bethell D, Wills B, Oneko M, Peto TE, von Seidlein L, Day NP, White NJ. AQUAMAT group. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet. 2010;376:1647–1657. doi: 10.1016/S0140-6736(10)61924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunyo S, Milligan P, Edwards T, Sutherland C, Targett G, Pinder M. Gametocytaemia after drug treatment of asymptomatic Plasmodium falciparum. PLoS Clinical Trials. 2006;1:e20. doi: 10.1371/journal.pctr.0010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraisingh MT, Drakeley CJ, Muller O, Bailey R, Snounou G, Targett GAT, Greenwood BM, Warhurst DC. Evidence for selection for the tyrosine-86 allele of the pfmdr 1 gene of Plasmodium falciparum by chloroquine and amodiaquine. Parasitology. 1997;114:205–211. doi: 10.1017/s0031182096008487. [DOI] [PubMed] [Google Scholar]
- Duraisingh MT, Jones P, Sambou I, von Seidlein L, Pinder M, Warhurst DC. The tyrosine-86 allele of pfmdr1 gene of the Plasmodium falciparum is associated with increased sensitivity to the anti-malarials mefloquine and artemisinin. Mol Biochem. Parasitol. 2000;108:13–23. doi: 10.1016/s0166-6851(00)00201-2. [DOI] [PubMed] [Google Scholar]
- El-Sayed B, El-Zaki SE, Babiker H, Gadalla N, Ageep T, Mansour F, Baraka O, Milligan P, Babiker A. A randomized open-label trial of artesunate-sulfadoxine-pyrimethamine with or without primaquine for elimination of sub-microscopic P. falciparum parasitaemia and gametocyte carriage in eastern Sudan. PLoS One. 2007;2:e1311. doi: 10.1371/journal.pone.0001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elamin SB, Awad AI, Eltayeb IB, Elmardi KA, Hassan AH, Mohamed AO, Malik EM, Mohamad TA. Descriptive study on the efficacy of artemether-lumefantrine in the treatment of uncomplicated Plasmodium falciparum malaria in Sudan. European Journal of Clinical Pharmacology. 2010;66:231–237. doi: 10.1007/s00228-009-0750-4. [DOI] [PubMed] [Google Scholar]
- Gadalla NB, Elzaki SE, Mukhtar E, Warhurst DC, El-Sayed B, Sutherland CJ. Dynamics of Pfcrt alleles CVMNK and CVIET in chloroquine-treated Sudanese patients infected with Plasmodium falciparum. Malaria Journal. 2010;9:74. doi: 10.1186/1475-2875-9-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosling RD, Drakeley CJ, Mwita A, Chandramohan D. Presumptive treatment of fever cases as malaria: help or hindrance for malaria control? Malaria Journal. 2008;7:132. doi: 10.1186/1475-2875-7-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett RL, Dunyo S, Ord R, Jawara M, Pinder M, Randall A, Alloueche A, Walraven G, Targett GAT, Alexander N, Sutherland CJ. Treatment of malaria in Gambian children with chloroquine plus sulphadoxine-pyrimethamine favours survival and transmission to mosquitoes of multi-drug-resistant. Plasmodium falciparum PLoS Clinical Trials. 2006;1:e15. doi: 10.1371/journal.pctr.0010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren G, Hamrin J, Svard J, Martensson A, Gil JP, Bjorkman A. Selection of pfmdr1 mutations after amodiaquine monotherapy and amodiaquine plus artemisinin combination therapy in East Africa. Infection, Genetics and Evolution. 2007;7:562–569. doi: 10.1016/j.meegid.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Humphreys GS, Merinopoulos I, Ahmed J, Whitty CJ, Mutabingwa TK, Sutherland CJ, Hallett RL. Amodiaquine and artemether-lumefantrine select distinct alleles of the Plasmodium falciparum mdr1 gene in Tanzanian children treated for uncomplicated malaria. Antimicrobial Agents and Chemotherapy. 2007;51:991–997. doi: 10.1128/AAC.00875-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayentao K, Maiga H, Newman RD, McMorrow ML, Hoppe A, Yattara O, Traore H, Kone Y, Guirou EA, Saye R, Traore B, Djimde A, Doumbo OK. Artemisinin-based combinations versus amodiaquine plus sulphadoxine-pyrimethamine for the treatment of uncomplicated malaria in Faladje, Mali. Malaria Journal. 2009;8:5. doi: 10.1186/1475-2875-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez F, Herrera S, Murrain B, Gutiérrez A, Moreno LA, Manzano M, Muñoz A, Plowe CV. Selection of antifolate-resistant Plasmodium falciparum by sulfadoxine-pyrimethamine treatment and infectivity to Anopheles mosquitoes. American Journal of Tropical Medicine & Hygiene. 2007;77:438–443. [PubMed] [Google Scholar]
