

Abstract
The viral factories of mammalian reovirus (MRV) are cytoplasmic structures that serve as sites of viral genome replication and particle assembly. A 721-aa MRV nonstructural protein, μNS, forms the factory matrix and recruits other viral proteins to these structures. In this report, we show that μNS contains a conserved C-proximal sequence (711-LIDFS-715) that is similar to known clathrin-box motifs and is required for recruitment of clathrin to viral factories. Clathrin recruitment by μNS occurs independently of infecting MRV particles or other MRV proteins. Ala substitution for a single Leu residue (mutation L711A) within the putative clathrin-binding motif of μNS inhibits clathrin recruitment, but does not prevent formation or expansion of viral factories. Notably, clathrin-dependent cellular functions, including both endocytosis and secretion, are disrupted in cells infected with MRV expressing wild-type, but not L711A, μNS. These results demonstrate μNS as a novel adaptor-like protein that recruits cellular clathrin to viral factories, disrupting normal functions of clathrin in cellular membrane trafficking. To our knowledge, this is the only viral or bacterial protein yet shown to interfere with clathrin functions in this manner. The results additionally establish a new approach for studies of clathrin functions, based in μNS-mediated sequestration.
Keywords: adaptor, clathrin, membrane trafficking, reovirus, viral factories
Viral genome replication and particle assembly commonly occur within virally induced cellular subcompartments, often associated with either intracellular membranes (1–3) or cytoskeletal elements (4–6). Members of the family Reoviridae, multisegmented dsRNA viruses, are believed to replicate and assemble within large cytoplasmic structures, called viral factories in the case of mammalian reovirus (MRV) from the genus Orthoreovirus (7–9).
MRV factories contain viral proteins, dsRNA and partially and fully assembled particles (7,9–11). They contain few if any ribosomes and are not membrane-associated (8–12). MRV nonstructural protein μNS has been shown to form the factory framework or “matrix” (10,11), as μNS expressed from plasmids in the absence of infection or other viral proteins forms morphologically similar factory-matrix structures when viewed by light microscopy (13–15). In addition, upon plasmid-based co-expression of the different proteins, μNS can recruit MRV nonstructural protein σNS and core proteins λ1, λ2, λ3 or σ2 (see next paragraph concerning MRV core protein μ2) to factory-matrix structures via non-overlapping regions within the N-terminal one-third of μNS (with the exception of λ3, whose binding sequences within the C-terminal one-third of μNS have not been finely mapped) (14,16–18). Transiently expressed, plasmid-derived μNS can also recruit entering MRV core particles to pre-formed factory-matrix structures (16,19). These observations have implicated μNS in the recruitment of both entering core particles and newly synthesized viral proteins to viral factories, and have led to the proposal that μNS may help to organize and coordinate these different components promoting viral genome replication and particle assembly (16).
MRV factories also contain cellular proteins, but their identities have remained largely unknown. Several ultrastructural studies have detected microtubules in viral factories (7,10,20), and more recent work has identified a strain-independent requirement for functional microtubules in factory expansion and perinuclear localization (9). In addition, strain-dependent differences in the ability of MRV core protein μ2 to interact with microtubules have been associated with differences in factory morphology (9,21). Most MRV strains examined to date (>30), including prototype strain Type 1 Lang (T1L), form filamentous factories that are anchored to and spread along stabilized microtubules; however, certain clonal isolates of prototype strain Type 3 Dearing (T3D), such as the one used in our lab (T3DN), instead form globular factories (9,22). Despite the unusual appearance of their factories, these viruses replicate normally (9). Factory-matrix structures formed by expressing plasmid-derived μNS alone, regardless of MRV strain origin, are globular and resemble factories formed during T3DN infection (13,14). Plasmid-based co-expression of μNS with T1L μ2 changes the morphology of factory-matrix structures from globular to filamentous, resembling factories that form during T1L infection (13). Plasmid-based co-expression of μNS with T3DN μ2 does not have this effect, and this μ2 is instead recruited to globular factory-matrix structures (13). These observations are consistent with the conclusion that μNS forms the factory matrix while μ2 of most MRV strains determines the filamentous morphology of factories by anchoring them to microtubules. The sequences involved in recruiting μ2 to factory-matrix structures are also found in the N-terminal one-third of μNS (13,16,19).
In experiments that led to the current study, we observed cellular clathrin colocalizing with viral factories in MRV-infected cells. Clathrin is organized as a triskelion-shaped oligomer comprising three molecules of clathrin heavy chain (HC), trimerized at their C-termini, and three molecules of clathrin light chain A or B (LCA or LCB), each in contact with one molecule of clathrin HC (reviewed in ref. 23). Its best-described roles are in protein sorting and membrane vesicle trafficking from the plasma membrane and the trans-Golgi network (TGN). During clathrin-mediated vesiculation, adaptor protein complexes recognize sorting signals in the cytoplasmic tails of cargo molecules and also interact with membrane phosphoinositides and clathrin. Ordered recruitment of additional adaptor and clathrin molecules to the sites of cargo capture assembles the clathrin coat around the newly forming vesicle, or clathrin-coated pit (CCP). Clathrin drives membrane vesiculation by forming an ordered polyhedral lattice around the membrane, inducing its invagination. The process is terminated by scission of the CCP from the plasma membrane by the GTPase dynamin, releasing into the cytosol a clathrin-coated vesicle (CCV) (reviewed in ref. 24). The clathrin scaffold is then removed from the vesicle during uncoating driven by heat shock cognate protein 70 (Hsc70) and its co-chaperone auxilin.
A number of cellular adaptor proteins have been described, and they typically display a short consensus sequence, termed the clathrin-box motif, that binds to the N-terminal, β-propeller domain (NTD) of clathrin HC via a peptide-in-groove interaction (25). Adaptor proteins AP1 and AP2 mediate CCV formation at the TGN and the plasma membrane, respectively (reviewed in refs. 26 and 27). AP1 and AP2 are heterotetrameric protein complexes consisting of γ, β1, μ1A and σ1 subunits in the case of AP1, and α, β2, μ2 and σ2 subunits in the case of AP2. The clathrin-box motifs of AP1 and AP2 reside within their respective β subunits. The μ2 protein discussed hereafter in this report is the one from MRV.
The best-known use of the clathrin machinery by pathogens is during their internalization into the host cell. Many viruses and bacteria usurp the clathrin-mediated endocytotic pathway to promote their uptake from the cell surface as part of the productive infection process (28–30; reviewed in ref. 31). The possibility that clathrin may play other roles in viral life cycles has become recently apparent in at least one notable example. Huang et al. (32) have identified a functional clathrin-binding motif within the large antigen protein (Ag-L) of hepatitis delta virus (HDV), through which cytoplasmically localized Ag-L associates with clathrin HC. Further experiments have demonstrated the requirement for interaction of clathrin HC with protein Ag-L in HDV particle assembly (33).
In this report, we newly demonstrate a clathrin adaptor-like function of the MRV μNS protein. μNS contains a conserved sequence near its C-terminus, allowing recruitment of clathrin to viral factories via the NTD of clathrin HC. Mutation of a single residue within this putative clathrin-binding motif of μNS inhibits association with clathrin, but does not prevent formation or expansion of viral factories. The resulting large cellular redistribution of clathrin to viral factories over the course of MRV infection is associated with inhibition of clathrin-dependent cellular membrane trafficking, impacting both endocytosis and secretion. To our knowledge, this is the only viral or bacterial protein yet shown to interfere with clathrin functions in this manner. The results additionally establish a new approach for studies of clathrin functions, based in μNS-mediated sequestration.
Results
Clathrin LCA/B and HC colocalize with μNS in MRV factories
We attached nonpurified infectious subvirion particles (ISVPs) obtained by protease digestion of purified T1L virions to BSC-1 cells on ice for 1 h and then fixed them either immediately (0 h post-infection (p.i.)) or after incubation at 37°C for 3, 7 or 12 h (3, 7 or 12 h p.i.). To detect infected cells, we used indirect immunofluorescence (IF) microscopy with serum antibodies against MRV nonstructural protein μNS, which is expressed after productive cell entry. We also stained the cells with either monoclonal antibody (MAb) CON.1 specific for clathrin LCA/B (34) or MAb X22 specific for clathrin HC (35) (Figures 1 and S1A). At 0 h p.i., we observed both clathrin LCA/B and clathrin HC distributed in accordance with their roles in vesicular transport: localized throughout the cell but concentrated in the perinuclear region and at the plasma membrane. At this time point, we saw essentially no μNS staining, as expected for a nonstructural protein, not present in infecting particles. By 3 h p.i., staining for μNS revealed small, globular viral factories. We observed clathrin LCA/B and HC colocalized with these structures in addition to their typical, strongly perinuclear localization. By 7 and 12 h p.i., as the factories grew in size and became more filamentous in morphology due to anchoring to microtubules through MRV protein μ2 (9), we observed a concomitant increase in the amount of clathrin LCA/B and HC associating with these structures. Given the stability of the clathrin triskelion and the affinity of clathrin LC for clathrin HC (reviewed in ref. 23), it is very likely that clathrin HC and LCA/B are recruited to viral factories as clathrin triskelia.
Figure 1. Both clathrin LCA/B and clathrin HC, but not AP1 or AP2, colocalize with μNS in MRV factories.
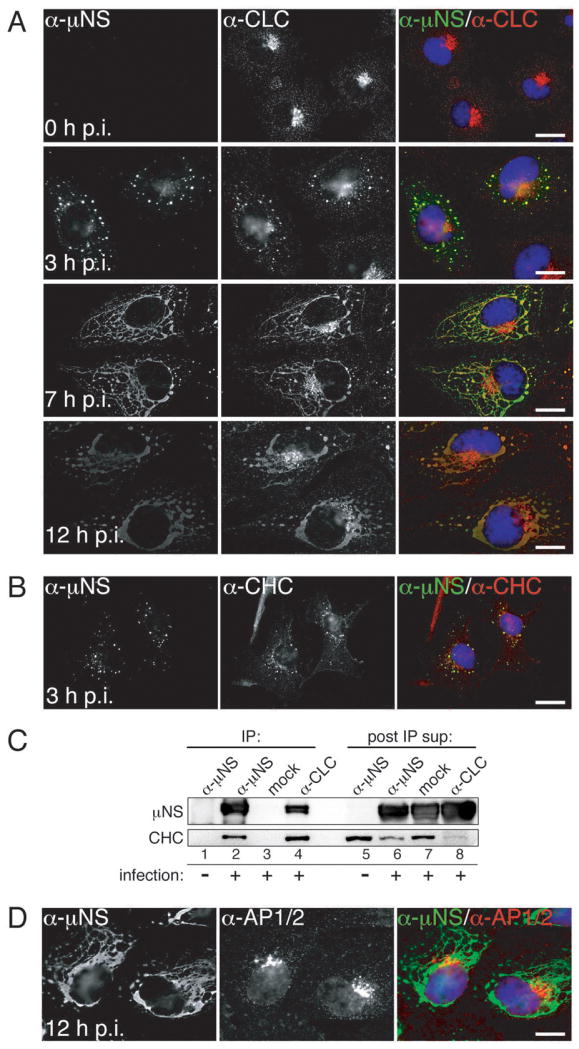
Cells were infected with T1L ISVPs and were either fixed for indirect IF microscopy at 0, 3, 7 or 12 h p.i. (A, B and D) or harvested for IP at 10 h p.i. (C). μNS-specific serum antibodies (α-μNS) and a monoclonal antibody (MAb) indicated below were used in IF (A, B and D). Merged images in A, B and D are shown at right (α-μNS, green; α-CLC, α-CHC and α-AP1/AP2, red). A) Cellular localizations of μNS and clathrin LCA/B (MAb CON.1). B) Cellular localizations of μNS and clathrin HC (MAb X22). C) Either μNS or clathrin LCA/B were immunoprecipitated from postnuclear supernatants using either μNS-specific serum antibodies (α-μNS) or MAb CON.1 (α-CLC). Immunoprecipitated proteins (left, lanes 1-4) as well as proteins remaining in the post-IP supernatants (right, lanes 5-8) were detected by SDS/PAGE and immunoblotting using μNS-specific serum antibodies (μNS) or MAb TD.1 (CHC). D) Cellular localizations of μNS and AP1/2 (MAb 10A). Scale bars, 20 μm.
We additionally observed recruitment of GFP-tagged clathrin LCA stably expressed in BSC-1 cells (LG2 cells) to cytoplasmic structures consistent with viral factories and reflecting a major redistribution of clathrin LCA within the cell at later times p.i. (e.g., 30 h) (Video S1). The observation that clathrin LCA redistributed to factories in the absence of antibody staining strongly argues that the observed co-localizations of clathrin LCA/B and HC with μNS are not an artifact of antibody cross-reactivity or fluorophore bleedthrough. Additional controls for genuine co-localizations are presented throughout the manuscript, and include absence of co-staining either between μNS and adaptor proteins AP1 and AP2 (see Figures 1D, S1B, 2A and 2B below and online) or between mutant μNS and clathrin (see Figures 3D,3E,4A, 4B and 5A below).
Figure 3. μNS contains a putative clathrin-binding motif required for clathrin recruitment to factory-matrix structures.
