


Abstract
During V(D)J recombination, recombination activating gene proteins RAG1 and RAG2 generate DNA double strand breaks within a paired complex (PC) containing two complementary recombination signal sequences (RSSs), the 12RSS and 23RSS, which differ in the length of the spacer separating heptamer and nonamer elements. Despite the central role of the PC in V(D)J recombination, little is understood about its structure. Here, we use fluorescence resonance energy transfer to investigate the architecture of the 23RSS in the PC. Energy transfer was detected in 23RSS substrates in which the donor and acceptor fluorophores flanked the entire RSS, and was optimal under conditions that yield a cleavage-competent PC. The data are most easily explained by a dramatic bend in the 23RSS that reduces the distance between these flanking regions from >160 Å in the linear substrate to <80 Å in the PC. Analysis of multiple fluorescent substrates together with molecular dynamics modeling yielded a model in which the 23RSS adopts a U shape in the PC, with the spacer located centrally within the bend. We propose that this large bend facilitates simultaneous recognition of the heptamer and nonamer, is critical for proper positioning of the active site and contributes to the 12/23 rule.
INTRODUCTION
V(D)J recombination assembles and diversifies the variable region of the antigen receptor genes of B and T lymphocytes. The process requires the recombination activating gene proteins RAG1 and RAG2 (together referred to as RAG), which interact with one another and perform DNA binding and cleavage functions to initiate V(D)J recombination (1–3). RAG specifically binds recombination signal sequences (RSSs) that flank the V (variable), D (diversity) and J (joining) coding gene segments that are the substrates of the reaction. RSSs consist of well conserved heptamer and nonamer elements separated by a less well conserved spacer region of 12 bp or 23 bp (referred to as the 12RSS or 23RSS, respectively) (Figure 1B).
Figure 1.

V(D)J recombination and the FRET assay for DNA bending. (A) Steps during V(D)J recombination. The complex containing RAG1, RAG2 and HMGB1/2 is represented as the green shape, V and J coding segments as rectangles and the 12RSS and 23RSS as red and blue triangles, respectively. (B) The RSS and DNA cleavage by RAG. The sequences of the consensus heptamer and nonamer elements and the structures of the nicked intermediate and the hairpin product are shown. (C) DNA bending can alter the FRET efficiency. Donor and acceptor fluorophores are represented as green and red shapes, respectively, and the two DNA strands are indicated with blue and black lines. (D) Schematic of a typical FRET DNA substrate, 23RSSdR2a. The heptamer and nonamer are represented as a blue rectangle and yellow oval, respectively, separated by a 23 bp spacer. The donor (green) is located at the ‘R’ position in the nonamer flank, while the acceptor (red) is located at the ‘2’ position in the coding flank, with distances indicated in bp.
The first step in V(D)J recombination is binding of RAG, probably together with high mobility group box protein (HMGB) 1 or HMGB2, to either a 12RSS or a 23RSS to form the 12 signal complex (12SC) or the 23SC, respectively (Figure 1A). The SC is then thought to capture a protein-free partner RSS (4,5) in a process known as synapsis to form the paired complex (PC). DNA cleavage takes place by a two-step nick-hairpin mechanism (Figure 1B) at each RSS, resulting in two double-strand breaks that separate the coding ends from the signal ends. Although the nicking step can occur in the SC before synapsis, hairpin formation is largely restricted to the PC. The DNA ends are subsequently processed and joined by factors of the nonhomologous end joining repair pathway (6,7). The generation of double strand breaks and the overall recombination reaction occur much more efficiently with a 12/23 RSS pair than with 12/12 or 23/23 RSS pairs, a preference known as the 12/23 rule. The 12/23 rule appears to be imposed at both the synapsis and hairpin formation steps of the reaction (1,2,8). HMGB1 or HMGB2, nonspecific DNA-binding proteins that are capable of stabilizing/inducing DNA bends (9), are vital in vitro for efficient formation of the PC and hence for hairpin formation. They are also important for formation of the 23SC but less so for formation of the 12SC (1,8).
RAG–RSS interactions have been studied in detail with footprinting, interference and photo-crosslinking methodologies [reviewed in (8)], revealing extensive interactions with the nonamer and nonamer-proximal spacer by RAG1 and in and around the heptamer, probably by both RAG1 and RAG2. RAG interactions with the phosphate backbone are detected primarily on one side of the DNA helix and are similar for the 12RSS and the 23RSS. HMGB1/2-RSS contacts, although less well defined, have been proposed to occur at the spacer/nonamer border, near the site of cleavage and within the 23RSS spacer (8,10,11). Consistent with the stable incorporation of a DNA bending protein into RAG–RSS complexes, several previous studies have provided evidence for DNA bends in RAG–RSS complexes, although there is little information about nature and magnitude of bends in the PC (12–16) (see Discussion).
Minimal ‘core’ regions required for DNA cleavage and recombination activity have been defined for murine RAG1 (amino acids 384–1008) and RAG2 (amino acids 1–387). The RAG1 core (RAG1c) contains heptamer and nonamer recognition domains as well as the active site for DNA cleavage. RAG2c appears to function as a cofactor for RAG1, enhancing DNA binding and playing a critical, although poorly understood, role in DNA cleavage. Little high-resolution structural information is available for the RAG core proteins, or for RAG–DNA complexes. The RAG1c nonamer binding domain (amino acids 389–464) is a tightly interwrapped dimer that binds to two nonamers, with the two DNA molecules oriented largely antiparallel and crossing one other at a 30-degree angle (17). Sequence analysis, modeling and mutagenesis has suggested that RAG2 core adopts a 6-bladed β-propellor structure (18,19). The stoichiometry of the RAG proteins in the SC and PC has not been firmly established, although the minimal unit needed for DNA cleavage in the PC is likely a heterotetramer consisting of a dimer of RAG1 and two molecules of RAG2 [discussed in (3,8)].
The factors that dictate the fidelity and efficiency of DNA binding, cleavage and repair during V(D)J recombination are not well understood but are likely important for preservation of the integrity of the genome. Roughly half of all childhood cancers derive from lymphoid cells, and ∼40% of these malignancies contain evidence of erroneous V(D)J recombination, such as chromosomal translocations (20). Proper pairing of RSSs in cis (on the same chromosome) and DNA cleavage in accordance with the 12/23 rule are likely to be steps at which improper recombination events are prevented, suggesting that the PC is an important control point during V(D)J recombination. However, neither the molecular basis of the 12/23 rule nor the architecture of the PC is well understood.
In a previous analysis, we examined the organization of the RSSs in the PC using fluorescence resonance energy transfer (FRET) and 12RSS and 23RSS oligonucleotide substrates in which the donor and acceptor fluorophores were placed in trans on the two different RSSs (13). The data suggested that the RSSs in the PC cross one another and are strongly bent, although we could not rule out a mixture of other, possibly less bent, configurations. Here, we set out to test the idea that the RSSs in the PC contain a large bend and to characterize in some detail the architecture of RSS DNA in this complex. Using FRET and numerous 23RSS oligonucleotide substrates labeled with both donor and acceptor fluorophores (in cis), we have obtained data that support a large bend of the 23RSS in the PC and which allow us to generate a working model for the trajectory of the 23RSS inside the PC.
MATERIALS AND METHODS
Oligonucleotide RSS substrates
Unlabeled and fluorescently labeled deoxyoligonucleotides were synthesized and HPLC purified by Integrated DNA Technologies Inc. (Coralville, IA). The double-stranded DNA RSS substrates, 5′ or internally fluorophore labeled, were assembled by annealing the following oligonucleotides with their complements: 12-RSS, 5′-GATCTGGCCTGTCTTACACAGTGATACAGACCTTAACAAAAACCTGCACTCGAGCGGAG-3′ and 23-RSS, 5′-GATCTGGCCTGTCTTACACAGTGATGGAAGCTCAATCTGAACTCTGACAAAAACCTCGAGCGGAG-3′. FAM (6-carboxy-fluorescein), Alexa 488, Alexa 594 and TAMRA-NHS (carboxytetramethylrhodamine ester) fluorophores were attached to the DNA base through a C6 methylene linker and were incorporated during synthesis using phosphoramidite-labeled nucleotides (dT for FAM and Alexa 488, and an amino-modified NHS ester nucleotide in the case of Alexa 594 and TAMRA). Annealing of oligonucleotides to generate double-stranded DNA was performed in binding buffer [10 mM Tris–HCl (pH 7.5), 50 mM NaCl, 5 mM MgCl2] by heating the complementary oligonucleotides mixed in equimolar amounts for five minutes at 95°C followed by slow cooling to room temperature.
Protein purification
Most experiments were performed with RAG1c (aa 384–1008) fused at its N-terminus to maltose binding protein (MBP) and tagged at its C-terminus with six histidine residues (MBP-RAG1c), and RAG2c (aa 1–387) fused at its N-terminus to glutathione S-transferase (GST-RAG2c). MBP-RAG1c and MBP-RAG1c-D708A were purified from bacteria, whereas GST-RAG2c was purified from HEK293T cells, as previously described (21). Murine polyhistidine–tagged HMGB1 was expressed and purified as described previously (22). In some experiments, MBP-RAG1c (aa 384–1040) and MBP-RAG2c (aa 1–387) proteins were coexpressed in and copurified from HEK293T cells as described by others (23).
