


Abstract
High-throughput sequencing is increasingly being used in combination with bisulfite (BS) assays to study DNA methylation at nucleotide resolution. Although several programmes provide genome-wide alignment of BS-treated reads, the resulting information is not readily interpretable and often requires further bioinformatic steps for meaningful analysis. Current post-alignment BS-sequencing programmes are generally focused on the gene-specific level, a restrictive feature when analysis in the non-coding regions, such as enhancers and intergenic microRNAs, is required. Here, we present Genome Bisulfite Sequencing Analyser (GBSA—http://ctrad-csi.nus.edu.sg/gbsa), a free open-source software capable of analysing whole-genome bisulfite sequencing data with either a gene-centric or gene-independent focus. Through analysis of the largest published data sets to date, we demonstrate GBSA’s features in providing sequencing quality assessment, methylation scoring, functional data management and visualization of genomic methylation at nucleotide resolution. Additionally, we show that GBSA’s output can be easily integrated with other high-throughput sequencing data, such as RNA-Seq or ChIP-seq, to elucidate the role of methylated intergenic regions in gene regulation. In essence, GBSA allows an investigator to explore not only known loci but also all the genomic regions, for which methylation studies could lead to the discovery of new regulatory mechanisms.
INTRODUCTION
DNA methylation is an epigenetic event essential for gene transcription regulation and generally accepted to be associated with gene repression. Aberrant DNA methylation profiles have been observed in cancers and other human diseases (1), highlighting the value for understanding its role in regulation of gene expression, as well as a wider range of biological and cellular processes, such as chromatin reorganization. In the past few years, DNA methylation profiling techniques have undergone a veritable revolution in tandem with the progress of massive parallel sequencing technologies (2). Sequencing has been coupled with several methods to replace microarrays for profiling DNA methylation at the genome-wide level. These methods include, enrichment-based methods that use either antibodies against 5-methylcytosine or methyl-binding domain protein to enrich for methylated DNA before sequencing. Such methods, known as (MeDIP-seq) (3) or methyl-binding domain-isolated genome sequencing (MiGS) (4), MethylCap-seq (5) have been proven to be cost-effective approaches to study and compare differentially methylated regions in a variety of organisms, tissues and cells of several developmental stages. Meanwhile, the sequencing of the methylated DNA after bisulfite (BS) conversion, which allows observation of methylation at nucleotide resolution, has also become feasible in the whole genomes by MethylC-Seq (6), bisulfite sequencing (BS-Seq) (7) as well as in the reduced complexity by reduced representation bisulfite sequencing (RRBS) (8,9). Until recently, bisulfite sequencing was mainly used for locus-specific analysis and several software packages were developed to address this issue (10–15). However, as the cost of the whole-genome sequencing has become more affordable, many laboratories are able to produce a snapshot of the entire methylome at nucleotide resolution. Sequencing usually entails mapping reads to a reference genome and much effort has been focused on developing various programmes to align BS-treated reads in the past few years (16–21) (Table 1). However, tools for post-alignment analysis that require further bioinformatics steps for a meaningful interpretation of the methylome are still lacking. Here, we present Genome Bisulfite Sequencing Analyser (GBSA), a software package that allows processing and analysis of aligned BS-reads. The stand-alone version of GBSA can be run on a personal computer and provides quality reports, scores and annotation of methylated domains. GBSA integrates two different analysis methods; the first is focused on genes and scores the methylation level according to several defined regions, such as promoters, transcription start site (TSS) regions or the gene body. The second method is less biased, as it first detects all methylated loci and then annotates them according to their position from the nearest TSS. All these results can be easily visualized via the built-in gene viewer or can be exported to third-party software.
Table 1.