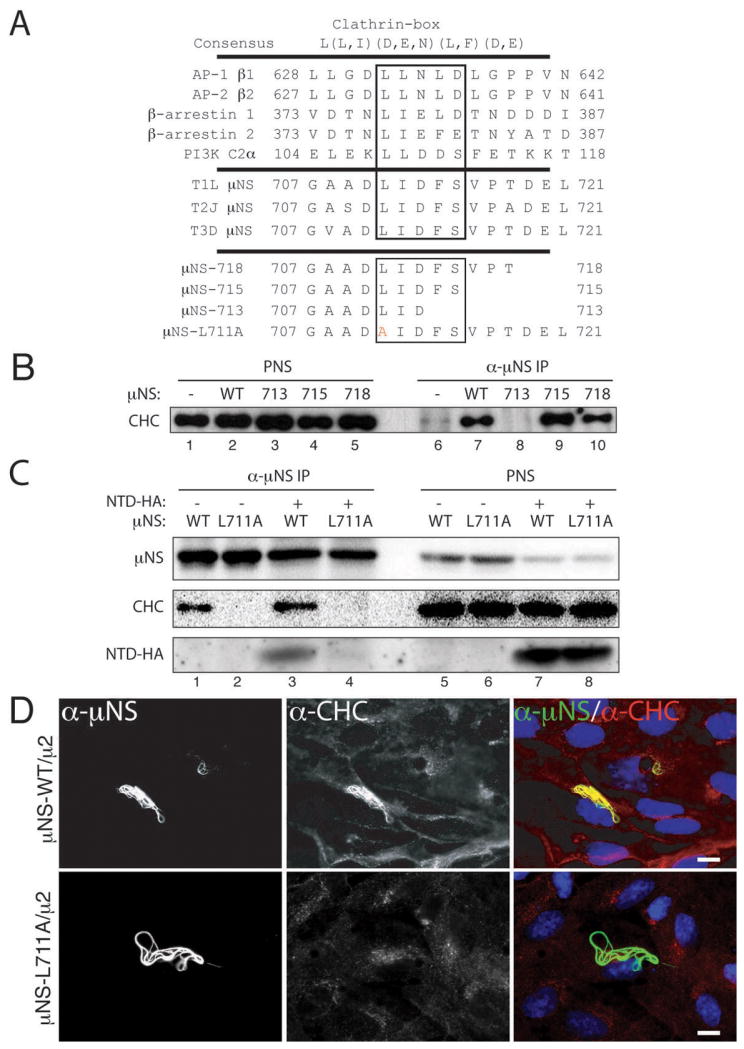
A) The following are shown: consensus clathrin-box motif; examples of proteins shown to contain functional clathrin-box motifs; C-terminal sequences of μN S of MRV strains T1L, T2J and T3D; and mutations engineered near the C-terminus of T1L μNS. Residues matching the clathrin-box consensus are boxed. Mutated residue in μNS-L711A is shown in red. B, C) μNS-WT or μNS-L711A was expressed alone (lanes 1, 2, 5 and 6) or in combination with NTD-HA (lanes 3, 4, 7 and 8) in CV-1 cells following plasmid transfection. Cells were harvested at 28 h p.t. and μNS was immunoprecipitated from postnuclear supernatants (PNS) using μNS-specific serum antibodies (α-μNS). Immunoprecipitated proteins (lanes 1 to 4) or proteins contained within PNS prior to immunoprecipitation (lanes 5 to 8) were detected by SDS/PAGE and immunoblotting using either μNS-specific serum antibodies (μNS) or MAb TD.1 (CHC) or a combination of MAb TD.1 and anti-HA (NTD-HA). D) μNS-WT or μNS-L711A was co-expressed with μ2 in CV-1 cells following plasmid transfection. At 38 h p.t., cellular localizations of μNS (α-μNS, left column) and clathrin HC (α-CHC, middle column) were visualized as in Figure 2. E) μNS-WT or μNS-L711A was expressed in CV-1 cells following plasmid transfection. At 24 h p.t., cellular localizations of μNS (α-μNS, left column) and clathrin HC (α-CHC, middle column) were visualized as in Figure 2. Arrow in the middle column points to residual staining for clathrin HC visible within one of the factory-matrix structures. F) μNS-WT and NTD-HA were co-expressed in CV-1 cells by plasmid-based transfection. At 24 h p.t., they were visualized by indirect IF microscopy using a combination of μNS-specific serum antibodies (α-μNS, left column) and MAb Mono HA.11 (α-HA, middle column). In D, E and F, merged images are shown at right (α-μNS, green; α-HA or α-CHC, red). Scale bars, 20 μm.
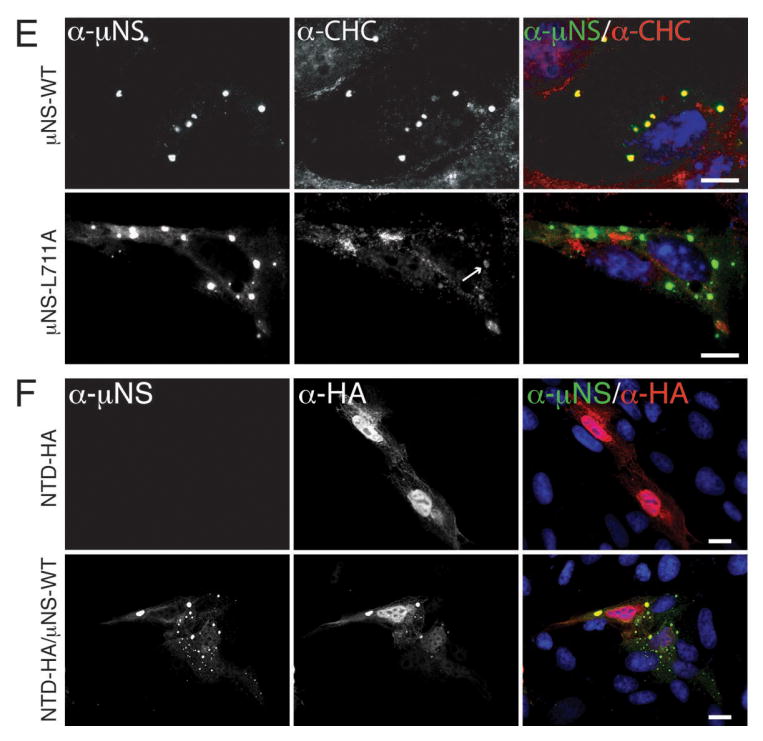
Figure 4. Clathrin recruitment to MRV factories is dispensable for their formation.
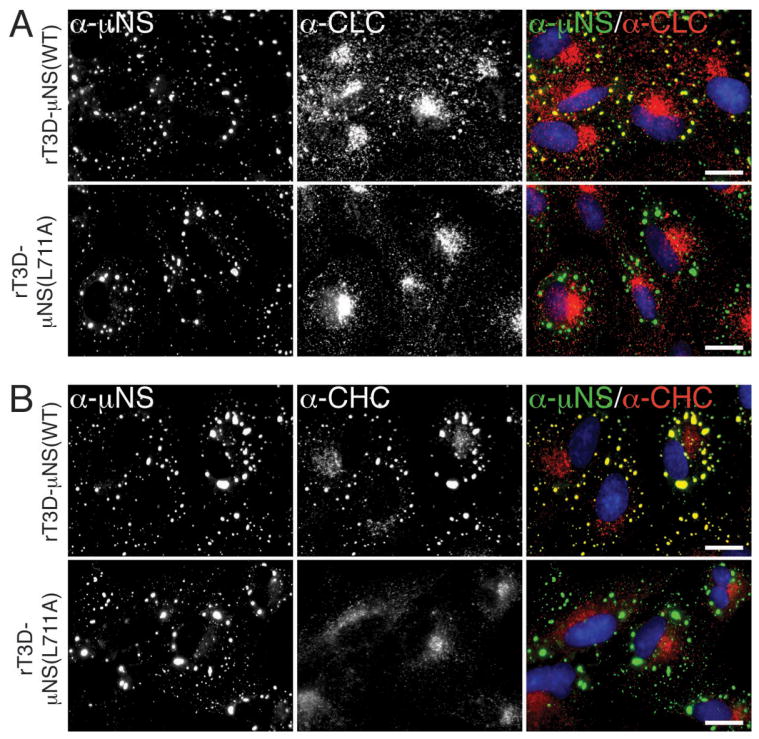
A, B) CV-1 cells were infected with second-passage stocks of rT3D-μNS(WT) or rT3D-μNS(L711A) at 5 PFU/cell. At 24 h p.i., cellular localizations of μNS (α-μNS, left column) and either clathrin LCA/B (α-CLC, middle column) (A) or clathrin HC (α-CHC, middle column) (B) were visualized as in Figure 2. In A and B, merged images are shown at right (α-μNS, green; α-CHC, red). Scale bars, 20 μm.
Figure 5. Sequestration of clathrin in MRV factories inhibits transferrin uptake.
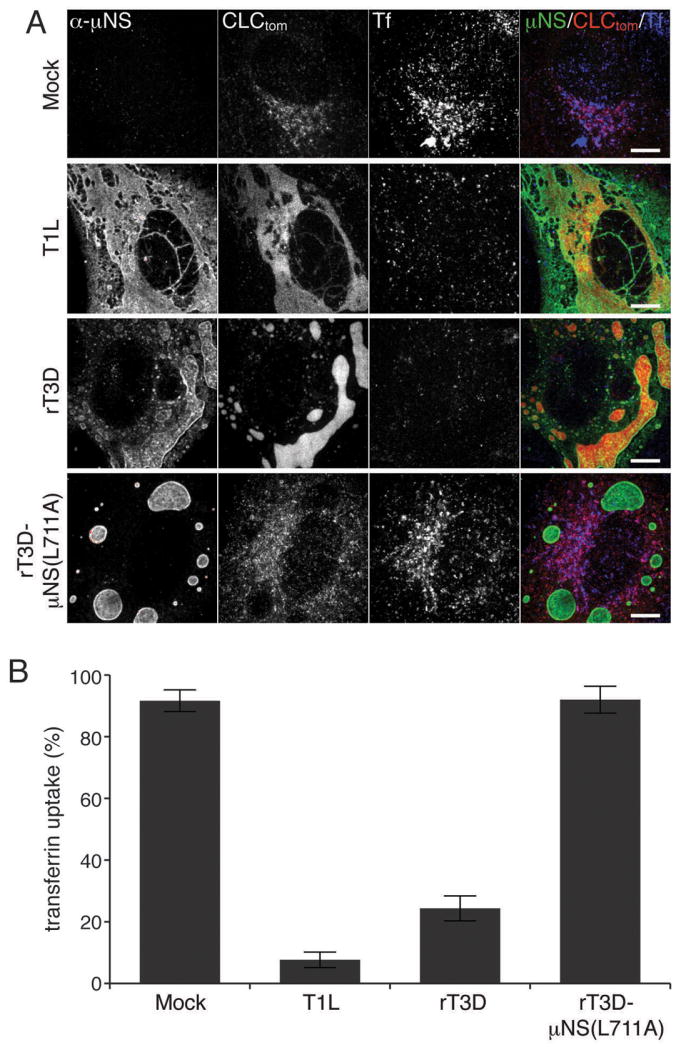
BSC-1 cells stably expressing clathrin LCA fused to tomato fluorescent protein (CLC-tom) were mock-infected or infected with either T1L, rT3D-μNS(WT) or rT3D-μNS(L711A) MRV. At 16h p.i. fluorescent-transferrin (Tf) uptake was performed as described in Materials and Methods and cells were fixed and immunostained using μNS-specific serum antibodies (α-μNS). A) Representative confocal images of mock- and MRV-infected cells. Merged images are shown at right (α-μNS, green; CLC-tom, red; Tf, blue). Scale bars, 10 μm. B) Bar plot depicting the percentage of cells showing perinuclear localization of the fluorescent Tf (Tf-uptake positive). The plot shows the average and the standard deviation of 3 independent experiments, each scoring at least 50, either mock- or MRV-infected (μNS-positive), cells.
Clathrin LCA/B and HC also colocalized with μNS in MRV factories in BSC-1 cells infected instead with ISVPs of strain T3DN or Type 3 Abney (T3A), as well as in CV-1, COS-7 or HeLa cells infected with T1L ISVPs (see Figure 1C for related results with CV-1 cells), suggesting a conservation of the observed phenotype across different MRV strains infecting different cell types. Additionally, clathrin HC colocalized with μNS in viral factories in BSC-1 cells infected with virions of strain T1L, T3DN, or T3A, indicating that this phenomenon reflects a general characteristic of MRV infection, not limited to infections with ISVPs.
Clathrin LCA/B and HC co-immunoprecipitate with μNS from infected-cell lysates
To provide further evidence for the association between clathrin and μNS in viral factories, we performed immunoprecipitation (IP) experiments. We incubated CV-1 cells with T1L ISVPs (infected) or buffer alone (mock infected) on ice for 1 h and then allowed infection to proceed for 10 h at 37°C. We next subjected post-nuclear supernatants to IP using either MAb CON.1 specific for clathrin LCA/B or μNS-specific serum antibodies. We analyzed both the immunoprecipitated proteins and those remaining in the post-IP supernatants by SDS/PAGE and immunoblotting using μNS-specific serum antibodies and MAb TD.1 specific for clathrin HC (34). CON.1 co-immunoprecipitated both μNS and clathrin HC along with clathrin LCA/B from infected-cell lysates (Figure 1C, lane 4). In the reverse IP by μNS-specific serum antibodies, clathrin HC co-immunoprecipitated with μNS (Figure 1C, lane 2). In sum, these results corroborate the conclusions from IF microscopy shown in Figure 1A and B. A notable decrease in the level of clathrin HC that remains in the post-IP supernatant following treatment of the infected-cell lysate with μNS-specific serum antibodies vs. mock treatment (Figure 1C, compare lanes 6 and 7), suggests that at these times p.i. a large proportion of clathrin HC exists in association with μNS. This finding is consistent with IF data in cells, where at similar times p.i. predominant staining for clathrin HC localized to viral factories (see related data for clathrin LCA in Figure 1A).
Adaptor proteins AP1 or AP2 do not colocalize with μNS in MRV factories
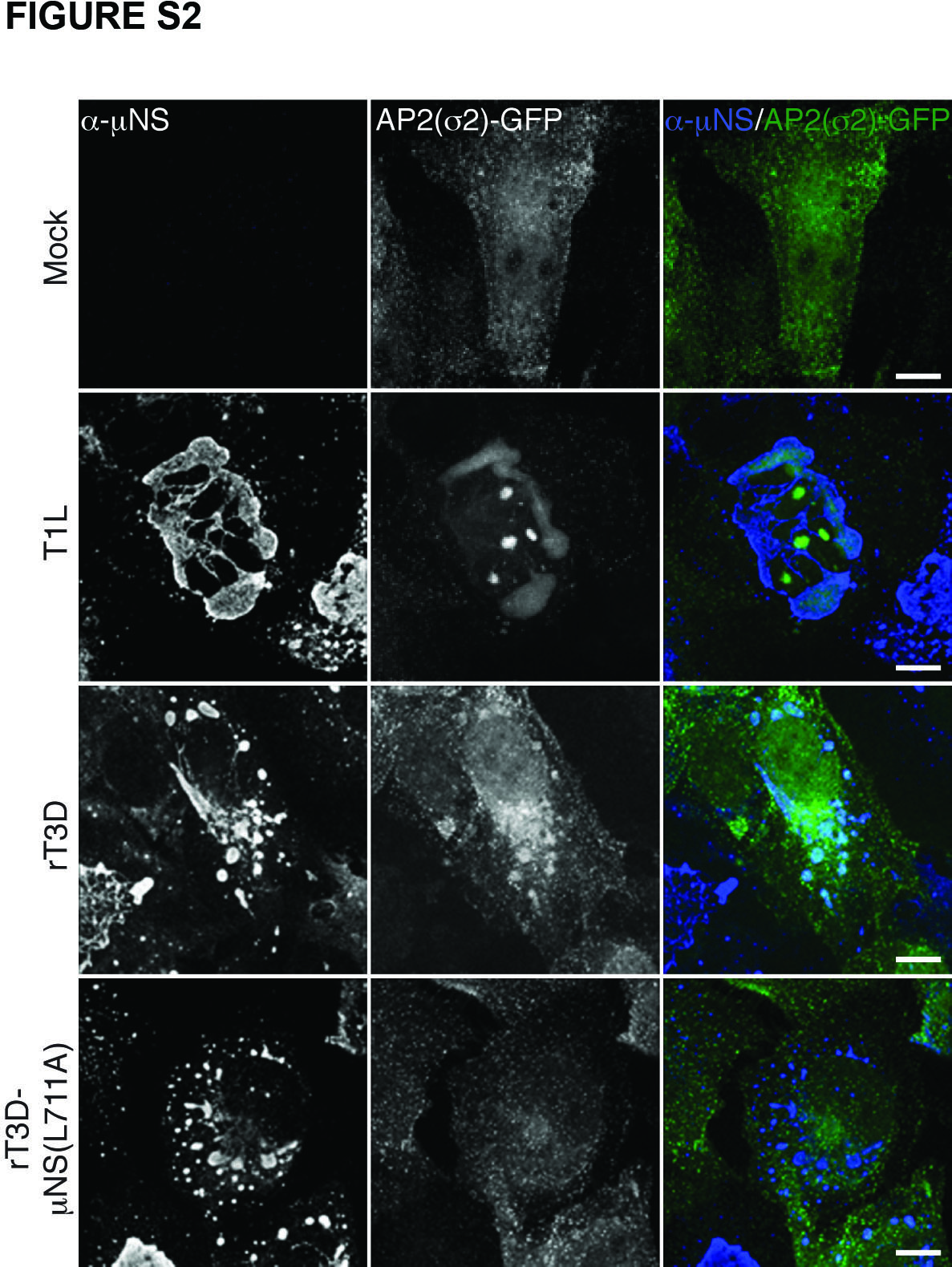
In the process of membrane vesiculation via the clathrin machinery, clathrin is recruited to the sites of vesiculation by interacting with adaptor proteins (28; reviewed in refs. 36 and 37). Adaptor proteins AP1 and AP2 are the canonical mediators of clathrin association with the membrane-trafficking machinery (26). How is clathrin recruited to MRV factories, which are devoid of membranes (8–12)? To address whether AP1 or AP2 might be present in factories, we infected BSC-1 cells with T1L ISVPs as described for Figure 1A, but then stained the cells with a combination of μNS-specific serum antibodies and MAb 10A specific for the β subunits of both AP1 and AP2. The staining pattern of 10A observed at 0 h p.i., reflective of the perinuclear localization of AP1 and plasma membrane localization of AP2, remained unchanged at 3, 7 and 12 h p.i. (Figures 1D and S1B). AP1 and AP2 were absent from factories not only in BSC-1 cells infected with T1L ISVPs but also from ones infected with ISVPs of strain T3DN or T3A. At very late times p.i. (e.g., 16 or 24 h in experiments presented in Figure 6 below) when factories nearly fill the cytoplasm, some level of stably expressed AP2(σ2)-GFP (in BSC-1 AP2(σ2)-GFP cells) (38) colocalized with these structures, showing that AP2 can be recruited to viral factories (Figure S2). Recruitment of AP2 largely lags behind that of clathrin, suggesting that it occurs as a result of the interaction with clathrin molecules and not the other way around. Combined, these data strongly argue against the requirement for AP2 in recruiting cellular clathrin to viral factories and suggest an alternative, non-AP1/2-dependent mode of clathrin recruitment to those structures.