In-solution fluorescence data acquisition and analysis
Fluorescence emission spectra were recorded on a PTI C-61 (Photon Technology International) T-format fluorometer equipped with a circulating water bath to control cell temperature. All fluorescence measurements were performed in 150 μl quartz cuvettes (Starna, Atascadero, CA) at 25°C. Fluorescence emission spectra were recorded with an excitation wavelength of 492 nm using an 8 nm band pass setting for both the excitation and emission monochromators. All fluorescence emission spectra were recorded between 510 and 650 nm (for FAM/TAMRA pairs) or 510 and 690 nm (for Alexa 488/Alexa 594 pairs), using 1 nm steps and 2 second integration times. Protein–DNA mixing for various samples was done in ice cold binding buffer followed by 10-min incubation at 25°C. After incubation, each sample mix was moved into the quartz cuvette for recording. In some cases, the first spectral recording was followed by a 20-min incubation at 25°C and a second recording. No photobleaching was detected between any of the two successive recordings. The standard complete reaction contained 15 nM doubly labeled 23RSS DNA, 45 nM unlabeled 12RSS, 125 nM RAG1 and 250 nM RAG2, and 197 nM HMGB1. Control reactions, in which the labeled DNA contains only the donor (FAM/Alexa 488) or the acceptor (TAMRA/Alexa 594) fluorophore, were otherwise identical in protein content and unlabeled RSS partner. Spectra were corrected for background created by lamp fluctuations and the wavelength dependence of the lamp and detector. If sample absorption at excitation wavelength exceeded 0.001, inner filter corrections were also applied according to (24).
For each FRET experiment for a particular configuration of donor and acceptor fluorophore probes, emission spectra (λex = 492 nm) were recorded for the following samples that lacked added proteins, (a) substrate labeled with donor RSS only, (b) singly labeled substrates with donor RSS only and acceptor RSS only (in trans), (c) substrate labeled with acceptor RSS, (d) substrate labeled in cis with donor and acceptor RSS, and for the following samples that contained added proteins, (e) substrate labeled with donor RSS only, (f) singly labeled substrates with donor RSS only and acceptor RSS only (in trans), (g) substrate labeled with acceptor RSS and (h) substrate labeled in cis with donor and acceptor RSS. Emission spectra collected for the donor RSS alone (no protein) and for the acceptor RSS alone (no protein) provided data used by the Felix software (Photon Technology International) to calculate correction coefficients. The emission spectra shown (e.g. Figure 2) were obtained by subtracting (c) from (d) and (c) from (b) to yield the ‘(d + a)’ and ‘(d) + (a)’ traces; or (g) from (f) and (g) from (h) to yield the ‘(d) + (a) + Proteins’ and ‘(d + a) + Proteins’ traces. The use of subtracted spectra corrects for the residual emission arising from direct excitation of the acceptor.
Figure 2.

Detection of energy transfer with the 23RSSdR2a substrate under conditions supporting formation of the PC. Representative steady state emission spectra (plotting fluorescence intensity in arbitrary units (AU) against emission wavelength) are shown for the doubly labeled 23RSSdR2a substrate with fluorophores in cis (A, C, E) or for a mixture of the singly labeled 23RSSdR and 23RSS2a substrates with the fluorophores in trans (B, D, F). The substrates are shown schematically as in Figure 1D. Solid black lines, a mixture of 23RSSdR and 23RSS2a in the absence of protein; red lines, 23RSSdR2a in the absence of protein; dashed black lines, 23RSSdR (donor only) in the presence of RAG + HMGB1 and 12RSS partner DNA; blue lines, 23RSSdR2a [panels (A), (C) and (E)], or 23RSSdR + 23RSS2a [panels (B), (D) and (F)] in the presence of RAG + HMGB1 and 12RSS partner DNA. (A) and (B) FAM/TAMRA-labeled substrates and individually expressed MBP-RAG1c and GST-RAG2c proteins. (C) and (D) FAM/TAMRA-labeled substrates and coexpressed MBP-RAG1c and MBP-RAG2c proteins. (E) and (F) Alexa 488/Alexa 594-labeled substrates and coexpressed MBP-RAG1c and MBP-RAG2c proteins.
Quantification of the energy transfer efficiency (E-FRET) was calculated from emission spectra by the acceptor sensitization method of Fairclough and Cantor (25), as described previously (13). Fluorophore to fluorophore (interfluorophore) distances for each doubly labeled 23RSS substrate in the PC (rcPC) were calculated based on the average E-FRET for that substrate using R0 = 55 Å for the FAM/TAMRA pair of fluorophores (which assumes a rotational diffusion randomized value of the dipole orientation factor κ2 = 2/3) as described previously (13).
The background in the FRET assay was estimated from the E-FRET values for DNA substrates in which the donor and acceptor fluorophores were separated by a large distance (calculated to be >160 Å) and hence no energy transfer was expected in the absence of protein. The 26 measurements made in the absence of protein for these four substrates (substrates 10–13 in Figure 6) were averaged together, yielding a background value of 1.9 ± 0.5% (SEM).
Figure 6.

FRET analysis of the 23RSS in the PC. (A) The 15 fluorophore-labeled substrates, depicted schematically as in Figure 1. (B) Average energy transfer efficiency for substrate in the absence of protein (E-FRET DNA), with the number of independent experiments and SEM in parentheses. ND, no energy transfer above background detected. (C) Distance between the donor and acceptor fluorophores in the substrate in the absence of protein (rsDNA), calculated from the E-FRET in (B), with the predicted interfluorophore distance in the DNA (pred. rs) in red in parentheses, derived from (32). (D) Average energy transfer efficiency for substrate in the complete reaction (E-FRET PC), with the number of independent determinations and SEM in parentheses. Statistical comparison of E-FRET PC versus E-FRET DNA: *P < 0.05; **P < 0.01, ***P < 0.001. (E) Distance between the donor and acceptor fluorophores in the substrate in the complete reaction (rcPC), calculated from the E-FRET in (D). (F) Interfluorophore distances in the model (Figure 7) of the 23RSS in the PC (rcModel, blue), estimated by measuring the distance between the 5′ carbons of the sugars of the two fluorophore-labeled nucleotides. Below (black), interfluorophore distance in the model minus the interfluorophore distance derived experimentally in the PC (rcM−rcPC).
Fluorescence correlation data
Measurements were made on a lab-built instrument based around an inverted microscope with a 488 nm diode-pumped solid state (DPSS) laser for excitation, as described previously (26). The laser power used for all experiments was ∼5 μW as measured before entering the microscope. The emitted fluorescence was collected through the objective and passed through a filter (500LP; Chroma) before being focused onto the aperture of a 50 μm diameter optical fiber directly coupled to an avalanche photo diode. Photon traces were measured for 10 seconds and autocorrelated using a Correlator.com correlator. For each measurement, 10 autocorrelation curves were collected and averaged, then the average curves were fit to an equation describing 3D diffusion of a single fluorescent species, providing a measure of the average diffusion time of all fluorescent species present (27). For each measurement, Alexa 488-labeled 23RSSdR DNA (5 nM) was incubated with MBP-RAG1c, GST-RAG2c, HMGB1 and 12RSS at a fixed molar ratio of 1:2:1.6:3 for 5 min at 25°C, followed by recording of photon traces for 2 min.
Molecular dynamics modeling
The model of the 23RSS in the PC was created using the calculated interfluorophore distances shown in Figure 6, column E, as the main constraints. The DNA was bent in silico in a two-step procedure involving Generalized Born Molecular Dynamics Annealing Simulation using the SANDER module of Amber (28). The first step involved curving the DNA so as to accommodate the constraint imposed by the data obtained with the substrate (23RSSdR5′upa) with the largest linear separation (∼210 Å) between donor and acceptor. To minimize departures from the structure of linear B form DNA, this bend was created by gradually imposing a series of small local changes in tilt and roll parameters of the DNA while blocking base pair openings, propeller twists and buckles as described in (29,30). Specifically, σ, ω and κ (parameters of base pair opening, propeller twist and buckle, respectively) were constrained and unequal constraints were imposed on the two opposite sides of the DNA. In linear B-DNA, the periodicity is 10.5 bases per turn and the distances between equivalent atoms of the two antiparallel strands are ∼20 and ∼14 Å along the major and minor grooves, respectively. The bend created in the first step was consistent with constraints that allowed these parameters to be larger by ∼10% on one side of the helix (residues n, n + 10, etc.) and smaller by ∼10% on the other side of the helix (residues n + 5, n + 15, etc.). In the second step, the initial model was refined by imposing the constraints dictated by the rest of the experimental data. To minimize departures from B form DNA, the constraints used in the first step were not completely eliminated but rather were relaxed by reducing both the energy penalty and the upper and lower boundaries of constraints. The fluorophore-linker moiety was modeled using xLEaP from Amber and Insight II from Accelrys. Molecular Dynamics simulations were further performed so as to sample locally the DNA configuration space consistent the experimental constraints assuming a water/Mg2+ environment at room temperature. Simulations were performed using Not just Another Molecular Dynamics program (NAMD) (31) with Amber force-field on a Bull NovaScale R422/R423 high performance computing cluster.
Statistical analysis
Average E-FRET values were compared with one another using a 2-tailed Student's t-test, and were tested to determine whether they were greater than the background of the FRET assay using a 1-tailed Student's t-test.
RESULTS
Design of the FRET DNA bending assay
We generated a series of consensus 23RSS oligonucleotide substrates containing a donor [6-carboxyfluoresceine (FAM)] and an acceptor [carboxytetramethylrhodamine (TAMRA)] fluorophore located at various positions relative to one another and to the 23RSS (depicted schematically in Figure 1C). In some cases, Alexa 488/Alexa 594 were used as the donor/acceptor fluorophore pair. Fluorophores were coupled to a DNA base using a C6 methylene linker. If RAG binding to the substrate bends or distorts the DNA in the region between the fluorophores, this can alter the interfluorophore distance and the efficiency of energy transfer. Depending on the location of the fluorophores and the magnitude and direction of the bend, the efficiency of energy transfer can increase or decrease compared with the free substrate (Figure 1C). For each substrate, we measured the efficiency of energy transfer (E-FRET) in the absence or presence of proteins and partner 12RSS (to allow PC formation) and used this information to calculate the change in interfluorophore distance that occurred as a result of complex formation. The proteins used in most experiments were individually expressed MBP-RAG1c (aa 384–1008), GST-RAG2c (aa 1–387) and full length HMGB1. In some experiments, copurified MBP-RAG1c (aa 384–1040) and MBP-RAG2c (aa 1–387) were used (see Materials and Methods). Fluorophore-labeled 23RSS (15 nM) and a 3-fold molar excess of unlabeled consensus 12RSS partner were incubated with MBP-RAG1c (125 nM), GST-RAG2c (250 nM) and HMGB1 (197 nM) (hereafter referred to as the full complement of proteins) in a buffer containing 5 mM Mg2+ (hereafter referred to as the complete reaction). These protein concentrations were chosen because they supported maximal RSS binding and cleavage in conventional mobility shift and cleavage assays (data not shown). DNA substrates were named according to the positions of the fluorophores, with the donor position specified with a capital letter preceded by ‘d’ and the acceptor by a number followed by ‘a’. For example, the 23RSSdR2a substrate depicted in Figure 1D has the donor at position R and the acceptor at position 2. The locations and names of the fluorophore positions studied are depicted in Supplementary Figure S1. The background of the FRET assay was calculated to be 1.9 ± 0.5% (see Materials and Methods).