GBSA in relation to related software
| Input files | Alignment algorithm | β-score calculation | Loci methylated scoring | Analysis scale | Genome annotation | GUI | Operating system | Output results | |
|---|---|---|---|---|---|---|---|---|---|
| BSmap (18) released 2009 | FASTA FASTQ BAM | Based on SOAP | No | No | Genome-wide | No | No | Linux | SAM (aligned reads) Tab-delimited text file (aligned reads) |
| BSseeker (19) released 2010 | FASTA FASTQ QSEQ | Based on Bowtie | No | No | Genome-wide, Loci digested by DpnI | No | No | Linux | SAM (aligned reads) Tab-delimited text file (aligned reads) Text file (QC reports) |
| GSNap (16) released 2010 | FASTA FASTQ | Based on GMAP | No | No | Genome-wide | No | No | Linux | SAM (aligned reads) |
| Tab-delimited text file (aligned reads) | |||||||||
| Bismark (17) released 2011 | FASTA FASTQ | Based on Bowtie | Yes | No | Genome-wide | No | No | Linux | Tab-delimited text file (aligned reads) bedgrapha (CpG β-scores) ausing third party script |
| MethylCoder (14) released 2011 | FASTA FASTQ | GSNap or Bowtie | Yes | No | Genome-wide | No | No | Linux | SAM (aligned reads) Tab-delimited text file (CpG β-scores) Text file (QC reports) |
| BIQ Analyser HT (15) released 2011 | FASTA BAM | based on Needleman–Wunch | Yes | Yes | ∼100 user-defined loci | No | Yes | Linux, Mac, Windows. | FASTA (Reads alignment for each loci) Graphics (QC reports, methylation profiles for each loci) |
| RRBSmap (20) released 2011 | FASTA FASTQ BAM | Based on SOAP | No | No | Loci digested by MspI | No | No | Linux | SAM (aligned reads) Tab-delimited text file (aligned reads) |
| GBSA released 2012 | BSmap RRBSmap BSseeker | NA | Yes | Yesa | Genome-wide | Yes | Yes | Linux, Mac, Windows. | bedgraph (Cytosine β-scores, Cytosine depth of coverage) Tab-delimited (gene/domains methylation scores) Graphics (QC reports, genes browser) |
NA, Not available; QC, Quality control. aGenes are scored according several customisable referential and, detects and scores genone-wide methylated domains.
MATERIALS AND METHODS
The aim of GBSA is to facilitate whole-genome methylation studies using BS sequencing by providing a comprehensive analysis and interpretable outputs. Briefly, the GBSA workflow consists of five steps: (i) data loading, aligned sequencing reads are loaded as input along with the experimental set-up; (ii) data processing; (iii) quality control reporting along with (iv) results, tables of the methylated loci associated with the genomic annotation; and (v) visualization, the methylome at the nucleotide resolution can be viewed directly in GBSA (Figure 1). Experiments are managed as projects that can be saved as files and reloaded at another time. Projects include all results, quality reports and details of user analysis set-up.
Figure 1.

The GBSA workflow. (A) Aligned reads are loaded as input along with several user-defined options. (B) Once loaded, aligned reads undergo four post-processing steps (i) duplicate reads removal; (ii) calculation of depth of strand specific coverage and ‘β score;’ (iii) methylation domain calling according to the two methods described earlier; and (iv) annotation of methylated domains. (C) Each post-processing step gives rise to a summarized quality control report. (D) The resulting output, which contains DNA methylation measurements, annotations and data quality, can be exported as tab-separated files for further in-depth analysis by spreadsheet or statistical software, such as R/Bioconductor and TM4-Mev. (E) Finally, data can be visualized graphically via the GBSA GUI or by a genome browser compatible with the ‘bedgraph’ format.
Input
GBSA accepts data from BSmap (18), BSseeker (19) and RRBSmap (20). Although multi-run experiments can be analysed separately as replicates, it is recommended to merge them into a single file to increase the depth of genome coverage and, consequently, increase data reliability. This operation is executed by the graphical user interface (GUI), alternatively users can easily use the linux ‘cat’ function.