Figure 6. Sequestration of clathrin in MRV factories increases the lifetime of clathrin-coated pits.
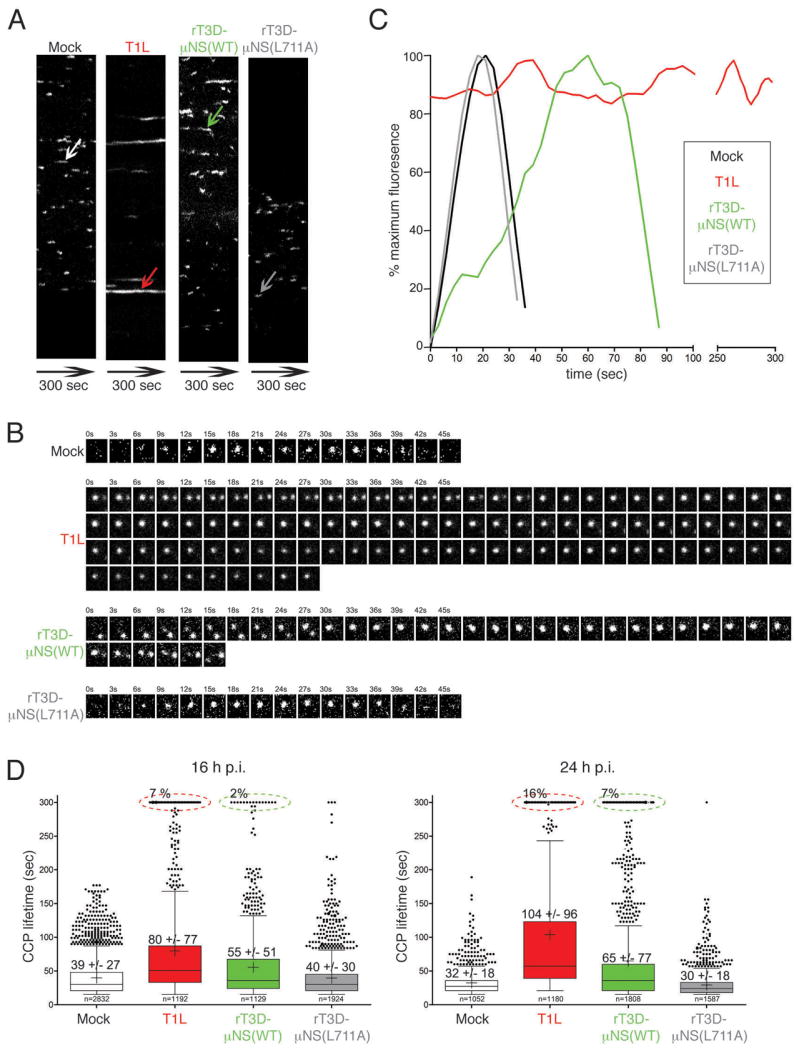
BSC-1 cells stably expressing AP2(σ2)-GFP were mock-infected or infected with either T1L, rT3D-μNS(WT) or rT3D-μNS(L711A) MRV. 16 h p.i. dynamics of clathrin-coated pits was monitored in live cells by taking time-lapse images every 3 sec for the total of 300 sec by confocal microscopy. The CCPs were identified and tracked over time as described in Materials and Methods. A) Kymographs of mock- and MRV-infected cells showing AP2(σ2)-GFP fluorescence projected over time (X-axes). Each fluorescent trace in the X-axis represents an individual CCP forming and disassembling at the membrane of the imaged cell, and the length of the trace reflects the CCP lifetime. The arrows point to a representative CCP analyzed further in B and C. B) Tile view of a representative CCP (shown by arrow in A) forming in either mock-infected, T1L-, rT3D-μNS(WT)- and rT3D-μNS(L711A)-infected cell. C) Fluorescence-intensity profiles of AP2(σ2)-GFP recruitment to CCPs shown by tile view in B. Weighted averages of smoothed fluorescent time trajectories are shown in each case. Mock-infected, black line; T1L, red line; rT3D-μNS(WT), green line; rT3D-μNS(L711A), grey line. D) Box-and-whisker plot comparing CCP lifetimes in mock-, T1L-, rT3D-μNS(WT)- and rT3D-μNS(L711A)-infected cells either at 16 h p.i. (left) or at 24 h p.i. (right). The bottom and top boundaries of each box represent the 25th and the 75th percentile, respectively, defining the interquartile range (IQR) between them. The horizontal line in each box marks the median, and the cross marks the average CCP lifetime. The whiskers on either side of the boxes represent the lowest and the highest data value still within 1.5 IQR of the lower and the upper quartiles. The dots represent the values outside the range encompassed by the whiskers. At least 1000 CCPs derived from three different cells were analyzed for each sample. Circled events (dashed lines) represent arrested CCPs.
Clathrin LCA/B and HC, but not AP1 or AP2, colocalize with μNS in factory-matrix structures
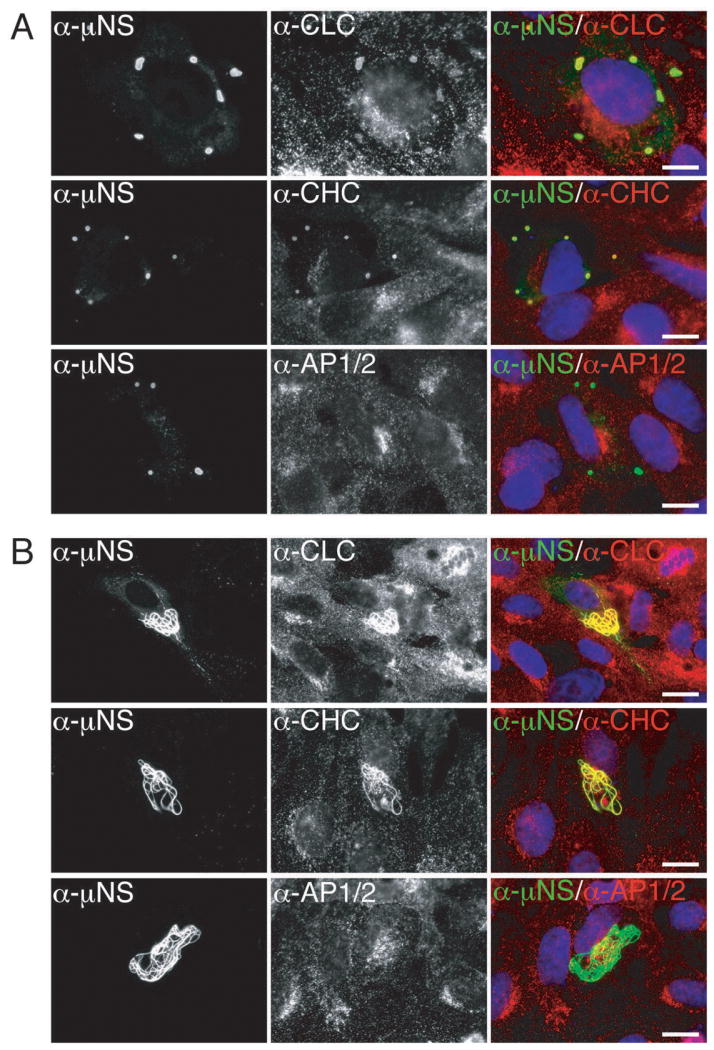
In all preceding experiments, the association between μNS and clathrin might have required a bridging viral molecule. Since expression of μNS alone yields globular factory-matrix structures in plasmid-transfected cells (13–15), we asked whether clathrin accumulated in these structures in the absence of other MRV proteins. We transfected CV-1 cells with a plasmid expressing T1L μNS and analyzed them at 24 h post-transfection (p.t.) by indirect IF microscopy using a combination of μNS-specific serum antibodies and one of three MAbs: CON.1 against clathrin LCA/B, X22 against clathrin HC and 10A against AP1/2. As in the case of infected cells, both clathrin LCA/B and clathrin HC, but not AP1 or AP2, colocalized with μNS in the factory-matrix structures (Figure 2A). These same results were obtained upon expression of T3DN μNS in plasmid-transfected CV-1 cells and upon expression of either T1L or T3DN μNS in plasmid-transfected BSC-1 cells. We conclude that μNS is sufficient for clathrin recruitment in the absence of any other MRV protein.
Figure 2. Clathrin LCA/B and clathrin HC, but not AP1 or AP2, colocalize with μNS in both globular and filamentous factory-matrix structures.
A) μNS-WT protein was expressed in CV-1 cells by plasmid-based transfection. At 24 h p.t, cellular localizations of μNS as well as clathrin LCA/B, clathrin HC or AP1/2 were visualized by indirect IF microscopy using a combination of μNS-specific serum antibodies (α-μNS) and MAb CON.1 (α-LC), X22 (α-HC) or 10A (α-AP), respectively, as labeled. B) μNS-WT and μ2 proteins were co-expressed in CV-1 cells by plasmid-based transfection. At 38 h p.t., cellular distributions of indicated proteins were visualized as in A. In A and B, merged images are shown at right (α-μNS, green; α-CLC, α-CHC or α-AP1/2, red). Scale bars, 20 μm.
When T1L μNS was co-expressed with the T1L microtubule-anchoring and μNS-associating protein μ2 (9,13), clathrin LCA/B and HC, but not AP1 or AP2, continued to colocalize with μNS in the resulting factory-matrix structures at 38 h p.t. The observed structures exhibited filamentous morphology (Figure 2B), similar to that seen at later times p.i. in T1L-infected cells (see Figure 1A) and as expected from previous co-expressions of μNS and μ2 (13,17,21). μNS association with clathrin in transfected cells can thus occur in the setting of either globular (absence of μ2) or filamentous (presence of μ2) factory-matrix structures.
μNS contains a putative clathrin-binding motif required for clathrin recruitment to factory-matrix structures
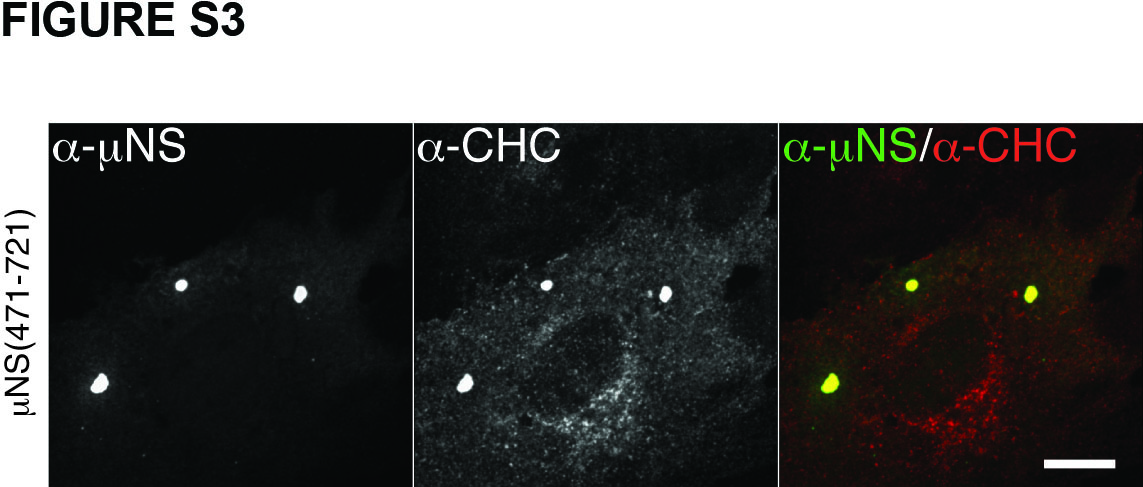
Clathrin binds directly to adaptor proteins such as AP1 or AP2, and more specifically, the NTD of clathrin HC binds directly to the clathrin-box motif in each adaptor (25,27). The clathrin-box motif is a five-residue sequence with the following consensus: L(L,I)(D,E,N)(L,F) (D,E) (27) (Figure 3A). The fifth position is most variable and in fact there are examples of functional clathrin-box sequences that lack the fifth residue altogether or contain an alternative residue such as Ser at this position (39–41). Upon inspection of the 721-aa μNS sequence, we identified a potential such clathrin-binding motif, LIDFS, just before the C-terminus (aa 711–715) of μNS of different MRV strains (Figure 3A). A similar motif, LFGLD, spans residues 191 to 195 of μNS. However, the observation that clathrin is recruited to factory-matrix structures by N-terminal truncation mutant μNS(471–721) (Figure S3), which lacks the 191–195 element, focused our attention instead on the C-terminal regions of μNS.