23RSSdR2a reveals a major RAG-induced DNA bend
Although evidence for DNA bending was obtained with many of the substrates examined (see below), the 23RSSdR2a substrate (Figure 1D) was particularly informative and was examined in greatest detail. In this substrate, the donor lies 3 bp 3′ of the nonamer and the acceptor 9 bp 5′ of the heptamer and are separated by a total of 51 bp, or a distance of 171–180 Å in B form DNA [calculated using data from (32)]. Because energy transfer is not detectable with these fluorophores at distances >∼90 Å, no energy transfer was expected in the free substrate DNA. Indeed, as shown in Figure 2A, C and E, the emission spectra for 23RSSdR2a [red line; indicated as ‘(d + a)’] was similar to that of a control reaction in which the donor and acceptor were in trans on different 23RSS substrates (an equimolar mixture of 23RSSdR and 23RSS2a substrates; solid black line; ‘(d) + (a)’). These spectra show a peak of emission at ∼520 nm, as expected, for the FAM (or Alexa 488) donor, with no evidence of acceptor emission or quenching of donor emission when the fluorophores are in cis as compared with in trans. Note that all spectra have been corrected for residual acceptor fluorescence (see Materials and Methods).
Addition of the full complement of proteins and unlabeled 12RSS to the 23RSSdR substrate that contains only the donor (dotted black line; ‘(d) + proteins’) resulted in substantial quenching of donor fluorescence (Figure 2A), which is due to interactions between the proteins and the FAM donor. When the doubly labeled 23RSSdR2a substrate was used (blue line; ‘(d + a) + Proteins’), we observed a further decrease in donor emission as well as acceptor sensitization (emission peak between 570 and 595 nm) (Figure 2A, compare blue and dotted black lines). These changes in the spectra, which are directly attributable to the sensitization of the acceptor, indicate that energy transfer is occurring between the two fluorophores. This was consistently observed in 17 independent experiments, quantitation of which (see Materials and Methods) yielded an average E-FRET of 18.0 ± 1.2%. This indicates that in the protein–DNA complex, the donor and acceptor are now separated by a distance <90Å. Using some simplifying assumptions (discussed below), we calculated the interfluorophore distance of 23RSSdR2a in the PC to be 71 Å ± 10Å. This could only occur if the DNA undergoes substantial bending/distortion in the region between the two fluorophores.
The experiments described above were performed with individually expressed MBP-RAG1c and GST-RAG2c proteins, and we wanted to determine if similar results would be obtained with coexpressed MBP-RAG1c and MBP-RAG2c, which are even more active in binding and cleavage assays (data not shown). The coexpressed MBP-RAG proteins yielded similar results, with an E-FRET of 19.7% (Figure 2C). To confirm that these results were not dependent on our choice of fluorophores, we tested a 23RSSdR2a substrate labeled with Alexa 488 (donor) and Alexa 594 (acceptor). This substrate, when assayed with the coexpressed MBP-RAG proteins, also yielded energy transfer (E-FRET = 28.3 ± 1.4%), with acceptor sensitization observed as a distinct peak between 610 and 650 nm (Figure 2E). We conclude that FRET is consistently observed with the 23RSSdR2a substrate under conditions that allow formation of the PC.
It was important to confirm that the observed energy transfer was occurring between the donor and acceptor on the same 23RSSdR2a substrate molecule (in cis), rather than by synapsis/aggregation of two (or more) 23RSSdR2a molecules, thereby allowing energy transfer in trans. 23/23 RSS synapsis was not anticipated to occur at substantial levels in our reactions given the preference of RAG/HMGB1 for 12/23 RSS synapsis and the 3-fold molar excess of the 12RSS. Nonetheless, numerous control reactions were performed to detect potential trans FRET using an equimolar mixture (7.5 nM each) of 23RSSdR and 23RSS2a under identical protein and partner 12RSS conditions. In no case was energy transfer detected in trans: this was true for reactions containing individually expressed MBP-RAG1c/GST-RAG2c (Figure 2B), and for reactions containing coexpressed MBP-RAG1c/MBP-RAG2c with either set of fluorophores (Figure 2D, F). These results argue that the energy transfer detected with the 23RSSdR2a substrate occurs in cis.
One caveat was that the cis and trans experiments of Figure 2 differed both in the concentration of the donor-labeled species and the proportion of 23RSS–23RSS interactions that could result in FRET. To make the cis and trans experiments more closely comparable, we repeated the cis experiments diluting the 23RSSdR2a substrate (with Alexa fluorophores) with unlabeled 23RSS substrate: using either an equimolar amount of the two 23RSSs (7.5 nM each), or a 2-fold excess of unlabeled substrate (5 nM 23RSSdR2a with 10 nM unlabeled 23RSS), a clear FRET signal was still detected (Supplementary Figure S2). We conclude that if a substantial fraction of the energy transfer were occurring in trans, the control reaction of Figure 2F would have detected it, and hence that much or all of the energy transfer we detect occurs between fluorophores located on the same DNA molecule. Further experiments, described below, provide additional evidence that FRET is occurring primarily or exclusively in cis.
Protein requirements for energy transfer with the 23RSSdR2a substrate
We next investigated the protein requirements for FRET with the 23RSSdR2a substrate. HMGB1 enhances RAG binding to the 23RSS, formation of the PC and coupled cleavage, and could reasonably be expected to generate and/or stabilize DNA bends, particularly in the 23RSS (8,33). Omission of HMGB1 from the complete reaction reduced FRET to background levels (Figure 3A), and energy transfer increased as HMGB1 was titrated into FRET reactions (Figure 3B). E-FRET reached a plateau at a HMGB1 concentration of 180 nM (Figure 3B), approximately the concentration used in the standard FRET reaction. Energy transfer was also not detectable in reactions lacking RAG1 (Figure 3E), consistent with the central role played by RAG1 in RSS binding (8). In contrast, omission of RAG2 reduced but did not eliminate energy transfer, with both donor quenching and acceptor sensitization detected (Figure 3C), yielding an average E-FRET value of 7.4 ± 1.0%, which was significantly above background (1-tailed t-test, P = 0.0006). We recently reported that RAG1 and HMGB1, in the absence of RAG2, are able to synapse two RSSs in a manner that is strongly dependent on the nonamer but not the heptamer (17), raising the possibility that the FRET detected with 23RSSdR2a in the absence of RAG2 was due to trans interactions. However, no evidence of energy transfer was detected in the absence of RAG2 when the fluorophores were located in trans (Figure 2D). Overall, the data indicate that FRET with the 23RSSdR2a substrate is strictly dependent on RAG1 and HMGB1, and optimal in the presence of RAG2. However, the detection of even weak energy transfer in the absence of RAG2 indicates that RAG1 and HMGB1 are capable of bending the 23RSS in such a way that the interfluorophore distance is reduced from 170–180 Å to <90 Å.
Figure 3.

The protein requirements for FRET with the 23RSSdR2a substrate. Steady state emission spectra displayed as in Figure 2 were gathered from reactions similar to those in Figures 2A, B, except that (A) HMGB1 was omitted from the reaction; (C) and (D) RAG2 was omitted from reactions with the fluorophores in cis (C) or in trans (D); (E) RAG1 was omitted from the reaction. (B) Plot of FRET efficiency for the 23RSSdR2a substrate as a function of HMGB1 concentration. All reactions contained RAG and 12RSS partner DNA and HMGB1 was added in increasing amounts. The experiment in (D) has been performed only once. The substrates are shown schematically as in Figure 1D.
Optimal energy transfer occurs under conditions that support coupled cleavage
The detection of FRET in the absence of RAG2 led us to measure energy transfer under other conditions where hairpin formation does not occur or is inefficient. Ca2+ supports SC and PC formation but prevents DNA catalysis (2,33); replacement of Mg2+ with Ca2+ in the complete reaction reduced E-FRET from 18.0% to 6.9 ± 0.2% (Figure 4, bar 1 versus 4). A similar reduction was observed when WT MBP-RAG1c was replaced with MBP-RAG1c-D708A (E-FRET of 7.2 ± 1.4%; Figure 4, bar 6), a mutant that supports SC and PC formation but no catalysis (21,34,35). Energy transfer was also reduced but not eliminated when the partner 12RSS was omitted from the reaction (E-FRET of 8.8 ± 2.8%; Figure 4, bar 5), a condition that allows 23SC formation and nicking but not 12/23 RSS synapsis or hairpin formation. In all three of these cases, as in the absence of RAG2, E-FRET was significantly reduced relative to the standard reaction but significantly greater than background (see Figure 4 legend for statistics). Omission of the 12RSS might also allow for increased synapsis of two 23RSSdR2a substrates compared with the complete reaction (4); the fact that E-FRET goes down, not up, in the absence of the 12RSS provides another argument against the idea that energy transfer in the complete reaction occurs in trans. Together, these data argue that energy transfer with the 23RSSdR2a substrate is optimal in a cleavage-competent PC.
Figure 4.