Quality control report
The quality of experiments can be assessed at the genome scale as well at the loci-specific level. First, GBSA removes reads that are more likely polymerase chain reaction duplicates and gives the proportion of usable unique reads. It then calculates the depth of coverage for each cytosine site of interest (CpG, CHG or CHH, where H can be A, C or T) and selects only those covered by a user defined minimum amount of reads (three by default) for further analysis. These selected cytosine sites are then clustered to calculate a methylation score at a given locus (hereafter named domains). Only domains with a minimum number of cytosine sites sequenced are selected. All domains are scored for their methylation level, and their quality is assessed based on the number of cytosine sequenced and the ratio of sequenced cytosine to the total amount of cytosine within the domain. Statistics on numbers of sequence reads, cytosine sequenced within a domain and methylation levels can be visualized at the genome scale via graphs and charts produced by the GUI (Supplementary Figure S1).
Methylation calling and scoring
GBSA focuses not only on CpG methylation, which is the most common biochemical modification in eukaryotic DNA, but also allows CHG and CHH analysis, which has been observed in plants (6,7) and human stem cells (22). GBSA scores the methylation at three levels: (i) cytosine sites using ‘β score’; (ii) annotated transcripts; and (iii) whole genome.
β score calculation
Input data are parsed to select only sites of cytosines in CpG, CHG and/or CHH sites. For each site within each read, the programme assigns the value of 1 to methylated cytosine and 0 to unmethylated cytosine. GBSA then calculates a ‘β score’ representing the proportion of methylated cytosines among reads of a given genomic position:
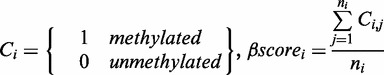 |
where, C denotes cytosine, n denotes the depth of coverage, i denotes a given cytosine position in a CpG, CHG and/or CHH site in a corresponding read j (ranging from 1 to ni).
At this step, GBSA is able to produce two ‘bed graph’ files; the first lists all sequenced cytosine with their respective strand specific ‘β scores’, whereas the second records the sequencing depth of coverage for each site. Both files can be viewed easily in genome browsers, such as IGB (23), IGV (24) or UCSC (25).
Gene-centric methylation scoring
GBSA was developed to address the need to increase our understanding of how methylation is associated with gene regulation. For example, recent genome-wide methylation studies have shown that the relationship between methylation level and gene repression depends not only on methylation status but also on the location of methylated loci within the gene (26). Indeed, it has been shown that the TSS region methylation seems to have a greater impact on the gene repression than gene body methylation (27,28). Consequently, GBSA is designed to score gene methylation levels according to several defined regions (promoter, TSS region, start of first exon to the end of the first intron or gene body) relative to all known RefSeq transcripts. The methylation score is the average ‘β score’ within a given region of interest, and its reliability can be assessed from the percentage of sequenced cytosine sites with a depth of coverage greater than the specified threshold.
Gene independent methylation detection
The advantage of sequencing technologies over microarrays is the ability to study methylation events throughout the genome, that is, within gene loci as well as across intergenic regions. For this purpose, GBSA is able to identify all methylated domains by scanning the whole genome through a configurable sliding window. According to the window set-up (specified length, minimum number of cytosines sequenced and average ‘β score’), GBSA allows detection of both medium- and highly methylated domains. For each detected methylated domains, the software reports several annotations, including the domain coordinates, the distance from the nearest TSS and its RefSeq ID and the percentage of sequenced cytosines. All these features should help users further classify and explore the methylome conveniently.
Visualization
Some BS sequencing alignment programme, such as BSmap and BSseeker, produce SAM or BAM files (29) where reads can be visualized in a genome browser. However, standard genome browsers are not adapted to interpret BS-treated reads, leading to the annotation of unmethylated cytosines (converted to uracil/thymine) as SNPs. GBSA addresses this issue by producing two ‘bedgraph’ files that include all sequenced cytosine of interest and their corresponding strand specific ‘β scores’ and sequence coverage (Supplementary Figure S2a and b). Moreover, for each gene, the DNA methylation pattern, reads coverage and scores can be visualized using the gene viewer implemented in the GUI. If the sliding window-based methylation calling is used, each gene is displayed in reference to its nearest intergenic methylated domains.