To address whether the putative clathrin-binding motif near the C-terminus of μNS is functional for recruiting clathrin to factory-matrix structures, we first made use of one previously engineered (Broering et al., 2005) and two newly engineered C-terminal truncation mutants of T1L μNS, which either preserved this motif intact (truncations μNS-718 and μNS-715) or deleted a portion of it (truncation μNS-713) (Figure 3A). Whereas wild-type μNS (μNS-WT), μNS-718 and μNS-715 associated with clathrin HC (Figure 3B, lanes 7, 9 and 10) in the IP assay after expression in plasmid-transfected CV-1 cells, μNS-713 did not (Figure 3B, lane 8). We next tested a mutant of T1L μNS in which one of the key large hydrophobic residues within the motif, Leu711, was changed to Ala (mutation L711A) (Figure 3A). Whereas μNS-WT associated with clathrin HC (Figure 3C, lane 1), μNS-L711A did not (Figure 3C, lane 2). Together, the experiments with mutants of μNS are consistent with the LIDFS sequence in μNS representing a functional clathrin-binding motif. Because C-terminal μNS truncations also delete important determinants of factory-matrix formation independent of clathrin interaction (e.g., μNS-715 and μNS-718 are defective at forming factory-matrix structures while retaining the ability to interact with clathrin), we used μNS-L711A for subsequent analyses.
To eliminate the possibility that the lack of association between μNS-L711A and clathrin HC was due to misfolding and to provide further evidence that this mutation disrupts association of clathrin LCA/B and HC with μNS, we separately co-expressed μ2 along with either μNS-WT or μNS-L711A by plasmid transfection. The mutant was effectively expressed and recruited to microtubules by μ2 (Figure 3D), suggesting that the mutation did not result in misfolding that abrogated its binding to μ2. Notably, the mutant did not recruit either clathrin LCA/B or clathrin HC (Figure 3D).
We next compared the abilities of μNS-WT and μNS-L711A to induce factory-matrix structures upon plasmid-based expression in CV-1 cells. We analyzed the cells at 24 h p.t. by indirect IF microscopy using a combination of μNS-specific serum antibodies and either MAb CON.1, specific for clathrin LCs or MAb X22, specific for clathrin HC (Figure 3E). Cells expressing μNS-L711A contained numerous factory-matrix structures with smooth perimeters, indistinguishable from those formed by μNS-WT (Figure 3E, compare top and bottom rows). As expected from the IP results, the intensity of CON.1 or X22 staining of the structures that formed upon expression of μNS-L711A was greatly reduced (Figure 3E). Together, the experiments presented in this section provide strong evidence that conserved residue Leu711 within the putative clathrin-binding motif of μNS is required for recruiting clathrin to factory-matrix structures. The data further suggest that clathrin does not serve an essential role in forming the factory matrix, in that factory-matrix structures continued to be formed in cells expressing μNS-L711A.
To test whether the NTD of clathrin HC (25) may be involved in recruitment to factory-matrix structures upon μNS expression, we expressed influenza virus hemagglutinin (HA)-tagged NTD (NTD-HA) alone or in combination with T1L μNS by plasmid transfection of CV-1 cells. In the absence of μNS, NTD-HA was distributed throughout the cell, though concentrating in the perinuclear region as expected for this protein that retains capacity to bind clathrin-box motifs (25) (Figure 3F, top row). Co-expression with μNS, in contrast, allowed recruitment of a subset of expressed NTD-HA to factory-matrix structures (Figure 3F, bottom row), and co-immunoprecipitation confirmed this association (Figure 3C, lanes 3 and 4). We conclude that the NTD of clathrin HC is sufficient for localization to factory-matrix structures.
Clathrin recruitment to viral factories is dispensable for their formation
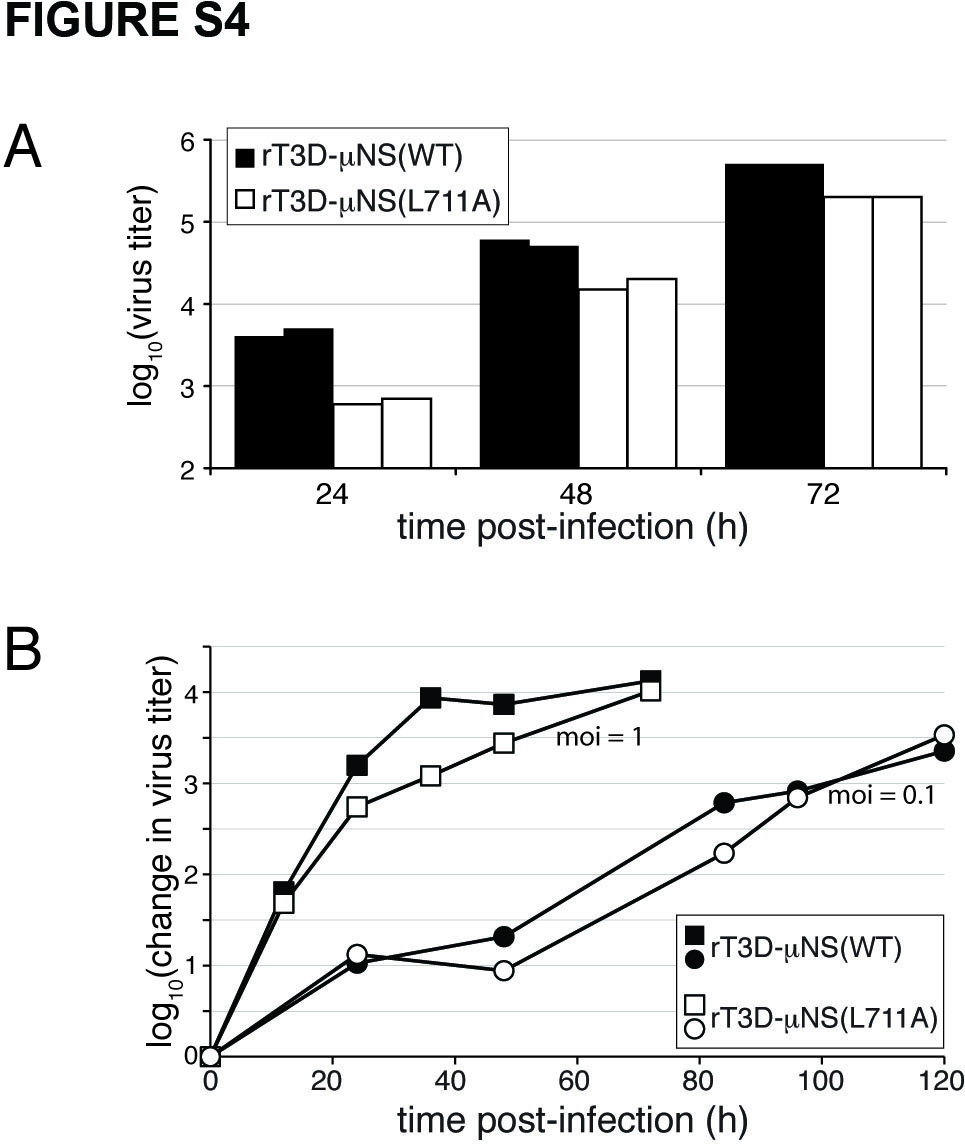
To address the requirement for clathrin recruitment to viral factories in the context of MRV infection, we attempted to rescue infectious, recombinant viruses encoding either μNS-WT or μNS-L711A in the T3D background (rT3D-μNS(WT) and rT3D-μNS(L711A), respectively) (see Materials and Methods for plasmid-based rescue protocol). Both recombinant viruses were indeed rescued, stably passaged into cell-lysate stocks, and used to generate purified virions with similar particle/PFU ratios and similar plaque sizes. We then compared the recombinant viruses for their abilities to form viral factories and to recruit clathrin to those structures. We infected CV-1 cells for 24 h and then analyzed the cells by indirect IF microscopy using a combination of μNS-specific serum antibodies and either MAb CON.1, specific for clathrin LC (Figure 4A) or MAb X22, specific for clathrin HC (Figure 4B). Consistent with the plasmid-based μNS-expression data in Figure 3E, rT3D-μNS(WT) and rT3D-μNS(L711A) were indistinguishable in their abilities to form globular viral factories, characteristic of infection by the T3D isolate from which they were derived. In contrast, rT3D-μNS(L711A) was distinct in failing to recruit either clathrin LCA/B or clathrin HC to the factories (Figure 4). From these results, we first conclude that conserved residue Leu711 within the putative, C-proximal clathrin-binding motif of μNS is required for clathrin recruitment even in the presence of other viral proteins in the setting of MRV infection. Based on this conclusion, as well as preceding evidence for the sufficiency of μNS for clathrin recruitment in transfected cells, we speculate that μNS is likely the sole MRV protein responsible for clathrin recruitment to viral factories. From the results in this section, we further conclude that clathrin recruitment to viral factories is dispensable for their formation in infected cells. Moreover, given that recombinant virus rT3D-μNS(L711A) was rescued and stably maintained in cultured cells, we conclude that clathrin recruitment to viral factories is not essential for MRV growth in culture. Results of experiments to compare infectious titers of rT3D-μNS(WT) and rT3D-μNS(L711A) at different times p.i. concurred with this conclusion, although a mild kinetic defect in the growth of rT3D-μNS(L711A) could not be ruled out (Figure S4).
In the following experiments, we turned our attention to address the effects of clathrin sequestration by μNS on normal clathrin functions in the cell.
Sequestration of clathrin in MRV factories inhibits transferrin uptake
Clathrin is an abundant cytoplasmic protein, which constitutes the building block of CCVs that selectively sort cargo at the plasma membrane, TGN and endosomal compartments for multiple membrane-trafficking pathways. To characterize the impact of clathrin redistribution to viral factories during MRV infection, we monitored the uptake of transferrin (Tf) into BSC-1 cells. Tf is an iron-binding glycoprotein, which is internalized into cells by clathrin-mediated endocytosis after engagement of its cellular receptor, TfR. We mock-infected BSC-1 cells stably expressing clathrin LCA tagged with tomato fluorescent protein (LCA-tom) (42) or infected them with one of the following types of ISVPs: T1L, rT3D-μNS(WT) or rT3D-μNS(L711A) for 16 h at 37°C. We then pulsed the cells with fluorescently labeled human Tf for 7 min at 37°C. After washing the excess Tf and removing the remaining plasma-membrane-bound Tf by acid wash, we fixed and immunostained the cells for μNS. Mock-infected cells efficiently internalized fluorescent Tf as revealed by extensive labeling of the endosomal compartment (Figure 5A, top row). Quantification of at least 50 mock-infected cells in each of the three independent experiments yielded an average of 92% as showing significant endosomal labeling (Tf-positive) (Figure 5B). We detected much less Tf inside the cells infected with either T1L or rT3D-μNS(WT) viruses (Figure 5A, second and third rows), yielding an average of 8 and 24% Tf-positive cells, respectively (Figure 5B). As expected, cells infected with rT3D-μNS(L711A), which fails to recruit clathrin to viral factories, efficiently internalized Tf and were indistinguishable from mock-infected cells both qualitatively (Figure 5A, compare top and bottom rows) and quantitatively, yielding an average of 92% Tf-positive cells (Figure 5B).
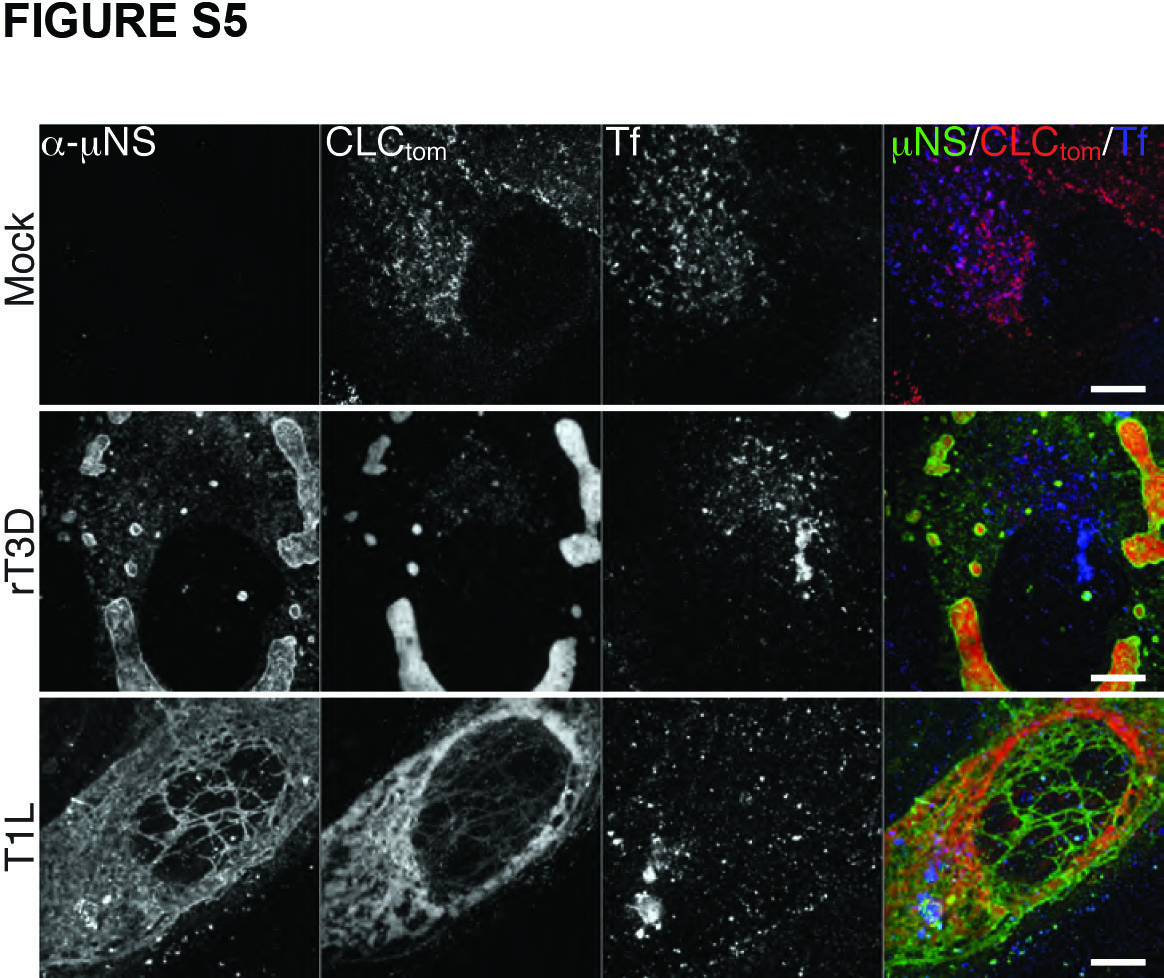
Inhibition of Tf uptake by infected cells was limited to late times p.i. (at least 16 h p.i.), when most of the cytoplasmic clathrin has been redistributed to viral factories (see Figures 1 and 5) in most cells. We observed significant Tf uptake up to 12 h p.i. with either T1L or rT3D-μNS(WT) viruses (Figure S5). There was a direct correlation between the size of factories, the amount of clathrin recruited to them and the efficiency of Tf uptake. In cells with small factories, even at 16 h p.i., the localization of most clathrin was unaffected and we observed no inhibition of Tf uptake. Infection with rT3D-μNS(WT) resulted in greater variation in factory size between different cells than did that with T1L, and as a result less overall inhibition of Tf uptake at the measured time point (16 h p.i.) (Figure 5B). Althougth rT3D-μNS(WT) and rT3D-μNS(L711A) were indistinguishable in factory size and morphology (see Figures 4 and 5A), clathrin-mediated endocytosis of Tf was unaffected in cells infected with rT3D-μNS(L711A) (Figure 5), arguing that the inhibition of clathrin-mediated endocytosis observed in rT3D-μNS(WT)-infected cells was not an indirect consequence of viral infection, but rather a direct effect of sequestering clathrin in viral factories. These data thus indicate that extensive sequestration of clathrin in viral factories inhibits clathrin-dependent endocytosis of a cellular receptor-ligand complex, Tf-TfR.