Energy transfer with the 23RSSdR2a substrate is optimal under conditions supporting formation of a cleavage-competent PC. Bars represent the average energy transfer efficiency, with error bars indicating the SEM. The average E-FRET value is listed in or above each bar, with the number of experiments in parentheses. Bar 1 shows data for the standard FRET reaction with the 23RSSdR2a substrate performed as in Figure 2A. The other bars represent experiments that differ from the standard reaction as follows: bar 2, FRET measured after an additional 20 min at 25°C; bar 3, use of a prenicked 23RSSdR2a substrate; bar 4, substitution of Ca2+ for Mg2+; bar 5, omission of the 12RSS partner DNA; bar 6, substitution of MBP-RAG1c D708A for MBP-RAG1c; bar 7, mutation of the 23RSSdR2a nonamer; bar 8, mutation of the 23RSSdR2a heptamer; bar 9, omission of HMGB1; bar 10, omission of RAG2; bar 11, omission of RAG1. Average E-FRET values were compared with that in the standard reaction (bar 1) using a 2-tailed t-test; *P < 0.01; **P < 0.001. All average E-FRET values were significantly above the background of 1.9 ± 0.5% except for bar 7, bar 9 (P = 0.62) and bar 11, using a 1-tailed t-test (P < 0.001 for bars 1, 2, 3, 4, 5, 6 and 10, and P = 0.01 for bar 8). The substrate is depicted schematically with the heptamer and nonamer as a dark gray rectangle and circle, respectively, and the donor and acceptor fluorophores as a light gray hexagon and circle, respectively.
This led us to consider the idea that DNA cleavage itself contributed to higher levels of energy transfer in our FRET assay. This seemed unlikely because our previous study demonstrated that nicking is weak (<2.5%) and hairpin formation is virtually undetectable at 10 minutes (the time point at which FRET is assessed) with MBP-RAG1c and GST-RAG2c (13), and we have confirmed these results with some of the preparations of proteins used in this study (data not shown). Furthermore, cleavage reactions were performed at 37°C and hence overestimate the amount of cleavage that occurs in FRET reactions (which take place at 25°C). Hence, levels of nicking and hairpin formation are likely to be extremely low when we assess energy transfer. However, we further investigated possible contributions of DNA cleavage to the observed FRET using a ‘prenicked’ 23RSSdR2a substrate (containing a single strand break at the position normally nicked by RAG) together with an intact 12RSS partner. This yielded a level of energy transfer similar to that observed with the intact 23RSSdR2a substrate (Figure 4, bars 1 and 3), indicating that nicking does not substantially influence FRET levels. We also assessed FRET in standard reactions allowed to incubate for an additional 20 minutes at room temperature, which substantially increases nicking and allows for a small amount of hairpin formation [(13) and data not shown]; this also did not cause a large change in the amount of energy transfer detected, although the decrease (to 15.8%) was statistically significant, in part because of the large number of measurements (Figure 4, bar 2). We conclude that nicking does not substantially alter the interfluorophore distance of the 23RSSdR2a substrate in the PC and that hairpin formation is not a relevant factor in our analysis because it occurs at low levels. This in turn argues that the reductions in E-FRET observed in Ca2+, with the D708A RAG1 mutant, and in the SC, are not due to alterations in catalysis itself, but rather to alterations in the structure and/or composition of the protein–DNA complexes formed.
Heptamer and nonamer contributions to 23RSSdR2a FRET
The nonamer is critical for stable RAG binding to the RSS (1,8), leading to the prediction that mutation of the nonamer would strongly reduce energy transfer. Indeed, mutation of three critical top strand A bases in the 23RSSdR2a nonamer (5′-ACAAAAACC changed to 5′-ACAAGTCCC) reduced E-FRET to background levels (Figure 4, bar 7). The heptamer also makes contributions to RAG binding, with most protein contacts in the vicinity of the 23RSS heptamer being RAG2 and HMGB1 dependent (8). Scrambling of the 23RSSdR2a heptamer (5′-CACAGTG changed to 5′-TGAATAC) reduced but did not eliminate energy transfer (E-FRET = 6.4 ± 1.5%; Figure 4, bar 8), with the residual level of energy transfer comparable with that seen in the absence of RAG2 or under conditions that interfere with cleavage (Figure 4, bars 4–6, 10). Thus, our data indicate that optimal energy transfer requires specific RAG-nonamer and RAG-heptamer contacts, but in the absence of the latter, as in the absence of RAG2, the substrate still undergoes a substantial bend that supports detectable energy transfer.
The consensus nonamer contains an A5 tract, which is predicted to have an intrinsic (protein-independent) bend (36–38). This could contribute to the bending we detect using the 23RSSdR2a substrate. We designed FRET substrates to test whether the nonamer A5 tract does indeed have an intrinsic bend. The 23RSSdR6a substrate contains donor and acceptor fluorophores flanking the nonamer (Supplementary Figure S3), positioned 14 bp apart on opposite strands so that they lie on the same side of the helix and so that the predicted intrinsic bend of the A5 tract would bring them closer together (36). As expected from the close proximity of the fluorophores (calculated to be ∼50–58 Å), the 23RSSdR6a substrate exhibits substantial energy transfer in the absence of protein (E-FRET = 39.4%; Supplementary Figure S3). Disruption of the A5 tract in the 23RSSdR6aNonMut substrate reduced E-FRET (28.9%; Supplementary Figure S3), as predicted for disruption of an intrinsic bend, although this decrease was not statistically significant (2-tailed t-test, P = 0.25). Although more experiments would be required to address this issue definitively, these results are consistent with the existence of an intrinsic bend in the nonamer, which is lost upon disruption of its A5 tract. Unfortunately, FRET experiments performed with the 23RSSdR6a substrate in the complete reaction were uninformative owing to strong quenching of both fluorophores; no such quenching (or other spectral changes) were observed with the 23RSSdR6aNonMut substrate (data not shown), suggesting that the quenching observed with 23RSSdR6a was due to protein–fluorophore interactions.
Characterization of RAG-HMGB1 interactions with the 23RSS by fluorescence correlation spectroscopy
As discussed above, experiments with donor and acceptor fluorophores positioned in trans argue that the energy transfer we detect with the 23RSSdR6a substrate is not a result of synapsis or aggregation of labeled 23RSS substrate molecules. To strengthen this conclusion further and search for evidence of a physical association of two (or more) 23RSSs using a different approach, we examined the behavior of the 23RSSdR substrate using fluorescence correlation spectroscopy (FCS). FCS assesses the dynamic diffusion properties of fluorescent molecules based on a statistical analysis of fluctuations of fluorescence emission. A confocal optical geometry is used to record changes in emission fluorescence in a small well-defined focal volume in the sample. The recorded fluorescence emission data are used to construct an autocorrelation function, from which one derives the diffusion time, the concentration and the ‘brightness’, or photon counts per molecule, of the fluorescent species. Photon emission traces were recorded from samples containing 5 nM Alexa 488-labeled 23RSSdR and increasing concentrations of MBP-RAG1c, GST-RAG2c, HMGB1 and 12RSS, maintained at a fixed molar ratio of 1:2:1.6:3. If substantial 23RSSdR–23RSSdR interactions occurred, we expected to see an increase in photon counts per molecule. Furthermore, aggregation would result in a spurious FCS signal that cannot generally be fit by the correlation model used for the analysis. Contrary to these predictions, we observed a steady decrease in photon counts per molecule as the concentrations of the other reactants were increased (Figure 5A) due to donor quenching that occurs on incorporation of the 23RSS into complexes, and the FCS signals could be fit by the model. Control reactions using a DNA molecule doubly labeled with Alexa 488 yielded signals twice as bright as the reactions with Alexa 488-labeled 23RSSdR, showing that if multiple DNAs were incorporated into complexes, we should have been able to detect this. In parallel with the decrease in brightness, the diffusion time increased (Figure 5B), as expected, for incorporation of increasing proportions of the DNA substrate into protein–DNA complexes, which are larger and diffuse more slowly than the free DNA. Both the counts per molecule and diffusion time begin to plateau at protein concentrations above 300/600/480 nM RAG1/RAG2/HMGB1, well above the concentrations used in the standard FRET reaction (125/250/185 nM). Hence, the protein concentrations that yield maximal incorporation of 23RSS into complexes are greater than those that are optimal for FRET and coupled DNA cleavage. This might be explained by the previous observation that high RAG concentrations inhibit PC formation and coupled cleavage, probably by driving high RAG occupancy of individual 12RSS and 23RSS substrates, which in turn inhibits synapsis (4). The FCS analysis provides no evidence for synapsis/aggregation of 23RSSs at any protein concentration, supporting the conclusion that in the FRET assays, most complexes contain only a single-labeled 23RSSdR2a substrate and hence that the energy transfer detected occurs in cis.
Figure 5.