- Muangnoicharoen S, Johnson DJ, Looareesuwan S, Krudsood S, Ward SA. Role of known molecular markers of resistance in the antimalarial potency of piperaquine and dihydroartemisinin in vitro. Antimicrobial Agents and Chemotherapy. 2009;53:1362–1366. doi: 10.1128/AAC.01656-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassir E, Abdel-Muhsin AM, Suliaman S, Kenyon F, Kheir A, Geha H, Ferguson HM, Walliker D, Babiker HA. Impact of genetic complexity on longevity and gametocytogenesis of Plasmodium falciparum during the dry and transmission-free season of eastern Sudan. International Journal of Parasitology. 2005;35:49–55. doi: 10.1016/j.ijpara.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM, Artemisinin Resistance in Cambodia 1 (ARC1) Study Consortium Evidence of artemisinin-resistant malaria in western Cambodia. New England Journal of Medicine. 2008;359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- Ord R, Alexander N, Dunyo S, Hallett RL, Jawara M, Targett GAT, Drakeley CJ, Sutherland CJ. Seasonal carriage of pfcrt and pfmdr1 alleles in Gambian Plasmodium falciparum implies reduced fitness of chloroquine-resistant parasites. Journal of Infectious Diseases. 2007;196:1613–1619. doi: 10.1086/522154. [DOI] [PubMed] [Google Scholar]
- Polley SD, Chokejindachai W, Conway DJ. Allele frequency-based analyses robustly map sequence sites under balancing selection in a malaria vaccine candidate antigen. Genetics. 2003;165:555–561. doi: 10.1093/genetics/165.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyburn H, Mbatia R, Drakeley C, Carneiro I, Mwakasungula E, Mwerinde O, Saganda K, Shao J, Kitua A, Olomi R, Greenwood BM, Whitty CJ. Overdiagnosis of malaria in patients with severe febrile illness in Tanzania: A prospective study. British Medical Journal. 2004;329:1212. doi: 10.1136/bmj.38251.658229.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa JM, Twu O. Protecting the malaria drug arsenal: halting the rise and spread of amodiaquine resistance by monitoring the PfCRT SVMNT type. Malaria Journal. 2010;9:374. doi: 10.1186/1475-2875-9-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sá JM, Twu O, Hayton K, Reyes S, Fay MP, Ringwald P, Wellems TE. Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proceedings of the National Academy of Science U S A. 2009;106:18883–18889. doi: 10.1073/pnas.0911317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekalaghe SA, ter Braak R, Daou M, Kavishe R, van den Bijllaardt W, van den Bosch S, Koenderink JB, Luty AJ, Whitty CJ, Drakeley C, Sauerwein RW, Bousema T. In Tanzania, hemolysis after a single dose of primaquine coadministered with an artemisinin is not restricted to glucose-6-phosphate dehydrogenase-deficient (G6PD A-) individuals. Antimicrobial Agents and Chemotherapy. 2010;54:1762–1768. doi: 10.1128/AAC.01135-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirima SB, Tiono AB, Gansané A, Diarra A, Ouédraogo A, Konaté AT, Kiechel JR, Morgan CC, Olliaro PL, Taylor WR. The efficacy and safety of a new fixed-dose combination of amodiaquine and artesunate in young African children with acute uncomplicated Plasmodium falciparum. Malaria Journal. 2009;8:48. doi: 10.1186/1475-2875-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisowath C, Ferreira PE, Bustamante LY, Dahlstrom S, Martensson A, Bjorkman A, Krishna S, Gil JP. The role of pfmdr1 in Plasmodium falciparum tolerance to artemether-lumefantrine in Africa. Tropical Medicine and International Health. 2007;12:736–742. doi: 10.1111/j.1365-3156.2007.01843.x. [DOI] [PubMed] [Google Scholar]