Sequestration of clathrin in MRV factories extends the lifetime of clathrin-coated pits
To demonstrate that sequestration of clathrin in MRV factories has a direct effect on CCP formation independent of a specific cargo molecule, we analyzed the dynamics of clathrin-mediated endocytosis in live BSC-1 cells stably expressing AP2(σ2)-GFP. We monitored the formation of CCPs at the plasma membrane by following the AP2(σ2)-GFP signal over time using fluorescent spinning-disk confocal microscopy. Previous work has demonstrated that in the course of CCV formation, the AP2(σ2)-GFP signal displays a characteristic pattern over time: appearance of a fluorescent spot, increase in fluorescence intensity over time, followed by sudden dissipation (28; reviewed in ref. 36). The increase in fluorescence results from incorporation of AP2(σ2)-GFP in the growing CCP. The abrupt dissipation is interpreted as uncoating of the CCV after its budding from the plasma membrane.
In the current experiments, we compared CCP lifetimes between mock-infected cells, and ones infected with either T1L, rT3D-μNS(WT) or rT3D-μNS(L711A) ISVPs at 16 h p.i. We confirmed infection of analyzed cells by observing viral factories by phase microscopy. We generated time-lapse videos recording the bottom surface of a BSC-1 AP2(σ2)-GFP cell by taking individual images every 3 sec for the total duration of 300 sec (see Videos S2–S5 online). The majority of the fluorescent AP-2 spots belonged to a single class of diffraction-limited objects, as expected for CCPs or CCVs, about 100–200 nm in diameter (28,30,42). To analyze the dynamics of CCP formation, we generated kymograph views comparing different samples (Figure 6A). Here, a single imaged axis showing AP2(σ2)-GFP fluorescence (Figure 6A, Y-axis) is projected over time (Figure 6A, X-axes). The length of each continuous fluorescent trace in the X-axis reflects the lifetime of each individual CCP. Qualitative comparison of kymographs reveals extension of CCP lifetimes in cells infected with either T1L or rT3D-μNS(WT) relative to either mock- or rT3D-μNS(L711A)-infected cells (Figure 6A).
This initial observation was corroborated by analysis of fluorescence-intensity profiles of individual CCPs over time (representative examples shown in Figure 6B and C). Mock-infected cells displayed the previously observed profile of AP-2(σ2)-GFP fluorescence over time (28,30). In T1L-infected cells, many CCPs had extended lifetimes and a portion of them remained stationary during the course of the experiment (one such stationary structure is shown in Figure 6B and C, and others in Figure 6D, below). In rT3D-μNS(WT)-infected cells, most of the pits were active, but their lifetimes were significantly longer compared to those in mock-infected cells. Finally, in rT3D-μNS(L711A)-infected cells, the CCP fluorescence-intensity profiles were indistinguishable from those in mock-infected cells.
To assess the effect of MRV infection on the dynamics of CCP formation more quantitatively, we compiled lifetime data from at least 1000 CCPs derived in each case from 3 cells either mock-infected or infected with one of the following types of ISVPs: T1L, rT3D-μNS(WT) or rT3D-μNS (L711A). We considered a fluorescent structure to be a CCP if this structure was dynamic, appeared and disappeared within the time window of the data series (300 sec), and had a lifetime of at least 15 sec. Objects that existed both at the beginning and the end of the data series were counted as arrested CCPs. Objects that existed at the beginning but disappeared during the experiment time window, or those that appeared during the experiment time window but that did not disappear before the end, were not counted. To compare the CCP-lifetime distributions in mock- to those in MRV-infected cells, we show the box-and-whisker plot for each condition (Figure 6D). The average CCP lifetime in mock-infected cells was 39 ± 27 sec, similar to previously published data (28,30,42). In T1L- and rT3D-μNS(WT)-infected cells at 16 h p.i., short-lived CCPs were under-represented, causing a shift in the population toward longer lifetimes, yielding averages of 80 ± 77 sec and 55 ± 51 sec, respectively (Figure 6D, left). Moreover, around 7% and 2% of the pits in T1L- and rT3D-μNS(WT)-infected cells, respectively, were completely arrested and present throughout the entire time series. As expected, in rT3D-μNS(L711A)-infected cells the CCP-lifetime distribution was statistically identical to that in mock-infected cells, with an average lifetime of 40 ± 30 sec.
As mentioned in preceding sections, we found a correlation between the size of MRV factories, consequent amount of clathrin sequestered in them (see Figure 1) and effect on clathrin-dependent Tf uptake (Figure 5). We hypothesized that at even later times p.i., when viral factories grow to occupy much of the cytoplasmic volume and sequester much of the cytoplasmic clathrin (see Video S1 online), the fraction of arrested CCPs would be larger. We hence additionally compared CCP lifetimes in an identical experiment to that above, but at 24 h p.i. The average CCP lifetimes increased to 104 ± 96 sec and 65 ± 77 sec in T1L- and rT3D-μNS(WT)-infected cells, respectively. The fraction of arrested CCPs followed this increase to 16% and 7%, respectively. The arrested CCPs were virtually absent from either mock- or rT3D-μNS(L711A)-infected cells, and again no statistical difference in average CCP lifetimes was observed between rT3D-μNS(L711A)- and mock-infected cells (30 ± 18 sec versus 32 ± 18 sec, respectively). Despite strain-specific differences in the extent of the measured effects (T1L vs. rT3D-μNS(WT)), all our analyses of CCP lifetimes strongly argue that MRV infection negatively affects the dynamics of CCP formation at the plasma membrane, independent of a specific endocytic cargo molecule and in a manner dependent on clathrin sequestration by μNS.
Sequestration of clathrin in MRV factories inhibits VSV-Gts-GFP protein secretion
To test whether clathrin sequestration in viral factories affects the cellular secretory pathway, we used a well-characterized assay monitoring secretion of GFP-tagged temperature-sensitive VSV-G (VSV-Gts-GFP) (43). At 40°C VSV-Gts-GFP misfolds and cannot exit the ER. Upon shifting to 32°C, VSV-Gts-GFP refolds and can then exit the ER to the Golgi, enabling its subsequent clathrin-dependent transport to endosomes that target it to the plasma membrane. To test the effect of MRV infection on this process, we transfected BSC-1 cells with the plasmid expressing VSV-Gts-GFP and incubated the cells at 40°C for 16 h to allow VSV-Gts-GFP expression, but to prevent its correct folding. We then mock-infected the cells or infected them with one of the following types of ISVPs: T1L, rT3D-μNS(WT) or rT3D-μNS(L711A) for 16 h at 40°C. At this point we either shifted the cells to 32°C or kept a duplicate sample at 40°C for 2 h more before fixing and immunostaining for μNS. In mock-infected cells kept at 40°C for the duration of the experiment, we observed VSV-Gts-GFP localized almost exclusively in the ER (Figure 7A, Mock, top row). Incubation at 32°C allowed for significant transfer of VSV-Gts-GFP to the Golgi, and its subsequent clathrin-dependent transport to the plasma membrane (Figure 7A, Mock, bottom row). Quantification of at least 50 mock-infected cells in each of the 3 experiments revealed an average of 10% VSV-Gts-GFP secretion at 40°C (i.e., 90% of cells had VSV-Gts-GFP strictly localized in the ER and only 10% showed some plasma membrane localization), whereas VSV-Gts-GFP secretion increased to 92% after the shift to 32°C (Figure 7B). All MRV-infected cells incubated at 40°C for the duration of the experiment displayed predominant ER localization of VSV-Gts-GFP indistinguishable from that in mock-infected cells incubated under the same conditions (Figure 7A and B). In cells infected with either T1L or rT3D-μNS(WT), VSV-Gts-GFP failed to efficiently relocalize to the plasma membrane after the shift to 32°C, showing only 9% and 38% VSV-Gts-GFP secretion, respectively (Figure 7). As expected, in cells infected with rT3D-μNS(L711A), in which μNS does not sequester clathrin, VSV-Gts-GFP efficiently relocalized to the plasma membrane after the shift to 32°C, a phenotype indistinguishable from mock-infected cells both qualitatively (Figure 7A) and quantitatively, yielding 88% of VSV-Gts-GFP secretion (Figure 7B).
Figure 7. Sequestration of clathrin in MRV factories blocks secretion of VSV-Gts-GFP.
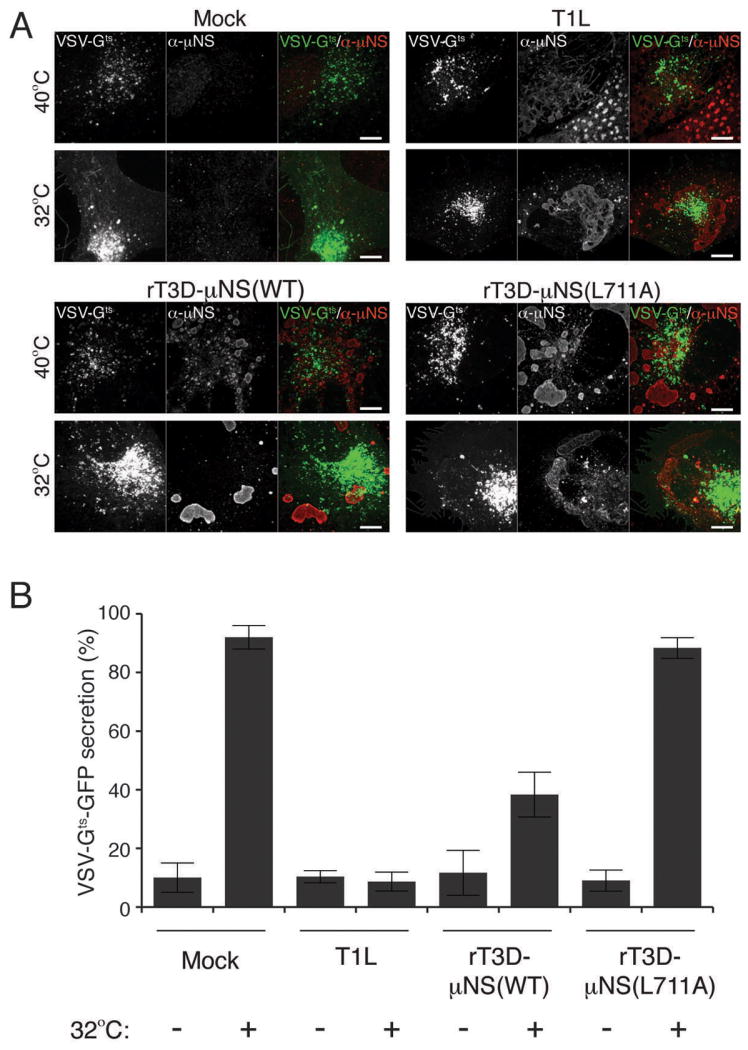
BSC-1 cells were transfected with VSV-Gts fused to GFP and incubated overnight at 40°C. Cells were then mock-infected or infected with either T1L, rT3D-μNS(WT) or rT3D-μNS(L711A) MRV and incubated at 40°C for 12-16 h. 2 h prior to fixation, cells were either kept at 40°C or transferred to 32°C to allow VSV-Gts-GFP protein refolding and secretion from the TGN to the plasma membrane. Fixed cells were immunostained using μNS-specific serum antibodies (α-μNS) and observed by confocal microscopy. A) Representative confocal images of mock- and MRV-infected cells, either kept at 40°C (top panels) or shifted to 32°C (bottom panels). Merged images are shown at right (VSV-Gts-GFP, green; α-μNS, red). Scale bars, 10 μm. B) Bar plot depicting the percentage of cells, which showed plasma-membrane localization of VSV-Gts-GFP in addition to the perinuclear staining after the shift to 32°C (VSV-Gts-GFP-secretion positive). The plot shows the average and the standard deviation of 3 independent experiments, each scoring at least 50 VSV-Gts-GFP-expressing, either mock- or MRV-infeceted (μNS-positive) cells.
Similar to results for Tf uptake (see Figure 5), we observed a correlation between the size of MRV factories, the level of cellular clathrin redistrbution and the inhibition of VSV-Gts-GFP secretion. In cells with small factories, even at 24 h p.i., the localization of most clathrin was unaffected, and VSV-Gts-GFP was efficiently transported to the plasma membrane. Infection with rT3D-μNS(WT) resulted in greater variation in factory size between different cells than that with T1L, and as a result less overall inhibition of VSV-Gts-GFP secretion at the measured time point (16 h p.i.) (Figure 7B). Althougth rT3D-μNS(L711A) and rT3D-μNS(WT) were indistinguishable in factory size and morphology (see Figures 4, 5A and 7A), clathrin-dependent VSV-Gts-GFP secretion was unaffected in cells infected with rT3D-μNS(L711A) (Figure 7), arguing that the inhibition of clathrin-dependent secretion of VSV-Gts-GFP observed in rT3D-μNS(WT)-infected cells was not an indirect consequence of viral infection, but rather a direct effect of clathrin sequestration by μNS. Together, the results in this section demonstrate that the effect of clathrin sequestration by μNS in viral factories during MRV infection is not limited to clathrin-mediated endocytosis, but includes interference with another clathrin-dependent process: protein secretion from TGN to plasma membrane.
Conclusions
Abundant literature describes utilization of clathrin-mediated endocytosis by both viruses and bacteria during internalization into cells (reviewed in refs. 31 and 45), including for MRV uptake into endocytic vesicles during cell entry (28; S. B., M. L. N. and T. K., manuscript in preparation). In this study, however, recruitment of clathrin to viral factories by μNS, which is not a component of entering MRV particles, suggests that clathrin plays an additional role in the MRV life cycle, apart from its role in endocytic uptake. Involvement of clathrin at a stage of MRV infection following cell entry is reminiscent of findings with HDV, for which clathrin HC has been shown to interact with HDV protein Ag-L, with important consequences for HDV particle assembly (32,33). On the other hand, there is no evidence to date that HDV disrupts clathrin functions. Thus, to our knowledge, μNS-mediated clathrin sequestration represents the only reported example of a viral or bacterial protein whose interaction with clathrin inside cells interferes with clathrin-dependent cellular functions.
A seemingly unusual aspect of the mechanism by which MRV interferes with clathrin-dependent membrane trafficking is that it depletes the most abundant component of this cellular machinery, i.e., clathrin itself. Although inefficient, this strategy has important consequences for the timing with which trafficking is disrupted: disruption does not become substantive until later in MRV infection, perhaps ensuring that cellular functions remain intact during the critical phases of virus multiplication. In this light, one simple reason for the virus to inhibit clathrin-dependent membrane trafficking at later times might be to prevent superinfection of a cell whose resources have by then been used up.