Results files
Both gene-centric and gene independent analyses produce a tabular file that lists methylated loci and the previously cited annotations and features. These files include RefSeq IDs for easy data integration and can be manipulated using Excel or R. The whole methylome at the nucleotide resolution is exported as a ‘bedgraph’ file which can be viewed via a genome browser. Along with this, GBSA produces another ‘bedgraph’ file describing the depth of coverage of all sequenced cytosines of interest.
Post-processing
GBSA output result files can be managed via the GUI as well as standard spreadsheet or statistical software, such as Excel or R. As all methylated regions are assigned to their closest RefSeq ID and gene name, data manipulation is easily facilitated in other standard genome analysis software. Post-GBSA genes methylation analysis can be merged into a single file to study differential gene methylation (e.g. multi-conditions) via standard third-party software, such as R/Bioconductor (http://www.bioconductor.org) or TM4-MeV (30).
Implementation
GBSA is programmed in Python 2.7 (http://python.org), and the GUI was designed using QT4 (http://qt.nokia.com). The script command-line release is multi-platform compatible, and the GUI version was compiled for Microsoft Windows vista/7. Source files are also released for compilation to other operating systems.
Availability
GBSA is freely available (General Public License) online at http://ctrad-csi.nus.edu.sg/gbsa. We provide two versions: the GUI release for convenient analysis and the Python command-line script to facilitate integration into custom pipelines.
RESULTS
To highlight the usefulness of our software, we have re-analysed whole-genome BS-seq data of the human embryonic stem cell line H1 (H1-hESC) reported by Lister et al. (22). This is one of the largest whole-genome bisulfite sequencing data sets available, which provides analysis and management challenges that any other data set would generate. The 57 SRR files obtained from 57 runs (NCBI SRA access number: SRX006789) were converted to Fastq and aligned against the hg19 genome assembly using BSseeker allowing two mismatches. After the alignment, all the analyses thereafter were performed by GBSA. The genome-aligned files were concatenated (325 million reads, 36 bp) and analysed using both methods (gene-centric and gene independent) with default parameters. The GBSA quality report on the H1-hESC bisulfite sequencing data revealed that 83% of aligned reads were not polymerase chain reaction duplicates. The gene-centric method showed that 33 968 and 31 585 transcripts met the required depth of coverage and amount of CpG sequenced in gene bodies and promoters, respectively. Finally, 89 803 methylated domains were identified using the gene-independent approach.
Methylated domain characteristics
The default parameters of GBSA allowed detection of both medium- and highly methylated regions. Among the 89 803 identified domains, a majority (92%) were highly methylated (score >0.8) with sizes ranging from 100 to 500 bp (Figure 2A). Using the unbiased gene independent approach, we found 63% of methylated domains in the H1-hESC data were scattered within the intergenic regions, 34% within gene bodies and only ∼3% in gene promoters (Figure 2B). This finding highlights the value of having programmes like GBSA to be able to annotate and analyse methylated domains beyond the commonly studied gene promoters and CpG islands (31). In other words, whole-genome methylation studies using BS sequencing allow the detection of methylated domains in unexpected genomic regions.
Figure 2.

Methylated domains characteristics. GBSA’s methylated domains results file records the coordinates, scores and annotations of the methylation domains and presents them for easy interpretation. Global methylation trends can be appreciated according to (A) the length and the methylation score of detected domains, and by (B) the annotation of these domains relative to their nearest TSS (histogram), the overlap with gene annotation (barplot) or predicted CpG island (pie chart). (C) Methylated domains can be further overlapped with published data sets, such as Encode DNAse clusters, Encode TFBS cluster (ChIP-seq data) or UCSC TFBS conserved (predicted TFBS), to find loci more susceptible for gene regulation. In this bar-plot, each bar represents the total number of methylated domains that overlap with these data sets. The methylated domains are colour coded according their genomic location (genic, promoter or intergenic region). Within each bar, the percentage represents the proportion of methylated domains within the indicated genomic location that overlaps the cited data set.