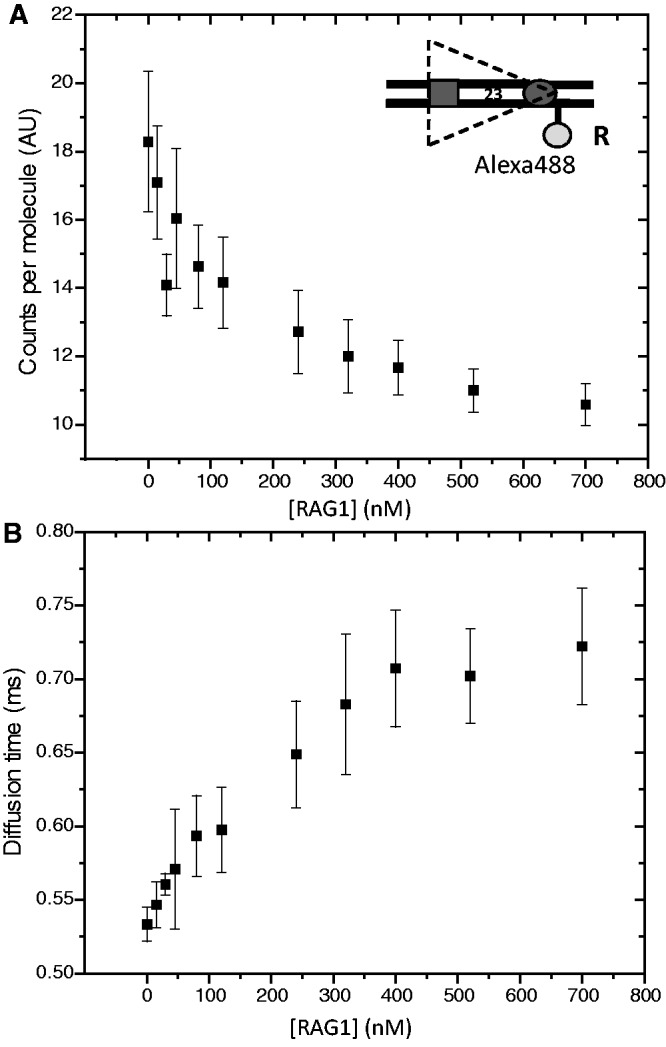
Analysis of RAG/HMGB1 binding by FCS. Photon emission traces were recorded from samples containing 5 nM Alexa 488-labeled 23RSSdR and increasing concentrations of MBP-RAG1c, GST-RAG2c, HMGB1 and 12RSS, maintained at a fixed molar ratio of 1:2:1.6:3. Each data point represents the average of three independent measurements (error bar = standard deviation). (A) Brightness, or photons per labeled DNA molecule, plotted against RAG1 concentration. (B) Diffusion time versus RAG1 concentration. The substrate is depicted schematically as in Figure 4.
Mapping 23RSS bending in the paired complex
Our data argue that the 23RSS bends in the PC so as to reduce the interfluorophore distance of 23RSSdR2a from ∼170 Å in free DNA to ∼70 Å in the PC. This large bend might result from the additive effect of multiple bends/distortions, or from a single large bend associated with extreme DNA distortions. To address this issue and gain a better understanding of the shape of the 23RSS in the PC, we performed FRET experiments with a collection of additional DNA substrates in which FAM and TAMRA fluorophores spanned a variety of intervals along the length of the 23RSS. Reactions were performed under the same PC conditions described above for 23RSSdR2a (e.g. Figure 2A). The results obtained, along with schematic diagrams of the substrates, are shown in Figure 6. The precise location of each fluorophore labeling position is shown in Supplementary Figure S1. Fluorophores were positioned to span the nonamer (Figure 6, substrates 1–3), portions of the spacer (substrates 4–6), the heptamer (substrates 7–9) or the entire RSS (substrates 10–13). Two substrates were also tested in which the donor and acceptor were directly across the DNA helix from one another (substrates 14–15). Columns B and D of Figure 6 show the E-FRET values measured in the absence or presence of proteins, respectively, whereas columns C and E show the interfluorophore distances calculated from these measurements. Column C also shows predicted interfluorophore distances calculated using a vector function described for double stranded B form DNA (32).
For the majority of the substrates examined, energy transfer was significantly altered in the PC relative to naked DNA (Figure 6, columns D and B). This included all of the substrates with fluorophores flanking the nonamer (substrates 1–3), two out of three substrates with fluorophores flanking the heptamer (substrates 7 and 9) and all substrates with fluorophores flanking the entire RSS (substrates 10–13). The latter results are of particular importance, demonstrating that in four different substrates in which the fluorophores are separated by at least 160 Å in the linear DNA, formation of the PC leads to clearly detectable energy transfer and a calculated interfluorophore separation of <80Å. This provides strong support for the conclusion that the 23RSS undergoes a major bend in the PC. In some substrates, E-FRET was similar in free DNA and the PC. This was true for the three substrates in which the fluorophores span some or most of the spacer (substrates 4–6; substrate 5 shows increased energy transfer in the PC relative to free DNA, and the change is not statistically significant). It was also the case for substrates in which the fluorophore pairs were directly across the helix from one another (substrates 14–15), as expected, as bends or distortions were unlikely to alter the interfluorophore distance in such cases.
Together, these results indicate the presence of DNA bends in the vicinity of both the heptamer and the nonamer, and suggest that multiple bends in different parts of the substrate combine to create a large overall bend in the 23RSS in the PC.
Molecular dynamics modeling of the 23RSS in the PC
We then used the calculated interfluorophore distances (Figure 6, column E) as the constraints for Molecular Dynamics modeling of the structure of the 23RSS in the PC. This involved the in silico introduction of a large bend in the DNA followed by refinement of the bend to maximize conformity with the experimental data and to minimize the energy and deviations from B form DNA of the final structure (see Materials and Methods). The resulting model shows a strongly bent U-shaped DNA molecule with the spacer occupying the base of the U and the heptamer/coding flank and nonamer/nonamer flank constituting its arms (Figure 7A; PDB file provided as Supplementary dataset 1). Multiple distinct bends exist in the model, with the four most prominent located near the coding flank/heptamer border (51°; external angle), the heptamer/spacer border (44°), the center of the spacer (49°) and the spacer/nonamer border (55°). Interestingly, when sites of protein–DNA backbone interactions (ethylation interference) in the 23SC (39) are mapped onto the model, most contacts lie on the inner (concave) surface of the DNA (Figure 7B; yellow). In contrast, sites of DNAse I hypersensitivity in the signal end complex (40), a postcleavage complex containing RAG, HMGB1, and two cleaved RSSs (41), map predominantly to the outer (convex) surface of the DNA, and notably, are located near the prominent bends in the spacer and at the spacer/nonamer border (Figure 7B; magenta).
Figure 7.

Model of the 23RSS DNA in the PC derived from molecular dynamics modeling of the data from Figure 6. DNA is depicted as a ball and stick model with N in blue, O in red, C in grey, P in orange, and H in white. (A) The locations of the heptamer (red), nonamer (purple), and the bases that are donor (green) and acceptor (yellow) fluorophore labeled the 23RSSdR2a substrate are indicated. (B) Sites of ethylation interference in the RAG1–RAG2–HMGB1–23RSS (23SC) complex (yellow), as determined by Swanson (10), and sites of DNAse I hypersensitivity in the signal end complex (magenta) (40).
To assess the fit between the model and the FRET data, it was necessary to calculate the interfluorophore distances predicted by the model. This was complicated by the fact that the fluorophores are covalently attached to the DNA through a flexible six-carbon methylene linker and hence are mobile. Interfluorophore distances in the model were therefore estimated by measuring the distance between the 5′ carbons of the sugars of the two fluorophore-labeled nucleotides (Figure 6, column F). Model analysis and molecular dynamics simulations of the fluorophore/linker configurations revealed that the average distance between the aromatic center of the fluorophore and the linker atom that attaches to the DNA base was 13–14 Å, placing the fluorophore (on average) farther from the attachment point on the base than the 5′ carbon of the sugar (which is 5.5 Å from the attachment point). This needs to be taken into account when comparing the values calculated from the FRET data (rcPC; Figure 6, column E) and those calculated from the model (rcModel; column F). The difference between rcModel and rcPC (column F) is ≤5Å for the substrates with fluorophores flanking the nonamer (Figure 6, rows 1–3), the substrate with fluorophores flanking the entire spacer (row 5) and for most of the substrates with fluorophores flanking the entire RSS (rows 10–13). The difference between the model and the experimental data is somewhat greater (5.9–10.7 Å) for substrates in which the fluorophores flank the heptamer (rows 7–9). The discrepancy becomes large (16–21 Å), and the model consistently underestimates the experimentally derived distances, for substrates in which the fluorophores are attached close to or directly opposite one another on the two DNA strands (rows 4, 6, 14 and 15). In these cases, the model fails to account for the large contribution of the C6 linkers to the interfluorophore separation and hence to the measured E-FRET values.
Testing predictions of the 23RSS model
The U shape of the 23RSS in our model leads to clear predictions for the results of additional FRET experiments. Specifically, substrates containing fluorophore pairs positioned on the inner aspect of the U should exhibit an increase in energy transfer on PC formation, whereas the opposite should occur for fluorophore pairs positioned appropriately on the outside of the U. We designed and tested one new 23RSS substrate of each type, selecting fluorophore positions flanking the central portion of the spacer (23RSSdJ10a and 23RSSdL11a; Figure 8). Focusing on this region allowed us to test the direction of bending in a critical location (the base of the U) and to augment the FRET data relating specifically to the spacer. In both substrates, the fluorophores were positioned 14 bp apart on opposite strands, and, as expected, their E-FRET values in the absence of protein were relatively similar (33.0 and 27.7%). However, the two substrates yielded distinct results upon PC assembly. In 23RSSdJ10a, where the fluorophores are predicted to lie on the inside of the bend, energy transfer increased significantly to 44.6% (Figure 8A; P = 0.008), whereas in 23RSSdL11a, where the fluorophores are predicted to lie on the outside of the bend, energy transfer decreased to 24.5% [Figure 8B; this change, while in the expected direction, was not statistically significant (P = 0.17)]. These results provide support for our model and strengthen the evidence for DNA bending in the 23RSS spacer in the PC.
Figure 8.

Testing the model of the 23RSS in the PC. (A) and (B) The locations of the bases that are donor (green) and acceptor (yellow) fluorophore labeled in the 23RSSdJ10a (A) and 23RSSdL11a (B) substrates are shown on the model of the 23RSS, depicted as in Figure 7A. The interfluorophore distance in the model (rcModel), calculated as in Figure 6, is indicated. (C) and (E) Histograms of E-FRET values (± SEM) obtained for the 23RSSdJ10a (C) and 23RSSdL11a (E) substrates, displayed as in Figure 4; *P = 0.008 for E-FRET PC versus E-FRET DNA. (D) Values for rsDNA, pred. rs, rcPC, rcModel, for the 23RSSdJ10a and 23RSSdL11a substrates, calculated and displayed as in Figure 6.