Although clathrin recruitment to viral factories is not essential for factory formation or MRV growth in cultured cells, the consequences of inhibiting clathrin functions seem likely to be more significant during growth in an animal host, where multiple rounds of virus multiplication occur in complex tissue settings and in parallel with host responses. Inhibition of clathrin-dependent membrane trafficking might, for example, reduce the cell's capacity for innate immune signaling or for presentation of viral antigens in MHC complexes. Given evidence that clathrin depletion in polarized cells has negative consequences for basolateral sorting (46), another interesting possibility is that MRV infection of polarized epithelial cells in the infected animal might, through clathrin sequestration by μNS and consequent effects on basolateral polarity, damage tight junctions and facilitate virus spread.
Another unusual aspect of clathrin recruitment by μNS is that this process takes place within structures, viral factories, that are largely devoid of membranes (8–12). As a parallel to μNS-clathrin association, binding of clathrin to kinetochore fibers during mitosis appears to be independent of membranes or the endocytic process (47), and adaptor proteins such as AP1 or AP2 are not detected at the sites of clathrin localization during mitosis (47,48). There clathrin seems to stabilize the kinetochore fibers either directly or indirectly and appears required for normal chromosome congression (47,49). Thus, MRV might also benefit from the effect that clathrin sequestration may have on cell-cycle progression.
μNS is an 80-kDa, 721-aa protein, of which the C-terminal one-third (aa 471–721) is necessary and sufficient for formation of the MRV factory matrix (15,44) and the N-terminal one-third (aa 1–221) is necessary and sufficient for recruitment of viral proteins σNS, λ1, λ2, μ2, and σ2 to the factory matrix (14,16–18). Deletion of the two most C-terminal residues of μNS, Glu720 and Leu721, prevents factory-matrix formation, resulting in diffuse cytoplasmic distribution of the protein (44). Here we identify a putative clathrin-binding motif near the C-terminus of μNS, spanning aa 711–715. Ala substitution for Leu at μNS aa 711, the only fully conserved residue among diverse clathrin-box motifs in other known clathrin-binding proteins (27), inhibits clathrin association with μNS, without an apparent effect on ability of μNS to form the factory matrix. Similarly, μNS with three- or six-residue C-terminal deletions that abolish formation of the factory matrix (μNS-715 and μNS-718) continues to co-immunoprecipitate with clathrin, consistent with the conclusion that these truncations retain the putative clathrin-binding motif, whereas μNS with an eight-residue C-terminal deletion (μNS-713) fails to co-immunoprecipitate with clathrin, consistent with the conclusion that this truncation lacks one or two C-terminal residues of the putative clathrin-binding motif. Thus, the determinant of factory-matrix formation located at the extreme C-terminus of μNS is separable from the C-proximal determinant of clathrin interaction, and the putative clathrin-binding motif spanning μNS residues 711 to 715 appears to be required for clathrin recruitment.
μNS-WT, but not μNS-L711A, recruits the NTD of clathrin HC, consistent with the clathrin-binding motif of μNS engaging the same general region of clathrin HC as is engaged by cellular proteins with bona fide clathrin-box motifs. However, defining the precise mode of binding between μNS and NTD, such as whether it involves the same site on NTD and the same type of peptide-in-groove interaction as seen with cellular adaptors (25), requires additional studies. Even though known single clathrin-box motifs displayed on peptides mediate low-affinity, dynamic interactions with the NTD (50,51), we believe that the interaction between viral factories or factory-matrix structures and clathrin is of apparently higher affinity at least in part because it results from NTD binding to a concentrated accretion of μNS molecules in those structures.
Recruitment of clathrin by μNS occurs in the context of either filamentous or globular viral factories, as respectively formed by MRV strains T1L and T3D. Infection of cells by T1L inhibits clathrin-dependent membrane trafficking to a greater extent than does rT3D (see Figures 5–7). The extent of inhibition of cellular clathrin functions is proportional to the size of factories in a given cell. We explain the smaller effect of rT3D infection by the presence of a greater range of factory sizes among infected cells, or a greater prevalence of cells with smaller factories, during rT3D relative to T1L infection. These observations may suggest existence of an additional viral determinant affecting the extent of inhibition of cellular clathrin functions, which we hypothesize to reside within microtubule-anchoring protein μ2 and to be related to factory morphology, either directly (more effective clathrin recruitment to spread-out structures) or indirectly (an effect related to the role of microtubules in factory expansion (9).
The only widely applied method for reducing clathrin function has been by RNA interference (46,47,52). One drawback of that approach is the long half-life of cytoplasmic clathrin (20–50 h) (52,53), requiring long incubations (at least 72 h) and often repeated transfections with inhibitory RNA, which increase the risk of nonspecific effects on cell viability. Sequestration of clathrin by μNS, by comparison, is more effective and allows experiments to be performed as early as 16 h p.i. (see Figures 5–7). Moreover, the ability to include mutant control virus expressing μNS-L711A in each experiment serves to address questions about treatment specificity.
Moskowitz et al. (54) have developed another alternative for inhibiting clathrin-dependent cellular traffic, employing a modified form of clathrin LC that can be crosslinked easily and reversibly upon addition of a cell-permeable crosslinker. The advantage of that method relative to both RNA interference and MRV-based approaches is that effective inhibition of clathrin functions occurs within minutes of the crosslinker treatment. However, that method is dependent on establishing cell lines that stably express the modified clathrin LC and on extensively replacing endogenous clathrin pools with the modified form. Because of broad cell tropism by MRV, the MRV-based clathrin depletion can in contrast be directly applied to many different non-engineered cell types and can furthermore affect a large fraction of the cells consequent to high levels of μNS expression, especially following infection with high multiplicities of MRV.
Utility of the μNS-based approach for knocking-down clathrin function might even extend to studies of other cellular machineries that involve long-lived components or for which alternative inhibitors are not available. The ability of μNS to form the factory matrix has been exploited to generate a platform for studies of protein–protein interactions (18). In that case, a fragment of μNS, which retains the ability to form factory-matrix structures (15), is expressed in fusion with a protein of interest and that protein's interaction with its binding partners is then visualized by the partners' recruitment to the factory-matrix structures. The results of the current study provide a precedence by which a variation of this method might allow for targeted depletion of any cellular binding partner one aims to study.
Materials and Methods
Virions and ISVPs
Virions of MRV strains T1L, T3DN and T3A, derived from stocks originally obtained from Dr. Bernard N. Fields, were grown and purified by the standard protocol (55) and stored in virion buffer (VB) (150 mM NaCl, 10 mM MgCl2, 10 mM Tris-HCl, pH 7.5). For T1L, ISVPs were obtained by digesting virions in VB at a concentration of 1 × 1013 particles/ml with 200 μg/ml chymotrypsin (Sigma–Aldrich) for 10–20 min at 32 or 37°C. For T3DN or T3A, the chymotrypsin digestions to make ISVPs were performed at a concentration of 1 × 1012 particles/ml at 32°C. Digestion was stopped with 2–5 mM phenylmethyl sulfonyl fluoride (Sigma–Aldrich) on ice.
Cells and antibodies
BSC-1, CV-1, COS-7, LG2, BSC-1 LCA-tom and BSC-1 AP2(σ2)-GFP cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented to contain 10% fetal bovine serum (FBS) (HyClone). Medium in which LG2 cells were grown additionally contained G418 (Cellgro) at 500 μg/ml. Generation of anti-μNS serum antibodies was previously described (56). Here they were used at 1:1,000 dilution for IF, 1:10,000 dilution for immunoblotting and 1:100 dilution for IP. MAbs against clathrin HC, X22 and TD.1, were obtained from the American Type Culture Collection (ATCC). X22 was used for IF at 1:1,000 dilution, and TD.1 was used for immunoblotting at 1:100 dilution. MAb CON.1 specific for clathrin LCA and LCB (LCA/B) was from ATCC and was used at 1:500 dilution for IF, 1:100 dilution for immunoblotting and 1:20 dilution for IP. MAb 10A specific for the β subunits of both adapter proteins AP1 and AP2 (AP1/2) was previously described (57) and was used at 1:2,000 dilution for IF. MAb specific for the influenza HA tag, Mono HA.11 (Covance), was used at 1:500 dilution for both IF and immunoblotting of the HA-tagged construct (see next section). Goat anti-mouse immunoglobulin G (IgG) and goat anti-rabbit IgG conjugated to Alexa 488 or Alexa 594 were obtained from Molecular Probes and used at 1:500 dilution for IF. Rabbit-specific donkey IgG conjugated to horseradish peroxidase (Jackson ImmunoResearch) was used in secondary immunoblot detection at 1:5,000 dilution. Peroxidase-labeled goat anti-mouse IgG (KPL) was used in secondary immunoblot detection at 1:2,000 dilution.
Generation of plasmids for protein expression
C-terminally tagged NTD-HA (aa 1–363) was constructed by PCR amplification from a template encoding the full-length clathrin HC (pCDNA3-rHC) (58). HA tag was engineered in the reverse primer. The amplified fragment was inserted into the mammalian expression vector pCI-neo (Promega). The subcloned region was sequenced to confirm correctness. Wild-type and mutant μNS proteins were expressed from genes cloned into the pCI-neo vector. pCI-M3(T1L) and pCI-M3(T3DN) to express μNS from MRV strains T1L or T3DN have been described previously (13). Constructs to express μNS-715 and μNS-718 were made in the same general manner as previously described for μNS-713 (15), except using distinct reverse primers to introduce novel premature stop codons at the desired positions in μNS (codons 716 and 719). To express T1L μNS containing the L711A mutation within the putative clathrin-binding motif, QuikChange site-directed mutagenesis (Stratagene) was performed using pCI-M3(T1L) as template. The region containing the desired mutation was subcloned into an equivalent vector that had not been subjected to PCR. The subcloned region was sequenced to confirm correctness.
Recombinant viruses
We generated wild-type (rT3D-μNS(WT)) and mutant (rT3D-μNS(L711A)) recombinant viruses by using the protocol and plasmid constructs of Kobayashi et al. (59). To make the new plasmid construct for mutant μNS, we used wild-type pT7-M3T3D construct (59) as template for QuikChange site-directed mutagenesis (Stratagene). Presence of the desired mutation and absence of any spurious mutations within the M3 segment of rT3D-μNS(L711A) virions were confirmed as follows. A monolayer of murine L929 fibroblast cells was infected with second-passage stock of rT3D-μNS(L711A). At 24 h p.i., total RNA was isolated with TRIzol reagent (Invitrogen), subjected to reverse transcription with SuperScript III reverse transcriptase (Invitrogen) according to manufacturer's instructions and PCR-amplified using M3-specific primers. Purified PCR products were then analyzed by sequencing. Virions of plasmid-rescued viruses were purified and treated in the same manner as native virions. Titration experiments—L929 or CV-1 cell monolayers were incubated for 1 h with purified virions of rT3D-μNS(WT) or rT3D-μNS(L711A) over a range of different multiplicities of infection (0.0001 to 2 PFU/cell) in 100 μl of attachment buffer. After removal of unabsorbed virus by two washes with attachment buffer, cells were incubated in normal media at 37°C for the indicated times. To determine virus yields at these times p.i., infected cells were lysed by freezing and thawing, and infectious titers in the resulting lysates were measured by plaque assay in L929 cells (60).
Infections and transfections
For IF imaging experiments, 1–4 × 104 cells per well of 24-well culture plates (or twice that cell number per well of 12-well culture plates) were plated on glass cover slips 1 day before the experiment. For IP experiments, 3–6 × 105 cells were plated per 100-mm culture dish 1 day before the experiment. For transferrin uptake, VSV-Gts-GFP protein secretion and live-cell imaging experiments, 2–5 × 105 cells per 25-mm glass cover slip in 6-well culture plates were plated 16 h before the experiment unless stated otherwise. Infections—ISVPs or virions of indicated strains diluted in attachment buffer (phosphate-buffered saline (PBS) with 2 mM MgCl2) were bound to cells at 1–5 × 104 particles/cell (≥50 PFU/cell) unless stated otherwise. The high multiplicity of infection was used in most experiments to increase the fraction of cells infected as well as the level of μNS expression per cell. To rule out any artifacts attributable to the high multiplicity, however, experiments were later performed at lower multiplicities (5, 0.5 and 0.05 PFU/cell) and yielded the same conclusions: recruitment of both clathrin HC and clathrin LC to viral factories in the case of rT3D-μNS(WT) but not rT3D-μNS(L711A) (see Figure 4 for results with 5 PFU/cell). ISVPs were used to initiate infections in most experiments because in our experience they infect cells more efficiently, quickly and synchronously than do virions. The attachment of virus particles to cells on cover slips was performed on ice in 50 μl of attachment buffer for the 24-well format, 100 μl for the 12-well format, and 200 μl for the 6-well format. The attachment of virus particles to cells in 100-mm plates was performed at room temperature in 600 μl of attachment buffer. Mock infections were performed equivalently, except that virus was omitted from the attachment buffer. After attachment, cells were fixed or harvested either immediately (time 0) or after incubating at 37°C in normal media for various times to allow infection to proceed. Transfections—Most transfections were performed using Lipofectamine 2000 (Invitrogen) following manufacturer's protocol, except that half of the recommended amounts of DNA and transfection reagent were used. Cells were fixed or harvested at 24–38 h p.t. as indicated in legends to figures. Transfections of a plasmid expressing VSV-Gts-GFP were performed using TransIT-LT transfection reagent (Mirus) according to manufacturer instructions.