Potential function of methylated domains
A possible strategy to elucidate the effects of DNA methylation domains on gene regulation is to investigate the roles of these domains in multiple cell types from published data sets. The UCSC browser provides an avenue for such an analysis. The ENCODE project track in UCSC browser (32) provides compilation of genome-wide DNAse hypersensitive areas as well as transcription factor-binding site (TFBS) compilations derived from a large collection of ChIPseq experiments on various cell lines. Additionally, the browser also provides predicted TFBS localization using genome conservation (UCSC tfbsConsSites track) (33) that allows prioritization if found in multiple species. Overlapping these tracks with methylated domains gives useful insights on the potential functions of these loci. For example, by using the UCSC tracks, we found that one third of the H1-hESC methylated domains identified by GBSA overlap with a known DNAse hypersensitive site. Around 10 000 are located in gene bodies, 1400 in promoters and >16 000 in intergenic regions, which represent 30.3% of methylated domains located in gene bodies, 49.9% located in promoters and 26.7% located in intergenic regions. We observed a similar pattern using the compilation of ChIPseq data as well as the UCSC predicted TFBS (Figure 2c). As expected for promoters, a significant amount of methylated domains co-localize with known DNAse hypersensitive sites and TFBS. Interestingly, this co-localization is also observed in a great proportion of methylated domains in gene bodies and intergenic regions. Altogether, these findings suggest that a large proportion of methylated domains are potential proximal, distal or intronic elements that influence gene regulation. This demonstrates the value of being able to observe methylation in both genic and intergenic regions in the genome.
Integration of methylation levels with expression data using GBSA output
As GBSA assigns each methylated domain to a RefSeq ID, it is logical to combine this information with data such as RNA-seq to test for direct evidence of methylation-associated gene repression. To illustrate how GBSA output can facilitate this, we integrated gene methylation levels in several defined regions with their corresponding gene expression level using RNA-seq data from Lister et al. (NCBI SRA access number: SRX026839). RNA-seq reads were aligned against the hg19 genome assembly by TopHat (allowing two mismatches) (34), RPKM expression scores were calculated using the Partek Genome Suite version 6.6 with default parameters (http://www.partek.com), and data integration was performed using R. As evidenced by previous studies (27,28), methylation on the TSS region (±1 kb) had the strongest association with gene repression (Figure 3). In contrast, gene body methylation had no correlation with the expression level. This demonstrates the importance of choosing an appropriate region for studying the functional effects of methylation in whole genome, and how GBSA can help to explore the relationships.
Figure 3.
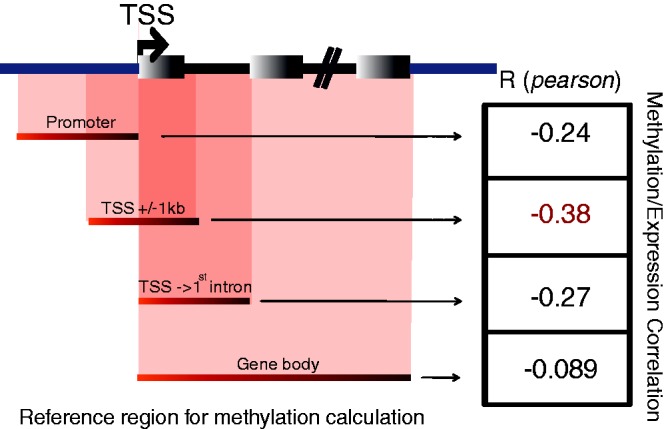
Methylation and gene expression correlation. The Pearson correlation scores between gene expression level and methylation levels found in different gene-based regions [promoter (−2 kb), TSS region (±1 kb), first exon–intron] shows different strengths of associations between locality of the methylation event and gene repression.