DISCUSSION
There are numerous reasons to think that DNA bending occurs during the formation of RAG–RSS complexes. The incorporation of DNA-bending proteins HMGB1 or HMGB2 into these complexes (8), the strong correlation between the DNA bending activity of HMGB1 and its function in RAG-mediated DNA cleavage (42,43), the particularly important role of HMGB1/2 in complexes involving the 23RSS (44,45) and the similarity of the protein-DNA contacts in the 12SC and 23SC despite the differences in spacer length (8), provide support for RSS bending. Experimental evidence for DNA bending in the SC (12,14) and the PC (16) have been reported. The lack of high-resolution structures of RAG–HMGB–DNA complexes, however, has left major uncertainties about the structure of the RSS in these complexes, particularly the PC. The results we report here begin to fill this gap by identifying a dramatic bend in the 23RSS, by defining the conditions for optimal formation of this bent structure, and by allowing construction of a detailed working model of the 23RSS in the PC.
Defining a large bend in the 23RSS
The most important support for the U-shaped structure we propose for the 23RSS in the PC derives from the detection of energy transfer in the complete reaction with four DNA substrates in which the donor and acceptor fluorophores lie in the coding and nonamer flanks and hence span the entire RSS (Figure 6, rows 10-13). The data strongly suggest that the fluorophores in these substrates, which are >160 Å apart in the free DNA, are separated by <∼80 Å in PC. For the 23RSSdR2a substrate, energy transfer was shown to occur with different preparations of the RAG proteins and with different fluorophores, and to require those elements (RAG1, HMGB1 and the nonamer) that would be expected to be critical for formation of stable RAG–RSS complexes. The data we obtained with a variety of other substrates, in which the fluorophores flank subregions of the 23RSS, argue that the large bend arises from the additive effect of multiple smaller bends.
In our previous study of the organization of the RSSs in the PC, we did not detect energy transfer between fluorophores positioned in cis at the 5′-ends of the two strands of the 23RSS (13), which contrasts with the detection of FRET in configurations 10–13 where one or both of the fluorophores is not at the end of the substrate. The calculated distance between the 5′-ends of the 23RSS in our model (Figure 7) is ∼77Å, which is compatible with the detection of energy transfer. Because our experimental conditions were subtly different in the two studies, we analysed the same 5′-end–labeled 23RSS substrate (d5′up-5′bta) as examined previously, but under the conditions used in the current study. Consistent with our previous results, no energy transfer was detected with this substrate (data not shown). It is possible that this is due to the fluorophore linkers being oriented away from one another in this substrate.
Critical to our conclusions is that the energy transfer we detect in the complete reaction occurs in cis (between the two fluorophores on a single 23RSS molecule) and not in trans (between fluorophores on different 23RSS molecules). Extensive evidence indicates that this is the case. No energy transfer was ever detected in numerous experiments in which donor and acceptor fluorophores were placed in trans on different 23RSS substrates. Dilution of the labeled 23RSSdR2a substrate with unlabeled 23RSS did not yield results compatible with substantial trans energy transfer (Supplementary Figure S2). Energy transfer actually decreased when the 12RSS was omitted from the complete reaction, despite the fact that doing so would have been expected to allow increased trans 23RSS interactions. FCS experiments detected no evidence for synapsis or aggregation of 23RSSs. And finally, our ability to incorporate data derived from numerous different substrates (Figure 6, 8) into a unifying structural model—an effort based on the assumption of energy transfer in cis—argues against a substantial contribution of energy transfer in trans.
Limitations of the FRET analysis and the 23RSS model
There are several features of our FRET assay that place limits on our conclusions and introduce uncertainty into our model. The calculation of interfluorophore separation in the PC (rcPC) assumes that all 23RSS molecules are incorporated into the PC in the FRET reaction. Although reaction conditions were chosen for maximal coupled DNA cleavage (presumably reflecting maximal formation of the active PC), this does not mean that all 23RSSs are in the PC. In fact, it is likely that optimal formation of the PC requires subsaturating protein concentrations to avoid incorporation of all available RSSs into SCs (4), and the results of the FCS analysis (Figure 5) argue that our reaction conditions are indeed subsaturating. We note that incorporation of <100% of 23RSSs into the PC should lead us to underestimate of the extent of energy transfer and overestimate rcPC. Our finding that optimal energy transfer with the 23RSSdR2a substrate requires RAG1, RAG2, HMGB1, the nonamer and heptamer, Mg2+ and partner 12RSS—everything known to be required for formation of a cleavage competent PC—argues that energy transfer derives primarily from active complexes.
Although the absolute amount of 23RSS incorporated into the PC in our FRET assays is not known, it is likely that the relative amount is similar in the assays used to derive (Figure 6) and test (Figure 8) our model. In all of these experiments, the sequence of the 23RSS and the reaction conditions are identical, the only variable being the locations of the fluorophores. We have avoided attaching fluorophores inside the heptamer or nonamer to minimize the possibility that they would interfere with RAG binding, and in the instances where we have measured DNA cleavage with fluorophore-labeled substrates (which includes the dR and 2a positions), we have not detected any influence of the fluorophore on cleavage efficiency [(13) and data not shown].
A second type of limitation arises from the possibility that the PC, and the DNA within it, exhibit substantial dynamic behavior. Our analysis yields an estimate only of the average position of the 23RSS in the PC and does not capture potential mobility of the DNA in the complex, or other sources of heterogeneity. The flexibility of the C6 linker attaching the fluorophores to the DNA is one source of uncertainty. In this regard, we cannot rule out the possibility that protein-fluorophore interactions (which occur with most of the substrates analysed) constrain fluorophore motion and thereby alter energy transfer in some substrates. We also note that modeling was performed under the constraint that all bases remain paired, which is unlikely to be the case given the substantial evidence for base unpairing near the site of cleavage before nicking and/or hairpin formation (46,47).
These considerations, combined with the limited number of FRET substrates that interrogate each subregion of the 23RSS, dictate that the 23RSS structure depicted in Figure 7 represents a working model and should not be regarded as providing high-resolution structural insight. The essential components of the model, and the central conclusions of our study, are that the 23RSS undergoes a dramatic bend in the PC such that the coding flank and nonamer flank are separated by <80 Å and that this bend arises from the additive effect of multiple smaller bends in several regions of the RSS. An appealing aspect of the model is that it makes a number of testable predictions, two of which we have verified (Figure 8).
Factors contributing to 23RSS bending in the PC
As noted above, optimal energy transfer with the 23RSSdR2a substrate requires all of the components known to be required for efficient coupled cleavage in vitro, but the various components contribute to different extents. The essential nature of RAG1, HMGB1 and the nonamer was expected and supports the biological relevance of our findings. RAG1 is the major RSS binding component and makes most or all of the protein contacts with the nonamer, the primary anchor point for RAG on the RSS. It is likely that in the absence of RAG1 or the nonamer, no stable RAG–DNA complexes are formed. HMGB1 likely contributes in multiple ways to the stability and structure of complexes containing the 23RSS. It enhances formation of the 23SC and PC and while not required for nonamer binding (8), HMGB1 is required for base-specific and DNA backbone interactions by RAG in the vicinity of the 23RSS heptamer (10). The lack of detectable energy transfer with the 23RSSdR2a substrate in the absence of HMGB1 is likely due to a reduction in 23SC and PC formation and a decrease in the formation of stable DNA bends in those complexes that do form.
Mutation of the heptamer or omission of RAG2 reduces to a similar extent, but does not eliminate, the detection of energy transfer with the 23RSSdR2a substrate. RAG2 is known to enhance RAG-DNA contacts in the vicinity of the heptamer (8,48,49), raising the possibility that a loss of heptamer contacts is a common mechanism underlying the decrease in E-FRET in these two cases. The detection of energy transfer in the absence of RAG2 or the heptamer is consistent with several prior observations. RAG1 displays sequence-specific interactions with the RSS (with the nonamer more important than the heptamer) (50–52), and interestingly, HMGB2 increases the affinity of RAG1 for the 23RSS, but not the 12RSS (52). Furthermore, RAG1 and HMGB1 are recruited in a synergistic manner to the 23RSS (A. Little and D.G.S., unpublished data), and are capable of mediating RSS synapsis that depends strongly on the nonamer but not the heptamer (17). And notably, atomic force microscopy (AFM) experiments revealed that RAG1 induces the same range of bend angles in a 12RSS as does RAG1 + RAG2 (14). Our detection of FRET in the absence of RAG2 or the heptamer, together with these prior observations, suggest that RAG1 and HMGB1 interactions with the nonamer and nonamer proximal spacer, likely together with interactions at other locations on the DNA, are able to induce a large bend in the DNA. Little is known about the stoichiometry of RAG1–HMGB1–RSS complexes, but available data are consistent with a minimum protein component of a dimer of RAG1 and an as yet undertermined number of HMGB1 molecules (52,53).
The FRET signal with the 23RSSdR2a substrate was also reduced but not eliminated by conditions that perturb the RAG active site and block catalysis (use of Ca2+ or MBP-RAG1c-D708A). This suggests that such perturbations of the active site are accompanied by changes in the structure of the PC that increase the average distance between the coding and nonamer flanks. We propose that altering the divalent cation, or coordination of the divalent cation, in the RAG1 catalytic center influences 23RSS structure by perturbing protein–DNA interactions near the site of cleavage. A similar drop in energy transfer is observed when partner 12RSS is omitted from the reaction, allowing formation of the 23SC (capable only of nicking) but not the PC (which supports both nicking and hairpin formation). This suggests a difference in the structure of 23RSS between the 23SC and the PC. Interestingly, DNAse I footprinting experiments reveal more extended protection of coding flank sequences in the PC than in the 12SC or 23SC (8,40,54), indicating that protein-DNA contacts change on RSS synapsis.