IF microscopy
Transfected or infected cells on cover slips were washed with PBS and then fixed with 4% paraformaldehyde in PBS (PFA 4%) for 10 min at room temperature. All experiments included relevant mock-infected or mock-transfected controls. PFA was removed by 3 washes in PBS, and cells were permeabilized in IF buffer (PBS, 1% bovine serum albumin, 0.5% Triton-X 100) for 15 min at room temperature. Antibodies were diluted in IF buffer, and incubations with both primary antibodies (anti-μNS serum, CON.1, X22, 10A or Mono HA.11) and secondary antibodies (goat anti-mouse IgG and goat anti-rabbit IgG conjugated to Alexa 488 or Alexa 594) were performed for 30–60 min at room temperature. 300 nM 4,6-diamidino-2-phenylindole (DAPI) (Molecular Probes) was included during the incubations with secondary antibodies to counterstain cell nuclei. Cells were washed three times with PBS both between and after antibody incubations. Cover slips were mounted on glass slides using Prolong antifade reagent (Molecular Probes) and examined in a Nikon TE-2000U inverted microscope. Images were collected digitally using a cooled, charge-coupled device camera (Hamamatsu) and processed using a combination of Metamorph 6.1 (Molecular Devices) and Adobe Photoshop CS2. For transferrin uptake and VSV-Gts-GFP secretion experiments, images were examined under an inverted spinning-disc confocal microscope (Zeiss) and processed using Slidebook 5 software (Intelligent Imaging Innovations, 3i)
IP
Infections—Infected cells from five 100-mm dishes were scraped and collected into a single tube. They were then pelleted at 500 × g for 5 min and lysed in 666 μl of Raf buffer (20 mM Tris, pH 7.9, 137 mM NaCl, 10% glycerol, 1% NP40) containing protease inhibitors (Roche Biomedicals) for 30 min on ice with occasional vortexing. Cell debris was then removed by centrifugation at 8,000 × g for 3 min, and supernatant was transferred to a new tube with 83 μl of protein G agarose beads (Sigma–Aldrich) for pre-clearing by rotary mixing at 4°C for 2–12 h. Beads were removed by pelleting, and infected-cell lysate was split into three aliquots. One aliquot was incubated with anti-μNS serum antibodies, the second with MAb CON.1, and the third without antibody (mock IP). The identical protocol was followed for mock-infected cells except that two dishes were used and volumes of all solutions and reagents were appropriately scaled down at each step. Mock-infected cell lysates were incubated with anti-μNS serum antibodies. Antibody/protein complexes were isolated by three sets of incubations with 20 μl of protein G beads (Dynal) (4–12 h each at 4°C). Beads and aliquots of the supernatant were boiled in 1× gel sample buffer (125 mM Tris, pH 8.0, 1% sodium dodecyl sulfate (SDS), 0.02% b-mercaptoethanol, 10% glycerol). Transfections—Cells from three 100-mm plates transfected with the same plasmid or a combination of plasmids were scraped and pooled into a single tube. They were then pelleted at 500 × g for 5 min and lysed in 400 μl of Raf buffer containing protease inhibitors for 30 min on ice with occasional vortexing. Cell debris was then removed by centrifugation at 16,000 × g for 3 min, and supernatant was transferred to a new tube with 50 μl of protein G agarose beads for pre-clearing by rotary mixing at 4°C for 2–12 h. Beads were separated from the lysate by pelleting, and two 30-μl aliquots of pre-cleared lysate were removed, boiled in gel sample buffer and saved for analysis of relative levels of expressed proteins. The remainder of the lysate was incubated with anti-μNS serum antibodies at 4°C overnight, and the antibody/protein complexes were isolated by three sets of incubations with 25 μl of protein G beads (Dynal) (4–12 h each at 4°C). Beads and aliquots of the supernatant were boiled in gel sample buffer.
Immunoblotting
A portion of immunoprecipitated protein sample (bead fraction; one-fourth for infected or singly transfected cells, four-fifths for doubly transfected cells), along with a portion of pre-IP or post-IP supernatant (one-twentieth for transfected cells and one-tenth for infected cells), was analyzed by SDS-polyacrylamide gel electrophoresis and transferred from the gel to a nitrocellulose membrane (BioRad) in transfer buffer (25 mM Tris, pH 8.3, 192 mM glycine). Antibody incubations were performed in 20 mM Tris-HCl (pH 7.5), 500 mM NaCl, 5% milk, 0.05% Tween-20. Binding of peroxidase-conjugated secondary antibodies was detected with Western Lightning chemiluminescence reagents (Perkin Elmer) and scanned on a Typhoon imager (GE Healthcare). For anti-HA blots, the membrane was instead exposed to fluorography film (Fujifilm SuperRX) for increased sensitivity.
Transferrin uptake and VSV-G protein secretion
Mock-infected or ISVP-infected BSC-1 LCA-tom cells on coverslips (12–16 h p.i.) were washed once with PBS and incubated with Alexa647-labeled Tf (50 μg/mL final) in DMEM supplemented with 10% FBS for 7 min at 37°C. Cells were washed twice with ice-cold PBS to remove unbound Tf, once with ice-cold PBS containing 10% acetic acid and 150mM NaCl to remove the cell-surface-bound Tf, then twice more with ice-cold PBS before fixing with PFA 4%. Cells were immunostained for μNS and imaged as described under IF Microscopy. To quantify Tf uptake, the localization of Alexa647-labeled Tf was scored in at least 50 cells: cells showing labeled Tf localized in the vicinity of the nucleus were considered positive for Tf uptake; those cells revealing no labeled Tf in their interior (with little to no labeled Tf at the plasma membrane) were considered negative for Tf uptake. To assay the effect of reovirus-induced clathrin redistribution on protein secretion, we used a modification of the previously described VSV-G protein secretion assay (43). Briefly, BSC-1 cells transfected with pVSV-Gts-GFP were incubated at 40°C for 16 h to allow VSV-Gts-GFP expression but to prevent its correct folding. Transfected cells were mock infected or infected with T1L, rT3D-μNS(WT) or rT3D-μNS(L711A) ISVPs. Infection was allowed to proceed for 12–16 h at 40°C, at which point the cells were shifted to 32°C for 2h to allow the re-folding of VSV-Gts-GFP. Cells were then fixed with PFA 4%, immunostained for μNS and imaged as described under IF Microscopy. To quantify VSV-Gts-GFP secretion, its localization in at least 50 cells was scored: cells showing strict localization of VSV-Gts-GFP in the ER and TGN were considered negative for VSV-Gts-GFP secretion; those cells additionally showing VSV-Gts-GFP localized at the plasma membrane were considered positive for VSV-Gts-GFP secretion.
Live-cell imaging and data analysis
BSC-1 AP2(σ2)-GFP cells on coverslips were mock-infected or infected with ISVPs of strains T1L, rT3D-μNS(WT) or rT3D-μNS(L711A) for 16 or 24 h at 37°C prior to imaging. Infected-cells on cover slips were placed into a perfusion chamber and maintained in phenol-red-free DMEM supplemented with 10% serum and 20mM Hepes pH 7.4. The temperature of the specimen was kept at 37°C using a Peltier-controlled sample holder (20/20 Technologies). The holding device and the microscope stage were maintained in an air-controlled environmental chamber. The air above the cells was maintained at 37°C, humidified and contained 5% CO2. All spinning disk confocal imaging experiments were conducted using the microscope setup recently described (42). A Spherical Aberration Correction unit (SAC; Intelligent Imaging 3i) was used to reduce the spherical aberration and increase the sensitivity. Images were captured using Slidebook 5. Images were then analyzed as described previously using semi-automated MATLAB routines (42). Briefly, to identify CCPs by monitoring the AP-2(σ2)-GFP signal, a sequence of 2D Gaussian and Laplacian filtering followed by thresholding were applied to the data set to create masks defining the fluorescent objects. The fluorescence-intensity profile of each object over time was measured and used to characterize the endocytic events. CCPs were selected according to the following criteria: (1) The selected objects displayed the fluorescence intensity profiles characteristic of endocytic events (28,42); (2) the objects appeared and disappeared within the time window of the data series unless stated otherwise; (3) the objects displayed limited movement (500 nm/lifetime) as expected for membrane-bound clathrin structures in the horizontal plane during their growth phase (28); and (4) the objects did not collide with each other during the time window of the data series. The CCP-lifetime distributions and the box and whisker graphs were plotted using GraphPad Prism software.
Supplementary Material
Figure S1. Clathrin HC, but not AP1 or AP2, colocalizes with μNS in MRV factories. Cells were infected with T1L ISVPs and were fixed for indirect IF microscopy at 0, 3, 7 or 12 h p.i. μNS-specific serum antibodies (α-μNS) and either MAb X22 (A) or MAb 10A (B) were used in IF. Merged images are shown at right (α-μNS, green; α-CHC or α-AP1/AP2, red). Scale bars, 20 μm.
{kind=link}
Figure S2. AP2(σ2)-GFP is recruited to MRV factories late in infection. BSC-1 cells stably expressing AP2(σ2)-GFP were mock-infected or infected with either T1L, rT3D-μNS(WT) or rT3D-μNS(L711A) MRV for 42 h. μNS-specific serum antibodies (α-μNS) and Alexa-647-conjugated secondary antibody were used in IF. Representative overlayed images of factories (blue) and AP2(σ2)-GFP (green) are shown. Scale bars, 10 μm.
{kind=link}
Figure S3. μNS(471-721) recruits clathrin to factory-matrix structures. μNS(471-721) protein was expressed in CV-1 cells by plasmid-based transfection. At 16 h p.t, cellular localizations of μNS as well as clathrin were visualized by indirect IF microscopy using a combination of μNS-specific serum antibodies (α-μNS) and MAb X22 (α-HC). Scale bar, 10 μm.
{kind=link}
Figure S4. Clathrin recruitment by μNS is not essential for virus growth in cultured cells. A) L929 cells were infected with two different preparations each of either purified rT3D-μNS(WT) (closed bars) or rT3D-μNS(L711A) (open bars) at 0.0001 PFU/cell, and infectious titers were determined at indicated times p.i. by plaque assay. Results of a representative experiment are shown. B) CV-1 cells were infected with purified rT3D-μNS(WT) (closed symbols) or rT3D-μNS(L711A) (open symbols) at 0.1 PFU/cell (circles) or 1 PFU/cell (squares), and infectious titers were determined at indicated times p.i. by plaque assay. Yields are expressed relative to paired 0-h samples for each virus. Results of a representative experiment are shown.
{kind=link}
Figure S5. Transferrin uptake is not inhibited at 12 h p.i. by MRV. BSC-1 cells stably expressing clathrin LCA fused to tomato fluorescent protein (CLC-tom) were mock-infected or infected with either T1L or rT3D-μNS(WT) MRV. At 12 h p.i. fluorescent-transferrin (Tf) uptake was performed as described in Materials and Methods, and cells were fixed and immunostained using μNS-specific serum antibodies (α-μNS). Representative confocal images of mock- and MRV-infected cells are shown, with merged images at right (α-μNS, green; CLC-tom, red; Tf, blue). Scale bars, 10 μm.
{kind=link}
Video S1: Redistribution of clathrin LCA to MRV factories late in infection. BSC-1 cells stably expressing clathrin LCA tagged with GFP (LG2 cells) were infected with MRV strain T1L and time-lapse video (3 sec per frame) was recorded at 30 h p.i. Video is shown at the speed of 10 frames per sec (actual time passage (hh:min:sec) is shown in the lower left corner).
Video S2: Dynamics of CCP formation in mock-infected cells. BSC-1 cells stably expressing AP2(σ2)-GFP were mock-infected 36 h prior to imaging. The movie is a 2-D time series acquired for 300 sec at 3-sec intervals from a single optical section obtained by spinning-disk confocal microscopy and using 100 msec exposures.
Video S3: Dynamics of CCP formation in T1L-infected cells. BSC-1 cells stably expressing AP2(σ2)-GFP were infected with MRV strain T1L 36 h prior to imaging. The movie has the same characteristics as Video S2.
Video S4: Dynamics of CCP formation in rT3D-μNS(WT)-infected cells. BSC-1 cells stably expressing AP2(σ2)-GFP were infected with MRV strain rT3D-μNS(WT) 36 h prior to imaging. The movie has the same characteristics as Video S2.
Video S5: Dynamics of CCP formation in rT3D-μNS(L711A)-infected cells. BSC-1 cells stably expressing AP2(σ2)-GFP were infected with MRV strain rT3D-μNS(L711A) 36 h prior to imaging. The movie has the same characteristics as Video S2.
Acknowledgments
We thank Elaine Freimont for technical support; other lab members for discussions; and Melina Agosto, Cathy Eichwald, Cathy Miller and John Parker for comments on an early manuscript. We also thank Jim Chappell, Terry Dermody and Takeshi Kobayashi (Vanderbilt University) for reagents and advice for plasmid-based rescue of MRV and Megan Stanifer for assistance in generating Figure S2. This research was supported by NIH grants R01 GM36548 to T. K. and R01 AI47904 and R56 AI067445 to M. L. N. Support was also provided by the Bill and Melinda Gates Foundation through a Collaboration for AIDS Vaccine Discovery grant to a Vaccine Discovery Consortium (including M. L. N.) headed by the International AIDS Vaccine Initiative. T. I. and M. M. A. received initial support from NIH training grant T32 AI07245., and M. E. was additionally supported by a Dorit fellowship from Harvard Medical School.