DISCUSSION
A common mechanism by which DNA methylation can interfere with gene expression is by obstructing TFs/DNA interaction (35). Besides the TFBS region, promoter regions are also key regulatory elements of genes. Their role is to recruit the basal transcriptional machinery, including the pre-initiation complex that interacts directly with RNA polymerase II and leads to the mRNA expression (36). In this context, it is relevant to test the correlation between methylation at these sites and gene repression. Nevertheless, recent findings have suggested that methylation in the proximal coding region (+1 kb from the TSS) or the first exon are also correlated with gene repression (26). Furthermore, distal regulatory elements can act as silencers as well as enhancers depending on recruited protein complexes. Methylation at the distal region can not only lead to gene silencing (in the case of a methylated enhancer) but also gene activation (methylated silencer) (37,38). This ‘methylation paradox’ (39) shows how the widely accepted thought that DNA methylation mediates gene repression should not be spuriously applied to all cases and highlights the need to accurately annotate methylated domains according to several features. Nucleotide resolution methylation analysis had provided new insight into gene regulatory mechanisms, particularly into roles and effects of methylated CpGs in chromatin states and DNA/protein interactions (40). Unlike microarray technologies, whole genome BS sequencing allows us to interrogate methylation beyond promoters and predicted CpG Islands. Therefore, it has rapidly gained popularity over microarray within the past few years. Indeed, this technology has led to much more comprehensive methylation studies and has revealed several new aspects of DNA methylation. However, as sequencing technologies have become more widely applied, efficient data processing and standardization has become a major challenge. GBSA was thus created to facilitate whole-genome methylation analysis of BS sequencing by providing a comprehensive analysis and interpretable output. Through the gene independent analysis function, GBSA allows systematic localization of genome-wide methylation domains based on sequence methylation levels without a priori knowledge of sequence composition (predicted CpG islands). These domains are scored and labelled in such a way that they can be ranked by methylation scores, sequencing reliability and genomic localization. Furthermore, gene-based studies can be performed using several reference points (promoter, first exon–intron, gene body) according to all known Refseq transcripts. Along with all output, GBSA provides functional data management, quality assessment and data visualization of large-scale genomic methylation experiments at nucleotide resolution. The usefulness of these functionalities was illustrated using one of the largest data sets published to date, and we have shown how GBSA makes genome-wide methylation investigations manageable and how results can be integrated with expression data and other epigenetic marks. To conclude, GBSA opens up new avenues in the analysis of genome-wide methylation events of coding and non-coding regions at the nucleotide resolution and provides an avenue for standardization.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1 and 2.
FUNDING
Funding for open access charge: National Medical Research Council of Singapore [IRF/1278/2010].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Mohd Feroz Mohd Omar and staff from the Centre for Translational Research and Diagnostics for their comments and suggestions.