A recent study using a single-molecule cyclization assay found that duplex DNA is remarkably flexible, able to adopt sharply bent configurations that allow for cyclization of molecules as short as 67 bp (55). It is possible that the RAG/HMGB1 proteins take advantage of this property of DNA to capture and stabilize a transiently bent 23RSS during formation of the 23SC and the PC.
Prior studies of DNA bending in RAG–RSS complexes
AFM images support the existence of DNA bends in the 12SC and the 23SC with a bias toward a bend angle of ∼60 degrees, although estimated bend angles were heterogeneous, particularly for the 23SC (40–140 degrees) (14). A bend angle of ∼60 degrees in the 12SC and 23SC is broadly consistent with the results of circular permutation gel shift assays (12), although there are some potential difficulties in the interpretation of the 23SC data, noted previously (8). Our ability to detect energy transfer with the 23RSSdR2a substrate in the absence of partner 12RSS suggests that the overall bend angle in the 23SC is considerably >60 degrees (12). Our data were gathered in solution, and do not require exposure of protein–DNA complexes to gel electrophoresis or deposition on a solid support matrix (as in the case of AFM). Furthermore, in all of the imaging studies, the shape and path of DNA in the vicinity of the RSSs was largely or completely obscured by the protein complex, with bend angles inferred from the exit points of the DNA from the complexes. DNA bending has been detected in the PC using AFM, and while bend angles were not quantitated, the images suggested that some molecules contained large (>90 degree) bends (16), which is consistent with our model. Finally, electron microscopy has been used to probe the structure of the signal end complex (15). This complex was found to be anchor-shaped and the DNA was modeled to contain a relatively small bend in the vicinity of the spacer. In conjunction with our model, this would suggest unbending of the 23RSS after cleavage and release of the coding ends.
In a previous study, we used FRET to examine the relative orientation of the two RSSs in the PC (13). The data argued against a single planar configuration of the RSSs in either parallel or antiparallel alignment, and instead led us to propose that the two RSSs in the PC are strongly bent and cross one another. We favored a model for the PC in which two U-shaped RSSs were organized around a protein core so as to allow equivalent protein-DNA contacts with the two RSSs (13). The 23RSS model that arises from our current study is entirely consistent with this, with the striking feature that most RAG-DNA contacts map to the inside surface of the bent 23RSS (Figure 7B). HMGB1/2 proteins bind in the minor groove on the outside of DNA bends, and footprinting data suggest that they interact on the opposite face of the DNA from the RAG proteins at the nonamer-spacer junction in the 23SC. This led to the suggestion that DNA in this region bends toward the RAG proteins with HMGB1/2 on the opposite, outside surface of the bend (8,10), an idea supported by the location of DNA backbone contacts in our model. We are currently investigating the structure of the 12RSS in the PC.
Implications of the 23RSS model
The locations of bends in our model are interesting in light of previous work in the field. One predicted bend lies near the nonamer-spacer junction, the same location where 23SC footprinting data suggest HMGB1 makes contact with the phosphate backbone (8,10). A second predicted bend is located near the heptamer-coding flank junction, where it has been proposed that HMGB1/2 interacts with distorted DNA structures thought to be created by RAG before hairpin formation (3,8,46,56). A third bend is predicted to occur near the heptamer-spacer junction, a region in which RAG-DNA backbone interactions and photocrosslinking to HMGB1 have been detected in the 23SC (8,10,11). Finally, substantial bending is predicted by our model near the center of the 23RSS spacer, another location of HMGB1 photocrosslinking in the 23SC (11). Interestingly, the center of this bend is surrounded by four residues (two on each strand) that exhibit DNAse I hypersensitivity in the signal end complex (40) (Figure 7B). These four residues are predominantly on the outer surface of the bend, away from the RAG binding surface. This location, together with their predicted proximity to the bend in the spacer, could render these residues preferred sites for nuclease cleavage. A DNAse I hypersensitive residue is also located near the bend at the nonamer-spacer junction (40) (Figure 7B). Bending in the 23RSS spacer is an attractive mechanism for dealing with the difference in spacer length between the 12RSS and 23RSS and allowing coordinated engagement of the 23RSS heptamer and nonamer by RAG (2,8,42–45). It is appealing to think that differential bending of the 12RSS and 23RSS, particularly in the spacer region, helps the RAG proteins distinguish between the two RSS types and contributes to the 12/23 rule, an idea proposed previously by others (11). Bending might also serve to store energy in the DNA to be used for DNA distortion/unwinding in the vicinity of the cleavage site or for conformational changes associated with synapsis (e.g. locking in the second DNA substrate) or postcleavage remodeling of the complex to allow for efficient end processing and repair (e.g. ejection of the coding ends so as to deliver them to the non-homologous end joining repair machinery). ‘Spring loading’ of DNA by integration host factor plays an important regulatory function in Tn10 transposition (57). It is plausible that the dramatic DNA bend created by RAG/HMGB1 in the 23RSS plays both an architectural role in assembly of a cleavage-competent PC and a regulatory role in ensuring the desired reaction outcome.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–3, and Supplementary Dataset 1.
FUNDING
National Institutes of Health [AI32524 to D.G.S.] and [F31 AG038110 to A.J.T.]; Ellison Medical Foundation (to E.R.); Agentia Nationala pentru Cercetarea Stiintifica (ANCS) [PN-II-ID-PCCE-2011-2-0024 to M.C.]; Romanian Academy [ANCS 3-0342-181/2011 and POSDRU/89/1.5/S/60746 program to A.J.P., L.N.S. and M.D.S.]. Funding for open access charge: NIH [AI32524].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank N. Grindley, L. Regan, and A. Miranker for generously providing access to their fluorometers. We would like to thank J. Banerjee and P. Koo for helpful discussions and suggestions in writing the manuscript, and an anonymous reviewer for insightful suggestions.
REFERENCES
- 1.Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu. Rev. Immunol. 2000;18:495–527. doi: 10.1146/annurev.immunol.18.1.495. [DOI] [PubMed] [Google Scholar]
- 2.Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu. Rev. Biochem. 2002;71:101–132. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- 3.Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu. Rev. Genet. 2011;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- 4.Jones JM, Gellert M. Ordered assembly of the V(D)J synaptic complex ensures accurate recombination. EMBO J. 2002;21:4162–4171. doi: 10.1093/emboj/cdf394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mundy CL, Patenge N, Matthews AGW, Oettinger MA. Assembly of the RAG1/RAG2 synaptic complex. Mol. Cell. Biol. 2002;22:69–77. doi: 10.1128/MCB.22.1.69-77.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rooney S, Chaudhuri J, Alt FW. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol. Rev. 2004;200:115–131. doi: 10.1111/j.0105-2896.2004.00165.x. [DOI] [PubMed] [Google Scholar]
- 7.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swanson PC. The bounty of RAGs: recombination signal complexes and reaction outcomes. Immunol. Rev. 2004;200:90–114. doi: 10.1111/j.0105-2896.2004.00159.x. [DOI] [PubMed] [Google Scholar]
- 9.Thomas JO, Travers AA. HMG1 and 2, and related 'architectural' DNA-binding proteins. Trends Biochem. Sci. 2001;26:167–174. doi: 10.1016/s0968-0004(01)01801-1. [DOI] [PubMed] [Google Scholar]
- 10.Swanson PC. Fine structure and activity of discrete RAG-HMG complexes on V(D)J recombination signals. Mol. Cell Biol. 2002;22:1340–1351. doi: 10.1128/mcb.22.5.1340-1351.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mo X, Bailin T, Noggle S, Sadofsky MJ. A highly ordered structure in V(D)J recombination cleavage complexes is facilitated by HMG1. Nucleic Acids Res. 2000;28:1228–1236. doi: 10.1093/nar/28.5.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aidinis V, Bonaldi T, Beltrame M, Santagata S, Bianchi ME, Spanopoulou E. The RAG1 homeodomain recruits HMG1 and HMG2 to facilitate recombination signal sequence binding and to enhance the intrinsic DNA-bending activity of RAG1-RAG2. Mol. Cell. Biol. 1999;19:6532–6542. doi: 10.1128/mcb.19.10.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciubotaru M, Kriatchko AN, Swanson PC, Bright FV, Schatz DG. Fluorescence resonance energy transfer analysis of recombination signal sequence configuration in the RAG1/2 synaptic complex. Mol. Cell Biol. 2007;27:4745–4758. doi: 10.1128/MCB.00177-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pavlicek JW, Lyubchenko YL, Chang Y. Quantitative analyses of RAG-RSS interactions and conformations revealed by atomic force microscopy. Biochemistry. 2008;47:11204–11211. doi: 10.1021/bi801426x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grundy GJ, Ramon-Maiques S, Dimitriadis EK, Kotova S, Biertumpfel C, Heymann JB, Steven AC, Gellert M, Yang W. Initial stages of V(D)J recombination: the organization of RAG1/2 and RSS DNA in the postcleavage complex. Mol. Cell. 2009;35:217–227. doi: 10.1016/j.molcel.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shlyakhtenko LS, Gilmore J, Kriatchko AN, Kumar S, Swanson PC, Lyubchenko YL. Molecular mechanism underlying RAG1/RAG2 synaptic complex formation. J. Biol. Chem. 2009;284:20956–20965. doi: 10.1074/jbc.M109.028977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin FF, Bailey S, Innis CA, Ciubotaru M, Kamtekar S, Steitz TA, Schatz DG. Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nat. Struct. Mol. Biol. 2009;16:499–508. doi: 10.1038/nsmb.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Callebaut I, Mornon JP. The V(D)J recombination activating protein RAG2 consists of a six-bladed propeller and a PHD fingerlike domain, as revealed by sequence analysis. Cell. Mol. Life Sci. 1998;54:880–891. doi: 10.1007/s000180050216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez CA, Ptaszek LM, Villa A, Bozzi F, Sobacchi C, Brooks EG, Notarangelo LD, Spanopoulou E, Pan ZQ, Vezzoni P, et al. Mutations in conserved regions of the predicted RAG2 kelch repeats block initiation of V(D)J recombination and result in primary immunodeficiencies. Mol. Cell Biol. 2000;20:5653–5664. doi: 10.1128/mcb.20.15.5653-5664.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raghavan SC, Lieber MR. Chromosomal translocations and non-B DNA structures in the human genome. Cell Cycle. 2004;3:762–768. [PubMed] [Google Scholar]