References
- 1.Restrepo-Hartwig MA, Ahlquist P. Brome mosaic virus helicase- and polymerase-like proteins colocalize on the endoplasmic reticulum at sites of viral RNA synthesis. J Virol. 1996;70:8908–8916. doi: 10.1128/jvi.70.12.8908-8916.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van der Meer Y, Snijder EJ, Dobbe JC, Schleich S, Denison MR, Spaan WJM, Locker JK. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J Virol. 1999;73:7641–7657. doi: 10.1128/jvi.73.9.7641-7657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kujala P, Heimonen AI, Ehsani N, Vinihen H, Auvinen P, Kaarianinen L. Biogenesis of the Semliki Forest virus RNA replication complex. J Virol. 2001;75:3873–3884. doi: 10.1128/JVI.75.8.3873-3884.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupta S, De BP, Drazba JA, Banerjee AK. Involvement of actin microfilament in the replication of human parainfluenza virus type 3. J Virol. 1998;72:2655–2662. doi: 10.1128/jvi.72.4.2655-2662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heath CM, Windsor M, Wileman T. Aggresomes resemble sites specialized for virus assembly. J Cell Biol. 2001;153:449–455. doi: 10.1083/jcb.153.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cabral-Romero C, Padilla-Noriega L. Association of rotavirus viroplasms with microtubules through NSP5 and NSP2. Mem Inst Oswaldo Cruz Rio de Janeiro. 2006;101:603–611. doi: 10.1590/s0074-02762006000600006. [DOI] [PubMed] [Google Scholar]
- 7.Fields BN, Raine CS, Baum SG. Temperature-sensitive mutants of reovirus type 3: defects in viral maturation as studied by immunofluorescence and electron microscopy. Virology. 1971;43:569–578. doi: 10.1016/0042-6822(71)90282-0. [DOI] [PubMed] [Google Scholar]
- 8.Sharpe AH, Chen LB, Fields BN. The interaction of mammalian reoviruses with the cytoskeleton of monkey kidney CV-1 cells. Virology. 1982;120:399–411. doi: 10.1016/0042-6822(82)90040-x. [DOI] [PubMed] [Google Scholar]
- 9.Parker JSL, Broering TJ, Kim J, Higgins DE, Nibert ML. Reovirus core protein μ2 determines the filamentous morphology of viral inclusion bodies by interacting with and stabilizing microtubules. J Virol. 2002;76:4484–4496. doi: 10.1128/JVI.76.9.4483-4496.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tournier P, Plissier M. The intracellular development of the reovirus studied with the electron microscope. Presse Med. 1960;68:683–688. [PubMed] [Google Scholar]
- 11.Rhim JS, Jordan LE, Mayor HD. Cytochemical, fluorescent antibody and electron microscopic studies on the growth of reovirus (ECHO 10) in tissue culture. Virology. 1962;17:342–355. doi: 10.1016/0042-6822(62)90125-3. [DOI] [PubMed] [Google Scholar]
- 12.Mayor HD. Studies on reovirus 3. A labile, single-stranded ribonucleic acid associated with the late stages of infection. J Natl Cancer Inst. 1965;35:919–925. [PubMed] [Google Scholar]
- 13.Broering TJ, Parker JSL, Joyce PL, Kim J, Nibert ML. Mammalian reovirus nonstructural protein μNS forms large inclusions and colocalizes with reovirus microtubule-associated protein μ2 in transfected cells. J Virol. 2002;76:8285–8297. doi: 10.1128/JVI.76.16.8285-8297.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becker MM, Peters TR, Dermody TS. Reovirus σNS and μNS proteins form cytoplasmic inclusion structures in the absence of viral infection. J Virol. 2003;77:5948–5963. doi: 10.1128/JVI.77.10.5948-5963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broering TJ, Arnold MM, Miller CL, Hurt JA, Joyce PL, Nibert ML. Carboxyl-proximal regions of reovirus nonstructural protein μNS necessary and sufficient for forming factory-like inclusions. J Virol. 2005;79:6194–6206. doi: 10.1128/JVI.79.10.6194-6206.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Broering TJ, Kim J, Miller CL, Piggott CD, Dinoso JB, Nibert ML, Parker JSL. Reovirus nonstructural protein μNS recruits viral core surface proteins and entering core particles to factory-like inclusions. J Virol. 2004;78:1882–1892. doi: 10.1128/JVI.78.4.1882-1892.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller CL, Broering TJ, Parker JSL, Arnold MM, Nibert ML. Reovirus σNS protein localizes to inclusions through an association requiring the μNS amino terminus. J Virol. 2003;77:4566–4576. doi: 10.1128/JVI.77.8.4566-4576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller CL, Arnold MM, Broering TJ, Eichwald C, Kim J, Dinoso JB, Nibert ML. Virus-derived platforms for visualizing protein associations inside cells. Mol Cell Proteomics. 2007;6:1027–1038. doi: 10.1074/mcp.M700056-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Miller CL, Arnold MM, Broering TJ, Hastings CE, Nibert ML. Localization of mammalian orthoreovirus proteins to cytoplasmic factory-like structures via nonoverlapping regions of μNS. J Virol. 2010;84:867–882. doi: 10.1128/JVI.01571-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spendlove RX, Lennette EH, John AC. The role of the mitotic apparatus in the intracellular location of reovirus antigen. J Immunol. 1963;90:554–560. [PubMed] [Google Scholar]
- 21.Miller CL, Parker JSL, Dinoso JB, Piggott CDS, Perron MJ, Nibert ML. Increased ubiquitination and other covariant phenotypes attributed to a strain- and temperature-dependent defect of reovirus core protein μ2. J Virol. 2004;78:10291–10302. doi: 10.1128/JVI.78.19.10291-10302.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin P, Keirstead ND, Broering TJ, Arnold MM, Parker JSL, Nibert ML, Coombs KM. Comparison of the M1 genome segments and encoded μ2 proteins of different reovirus isolates. Virol J. 1:6. doi: 10.1186/1743-422X-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirchhausen T. Clathrin. Annu Rev Biochem. 2000;69:699–727. doi: 10.1146/annurev.biochem.69.1.699. [DOI] [PubMed] [Google Scholar]
- 24.Kirchhausen T. Imaging endocytic clathrin structures in living cells. Trends Cell Biol. 2009;19:596–605. doi: 10.1016/j.tcb.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.ter Haar E, Harrison SC, Kirchhausen T. Peptide-in-groove interactions link target proteins to the β-propeller of clathrin. Proc Natl Acad Sci USA. 2000;97:1096–1100. doi: 10.1073/pnas.97.3.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brodsky FM, Chen CY, Knuehl C, Towler MC, Wakeham DE. Biological basket weaving: formation and function of clathrin-coated vesicles. Annu Rev Cell Dev Biol. 2001;17:517–568. doi: 10.1146/annurev.cellbio.17.1.517. [DOI] [PubMed] [Google Scholar]
- 27.Lafer EM. Clathrin-protein interactions. Traffic. 2002;3:513–520. doi: 10.1034/j.1600-0854.2002.30801.x. [DOI] [PubMed] [Google Scholar]
- 28.Ehrlich M, Boll W, van Oijen A, Hariharan R, Chandran K, Nibert ML, Kirchhausen T. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell. 2004;118:591–605. doi: 10.1016/j.cell.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 29.Rust MJ, Lakadamyali LM, Zhang F, Zhuang X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nature Struct Mol Biol. 2004;11:567–573. doi: 10.1038/nsmb769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan SPJ. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 2009;5:e1000394. doi: 10.1371/journal.ppat.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mercer J, Schelhaas M, Helenius A. Virus entry by endocytosis. Annu Rev Biochem. 2010;79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 32.Huang C, Chang SC, Yu IC, Tsay YG, Chang MF. Large hepatitis delta is a novel clathrin adaptor-like protein. J Virol. 2007;81:5985–5994. doi: 10.1128/JVI.02809-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang C, Chang SC, Yang HC, Chien CL, Chang MF. Clathrin-mediated post-Golgi membrane trafficking in the morphogenesis of hepatitis delta virus. J Virol. 2009;83:12314–12324. doi: 10.1128/JVI.01044-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nathke IS, Heuser J, Lupas A, Stock J, Turck CW, Brodsky FM. Folding and trimerization of clathrin subunits at the triskelion hub. Cell. 1992;68:899–910. doi: 10.1016/0092-8674(92)90033-9. [DOI] [PubMed] [Google Scholar]
- 35.Brodsky FM. Clathrin structure characterized with monoclonal antibodies. I. Analysis of multiple antigenic sites. J Cell Biol. 1985;101:2047–2054. doi: 10.1083/jcb.101.6.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirchhausen T. Adaptors for clathrin-mediated traffic. Annu Rev Cell Dev Biol. 1999;15:705–732. doi: 10.1146/annurev.cellbio.15.1.705. [DOI] [PubMed] [Google Scholar]
- 37.Traub LM. Tickets to ride: selecting cargo for clathrinregulated internalization. Nat Rev Mol Cell Biol. 2009;10:583–596. doi: 10.1038/nrm2751. [DOI] [PubMed] [Google Scholar]
- 38.Boucrot E, Saffarian S, Massol R, Kirchhausen T, Ehrlich M. Role of lipids and actin in the formation of clathrin-coated pits. Exp Cell Res. 2006;312:4036–4048. doi: 10.1016/j.yexcr.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Pietro SM, Cascio D, Feliciano D, Bowie JU, Payne GS. Regulation of clathrin adaptor function in endocytosis: novel role for SAM domain. EMBO J. 2010;29:1033–1044. doi: 10.1038/emboj.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wendland B, Steece KE, Emr SD. Yeast epsins contain an essential N-terminal ENTH domain, bind clathrin and are required for endocytosis. EMBO J. 1999;18:4383–4393. doi: 10.1093/emboj/18.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaidarov I, Smith MEK, Domin J, Keen JH. The class II phophoinositide 3-Kinase C2a is activated by clathrin and regulates clathrin-mediated membrane trafficking. Mol Cell. 2001;7:443–449. doi: 10.1016/s1097-2765(01)00191-5. [DOI] [PubMed] [Google Scholar]
- 42.Saffarian S, Cocucci E, Kirchhausen T. Distunct dynamics of endocytic clathrin-coated pits and coated plaques. PLoS Biol. 2009;7:e1000191. doi: 10.1371/journal.pbio.1000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arnheiter H, Dubois-Dalcq M, Lazzarini RA. Direct visualization of protein transport and processing in the living cell by microinjection of specific antibodies. Cell. 1984;39:99–109. doi: 10.1016/0092-8674(84)90195-8. [DOI] [PubMed] [Google Scholar]
- 44.Arnold MM, Murray KE, Nibert ML. Formation of the factory matrix is an important, though not a sufficient function of nonstructural protein μNS during reovirus infection. Virology. 2008;375:412–423. doi: 10.1016/j.virol.2008.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cossart P, Veiga E. Non-classical use of clathrin during bacterial infections. J Microsc. 2008;231:524–528. doi: 10.1111/j.1365-2818.2008.02065.x. [DOI] [PubMed] [Google Scholar]
- 46.Deborde S, Perret E, Gravotta D, Deora A, Salvarezza S, Schreiner R, Rodriguez-Boulan E. Clathrin is a key regulator of basolateral polarity. Nature. 2008;452:719–723. doi: 10.1038/nature06828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Royle SJ, Bright NA, Lagnado L. Clathrin is required for the function of the mitotic spindle. Nature. 2005;434:1152–1157. doi: 10.1038/nature03502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okamoto CT, McKinney J, Jeng YY. Clathrin in mitotic spindles. Am J Physiol Cell Physiol. 2000;279:369–374. doi: 10.1152/ajpcell.2000.279.2.C369. [DOI] [PubMed] [Google Scholar]
- 49.Royle SJ, Lagnado L. Trimerisation is important for the function of clathrin at the mitotic spindle. J Cell Sci. 2006;119:4071–4078. doi: 10.1242/jcs.03192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drake MT, Downs MA, Traub LM. Epsin binds to clathrin by associating directly with the clathrin-terminal domain. J Biol Chem. 2000;275:6478–6489. doi: 10.1074/jbc.275.9.6479. [DOI] [PubMed] [Google Scholar]
- 51.Miele AE, Watson PJ, Evans PR, Traub LM, Owen DJ. Two distinct interaction motifs in amphiphysin bind two independent sites on the clathrin terminal domain β-propeller. Nat Struct Molec Biol. 2004;11:242–248. doi: 10.1038/nsmb736. [DOI] [PubMed] [Google Scholar]
- 52.Hinrichsen L, Harborth J, Andrees L, Weber K, Ungelwickell EJ. Effect of clathrin heavy chain- and alpha-adaptin-specific small inhibitory RNAs on endocytic accessory proteins and receptor trafficking in HeLa cells. J Biol Chem. 2003;278:45160–45170. doi: 10.1074/jbc.M307290200. [DOI] [PubMed] [Google Scholar]
- 53.Acton SL, Brodsky FM. Predominance of clathrin light chain LCb correlates with the presence of a regulated secretory pathway. J Cell Biol. 1990;111:1419–1426. doi: 10.1083/jcb.111.4.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moskowitz HS, Heuser J, McGraw TE, Ryan TA. Targeted chemical disruption of clathrin function in living cells. Mol Biol Cell. 2003;14:4437–4447. doi: 10.1091/mbc.E03-04-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Furlong DB, Nibert ML, Fields BN. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol. 1988;62:246–256. doi: 10.1128/jvi.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Broering TJ, McCutcheon AM, Centonze VE, Nibert ML. Reovirus nonstructural protein μNS binds to core particles but does not inhibit their transcription and capping activities. J Virol. 2000;74:5516–5524. doi: 10.1128/jvi.74.12.5516-5524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Massol RH, Boll W, Griffin AM, Kirchhausen T. A burst of auxilin recruitment determines the onset of clathrin-coated vesicle uncoating. Proc Natl Acad Sci USA. 2006;103:10265–10270. doi: 10.1073/pnas.0603369103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Musacchio A, Smith CJ, Roseman AM, Harrison SC, Kirchhausen T, Pearse BMF. Functional organization of clathrin in coats: combining electron cryomicroscopy and X-ray crystallography. Mol Cell. 1999;3:761–770. doi: 10.1016/s1097-2765(01)80008-3. [DOI] [PubMed] [Google Scholar]
- 59.Kobayashi T, Antar AAR, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. Plasmid-based reverse genetics for animal double-stranded RNA viruses: manipulation of the viral genome and development of a novel gene-transduction system. Cell Host Microbe. 2007;1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Middleton JK, Agosto MA, Severson TF, Yin J, Nibert ML. Thermostabilizing mutations in reovirus outer-capsid protein μ1 selected by heat inactivation of infectious subvirion particles. Virology. 2007;361:412–425. doi: 10.1016/j.virol.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Clathrin HC, but not AP1 or AP2, colocalizes with μNS in MRV factories. Cells were infected with T1L ISVPs and were fixed for indirect IF microscopy at 0, 3, 7 or 12 h p.i. μNS-specific serum antibodies (α-μNS) and either MAb X22 (A) or MAb 10A (B) were used in IF. Merged images are shown at right (α-μNS, green; α-CHC or α-AP1/AP2, red). Scale bars, 20 μm.
Figure S2. AP2(σ2)-GFP is recruited to MRV factories late in infection. BSC-1 cells stably expressing AP2(σ2)-GFP were mock-infected or infected with either T1L, rT3D-μNS(WT) or rT3D-μNS(L711A) MRV for 42 h. μNS-specific serum antibodies (α-μNS) and Alexa-647-conjugated secondary antibody were used in IF. Representative overlayed images of factories (blue) and AP2(σ2)-GFP (green) are shown. Scale bars, 10 μm.
Figure S3. μNS(471-721) recruits clathrin to factory-matrix structures. μNS(471-721) protein was expressed in CV-1 cells by plasmid-based transfection. At 16 h p.t, cellular localizations of μNS as well as clathrin were visualized by indirect IF microscopy using a combination of μNS-specific serum antibodies (α-μNS) and MAb X22 (α-HC). Scale bar, 10 μm.
Figure S4. Clathrin recruitment by μNS is not essential for virus growth in cultured cells. A) L929 cells were infected with two different preparations each of either purified rT3D-μNS(WT) (closed bars) or rT3D-μNS(L711A) (open bars) at 0.0001 PFU/cell, and infectious titers were determined at indicated times p.i. by plaque assay. Results of a representative experiment are shown. B) CV-1 cells were infected with purified rT3D-μNS(WT) (closed symbols) or rT3D-μNS(L711A) (open symbols) at 0.1 PFU/cell (circles) or 1 PFU/cell (squares), and infectious titers were determined at indicated times p.i. by plaque assay. Yields are expressed relative to paired 0-h samples for each virus. Results of a representative experiment are shown.
Figure S5. Transferrin uptake is not inhibited at 12 h p.i. by MRV. BSC-1 cells stably expressing clathrin LCA fused to tomato fluorescent protein (CLC-tom) were mock-infected or infected with either T1L or rT3D-μNS(WT) MRV. At 12 h p.i. fluorescent-transferrin (Tf) uptake was performed as described in Materials and Methods, and cells were fixed and immunostained using μNS-specific serum antibodies (α-μNS). Representative confocal images of mock- and MRV-infected cells are shown, with merged images at right (α-μNS, green; CLC-tom, red; Tf, blue). Scale bars, 10 μm.
Video S1: Redistribution of clathrin LCA to MRV factories late in infection. BSC-1 cells stably expressing clathrin LCA tagged with GFP (LG2 cells) were infected with MRV strain T1L and time-lapse video (3 sec per frame) was recorded at 30 h p.i. Video is shown at the speed of 10 frames per sec (actual time passage (hh:min:sec) is shown in the lower left corner).
Video S2: Dynamics of CCP formation in mock-infected cells. BSC-1 cells stably expressing AP2(σ2)-GFP were mock-infected 36 h prior to imaging. The movie is a 2-D time series acquired for 300 sec at 3-sec intervals from a single optical section obtained by spinning-disk confocal microscopy and using 100 msec exposures.
Video S3: Dynamics of CCP formation in T1L-infected cells. BSC-1 cells stably expressing AP2(σ2)-GFP were infected with MRV strain T1L 36 h prior to imaging. The movie has the same characteristics as Video S2.
Video S4: Dynamics of CCP formation in rT3D-μNS(WT)-infected cells. BSC-1 cells stably expressing AP2(σ2)-GFP were infected with MRV strain rT3D-μNS(WT) 36 h prior to imaging. The movie has the same characteristics as Video S2.
Video S5: Dynamics of CCP formation in rT3D-μNS(L711A)-infected cells. BSC-1 cells stably expressing AP2(σ2)-GFP were infected with MRV strain rT3D-μNS(L711A) 36 h prior to imaging. The movie has the same characteristics as Video S2.