- Sisowath C, Petersen I, Veiga MI, Martensson A, Premji Z, Bjorkman A, Fidock DA, Gil JP. In vivo selection of Plasmodium falciparum parasites carrying the chloroquine susceptible Pfcrt K76 allele after treatment with artemether-lumefantrine in Africa. Journal of Infectious Diseases. 2009;199:750–757. doi: 10.1086/596738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithuis F, Kyaw MK, Phe O, Win T, Aung PP, Oo AP, Naing AL, Nyo MY, Myint NZ, Imwong M, Ashley E, Lee SJ, White NJ. Effectiveness of five artemisinin combination regimens with or without primaquine in uncomplicated falciparum malaria: an open-label randomised trial. Lancet Infect Dis. 2010;10:673–81. doi: 10.1016/S1473-3099(10)70187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland CJ, Ord R, Dunyo S, Jawara M, Drakeley CJ, Alexander N, Coleman R, Pinder M, Walraven G, Targett GAT. Reduction of malaria transmission to Anopheles mosquitoes with a six-dose regimen of co-artemether. PLoS Medicine. 2005;2:e92. doi: 10.1371/journal.pmed.0020092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland CJ, Polley SD. Genomic insights into the past, current and future evolution of human parasites of the genus Plasmodium. In: Tibayrenc M, editor. Genetics and Evolution of Infectious Diseases. Elsevier; London: 2011. 2011. ISBN: 978-0-12-384890-1. [Google Scholar]
- Sutherland CJ, Drakeley CJ, Schellenberg D. How is childhood development of immunity to Plasmodium falciparum enhanced by certain antimalarial interventions? Malaria Journal. 2007;6:161. doi: 10.1186/1475-2875-6-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursing J, Kofoed PE, Rodrigues A, Rombo L, Gil JP. Plasmodium falciparum genotypes associated with chloroquine and amodiaquine resistance in Guinea-Bissau. American Journal of Tropical Medicine and Hygiene. 2007;76:844–848. [PubMed] [Google Scholar]
- Ursing J, Kofoed PE, Rodrigues A, Blessborn D, Thoft-Nielsen R, Björkman A, Rombo L. Similar efficacy and tolerability of double-dose chloroquine and artemether-lumefantrine for treatment of Plasmodium falciparum infection in Guinea-Bissau: a randomized trial. Journal of Infectious Diseases. 2011;203:109–116. doi: 10.1093/infdis/jiq001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Seidlein L, Walraven G, Milligan PJ, Alexander N, Manneh F, Deen JL, Coleman R, Jawara M, Lindsay SW, Drakeley C, De Martin S, Olliaro P, Bennett S, Schim van der Loeff M, Okunoye K, Targett GA, McAdam KP, Doherty JF, Greenwood BM, Pinder M. The effect of mass administration of sulfadoxine-pyrimethamine combined with artesunate on malaria incidence: a double-blind, community-randomized, placebo-controlled trial in The Gambia. Transactions of the Royal Society for Tropical medicine and Hygiene. 2003;97:217–225. doi: 10.1016/s0035-9203(03)90125-8. [DOI] [PubMed] [Google Scholar]
- Weedall GD, Preston BMJ, Thomas AW, Sutherland CJ, Conway DJ. Differential evidence of natural selection on two leading sporozoite stage malaria vaccine candidate antigens. International Journal of Parasitology. 2007;37:77–85. doi: 10.1016/j.ijpara.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Wong RP, Karunajeewa H, Mueller I, Siba P, Zimmerman PA, Davis TM. Molecular assessment of Plasmodium falciparum resistance to antimalarial drugs in Papua New Guinea using an extended ligase detection reaction fluorescent microsphere assay. Antimicrobial Agents and Chemotherapy. 2011;55:798–805. doi: 10.1128/AAC.00939-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwang J, Olliaro P, Barennes H, Bonnet M, Brasseur P, Bukirwa H, Cohuet S, D’Alessandro U, Djimdé A, Karema C, Guthmann JP, Hamour S, Ndiaye JL, Mårtensson A, Rwagacondo C, Sagara I, Same-Ekobo A, Sirima SB, van den Broek I, Yeka A, Taylor WR, Dorsey G, Randrianarivelojosia M. Efficacy of artesunate-amodiaquine for treating uncomplicated falciparum malaria in sub-Saharan Africa: a multi-centre analysis. Malaria Journal. 2009;8:203. doi: 10.1186/1475-2875-8-203. [DOI] [PMC free article] [PubMed] [Google Scholar]