REFERENCES
- 1.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 2.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 3.Ruike Y, Imanaka Y, Sato F, Shimizu K, Tsujimoto G. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genomics. 2010;11:137. doi: 10.1186/1471-2164-11-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;38:391–399. doi: 10.1093/nar/gkp992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brinkman AB, Simmer F, Ma K, Kaan A, Zhu J, Stunnenberg HG. Whole-genome DNA methylation profiling using MethylCap-seq. Methods. 2010;52:232–236. doi: 10.1016/j.ymeth.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 6.Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics. 2005;21:4067–4068. doi: 10.1093/bioinformatics/bti652. [DOI] [PubMed] [Google Scholar]
- 11.Xu YH, Manoharan HT, Pitot HC. CpG PatternFinder: a Windows-based utility program for easy and rapid identification of the CpG methylation status of DNA. Biotechniques. 2007;43:334, 336–340, 342. doi: 10.2144/000112537. [DOI] [PubMed] [Google Scholar]
- 12.Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res. 2008;36:W170–W175. doi: 10.1093/nar/gkn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohde C, Zhang Y, Reinhardt R, Jeltsch A. BISMA—fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics. 2010;11:230. doi: 10.1186/1471-2105-11-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedersen B, Hsieh T-F, Ibarra C, Fischer RL. MethylCoder: software pipeline for bisulfite-treated sequences. Bioinformatics. 2011;27:2435–2436. doi: 10.1093/bioinformatics/btr394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lutsik P, Feuerbach L, Arand J, Lengauer T, Walter J, Bock C. BiQ Analyzer HT: locus-specific analysis of DNA methylation by high-throughput bisulfite sequencing. Nucleic Acids Res. 2011;39:W551–W556. doi: 10.1093/nar/gkr312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 17.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen PY, Cokus SJ, Pellegrini M. BS Seeker: precise mapping for bisulfite sequencing. BMC Bioinformatics. 2010;11:203. doi: 10.1186/1471-2105-11-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xi Y, Bock C, Müller F, Sun D, Meissner A, Li W. RRBSMAP: a fast, accurate and user-friendly alignment tool for reduced representation bisulfite sequencing. Bioinformatics. 2011;28:430–432. doi: 10.1093/bioinformatics/btr668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krueger F, Kreck B, Franke A, Andrews SR. DNA methylome analysis using short bisulfite sequencing data. Nat. Methods. 2012;9:145–151. doi: 10.1038/nmeth.1828. [DOI] [PubMed] [Google Scholar]
- 22.Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O’Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;470:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicol JW, Helt GA, Blanchard SG, Raja A, Loraine AE. The integrated genome browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009;25:2730–2731. doi: 10.1093/bioinformatics/btp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, Scandura JM. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One. 2011;6:e14524. doi: 10.1371/journal.pone.0014524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodges E, Molaro A, Dos Santos CO, Thekkat P, Song Q, Uren PJ, Park J, Butler J, Rafii S, McCombie WR, et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol. Cell. 2011;44:17–28. doi: 10.1016/j.molcel.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, Zheng H, Yu J, Wu H, Sun J, et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Boil. 2010;8:e1000533. doi: 10.1371/journal.pbio.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- 31.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 32.Rosenbloom KR, Dreszer TR, Long JC, Malladi VS, Sloan CA, Raney BJ, Cline MS, Karolchik D, Barber GP, Clawson H, et al. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res. 2012;40:D912–D917. doi: 10.1093/nar/gkr1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39:D876–D882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curradi M, Izzo A, Badaracco G, Landsberger N. Molecular mechanisms of gene silencing mediated by DNA methylation. Mol. Cell. Biol. 2002;22:3157–3173. doi: 10.1128/MCB.22.9.3157-3173.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koch F, Jourquin F, Ferrier P, Andrau JC. Genome-wide RNA polymerase II: not genes only! Trends Biochem. Sci. 2008;33:265–273. doi: 10.1016/j.tibs.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 37.Tatetsu H, Ueno S, Hata H, Yamada Y, Takeya M, Mitsuya H, Tenen DG, Okuno Y. Down-regulation of PU.1 by methylation of distal regulatory elements and the promoter is required for myeloma cell growth. Cancer Res. 2007;67:5328–5336. doi: 10.1158/0008-5472.CAN-06-4265. [DOI] [PubMed] [Google Scholar]
- 38.Lai AY, Fatemi M, Dhasarathy A, Malone C, Sobol SE, Geigerman C, Jaye DL, Mav D, Shah R, Li L, et al. DNA methylation prevents CTCF-mediated silencing of the oncogene BCL6 in B cell lymphomas. J. Exp. Med. 2010;207:1939–1950. doi: 10.1084/jem.20100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones PA. The DNA methylation paradox. Trends Genet. 1999;15:34–37. doi: 10.1016/s0168-9525(98)01636-9. [DOI] [PubMed] [Google Scholar]
- 40.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.