- 21.Fugmann SD, Villey IJ, Ptaszek LM, Schatz DG. Identification of two catalytic residues in RAG1 that define a single active site within the RAG1/RAG2 protein complex. Mol. Cell. 2000;5:97–107. doi: 10.1016/s1097-2765(00)80406-2. [DOI] [PubMed] [Google Scholar]
- 22.Eastman QM, Villey IJ, Schatz DG. Detection of RAG protein-V(D)J recombination signal interactions near the site of DNA cleavage by UV cross-linking. Mol. Cell Biol. 1999;19:3788–3797. doi: 10.1128/mcb.19.5.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergeron S, Anderson DK, Swanson PC. RAG and HMGB1 proteins: purification and biochemical analysis of recombination signal complexes. Methods Enzymol. 2006;408:511–528. doi: 10.1016/S0076-6879(06)08032-3. [DOI] [PubMed] [Google Scholar]
- 24.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3rd edn. New York, NY: Springer Science Publishers; 2006. [Google Scholar]
- 25.Fairclough RH, Cantor CR. The use of singlet-singlet energy transfer to study macromolecular assemblies. Methods Enzymol. 1978;48:347–379. doi: 10.1016/s0076-6879(78)48019-x. [DOI] [PubMed] [Google Scholar]
- 26.Middleton ER, Rhoades E. Effects of curvature and composition on alpha-synuclein binding to lipid vesicles. Biophys. J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hess ST, Huang S, Heikal AA, Webb WW. Biological and chemical applications of fluorescence correlation spectroscopy: a review. Biochemistry. 2002;41:697–705. doi: 10.1021/bi0118512. [DOI] [PubMed] [Google Scholar]
- 28.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saenger W. Principles of Nucleic Acid Structure. New York, NY: Springer-Verlag; 1984. [Google Scholar]
- 30.Dickerson RE. DNA structure from A to Z. Methods Enzymol. 1992;211:67–111. doi: 10.1016/0076-6879(92)11007-6. [DOI] [PubMed] [Google Scholar]
- 31.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clegg RM, Murchie AI, Zechel A, Lilley DM. Observing the helical geometry of double-stranded DNA in solution by fluorescence resonance energy transfer. Proc. Natl Acad. Sci. USA. 1993;90:2994–2998. doi: 10.1073/pnas.90.7.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiom K, Gellert M. Assembly of a 12/23 paired signal complex: A critical control point in V(D)J recombination. Mol. Cell. 1998;1:1011–1019. doi: 10.1016/s1097-2765(00)80101-x. [DOI] [PubMed] [Google Scholar]
- 34.Kim DR, Dai Y, Mundy CL, Yang W, Oettinger MA. Mutations of acidic residues in RAG1 define the active site of the V(D)J recombinase. Genes Dev. 1999;13:3070–3080. doi: 10.1101/gad.13.23.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landree MA, Wibbenmeyer JA, Roth DB. Mutational analysis of RAG1 and RAG2 identifies three catalytic amino acids in RAG1 critical for both cleavage steps of V(D)J recombination. Genes Dev. 1999;13:3059–3069. doi: 10.1101/gad.13.23.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koo HS, Wu HM, Crothers DM. DNA bending at adenine-thymine tracts. Nature. 1986;320:501–506. doi: 10.1038/320501a0. [DOI] [PubMed] [Google Scholar]
- 37.Nelson HC, Finch JT, Luisi BF, Klug A. The structure of an oligo(dA).oligo(dT) tract and its biological implications. Nature. 1987;330:221–226. doi: 10.1038/330221a0. [DOI] [PubMed] [Google Scholar]
- 38.Nadeau JG, Crothers DM. Structural basis for DNA bending. Proc. Natl. Acad. Sci. USA. 1989;86:2622–2626. doi: 10.1073/pnas.86.8.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swanson PC. A RAG-1/RAG-2 tetramer supports 12/23-regulated synapsis, cleavage, and transposition of V(D)J recombination signals. Mol. Cell Biol. 2002;22:7790–7801. doi: 10.1128/MCB.22.22.7790-7801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagawa F, Kodama M, Nishihara T, Ishiguro K, Sakano H. Footprint analysis of recombination signal sequences in the 12/23 synaptic complex of V(D)J recombination. Mol. Cell Biol. 2002;22:7217–7225. doi: 10.1128/MCB.22.20.7217-7225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agrawal A, Schatz DG. RAG1 and RAG2 form a stable postcleavage synaptic complex with DNA containing signal ends in V(D)J recombination. Cell. 1997;89:43–53. doi: 10.1016/s0092-8674(00)80181-6. [DOI] [PubMed] [Google Scholar]
- 42.Bergeron S, Madathiparambil T, Swanson PC. Both High Mobility Group (HMG)-boxes and the acidic tail of HMGB1 regulate recombination-activating Gene (RAG)-mediated recombination signal synapsis and cleavage in vitro. J. Biol. Chem. 2005;280:31314–31324. doi: 10.1074/jbc.M503063200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dai Y, Wong B, Yen YM, Oettinger MA, Kwon J, Johnson RC. Determinants of HMGB proteins required to promote RAG1/2-recombination signal sequence complex assembly and catalysis during V(D)J recombination. Mol. Cell. Biol. 2005;25:4413–4425. doi: 10.1128/MCB.25.11.4413-4425.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sawchuk DJ, Weis-Garcia F, Malik S, Besmer E, Bustin M, Nussenzweig MC, Cortes P. V(D)J recombination: modulation of RAG1 and RAG2 cleavage activity on 12/23 substrates by whole cell extract and DNA-bending proteins. J. Exp. Med. 1997;185:2025–2032. doi: 10.1084/jem.185.11.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Gent DC, Hiom K, Paull TT, Gellert M. Stimulation of V(D)J cleavage by high mobility group proteins. EMBO J. 1997;16:2665–2670. doi: 10.1093/emboj/16.10.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bischerour J, Lu C, Roth DB, Chalmers R. Base flipping in V(D)J recombination: insights into the mechanism of hairpin formation, the 12/23 rule, and the coordination of double-strand breaks. Mol. Cell Biol. 2009;29:5889–5899. doi: 10.1128/MCB.00187-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swanson PC, Kumar S, Raval P. Early steps of V(D)J rearrangement: insights from biochemical studies of RAG-RSS complexes. Adv. Exp. Med. Biol. 2009;650:1–15. doi: 10.1007/978-1-4419-0296-2_1. [DOI] [PubMed] [Google Scholar]
- 48.Akamatsu Y, Oettinger MA. Distinct roles of RAG1 and RAG2 in binding the V(D)J recombination signal sequences. Mol. Cell. Biol. 1998;18:4670–4678. doi: 10.1128/mcb.18.8.4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swanson PC, Desiderio S. V(D)J recombination signal recognition—distinct, overlapping DNA-protein contacts in complexes containing RAG1 with and without RAG2. Immunity. 1998;9:115–125. doi: 10.1016/s1074-7613(00)80593-2. [DOI] [PubMed] [Google Scholar]
- 50.Difilippantonio MJ, McMahan CJ, Eastman QM, Spanopoulou E, Schatz DG. RAG1 mediates signal sequence recognition and recruitment of RAG2 in V(D)J recombination. Cell. 1996;87:253–262. doi: 10.1016/s0092-8674(00)81343-4. [DOI] [PubMed] [Google Scholar]
- 51.Spanopoulou E, Zaitseva F, Wang F-H, Santagata S, Baltimore D, Panayotou G. The homeodomain of Rag-1 reveals the parallel mechanisms of bacterial and V(D)J recombination. Cell. 1996;87:263–276. doi: 10.1016/s0092-8674(00)81344-6. [DOI] [PubMed] [Google Scholar]
- 52.Rodgers KK, Villey IJ, Ptaszek L, Corbett E, Schatz DG, Coleman JE. A dimer of the lymphoid protein RAG1 recognizes the recombination signal sequence and the complex stably incorporates the high mobility group protein HMG2. Nucl. Acids Res. 1999;27:2938–2946. doi: 10.1093/nar/27.14.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciubotaru M, Ptaszek LM, Baker GA, Baker SN, Bright FV, Schatz DG. RAG1-DNA binding in V(D)J recombination. Specificity and DNA-induced conformational changes revealed by fluorescence and CD spectroscopy. J. Biol. Chem. 2003;278:5584–5596. doi: 10.1074/jbc.M209758200. [DOI] [PubMed] [Google Scholar]
- 54.Nagawa F, Hirose S, Nishizumi H, Nishihara T, Sakano H. Joining mutants of RAG1 and RAG2 that demonstrate impaired interactions with the coding-end DNA. J. Biol. Chem. 2004;279:38360–38368. doi: 10.1074/jbc.M405485200. [DOI] [PubMed] [Google Scholar]
- 55.Vafabakhsh R, Ha T. Extreme bendability of DNA less than 100 base pairs long revealed by single-molecule cyclization. Science. 2012;337:1097–1101. doi: 10.1126/science.1224139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishihara T, Nagawa F, Imai T, Sakano H. RAG-heptamer interaction in the synaptic complex is a crucial biochemical checkpoint for the 12/23 recombination rule. J. Biol. Chem. 2008;283:4877–4885. doi: 10.1074/jbc.M709890200. [DOI] [PubMed] [Google Scholar]
- 57.Chalmers R, Guhathakurta A, Benjamin H, Kleckner N. IHF modulation of Tn10 transposition: sensory transduction of supercoiling status via a proposed protein/DNA molecular spring. Cell. 1998;93:897–908. doi: 10.1016/s0092-8674(00)81449-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.